
Reactivity of quinone methides with carbenes generated from α-diazocarbonyl compounds and related compounds
Sharma
Happy†
,
Mohammad
Junaid†
and
Dongari
Yadagiri
 *
*
Department of Chemistry, Laboratory of Organic Synthesis & Catalysis, Indian Institute of Technology Roorkee, Roorkee-247667, Uttarakhand, India. E-mail: yadagiri.dongari@cy.iitr.ac.in; Web: https://yadagiridongari.wixsite.com
First published on 8th December 2022
Abstract
Over the years, quinone methides have broadly been applied in synthesis and biological systems for synthesizing heterocyclic compounds and biologically active molecules. In this feature article, we have discussed the novel and uncovered reactivity of o-quinone methides, p-quinone methides, aza-o-quinone methides, and indolyl-2-methides with carbenes generated from α-diazocarbonyl compounds and related compounds. Two in situ-generated transient intermediates undergo cycloannulation reactions, metathesis-type reactions, 1,6-conjugate addition reactions, cyclopropanation reactions, and many other transformations to access nitrogen- and oxygen-containing heterocyclic compounds and beyond. The reactivity of quinone methides and carbenes is observed in various metal catalysts, Brønsted-acids, Lewis acids, phase transfer catalysts, additives, and visible-light-induced transformations.
 Sharma Happy | Sharma Happy was born in 1999 in Jammu and Kashmir. He did his BSc(H) in Chemistry at Kirori Mal College, the University of Delhi, in 2019. Then, he received his MSc (Chemistry) degree from the Indian Institute of Technology, Roorkee, in 2021. Currently, he is working as a PhD student under the supervision of Prof. Dongari Yadagiri at the Indian Institute of Technology, Roorkee. His research interests are the functionalization of C–H bonds via cooperative catalysis. |
 Mohammad Junaid | Mohammad Junaid was born in 1996 in Uttar Pradesh. He did his BSc at Zakir Hussain College, the University of Delhi, in 2018. Then, he received his MSc (Chemistry) degree from the Indian Institute of Technology, Indore, in 2020. Currently, he is working as a PhD student under the supervision of Prof. Dongari Yadagiri at the Indian Institute of Technology, Roorkee. His research interests are the functionalization of unactivated C–H bonds. |
 Dongari Yadagiri | Dongari Yadagiri received his PhD in 2016 from the Indian Institute of Technology (IIT) Madras, India, with Prof. P. Anbarasan. He was then a postdoctoral researcher (2017–2020) with Prof. V. Gevorgyan at the University of Illinois at Chicago. Later, he moved to the University of Texas at Dallas with his advisor. In 2021, he began his independent research career as Assistant Professor at IIT Roorkee in the Department of Chemistry. His research interests are focused on organic synthesis and catalysis. |
1. Introduction
Quinone methides (QMs) are versatile synthons in synthetic chemistry and have been used widely as intermediates in several biological and chemical syntheses. They are mostly extracted from organic materials, including wood, insect pigments, and fungus metabolites.1 Thus, a common structural pattern of quinone methides exists in numerous biologically active natural products. The structural assembly of QMs consists of a cyclohexadiene ring and an exo-methylene moiety with a reactive carbonyl group present either at the ortho (o-QMs) or para position (p-QMs), and they are similar to quinones, which have two carbonyl groups, and quinone dimethides, which have two methylene groups.2 QMs are very short-lived, polar, and highly reactive intermediates compared with their counterparts. The synthetic potential and properties of ortho- and para-QMs have been widely explored over the past few decades because of their potential applications in pharmaceutical as well as natural product synthesis.3Fries first generated o-QMs in 1907, and these have been widely used as a synthetic toolbox to construct bioactive molecules and complex molecular entities.4 Due to their potential applications and high reactivity, o-QMs can be generated using different methodologies, which include thermolysis,5 basic,6 photochemical,7 oxidation,8 and neutral conditions (Fig. 1), and the most common method is the acid-catalyzed dehydration of ortho-hydroxy benzhydryl alcohols to yield o-QMs.9 These innately reactive intermediates exist in zwitterionic or biradical form and in E/Z configurations, which depend on the steric hindrance of neighboring groups.
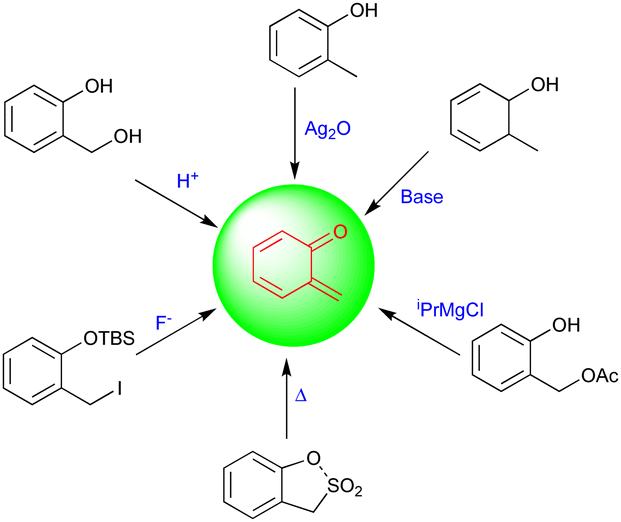 | ||
| Fig. 1 Generation of o-QMs. | ||
A unique method for synthesizing p-QMs is the condensation reaction of aromatic aldehydes with phenols in the presence of a base. By contrast, alcohol and 2,2-dichloroacetate chloride have been used to access transient p-QMs10 (Fig. 2). According to Soucek's group, benzyl alcohols can be transformed into sulfone intermediates by refluxing with PhSO2Na in aqueous acetic acid, producing p-QMs upon treatment with NaOH.11 In 2009, Mayr revealed a method that used ethereal HBF4 in dichloromethane at 0 °C to produce a cationic intermediate that yielded the desired product after treatment with triethylamine,12 and there are many other ways of synthesizing p-QMs.1f
