
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuasilinear polyglycidols by triethyborane-controlled anionic polymerization of unprotected glycidol†
Prakash
Alagi
,
Yves
Gnanou
 * and
Xiaoshuang
Feng
*
* and
Xiaoshuang
Feng
*
Physical Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955, Saudi Arabia. E-mail: yves.gnanou@kaust.edu.sa; fxs101@gmail.com
First published on 15th February 2023
Abstract
In this study, quasilinear polyglycidols (PG)s with ultralow degree of branching (DB) could be synthesized through anionic polymerization of glycidol carried out in the presence of triethylborane (TEB). PGs with DB ≤ 0.10, and molar masses up to 40 kg mol−1 could be indeed obtained using mono- or trifunctional ammonium carboxylates as initiator and under slow monomer addition conditions. The synthesis of degradable PGs through ester linkages obtained by copolymerization of glycidol with anhydride is also described. PG-based amphiphilic di- and triblock quasilinear copolymers were also derived. The role played by TEB is discussed and a polymerization mechanism is proposed.
Poly(ethylene glycol) (PEG) remains the most widely used polymer and the gold standard in biomedical applications due to its unique properties such as stealth effect, chemical stability, hydrophilicity, biocompatibility.1 However, the drawbacks of PEG such as low functionality, non-biodegradability and some toxicity2 (up to 400 g mol−1) due to enzyme-catalyzed oxidation processes cannot be overlooked. On the other hand, linear polyglycidol (LPG),3 a structural analogue to PEG, possesses multiple hydroxyls along its backbone for bioconjugation and exhibits a slightly better biocompatibility, lower intrinsic viscosity, longer in vivo circulation and shorter renal clearance time; it is thus viewed as a promising alternative to PEG for different biomedical applications.4
Invariably whether anionic or cationic means are used for the homopolymerization of glycidol, samples with high degree of branching, varying between 0.35 and 0.6, are generally produced.5 To obtain LPG the protection of glycidol hydroxyl group is required to avoid its participation in the initiation process; a deprotection step is then necessary after polymer synthesis. Different protecting group and thus different protected monomers such as trimethylsilyl glycidyl ether (TMSGE), ethoxyethyl glycidyl ether (EEGE), tert-butyl glycidyl ether (tBGE), allyl glycidyl ether (AGE), and isopropylidene glyceryl glycidyl ether (IGG) were reported in the literature for the preparation of LPG,3b,6 EEGE being the most frequently used protected glycidol due to the easy removal of its protective group under mild acidic conditions. To reduce the degree of branching and fine tune the extent of its incorporation, glycidol was copolymerized with monomers such as EO, AGE and EEGE,7 but overall the synthesis of PG with low DB or no branching at all directly from the ring-opening polymerization of unprotected glycidol by ROP is a major challenge. Few attempts were made to target PGs of relatively low DB. For instance, Harth et al. reported PGs with DB between 0.15 and 0.24 using tin(II) trifluoromethanesulfonate [Sn(OTf)2] as catalyst; they derived protein PG bioconjugates by using a protein (BSA) as initiator of glycidol by cationic polymerization.8 Subsequently, the same group successfully synthesized PGs with DB of 0.23 without any catalyst by aqueous ring-opening polymerization of glycidol in a phosphate buffer solution (pH = 3.75–8) at 80 °C with a modest yield.9 Similarly, Haag and his coworkers polymerized cationically glycidol with citric acid, the latter playing the double role of monomer protonating agent and of initiator, which resulted in the formation of PGs with DB of 0.32–0.54.10 Recently Kim et al. obtained PGs with a DB of 0.26–0.27 and used for that double metal cyanide as catalyst. In that contribution the authors showed that batch addition of glycidol produced PGs with low DB (<0.3), in contrast slow glycidol addition resulted in hyperbranched PGs with higher DB (>0.5).11 In all aforementioned reports the molar masses of resulting PGs were limited to 1000–2000 g mol−1 with the lowest DB close to 0.20 whether the polymerizations of glycidol were carried out through either cationic or coordinative mechanism. In this communication, we want to report how PGs with DB as low as 0.03 to 0.1 can be obtained by anionic ring-opening polymerization of unprotected glycidol with controlled molar masses up to 40 kg mol−1.