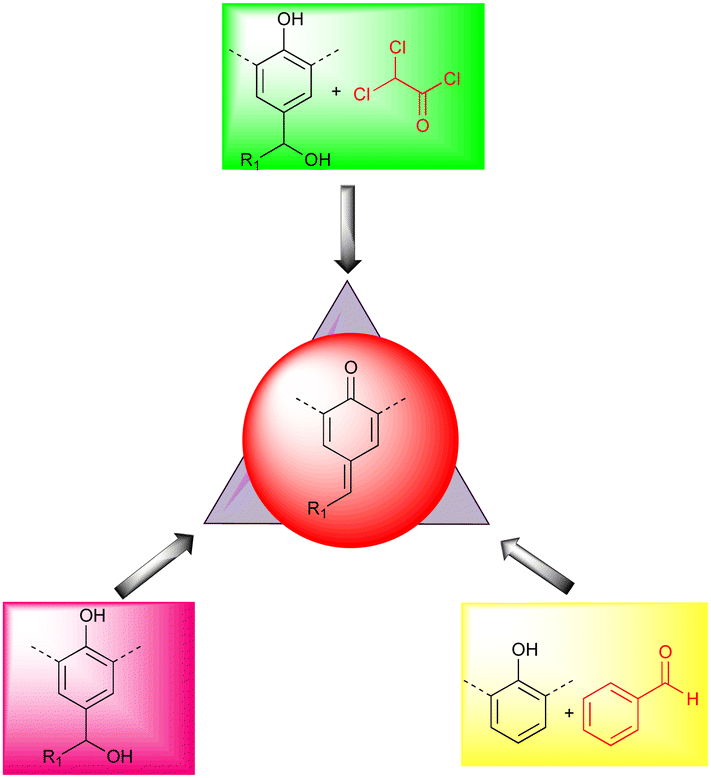 | ||
| Fig. 2 Synthesis of p-QMs. | ||
2. Reactivity of QMs
Typically, o-QMs exist as three main types, viz., biradical, neutral, and zwitterionic (Fig. 3). These forms are primarily responsible for their different synthetic uses. o-QMs have shown broad applications in synthetic chemistry including [6+4]-, [4+4]-, [4+3]-, [4+2]-, and [4+1]-cycloaddition reactions (Fig. 3). They also undergo nucleophilic addition reactions to construct interesting pharmaceutical molecules (Fig. 3). The re-establishment of aromaticity is what causes the reactions of o-QMs to occur. A detailed study of the reactivity of o-QMs for cycloaddition reactions and many other transformations is summarized elsewhere.13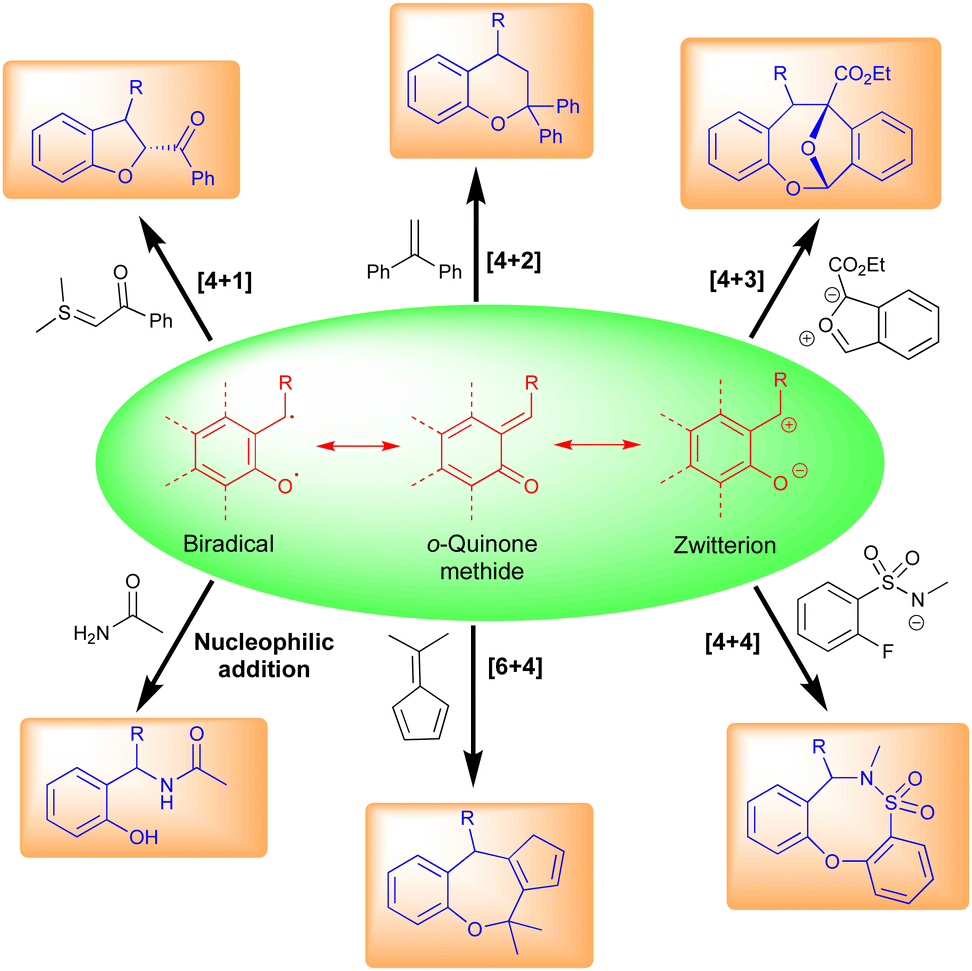 | ||
| Fig. 3 Reactivity of o-QMs. | ||
p-QMs also exist in neutral and zwitterionic forms through resonance (Fig. 4). Therefore, they exhibit wide-ranging chemical reactivity, such as an acceptor for 1,6-conjugate addition, including Michael addition and radical addition. The carbonyl group at one end results in the formation of an aromatic zwitterion,14 and this interacts with nucleophiles to produce a more stable neutral aromatic form. The exocyclic methylidene site, which is exposed to nucleophiles, forms an electrophilic carbocation. As a result, p-QMs demonstrate a unique reactivity profile to accomplish highly regioselective 1,6-conjugate addition reactions with various nucleophiles.15 From the past decade, p-QMs have shown unique reactivity due to conjugate addition that provides unsymmetrical diaryl methane and tri-aryl methane derivatives. p-QMs also undergo [4+3]-, [4+1]-, [3+2]-, and [2+1]-cycloaddition reactions, similar to o-QMs, with different reacting partners resulting the formation of three-, five-, six-membered and polycyclic systems (Fig. 4).1f,13c,16
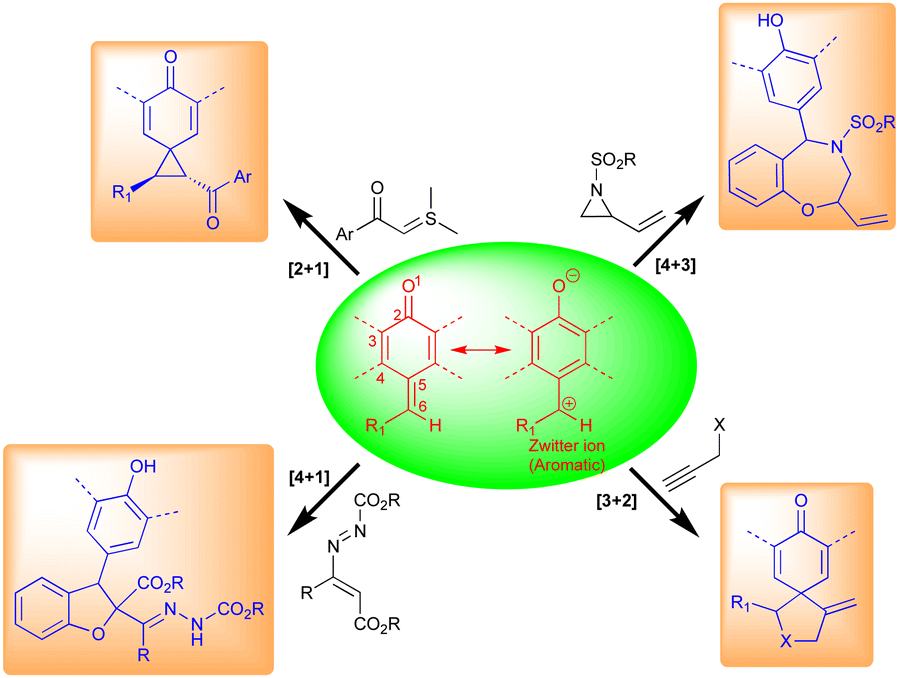 | ||
| Fig. 4 Reactivity of p-QMs. | ||
3. Reactivity of QMs with α-diazocarbonyl compounds and their precursors
The chemistry of ortho- and para-QMs has been widely explored, but their reactions with carbenes have not been extensively studied until recently. It was not until 2016 that the first reaction of o-QMs with carbenes was reported. Since then, this chemistry has expanded to synthesize N, O-containing heterocyclic compounds and beyond via cooperative catalysis and cycloannulation fashion. This review summarizes the reactivity of QMs with α-diazocarbonyl compounds and related compounds.In 2016, in the first report of its kind, the reactivity of o-QMs with α-diazocarbonyl compounds was reported by Schneider's group (Scheme 1),17 where an agile methodology was developed in the presence of rhodium/chiral phosphoric acid (CPA) as catalysts (Scheme 1) using a cooperative catalysis concept.18 They developed a highly enantioselective conjugate addition of an oxonium ylide to o-QMs that were generated in situ in the presence of a rhodium catalyst and CPA.1 (Scheme 1b). o-QM III was formed from the CPA.1-catalyzed dehydration of ortho-hydroxy benzhydryl alcohol 1 (Scheme 1b). By contrast, oxonium ylide II was generated by the reaction of water with rhodium carbene I generated in situ from the rhodium catalyst (Rh2(OAc)4) and α-diazo β-ketoester 2. Thus, the in situ generated oxonium ylide then undergoes conjugate addition to o-QM IIIvia a transition state stabilized by CPA.1, leading to the phenol intermediate 4, followed by subsequent cyclization onto the carbonyl group to deliver the desired chromane product 3 (Scheme 1b). Control experiments favorably supported the proposed mechanism. No reaction was observed when the authors carried out the reaction in the presence of 4 Å molecular sieves. However, when they added H2O as an additional reactant with ex situ generated o-QM, the reaction with α-diazo β-ketoester 2 under the optimized conditions delivered the product, confirming that H2O is essential for generation of the oxonium ylide II (Scheme 1b).
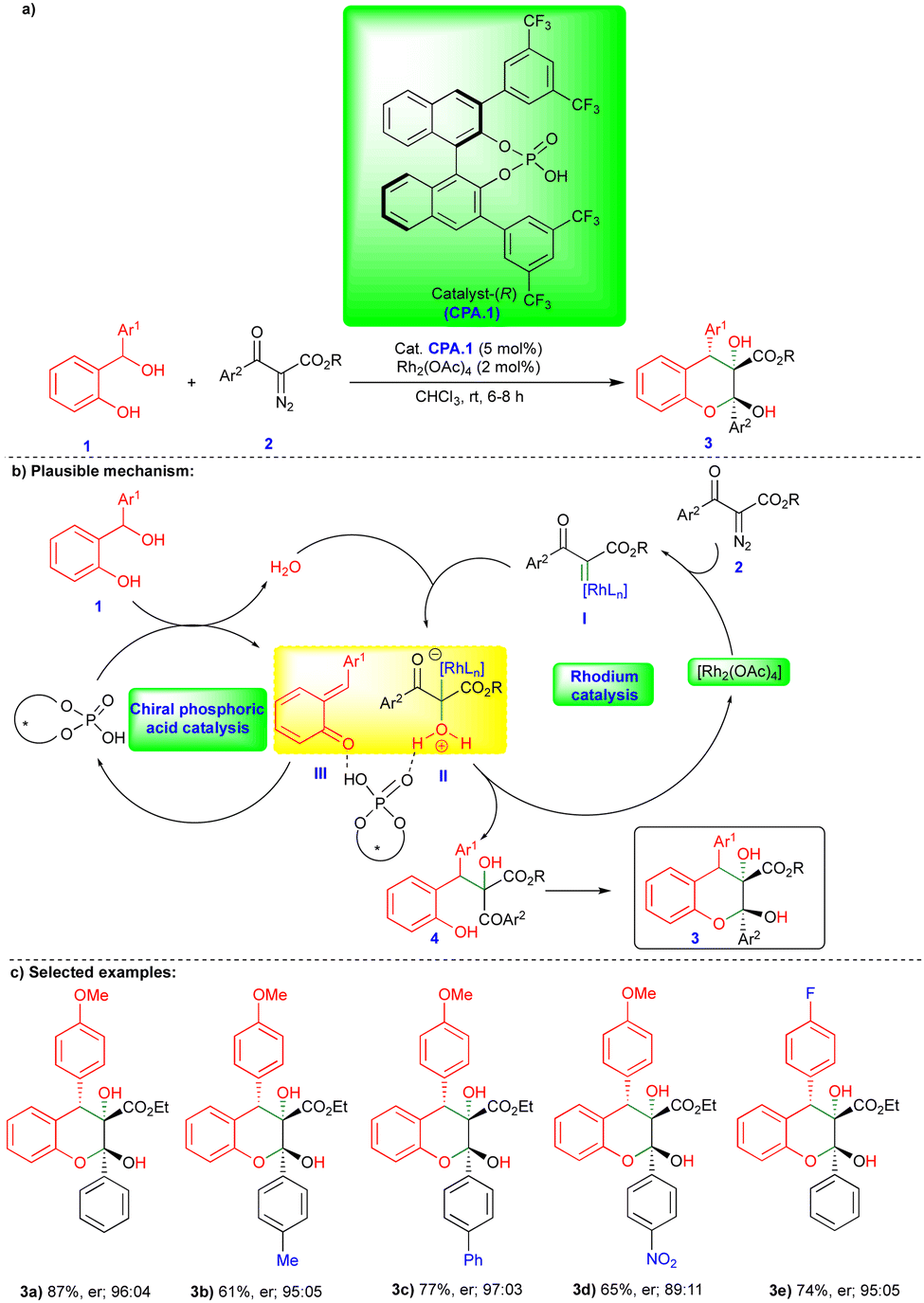 | ||
| Scheme 1 Cooperative catalysis enantioselective conjugate addition of oxonium ylides to o-QMs. | ||
Under the optimized conditions, a variety of ortho-hydroxy benzhydryl alcohols 1 and electronically different substituted α-diazo β-ketoester 2 derivatives reacted well in the presence of rhodium catalyst and chiral phosphoric acid, which has 3,5-bis(trifluoromethyl)phenyl group at the 3,3′ position (CPA.1), as the chiral catalyst, giving the corresponding products 3 good yields and in a highly diastereo- and enantioselective manner (Scheme 1c). However, the reaction did not take place when substitution of the α-diazo β-ketoester 2 was at the ortho position of the aryl group.
The reactions are categorized based on the reactivity of carbenes with QMs, for example, cycloannulation reactions, i.e., [4+1], [8+1], [4+3], [2+1], etc., and 1,6-conjugate addition reactions.
3.1 [4+1]-Cycloannulation
The [3+2]-cycloaddition reaction is the most common method for synthesizing 5-membered heterocyclic compounds. However, Ashfeld's group reported the phosphorus(III)-mediated formal [4+1]-cycloannulation of o-QM with carbene equivalents such as α-ketoester for the synthesis of 2,3-dihydrobenzofuran (Scheme 2a).19 Despite there being a few existing methods to synthesize 2,3-dihydrobenzofurans 7, a [4+1]-cycloannulation reaction provides a quick way to install a quaternary substitution at the C2 carbon. Ashfeld’s group employed a Kukhtin–Ramirez-like condensation method, in which phosphine adds to the α-ketoester 6a, generating an oxyphospholene 8 that is in equilibrium with zwitterion 9. Thus, carbene-like reactivity and reaction with o-QM was exhibited, which was generated in the presence of CsF from silyl ether 5a. By contrast, the o-QM could also be generated from the corresponding salicylaldehyde 5b and methyllithium or phenyllithium. The syn-stereochemistry of the product was explained by the favorable intermediates 10 and 13 (Scheme 2b), which were generated from the corresponding starting materials. Different substituted 2,3-dihydrobenzofurans were synthesized from the corresponding starting materials with low diastereoselectivity (Scheme 2c). Based on the observed results, the authors assumed that either the unselective formation of o-QMs or the rapid isomerization of the E-isomer to the Z-isomer mediated by P(NMe2)3 was responsible for the low diastereoselectivity of the products. However, o-QMs derived from 2-hydroxynaphthaldehyde derivatives formed the Z-alkylidene and gave product 7d with a high diastereoselectivity of up to ≥20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Scheme 2c).
:1 (Scheme 2c).
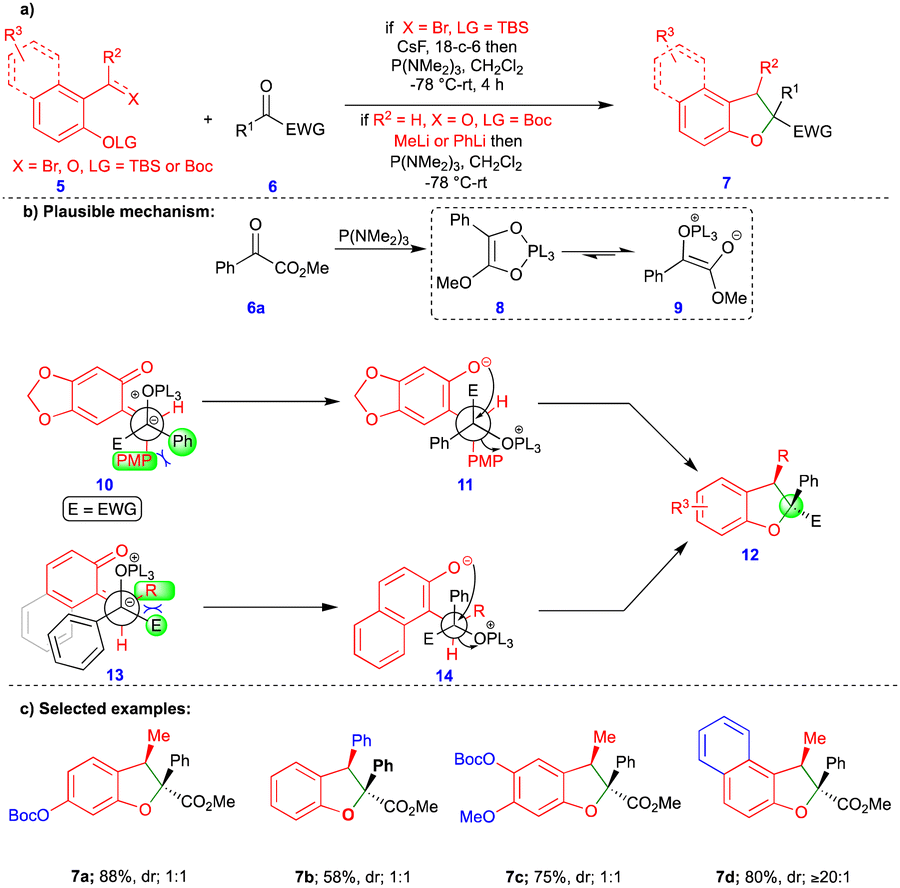 | ||
| Scheme 2 Phosphorus(III)-mediated formal [4+1]-cycloannulation of o-QMs with 1,2-dicarbonyl derivatives. | ||
Schneider's group reported the CPA.2-catalyzed [4+1]-cycloannulation reaction of o-QMs with α-diazoketones for the enantioselective synthesis of cis-2,3-dihydrobenzofuran (Scheme 3).20 Surprisingly, the obtained product 16 showed an inverted substitution pattern. The PMP (p-methoxyphenyl) group of o-QMs appeared in the 2nd position instead of the 3rd position, and the aryl group of the aryl ketone 15 moiety appeared in the 3rd rather than in the 2nd position. The broad spectrum of the cis-2,3-dihydrobenzofuran derivatives 16 was synthesized under optimized conditions with excellent yields and high stereoselectivity (Scheme 3b). The authors found that MgSO4 is essential for improving the yield and stereoselectivity via this transformation, and acts as a dehydrating agent.
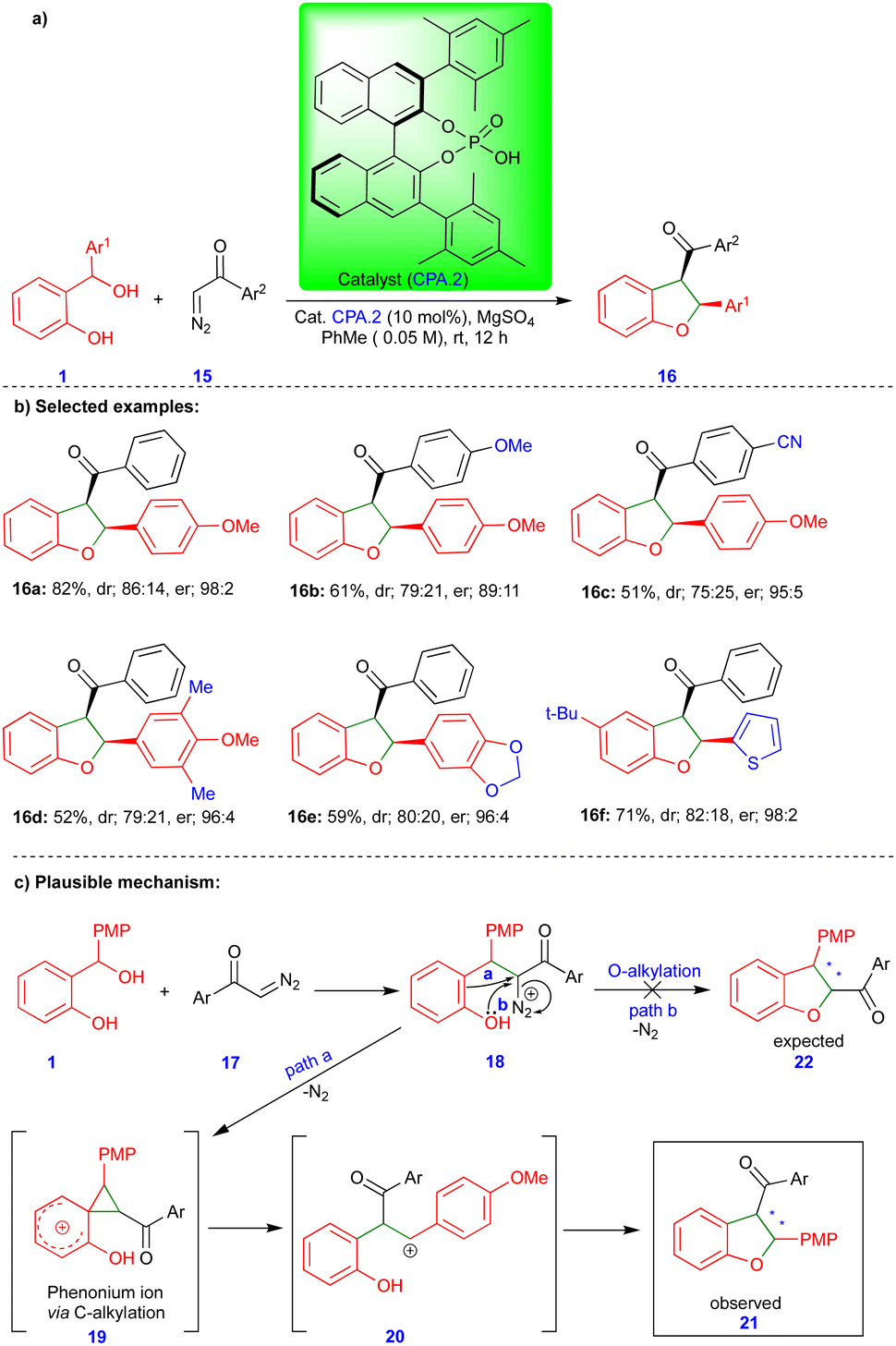 | ||
| Scheme 3 Chiral phosphoric acid catalyzed [4+1]-cycloannulation of diazoketone with o-QMs. | ||
The same authors suggested a mechanism for synthesizing cis-2,3-dihydrobenzofuran 16, as shown in Scheme 3c. The CPA.2-catalyzed dehydration of ortho-hydroxy benzhydryl alcohol yields the o-QM (not shown in the scheme) followed by the conjugate addition of α-diazoketone 17 deliver the phenol intermediate 18. Instead of O-alkylation (path b), the electron-rich phenol undergoes C-alkylation (path a), which means that it acts as a nucleophile and forms the spirocyclic cyclopropane phenonium ion 19 upon the elimination of nitrogen gas. Thus, intermediate 19 undergoes ring-opening to form a stable carbocation 20 (Scheme 3c), which reacts with phenol to form the observed product 21 (or 16) with an inverted substitution pattern. The proposed mechanism is well-supported by control experiments.