In their seminal contributions on the oxyanionic ring-opening polymerization of glycidol, Frey and coworkers established that a latent AB2 monomer such as glycidol produces upon polymerization hyperbranched polyether polyols.5a,b,12 The hyperbranched PGs formed comprise different types of repeating units, namely two types of linear units (L13 and L14) and dendritic (D) ones, in addition to terminal (T1 and T2) units (Scheme 1). In such multibranching polymerization the growing oxyanionic species has the option to add monomer and thus propagate or to undergo an inter/intramolecular transfer to hydroxyls carried either by the monomer or by the grown chains (see mechanism Scheme S1 in ESI†). Because of transfer reactions to chains the experimental degree of branching of the polymer formed increases with the degree of polymerization before reaching a value that plateaus between 0.55 and 0.59. As for transfers to the monomer, they are responsible for the loss of control of the sample molar mass.
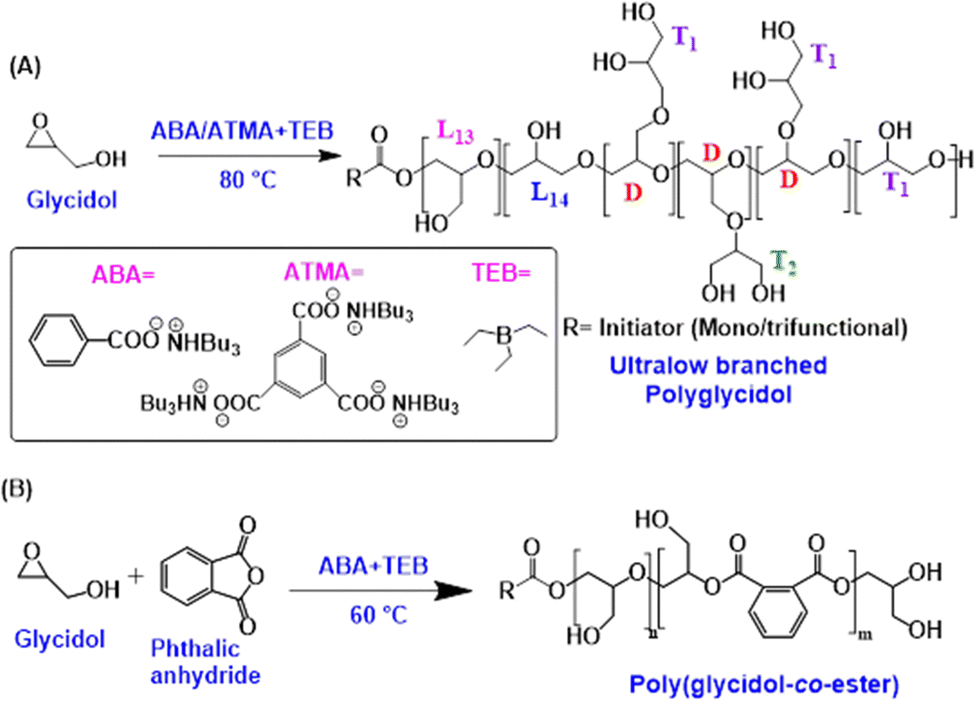 | ||
| Scheme 1 (A) Synthesis of ultralow branched polyglycidol in the presence of ABA/ATMA as initiators and TEB as activator; (B) synthesis of degradable poly(glycidol-co-ester) using ABA as initiator and TEB as activator. | ||
From our previous works on the anionic polymerization of epoxides fitted with electron-withdrawing groups and on the copolymerization of epoxides with CO2 or with anhydrides, we learnt that in the presence of a Lewis acid such as triethylborane (TEB) propagation is significantly favored over all possible side reactions. For instance in the synthesis of polycarbonates by copolymerization of epoxides with CO2 the formation of cyclic carbonates due to back-biting could be entirely eliminated and only well-defined linear polycarbonates were formed in the presence of TEB.13
Upon resorting to the same oxyanionic polymerization of glycidol as that used by Frey's group and applying their condition of slow monomer addition, we observed a totally different outcome from theirs when TEB was intentionally added to the reaction medium. In this work we show that the anionic polymerization of glycidol, when carried out in the presence of TEB, affords quasi-linear PG samples with low to ultralow degree of branching (0.03 ≤ DB ≤ 0.1) and controlled molar masses up to 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1. We also demonstrated that degradable and yet quasilinear poly(ether-co-ester) samples can be obtained through anionic copolymerization of glycidol with phthalic anhydride in the presence of TEB (Scheme 1). We also took advantage of the beneficial role played by TEB to derive PG-based amphiphilic di- and triblock copolymers by sequential polymerizations of propylene oxide and glycidol.