As discussed in Scheme 3, the cis-2,3-dihydrobenzofurans 16 synthesized contain an inverted substitution pattern, which was observed by Schneider's group. Later, Sun's group reported the borane-catalyzed synthesis of 2,3-dihydrobenzofuran derivatives via the formal [4+1]-cycloannulation of alkyne-tethered o-QMs and α-diazoacetates, with a normal substitution pattern of the 2,3-dihydrobenzofuran derivatives 25 (Scheme 4).21 They employed B(C6F5)3, a very strong Lewis acid, for this transformation. B(C6F5)3 plays a dual role in the generation of the o-QM and in activating the diazo moiety, resulting in a higher selectivity and reactivity. Furthermore, adding 4 Å molecular sieves increased the regioselectivity and diastereoselectivity and helped to trap the in situ formed water. The authors also observed a small amount of the inverted substitution pattern, as in Scheme 3.
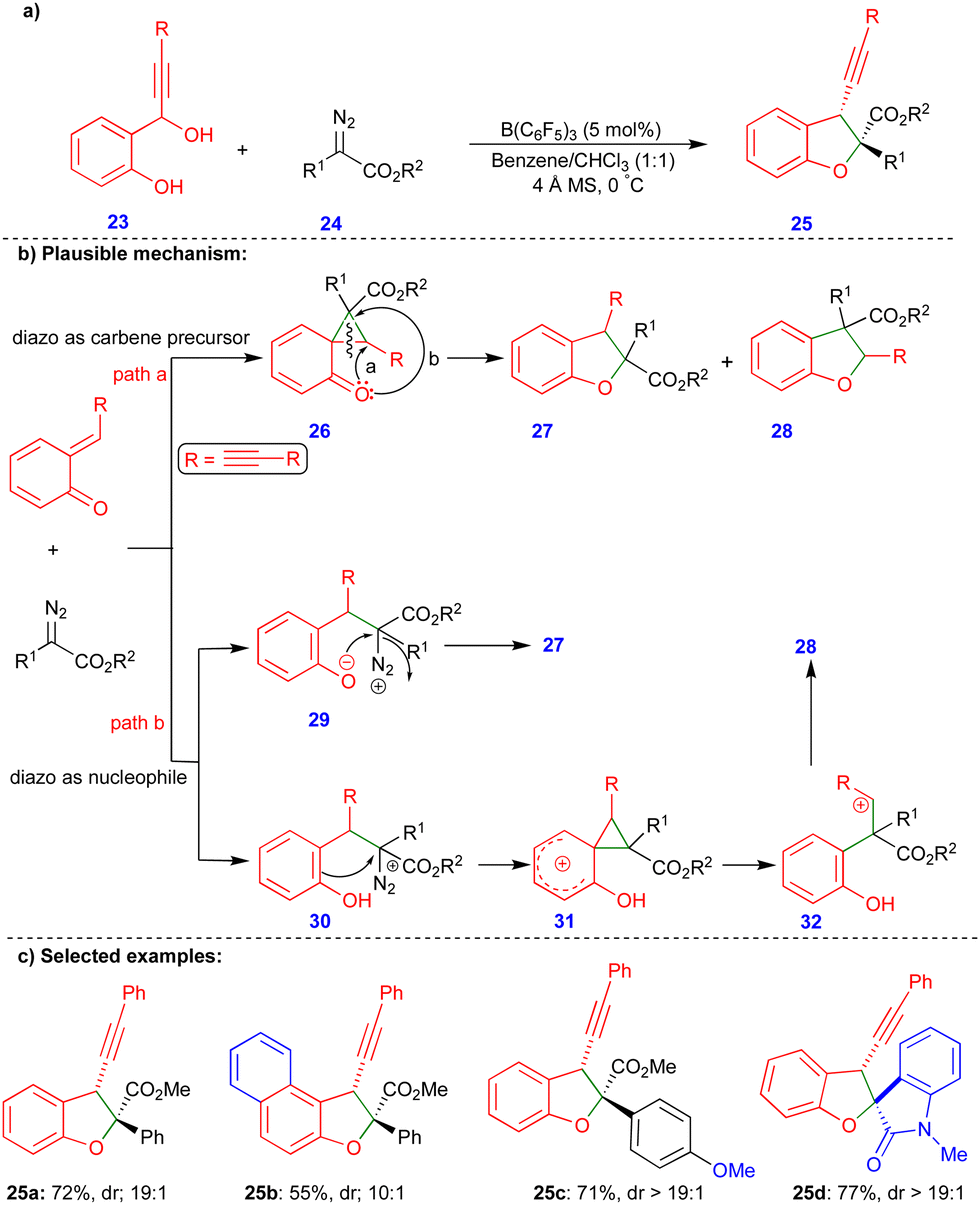 | ||
| Scheme 4 B(C6F5)3-catalyzed formal [4+1]-cycloannulation. | ||
Based on the observed products, two major reaction pathways were proposed, which led to a mixture of products 27 and 28. First, the diazo compound acts as a carbene precursor (path a, Scheme 4b). Interaction of the o-QM with the electrophilic carbene species to produce the cyclopropane intermediate 26, which then undergoes intramolecular ring-opening to produce a mixture of 27 and 28. Alternatively, the diazo compound can behave as a nucleophile (path b, Scheme 4b), adding to the o-QM to produce intermediate 29 or 30. To deliver compound 27, the intramolecular O-alkylation of 29 may occur. On the other hand, phenol intermediate 32 can be obtained by cyclopropanation of intermediate 30, and a ring-opening reaction of the phenol intermediate 31 followed by subsequent nucleophilic addition of 32 can deliver product 28 (Scheme 4b). Alkynyl tethered 2,3-dihydrobenzofuran 25 derivatives were synthesized with moderate to good yields and diastereoselectivity (Scheme 4c) from the corresponding alkynyl tethered ortho-hydroxy benzhydryl alcohol and α-diazoacetates via formal [4+1]-cycloannulation chemistry. Interestingly, isatin-based diazo compounds reacted well under the optimized conditions, to give the corresponding product 25d with good yield and diastereoselectivity. However, the corresponding products were not obtained via the reaction using a phenyl substituted ortho-hydroxy benzhydryl alcohol instead of alkynyl tethered reactant.
Recently, Ma and Du's group reported the palladium-catalyzed tandem [4+1]-cycloannulation of N-tosylhydrazones with 4-vinyl-3,1-benzoxazin-2-one compounds to synthesize indoline derivatives (Scheme 5).22N-Tosylhydrazone 34 is a precursor of diazo compounds in the presence of a base, and is equivalent to carbene 39 (Scheme 5b). Palladium-catalyzed decarboxylation of the 4-vinyl-3,1-benzoxazin-2-one 36 produces the zwitterionic π-allylpalladium intermediate 37, which is in equilibrium with the aza-o-QMs 38 upon elimination of Pd(0). Next, the 1,4-addition of the carbene to the reactive aza-o-QM 38 (Scheme 5b) produces two diastereoisomers 40 and 41. The diastereoselectivity originates from the more favorable intermediate 41via an intramolecular SN2 reaction, to deliver the corresponding product 35 (Scheme 5). Alternatively, the formation of product 35 can be explained via a tandem Pd–π-allyl carbene migratory insertion and reductive elimination pathway. Ashfeld's group proposed a similar mechanism via a non-aza-o-QM intermediate.23 The palladium-catalyzed tandem synthesis of indoline frameworks was obtained with good yields and excellent diastereoselectivities from 4-vinyl-3,1-benzoxazin-2-one derivatives 33 and N-tosylhydrazone 34 (Scheme 5c).
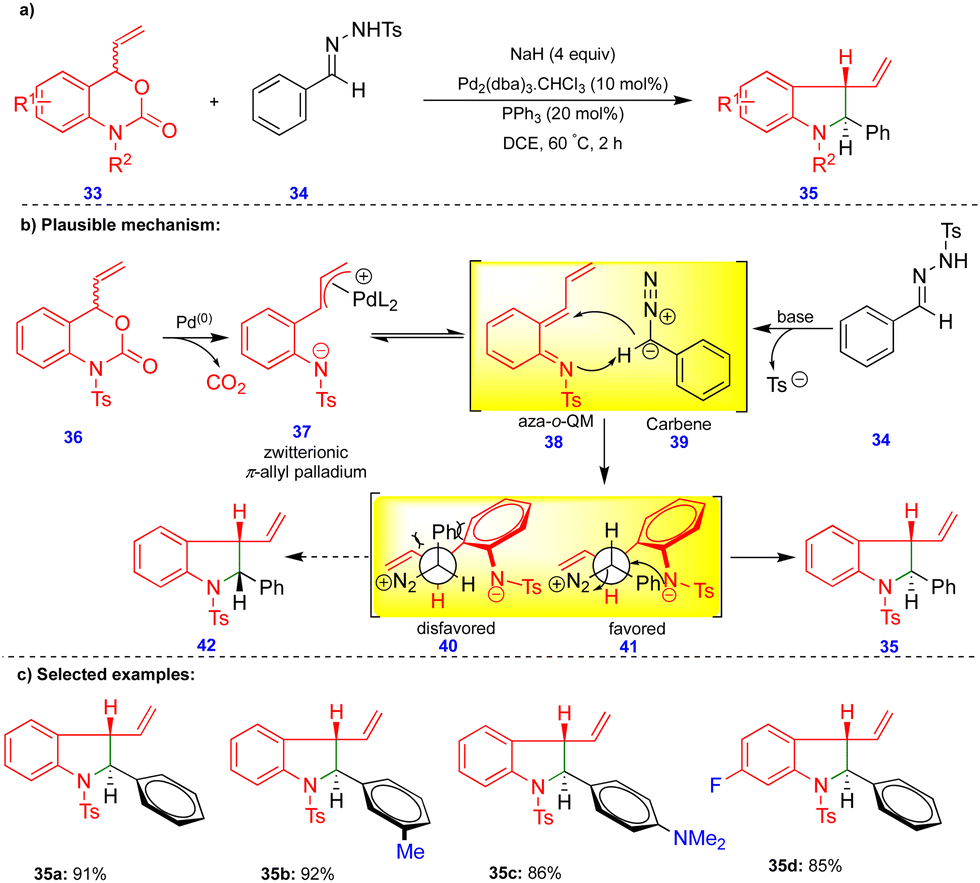 | ||
| Scheme 5 Palladium-catalyzed diastereoselective [4+1]-cycloannulation. | ||
2,3-Dihydrobenzofurans can also be easily synthesized from p-QMs. Li and Xuan's group reported that a visible-light-induced base promoted a one-pot, two-step protocol for synthesizing 2,3-dihydrobenzofuran compounds from p-QMs and aryldiazoacetates (Scheme 6).24 Wide range of 2,3-dihydrobenzofuran derivatives 45 were obtained with good yields and low diastereoselectivities (Scheme 6c) via sequential steps. First, the O–H insertion of 43 with carbene, generated from aryldiazoacetate 44 in the presence of blue LEDs and upon the elimination of molecular nitrogen, could deliver the O–H insertion product 46 (Scheme 6b). Subsequently, its base-mediated transformation delivered the carbanion intermediate 47, and cyclization (48) followed by protonation delivered the 2,3-dihydrobenzofuran 45 (Scheme 6b).
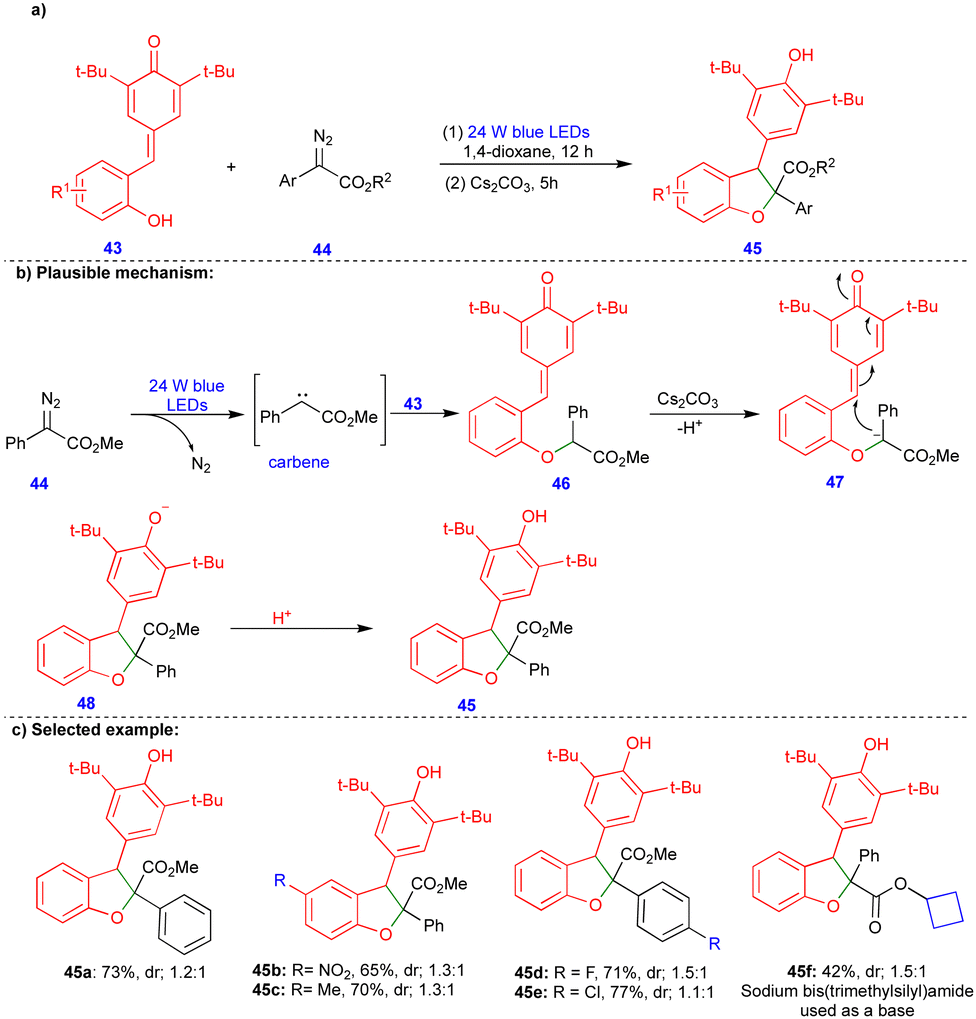 | ||
| Scheme 6 Visible-light-induced base mediated synthesis of 2,3-dihydrobenzofurans. | ||
In 2021, Cui and Chen's group described the asymmetric synthesis of spiro heterocyclic compounds such as spiro dihydrobenzofuran derivatives via the CPA.3-catalyzed [4+1]-cycloannulation of diazo heterocyclic compounds with p-QMs (Schemes 7 and 8).25 The CPA.3-catalyzed [4+1]-cycloannulation of p-QM 50 with electronically diverse 3-diazooxiindole compounds 49 reacted very well, giving the dihydrobenzofuran containing oxindole derivatives 51 in good yield, diastereoselectivity, and enantioselectivity (Scheme 7b). However, in the case of chlorine substitution at the C4 position of the 3-diazooxindole, no reaction was observed due to steric factors. This concept with p-QM 50 also works very well with other heterocyclic diazo compounds, i.e., 4-diazooxisoquinoline derivatives 52, under slightly modified conditions in the presence of the same catalyst (Scheme 8). However, a lower reactivity and stereoselectivity were observed (Scheme 8b).
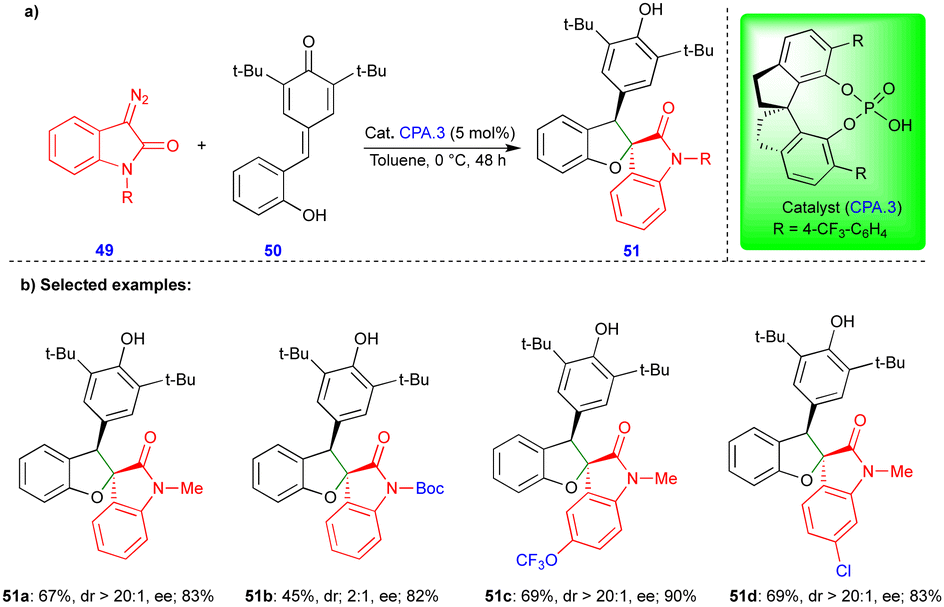 | ||
| Scheme 7 Reaction of diazo-oxindole compounds with p-QMs. | ||
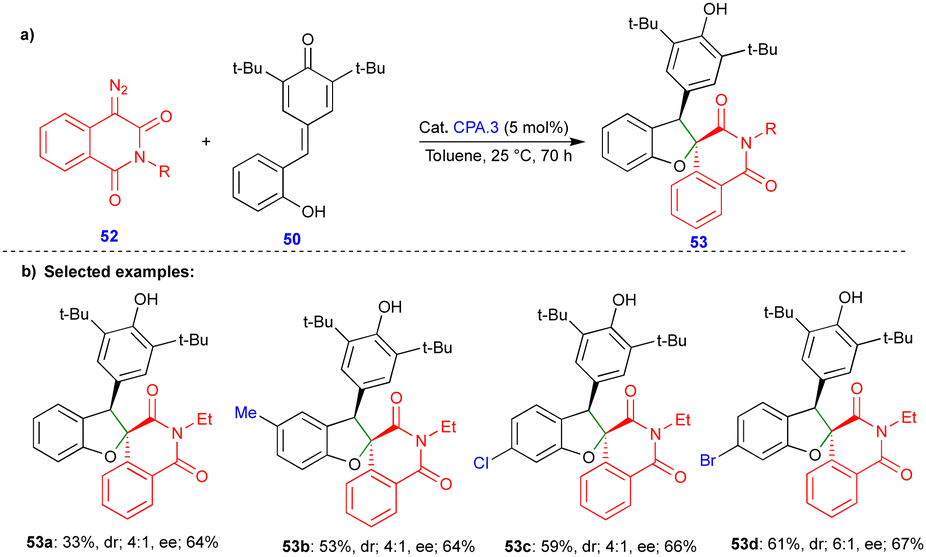 | ||
| Scheme 8 Reaction of diazooxisoquinoline derivatives with p-QMs. | ||
Based on control experiments, the same authors proposed the mechanism, which proceeds via proton transfer from the ortho-hydroxyphenyl group to the quinone methide oxygen of p-QM 50, i.e., p-QM 50 is isomerized to o-QM 56 (Scheme 9), which is the key factor for the reaction to proceed. Moreover, heterocyclic diazo compound 54 exists in equilibrium with ionic form 55 (Scheme 9). Being in situ generated, these intermediates are stabilized through hydrogen-bonding with CPA.3 (57) followed by intramolecular oxygen attack of 55 at the C3/C4 position via the elimination of molecular nitrogen and cyclization to deliver the enantioenriched products (Scheme 9).