000 g mol−1. We also demonstrated that degradable and yet quasilinear poly(ether-co-ester) samples can be obtained through anionic copolymerization of glycidol with phthalic anhydride in the presence of TEB (Scheme 1). We also took advantage of the beneficial role played by TEB to derive PG-based amphiphilic di- and triblock copolymers by sequential polymerizations of propylene oxide and glycidol.
As indicated in Table 1 we chose two organic carboxylate salts, one monofunctional -tributyl ammonium salt of benzoic acid (ABA)-, and a second trifunctional one -tributyl ammonium salt of trimesic acid- as initiators for these TEB-controlled ROP of glycidol. Through the variation of parameters such as the ratio of [Monomer]/[Initiator], that of [TEB/[Initiator], the rate of monomer addition and the temperature of reaction, experiments were then designed with a view of uncovering the role played by TEB in such polymerizations and its effect on the degree of branching of the samples eventually obtained. Experiments carried out at four different temperatures (Table 1 and Table S2, and Fig. S1, ESI†) show that the degree of branching increases marginally from 60 °C (DB = 0.06) to 90 °C (DB = 0.1) and that molar mass control is better achieved between 80 °C and 90 °C. In the same Table 1 which gathers information about the relation between the ratio of [Monomer]/[Initiator] and the DB of samples, one can point to the marginal degree of branching (DB = 0.035) for samples of low DPn (PGM-5, Table 1). As the targeted molar mass increases, DB progressively increases as well and plateaus at around a value of 0.10 (Fig. S2 and Table S2, ESI†). As for the control of the molar masses of the samples synthesized, it could be actually achieved upon following Frey's recommendation to slowly add monomer (0.1 mL hour−1) as shows the close agreement between the expected and experimental values of molar mass, whether samples were grown from mono or trifunctional initiators (Table 1 and Fig. S3–S5, ESI†). When the monomer was introduced in the reaction medium at a more rapid pace (0.3 mL hour−1) or at once (Tables S1, S2 and Fig. S6, S7, ESI†), then glycidol acted as an inimer, initiating chains through its hydroxyl and contributing to their growth by ring-opening. In the latter cases lower than expected molar masses were observed, entailing a loss of control of the sample molar masses. The amount of TEB used in these experiments with respect to that of the initiator also matters (Table S1, ESI†): an excess of [TEB] over [I] is necessary to favor monomer addition and thus propagation over monomer transfer and both intra- and intermolecular transfers that affect this kind of polymerizations.