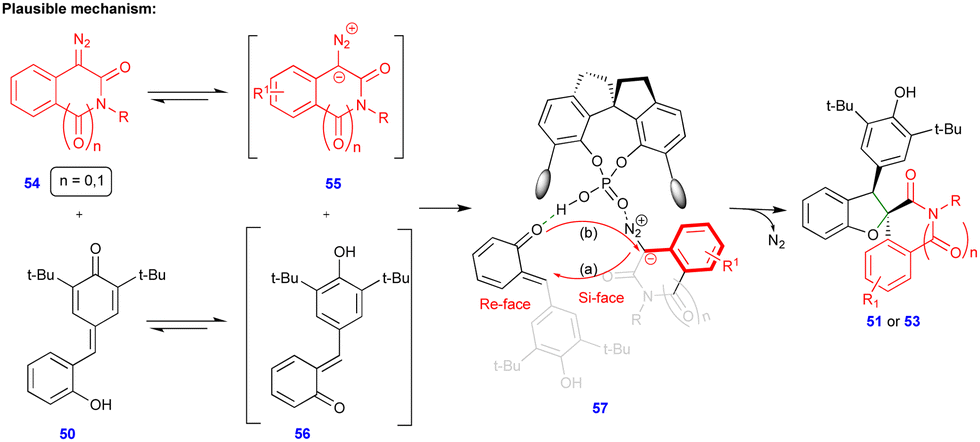 | ||
| Scheme 9 Proposed mechanism of 1,4-addition of p-QMs. | ||
Recently, Jiang and Yang's group reported the synthesis of gem-difluorinated oxygen-containing heterocyclic compounds via the [4+1] cycloannulation reactions of heteroconjugated alkenes such as o-QMs with difluorocarbenes (Scheme 10).26gem-Difluorinated 2,3-dihydrobenzofuran compounds 60 (Scheme 10c) were obtained in excellent yield through the [4+1]-cycloannulation of o-QMs with difluorocarbene generated in situ from TMSCF2Br (59) in the presence of tetrabutylammonium bromide (TBAB) as an additive. A free radical pathway was ruled out via radical quenching and carbene trapping experiments. Difluorocarbene 61 was generated from the thermal decomposition of TMSCF2Br (59) upon treatment TBAB (Scheme 10b), which then reacted with the O atoms of o-QM 58 to form zwitterionic intermediates 62. These intermediates then underwent an intramolecular 1,4-addition reaction to form the gem-difluorinated 2,3-dihydrofuran derivatives 60 (Scheme 10c).
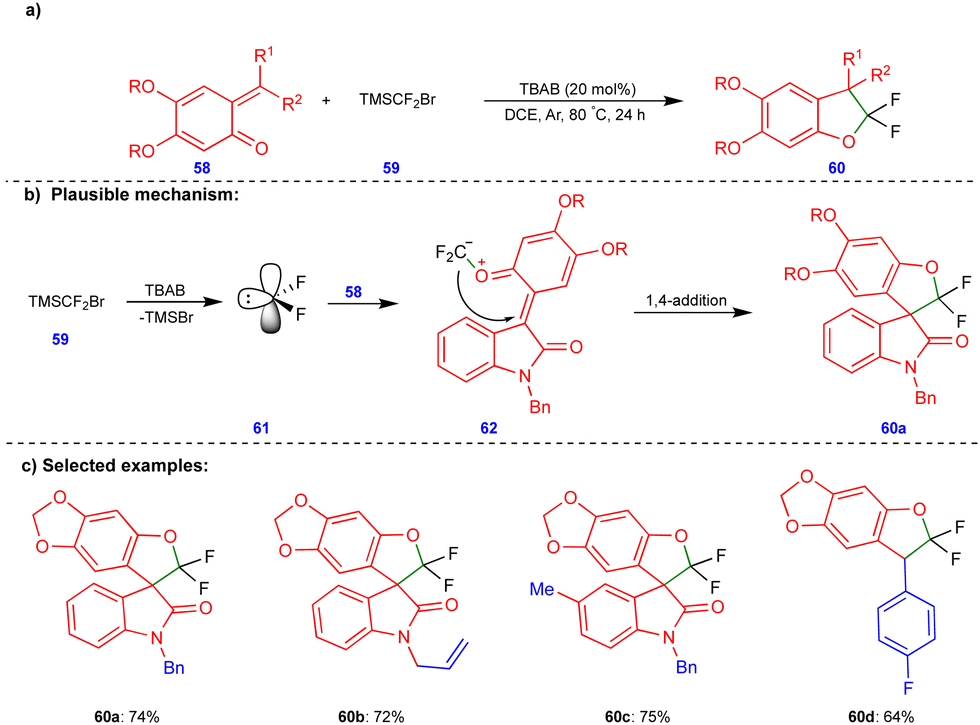 | ||
| Scheme 10 Synthesis of gem-difluorinated 2,3-dhydrobenzofurans. | ||
As noted above, Schneider's group reported the CPA.2-catalyzed enantioselective synthesis of 2,3-dihydrobenzofurans with an inverted substitution pattern (Scheme 3).20 Ryu's group observed a similar product with o-QMs in the presence of a Lewis acid-catalyst (Scheme 11).27 The enantioselective synthesis of 2-aryl-2,3-dihydrobenzofuran 65 was achieved via a one-pot, two-step protocol involving initial cyclopropanation via the [2+1]-cycloannulation of o-QMs with a α-diazoester followed by intramolecular rearrangement (Scheme 11b). Based on the observed stereochemistry, the authors proposed the initial asymmetric cyclopropanation step, stabilized by the pre-transition-state in the presence of the chiral oxazaborolidinium ion (COBI.1) as a catalyst through hydrogen bonding (69 and 70), followed by nucleophilic addition, to deliver intermediate 71 (Scheme 11c). Then, the ring-opening of cyclopropane 71, which is in equilibrium with zwitterionic intermediate 72, is stabilized by the electron-donating group on Ar1. Finally, the oxygen nucleophile attacks carbocation C1 to produce the 2-aryl-2,3-dihydrobenzofuran compound 65 (Scheme 11c).
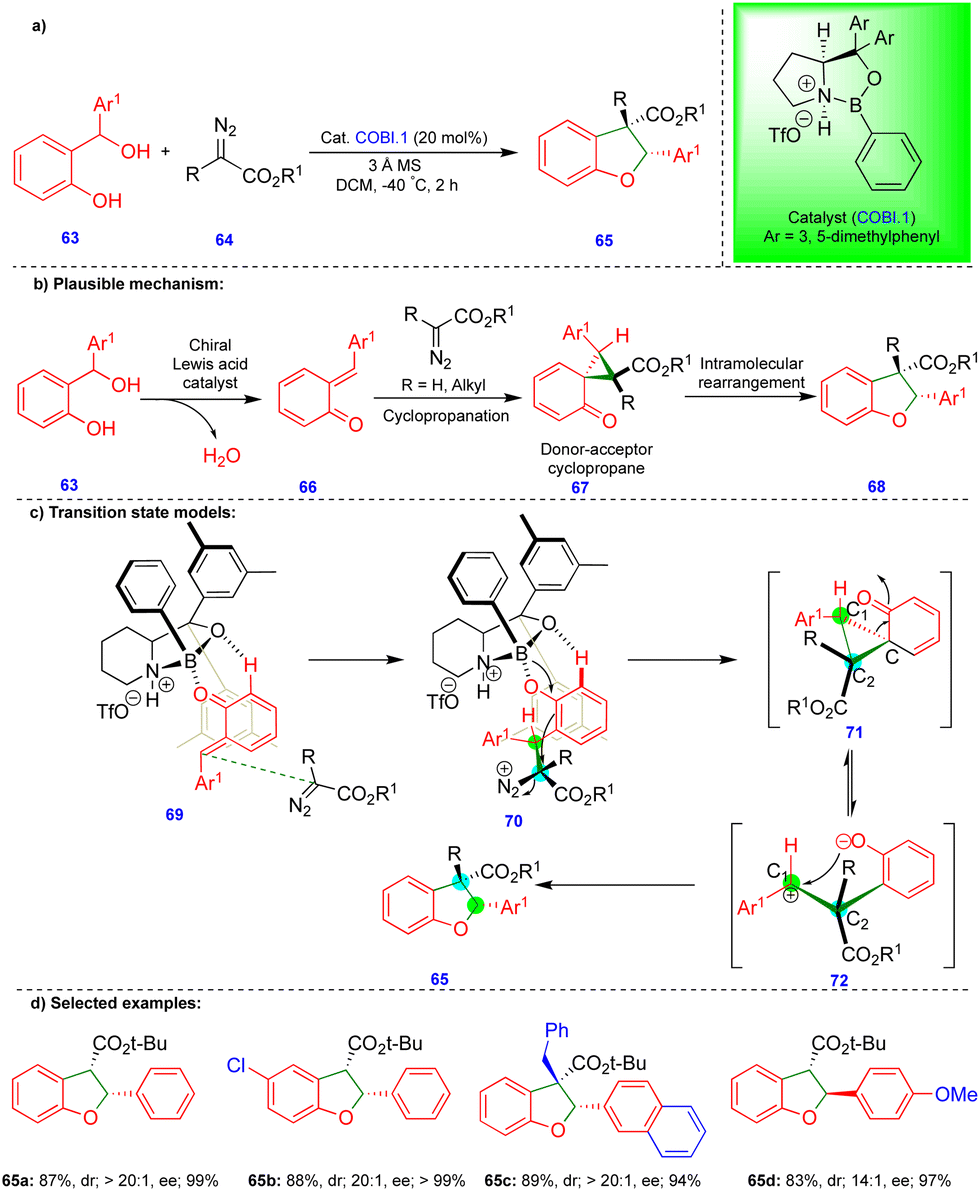 | ||
| Scheme 11 Asymmetric synthesis of 2-aryl-2,3-dihydrobenzofuran compounds using a chiral Lewis-acid catalyst. | ||
Electronically diverse o-QMs with α-diazoester compounds reacted well, giving the cis-2-aryl-2,3-dihydrobenzofuran as the major product with excellent yield and stereoselectivity in the presence of COBI.1 (Schemes 11d), using 3 Å molecular sieves as a dehydrating agent. Interestingly, an electron-rich group substituted on β-methide afforded trans-65d as the major product (Scheme 11d).
3.2 [8+1]-Cycloannulation
Scheme 10 discussed the [4+1]-cycloannulation of oxygen-containing heterocyclic compounds with difluorocarbenes reported by the group of Jiang and Yang. Similarly, the same group extended their work to [8+1]-cycloannulation with nitrogen-containing heterocyclic compounds such as azaheptafulvenes with difluorocarbenes for synthesizing gem-difluorinated azetidines (Scheme 12).26 The thermal decomposition of Ph3P+CF2CO2− (75) generates the difluorocarbene 61, which then reacts with azaheptafulvene 73 to form the zwitterionic intermediate 76. Then, it underwent a 1,8-addition reaction to form azetidine 74 (Scheme 12b). A wide variety of gem-difluorinated azetidines were synthesized in excellent yield (Scheme 12c).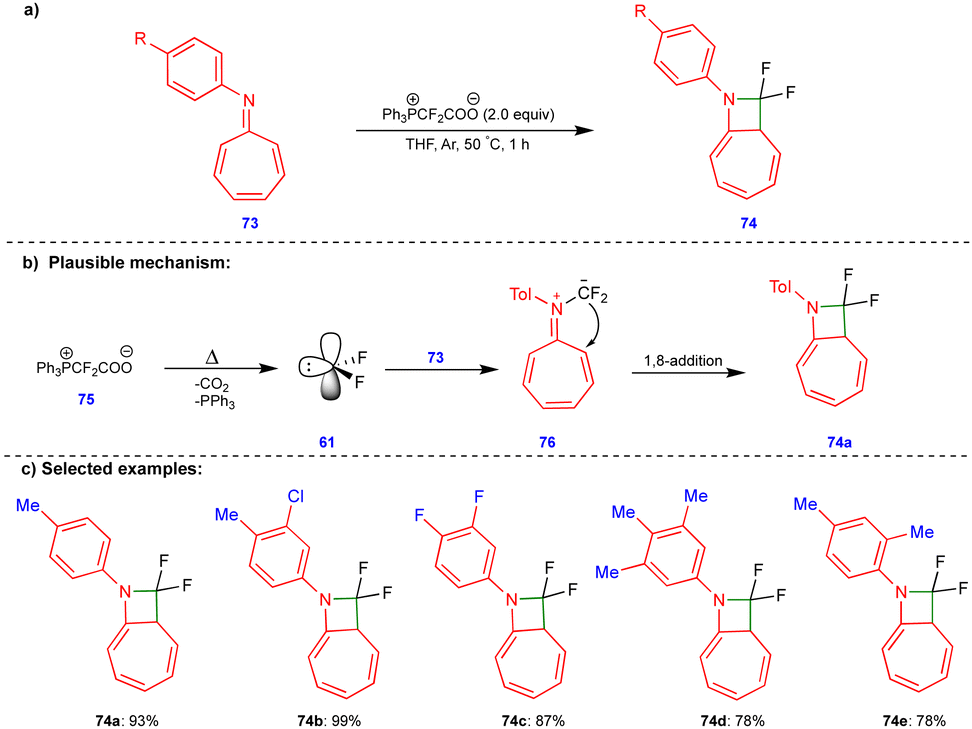 | ||
| Scheme 12 Synthesis of gem-difluorinated azetidines. | ||
3.3 [4+3]-Cycloannulation
Lautens's group reported the transition-metal-free diastereoselective synthesis of oxo-bridged oxazocine scaffolds via the o-QM and isomünchnone transient intermediates generated in situ.28 Isomünchnone 80 was generated from diazoimide 77via intramolecular amide carbonyl attack on the diazo compound, and o-QM 81 was generated in the presence of (+)-camphorsulfonic acid (CSA) as a catalyst from a naphthol-based or phenol-based o-QM precursor 78 (Scheme 13). Intermediates 80 and 81 reacted via a [4+3]-cycloannulation reaction to give the oxo-bridged oxazocine 79 with a single diastereomer.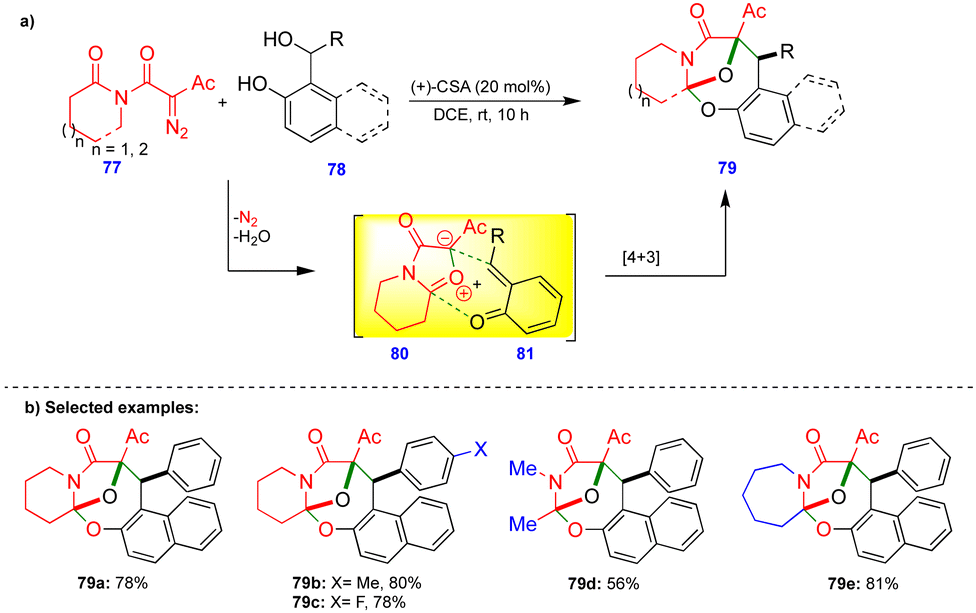 | ||
| Scheme 13 [4+3]-Cycloannulation of o-QMs with isomünchnones. | ||
Electronically diverse diazoimide 77 derivatives and naphthol-based o-QMs reacted smoothly, giving the oxo-bridged oxazocine derivatives as a single diastereomer in good yields (Scheme 13b). Thiophene-substituted naphthol-based o-QMs also gave the corresponding product. However, pyridine-substituted naphthol-based o-QMs failed to react. In the case of o-QMs derived from phenol-based substrates, lower product yields were observed, possibly due to the rapid decomposition of o-QMs. Interestingly, acyclic and seven-membered lactam-based diazoimides also react well under the optimized conditions, giving the corresponding products 79d and 79e in moderate to good yields (Scheme 13b).
Schneider's group developed an excellent stereoselective synthesis for oxa-bridged dibenzooxacines via [4+3]-cycloannulation using cooperative catalysis.29o-QMs 84 was generated from the ortho-hydroxy benzhydryl alcohol 1 in the presence of chiral-phosphoric acid (CPA.4), which was stabilized through hydrogen-bonding. Similarly, the carbonyl ylide 85 was formed from carbonyl-tethered α-diazoester 82 in the presence of a rhodium catalyst (Rh2(OAc)4) (Scheme 14b). The two transient intermediates reacted stereoselectively to produce the oxa-bridged dibenzooxacine compounds 83via [4+3]-cycloannulation. Control experiments revealed that chiral phosphoric acid (CPA.4) is essential for inducing enantioselectivity.
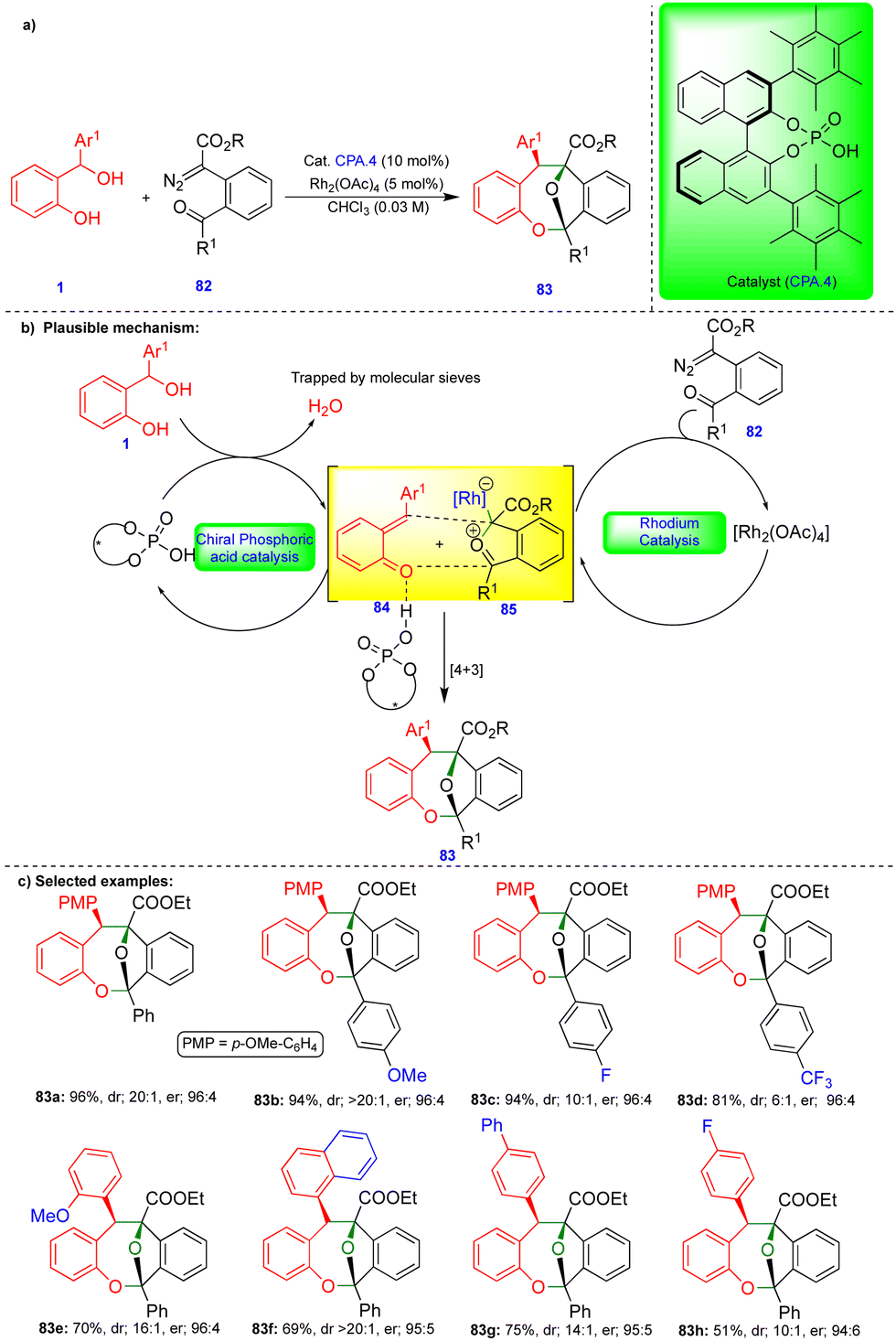 | ||
| Scheme 14 [4+3]-Cycloannulation of o-QMs and carbonyl ylides. | ||
Electron-donating-group-substituted α-diazoesters gave dibenzooxacine derivatives in excellent yield, diastereoselectivity, and enantioselectivity (Scheme 14c). In the case of electron-withdrawing groups, the substituted α-diazoesters produced dibenzooxacine derivatives with a slightly diminished diastereoselectivity. ortho-Substituted aryl groups affect both the diastereoselectivity and enantioselectivity. A wide variety of dibenzooxacine derivatives were synthesized from electronically different substituted ortho-hydroxy benzhydryl alcohols 1 under slightly elevated conditions. Electronically poor substituted substrates delivered dibenzooxacine derivatives with moderate yields (Scheme 14c). However, alkyl-substituted ortho-hydroxy benzhydryl alcohol failed to deliver the product, which may be due to the instability of o-QM towards [4+3]-cycloannulation.