| Entry | [ABA(ATMA)]/[TEB]/[Gly] | Temp. (°C) | M n(theo) | M n(NMR) | M n(GPC)/PDId | DBe | L13f (%) | L14g (%) | Dh (%) | Ti (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Samples PGM-1-PGM-8 were obtained using ABA as monofunctional initiator; star-shaped PGT-9-PGT-11 were obtained from ATMA trifunctional initiator. b Theoretical molar mass calculated assuming linear PG structure, Mn(theoretical) = [(Glycidol/Initiator) × 74.08 × conversion + Mw. of initiator acid]. c Calculated from 1H NMR spectra by comparison of repeating unit signal intensity to the initiator signal intensity (for all PG samples the conversion of glycidol is 100%). d Determined via GPC-RI in DMF using linear PMMA standards. e Degree of branching were calculated according to equation described by Frey et al; (DB = 2D/(2D + L13 + L14). f Content of linear 1–3 units. g Content of linear 1-4 units. h Content of dendritic units. i Content of terminal unit. | ||||||||||
| PGM-1 | 1/5/200 | 60 | 15000 |
13200 |
20200/1.59 |
0.06 | 44.34 | 44.12 | 2.66 | 8.87 |
| PGM-2 | 1/5/200 | 70 | 15000 |
13700 |
19600/1.49 |
0.08 | 43.20 | 43.84 | 3.89 | 9.07 |
| PGM-3 | 1/5/200 | 80 | 15000 |
14200 |
19000/1.43 |
0.09 | 42.55 | 42.55 | 4.26 | 10.64 |
| PGM-4 | 1/5/200 | 90 | 15000 |
15700 |
21600/1.68 |
0.10 | 40.32 | 44.36 | 4.44 | 10.89 |
| PGM-5 | 1/5/20 | 80 | 1600 | 1650 | 3000/1.20 | 0.04 | 38.24 | 49.52 | 1.53 | 10.71 |
| PGM-6 | 1/5/50 | 80 | 3800 | 3700 | 6100/1.45 | 0.08 | 40.16 | 42.97 | 4.02 | 12.85 |
| PGM-7 | 1/5/500 | 80 | 37200 |
29500 |
43100/1.43 |
0.12 | 39.68 | 43.65 | 5.95 | 10.71 |
| PGM-8 | 1/—/200 | 80 | 15000 |
22300 |
7600/1.85 | 0.57 | 9.01 | 28.38 | 24.41 | 38.20 |
| PGT-9 | 1/5/50 | 80 | 3900 | 4800 | 5300/1.50 | 0.07 | 31.55 | 47.00 | 3.12 | 18.30 |
| PGT-10 | 1/5/200 | 80 | 15000 |
19300 |
13200/1.75 |
0.09 | 39.22 | 44.71 | 4.31 | 11.76 |
| PGT-11 | 1/5/500 | 80 | 37300 |
38400 |
26800/1.60 |
0.11 | 41.93 | 42.98 | 5.45 | 9.64 |
When examining more closely the various parameters considered above and comparing the results obtained with those published by Frey and coworkers, it is manifest that TEB has a beneficial and essential role in the formation of quasilinear PGs (Scheme 2). First, by forming with growing oxyanionic species an ate complex of lower reactivity TEB helps to curb both intra- and intermolecular transfer reactions with hydroxyls carried either by the monomer or PG chains. Second, the excess of TEB that was systematically used served to activate glycidol (Fig. S8, ESI†), thus boosting specifically propagation over all other chemical events. This double role played by TEB (mitigating the reactivity of growing oxyanionic species through the formation of 1:1 ate complex with the latter anions and at the same time activating monomer through the excess used) thus upends all chemical events occurring in such polymerizations. The rate of polymerization can now be written as: Rp = kp,TEB [I-TEB] × [M*] where kp,TEB represents the rate constant of propagation in the presence of TEB, [I-TEB] the concentration of ate complex which is equal to the concentration of initiator [I]0, [M*] being the concentration of activated glycidol which can be assumed equal to the concentration of available TEB: [TEB]a = [TEB]0 – [I]0. Because of monomer activation by TEB, the rate of polymerization (Rp) dominates all rates of transfer, that to the monomer: Rt,M =k1,TEB [I-TEB] × [M] as well as that to the hydroxyls carried by the chains: Rt,L =k2,TEB [I-TEB] × ([L13] + [L14]), allowing propagation to prevail over all possible transfer reactions. In absence of TEB, such as the situation previously described by Frey et al., propagation and transfer reactions occur at similar rates as indicate the comparable proportion of linear and dendritic units (see Scheme S1 in ESI†).
 | ||
| Scheme 2 Plausible mechanism for the anionic polymerization of glycidol carried out in the presence of TEB. | ||
To illustrate the benefits imparted by the presence of TEB, representative interpretation of 13C IG NMR of PGs obtained in the presence and absence of TEB (PGM-4 and 8, Fig. 1A) clearly indicates the relative intensity difference of peaks at 78–79 ppm and 63–64 ppm corresponding to the D and T (dendritic and terminal) units due to remarkable different branch extents (PGM-4 = 0.09 DB and PGM-8 = 0.57). Furthermore, we carried out two experiments in all aspects identical, except that in the first case 5 equiv. of TEB was added with respect to the initiator (PGM-5) and in the second no TEB was introduced (PGMS-7, Table S1, ESI†). A low DP of 20 was intentionally targeted and ABA was used as initiator. MALDI-TOF analysis (Fig. 1C) of the first sample shows only one population of chains, matching the formula: 74.1n + 121.2 (ABA) + 23 (Na+) and thus indicating that PGs chains were initiated in the latter case by ABA initiator. The molar mass obtained corresponds to the ratio of glycidol to ABA times the conversion. Characterization by IG NMR hardly shows the presence of any branching which appears very limited (DB = 3.5%) (Fig. S2A, ESI†). In contrast, more than three populations were observed in absence of TEB (Fig. 1D, PGMS-7): one population obviously corresponds to PGs initiated by ABA, two other populations: 74.1n + 18 (H2O) + 23 (Na+), 74.1n + 23 (Na+) are self-initiated PGs with end-standing hydroxyls and cyclic structures. In the latter case, the DB reached a value of 0.57 and the molar masses of the chains obtained were lower than the expected ones (similar with sample PGM-8 in Table 1 and Fig. 1A). When characterized by differential scanning calorimetry (DSC) (Fig. 1B), a Tg of −45 °C was measured for the highly branched sample PGM-8 and a much higher value of Tg = −21 °C for the quasilinear sample PGM-4. Similar results were obtained for PGT star-shaped samples (Fig. S4C, ESI†). Tg values ranging from −8 °C to −27 °C are mentioned in the literature for LPGs of various molar masses.7,14
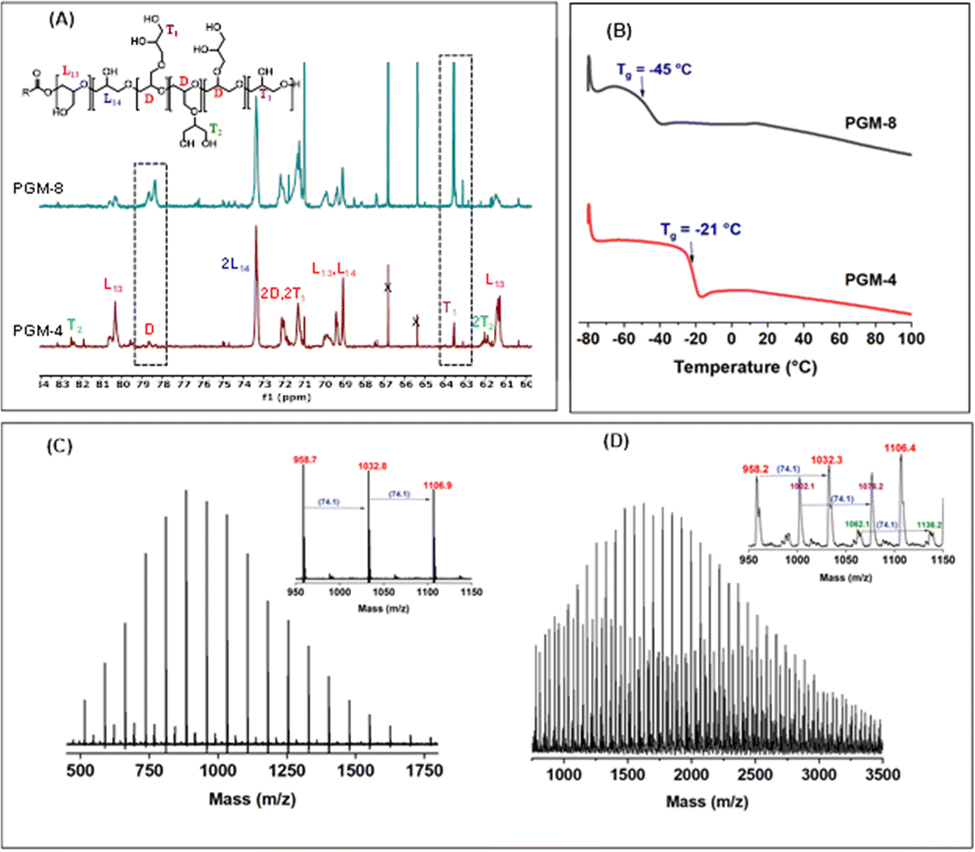 | ||
| Fig. 1 (A) Inverse gated 13C NMR spectra for PGM-4 and PGM-8, (B) DSC curves for PGM-4 and PGM-8 (Table 1), (C) MALDI-TOF mass spectra of PGs (DP = 20) with TEB (PGM-5), and (D) MALDI-TOF mass spectra of PGs (DP = 20) without TEB (PGMS-7, Table S1, ESI†). | ||
Quasilinear PG samples obtained by homopolymerization of unprotected glycidol in the presence of TEB opens up applicative opportunities. For instance the copolymerization of epoxides with anhydrides in the presence of TEB produces poly(ether-co-ester) with randomly distributed degradable ester linkages.15 We thus thought of incorporating ester linkages within PGs of ultralow DB to impart degradability. To this end, phthalic anhydride (PA) was mixed with glycidol and both monomers were added at 60 °C to the reaction medium containing TEB and ABA used as initiator. (Table S3 and Fig. S9, ESI†). The incorporation of ester linkages within PG chains did occur, producing poly(glycidol-co-ester) (PGE) copolymers with low DB (0.11). The GPC traces exhibit a unimodal and narrowly dispersed distribution of chains. Upon changing the ratio of ([glycidol] + [PA])/[ABA], PGEs of different molar masses and low DB could be easily generated (Table S3 and Fig. S10, RI and UV detector, ESI†). The degradation of PGE-3 using a 0.5 M solution of NaOH was straightforward. Within 24 h, the GPC traces of an aliquot shifted to lower molar mass region, the average molar mass value reducing from 16.3 kg mol−1 to 3 kg mol−1 (Fig. S11, ESI†).
We also prepared amphiphilic di- and triblock copolymers made of poly(propylene oxide) and quasilinear PG (Scheme S2, PGB 1–2 and Table S4, ESI†). Using either a mono- or difunctional ammonium carboxylate as initiators both di- and triblock copolymers with low DB could be synthesized in one-pot upon sequential polymerization of propylene oxide and glycidol in the presence of TEB (Fig. S12A, ESI†). The crude copolymer samples were then dialyzed for the diblock and washed with toluene for the triblock to remove residual homopolymers. The GPC (Fig. S12B and C, ESI†) and diffusion-ordered spectroscopy (Dosy NMR) (Fig. S13, ESI†) characterizations of these PGB samples confirm the block structure of the copolymers formed.
In summary, we report an efficient method to prepare quasilinear PGs with low to ultralow degree of branching (0.03 ≤ DB ≤ 0.1) through AROP of unprotected glycidol in the presence of TEB. TEB plays a crucial role in such AROP as it activates glycidol on one hand and forms ate complexes with growing alkoxides on the other hand; ate complexes being less prone to transfer compared to alkoxides, branching could be mitigated and even reduced to very low degrees. Quasilinear PGs including degradable ester linkages and well-defined PG-based amphiphilic block copolymers that can serve as surfactants were accordingly derived.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. A. D’Souza and R. Shegokar, Expert Opin. Drug Delivery, 2016, 13, 1257–1275 CrossRef PubMed.
- D. A. Herold, K. Keil and D. E. Bruns, Biochem. Pharmacol., 1989, 38, 73–76 CrossRef CAS PubMed.
- (a) M. Bochenek, N. Oleszko-Torbus, W. Wałach, D. Lipowska-Kur, A. Dworak and A. Utrata-Wesołek, Polym. Rev., 2020, 60, 717–767 CrossRef CAS; (b) R. K. Kainthan, J. Janzen, E. Levin, D. V. Devine and D. E. Brooks, Biomacromolecules, 2006, 7, 703–709 CrossRef CAS PubMed.
- J. N. Lockhart, T. J. Spoonmore, M. W. McCurdy, B. R. Rogers, S. A. Guelcher and E. Harth, ACS Appl. Mater. Interfaces, 2018, 10, 4050–4056 CrossRef CAS PubMed.
- (a) A. Sunder, R. Hanselmann, H. Frey and R. Mülhaupt, Macromolecules, 1999, 32, 4240–4246 CrossRef CAS; (b) D. Wilms, F. Wurm, J. Nieberle, P. Böhm, U. Kemmer-Jonas and H. Frey, Macromolecules, 2009, 42, 3230–3236 CrossRef CAS; (c) S. E. Kim, Y.-R. Lee, M. Kim, E. Seo, H.-J. Paik, J. C. Kim, J.-E. Jeong, Y. I. Park, B.-S. Kim and S.-H. Lee, Polym. Chem., 2022, 13, 1243–1252 RSC; (d) S. E. Kim, H. J. Yang, S. Choi, E. Hwang, M. Kim, H.-J. Paik, J.-E. Jeong, Y. I. Park, J. C. Kim, B.-S. Kim and S.-H. Lee, Green Chem., 2022, 24, 251–258 RSC.
- (a) M. Gervais, A.-L. Brocas, G. Cendejas, A. Deffieux and S. Carlotti, Macromolecules, 2010, 43, 1778–1784 CrossRef CAS; (b) F. Wurm, J. Nieberle and H. Frey, Macromolecules, 2008, 41, 1909–1911 CrossRef CAS.
- C. Schubert, M. Schömer, M. Steube, S. Decker, C. Friedrich and H. Frey, Macromol. Chem. Phys., 2018, 219, 1700376 CrossRef.
- B. R. Spears, J. Waksal, C. McQuade, L. Lanier and E. Harth, Chem. Commun., 2013, 49, 2394–2396 RSC.
- B. R. Spears, M. A. Marin, J. R. Montenegro-Burke, B. C. Evans, J. McLean and E. Harth, Macromolecules, 2016, 49, 2022–2027 CrossRef CAS.
- E. Mohammadifar, A. Bodaghi, A. Dadkhahtehrani, A. Nemati Kharat, M. Adeli and R. Haag, ACS Macro Lett., 2017, 6, 35–40 CrossRef CAS PubMed.
- C. H. Tran, M. W. Lee, S. A. Kim, H. B. Jang and I. Kim, Macromolecules, 2020, 53, 2051–2060 CrossRef CAS.
- D. Wilms, S.-E. Stiriba and H. Frey, Acc. Chem. Res., 2010, 43, 129–141 CrossRef CAS PubMed.
- (a) P. Alagi, G. Zapsas, N. Hadjichristidis, S. C. Hong, Y. Gnanou and X. Feng, Macromolecules, 2021, 54, 6144–6152 CrossRef CAS; (b) M. Jia, D. Zhang, G. W. de Kort, C. H. R. M. Wilsens, S. Rastogi, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2020, 53, 5297–5307 CrossRef CAS PubMed; (c) N. G. Patil, S. K. Boopathi, P. Alagi, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2019, 52, 2431–2438 CrossRef CAS; (d) D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, J. Am. Chem. Soc., 2016, 138, 11117–11120 CrossRef CAS PubMed.
- (a) F. Wurm, J. Nieberle and H. Frey, Macromolecules, 2008, 41, 1184–1188 CrossRef CAS; (b) C. Osterwinter, C. Schubert, C. Tonhauser, D. Wilms, H. Frey and C. Friedrich, Macromolecules, 2015, 48, 119–130 CrossRef CAS.
- (a) V. K. Chidara, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2021, 54, 2711–2719 CrossRef CAS; (b) V. K. Chidara, Y. Gnanou and X. Feng, Molecules, 2022, 27, 466 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, NMR, GPC, Tables S1–S4, Figures S1–S13. See DOI: https://doi.org/10.1039/d3cc00153a |
| This journal is © The Royal Society of Chemistry 2023 |