3.4 [2+1]-Cycloannulation followed by ring-opening
In 2016, Cui's group reported the Lewis acid (TiCl4) activated metathesis type reaction between p-QMs and α-diazoesters for the synthesis of tetrasubstituted alkenes and quinolinones.30 Here, the p-QMs act as Michael acceptors and react with nucleophilic reagents such as α-diazoesters. Tetrasubstituted alkenes were synthesized with good to moderate yields 88 (Scheme 15) from the same substitution pattern on the aromatic rings of the p-QMs 86 and the α-diazoester 87. By contrast, unsymmetrically substituted compounds gave mixtures of E/Z isomers. Interestingly, when the diazo compounds were switched from α-diazoesters 87 to diazooxindoles 89, quinolinones 90 were produced (Scheme 16) via a two-step, one-pot procedure. Initially, TiCl4-mediated cyclopropanation, subsequent chlorination, and AlCl3-mediated de-tert-butylation followed by ring expansion delivered a variety of quinolinones 90 with moderate yields (Scheme 16).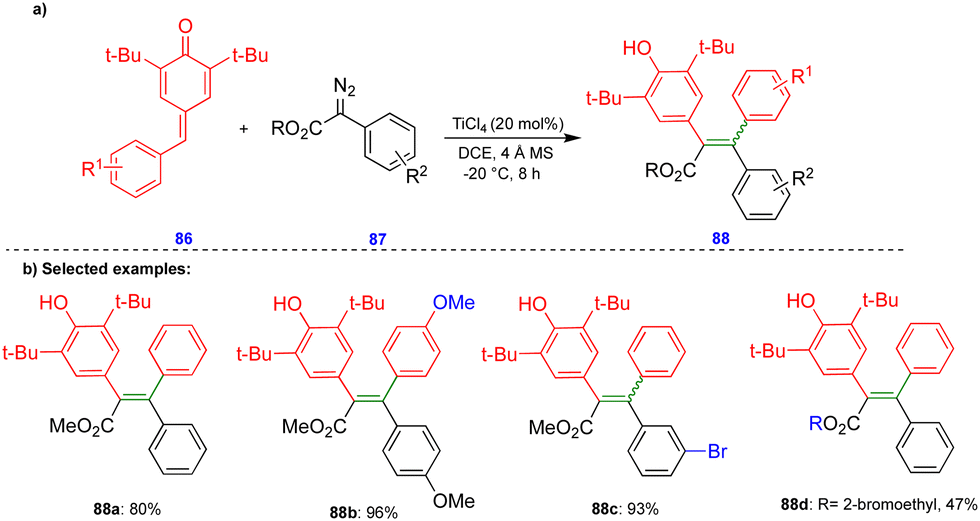 | ||
| Scheme 15 TiCl4-catalyzed synthesis of tetrasubstituted alkenes. | ||
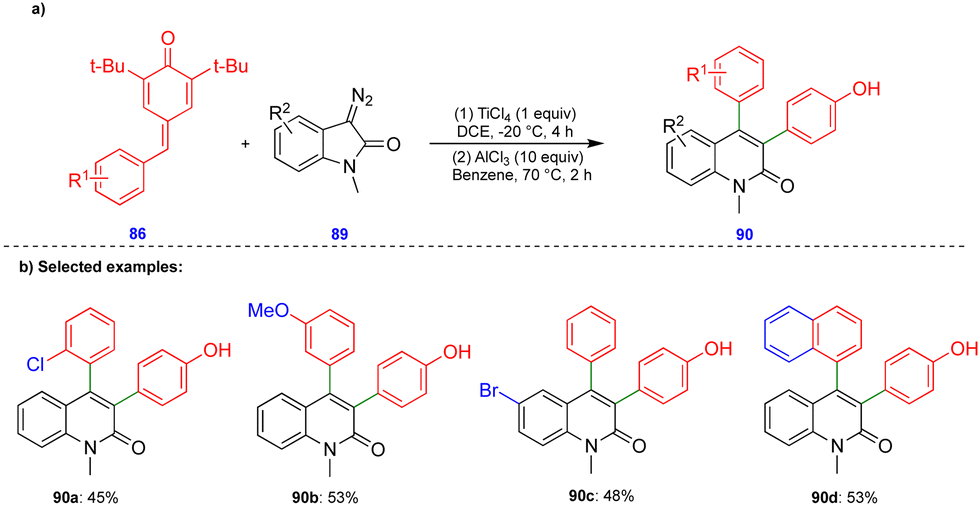 | ||
| Scheme 16 Lewis-acid mediated synthesis of quinolinones. | ||
Based on the 13C-labelling experiments, the catalytic amount of TiCl4 activated the p-QM 86 to form the benzyl carbocationic intermediate 91, which then reacted with α-diazoester 87 or diazooxindole 89 to produce the cyclopropane intermediate 92 (Scheme 17) in a [2+1]-cycloaddition fashion via the elimination of molecular nitrogen. TiCl4 further activated the cyclopropane intermediate to form another benzyl carbocationic intermediate 93. Intermediate 93 derived from the α-diazoester would then undergo aryl group migration of the α-diazoester, and subsequent elimination of a proton would furnish the alkene compound 95 (Scheme 17). Finally, protonation would deliver the corresponding tetrasubstituted alkene 88. If the TiCl4-mediated benzyl carbocationic intermediate 93 was derived from the diazooxindole, it would undergo chlorination at the benzyl cationic position, giving 96, with subsequent protonation to produce compound 97 (Scheme 17). Compound 97, upon treatment with AlCl3, proceeded via Friedel–Crafts type alkylation and ring expansion followed by the elimination of tert-butyl groups to produce the corresponding quinolinone 90 (Scheme 17).
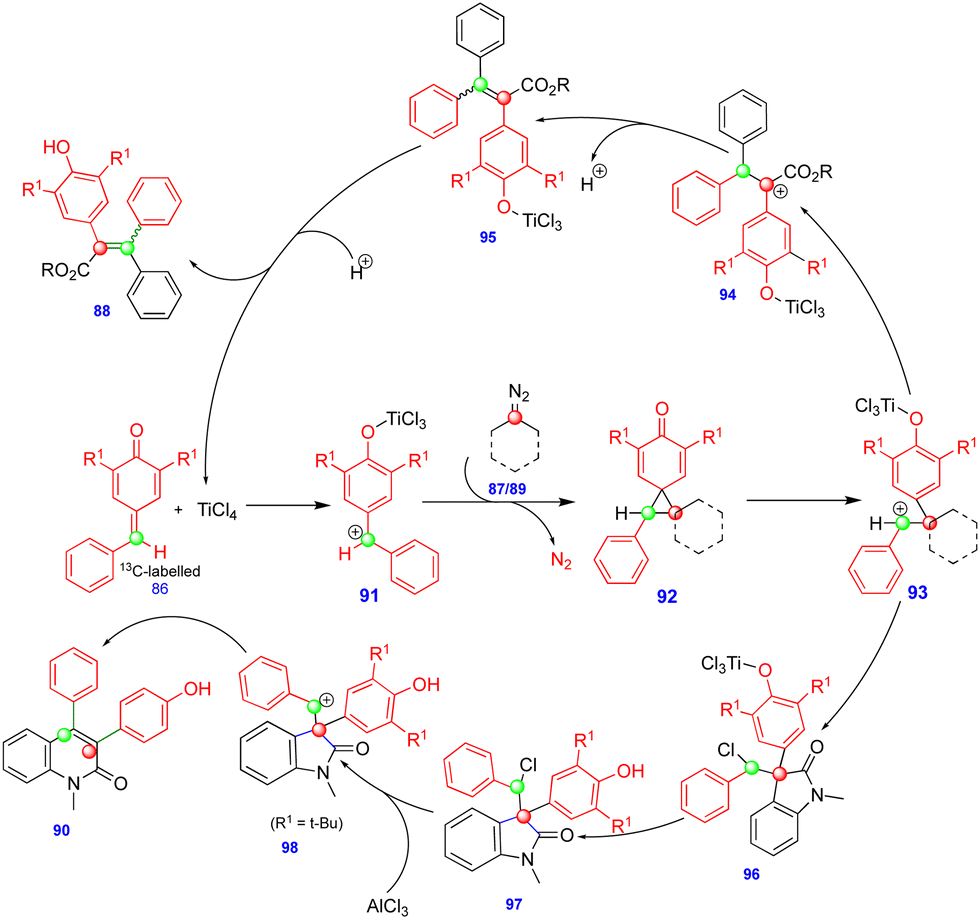 | ||
| Scheme 17 Synthesis of tetrasubstituted alkenes and quinolinones. | ||
Ryu's group reported the enantioselective synthesis of the Rauhut–Currier product from the α-alkyl diazoesters and o-QMs via [2+1]-cyclopropanation followed by a [1,5]-hydrogen shift in the presence of chiral oxazaborolidinium ion (COBI.1).31 Based on their previous results (Scheme 11)27 and control experiments, they revealed that the asymmetric cyclopropane intermediate 104 (Scheme 18b) is formed via a dipole–dipole interaction between the carbonyl groups of α-alkyl diazoesters and o-QMs. The hydrogen at C1 migrates to the carbonyl oxygen to give a six-membered transition state 105 in a concerted manner. The (Z)-selective product was obtained by concerted [1,5]-hydrogen shift through a six-membered transition state 105 (Scheme 18b) having ester group at C2 and R2 group at C1 in cis relation. Highly functionalized and enantioenriched (Z)-β-substituted Rauhut–Currier products were synthesized from α-alkyl diazoester derivatives 99 with in situ generated o-QMs in good to excellent yields (Scheme 18c) via a tandem cyclopropanation/[1,5]-hydrogen shift in the presence of a Lewis acid.
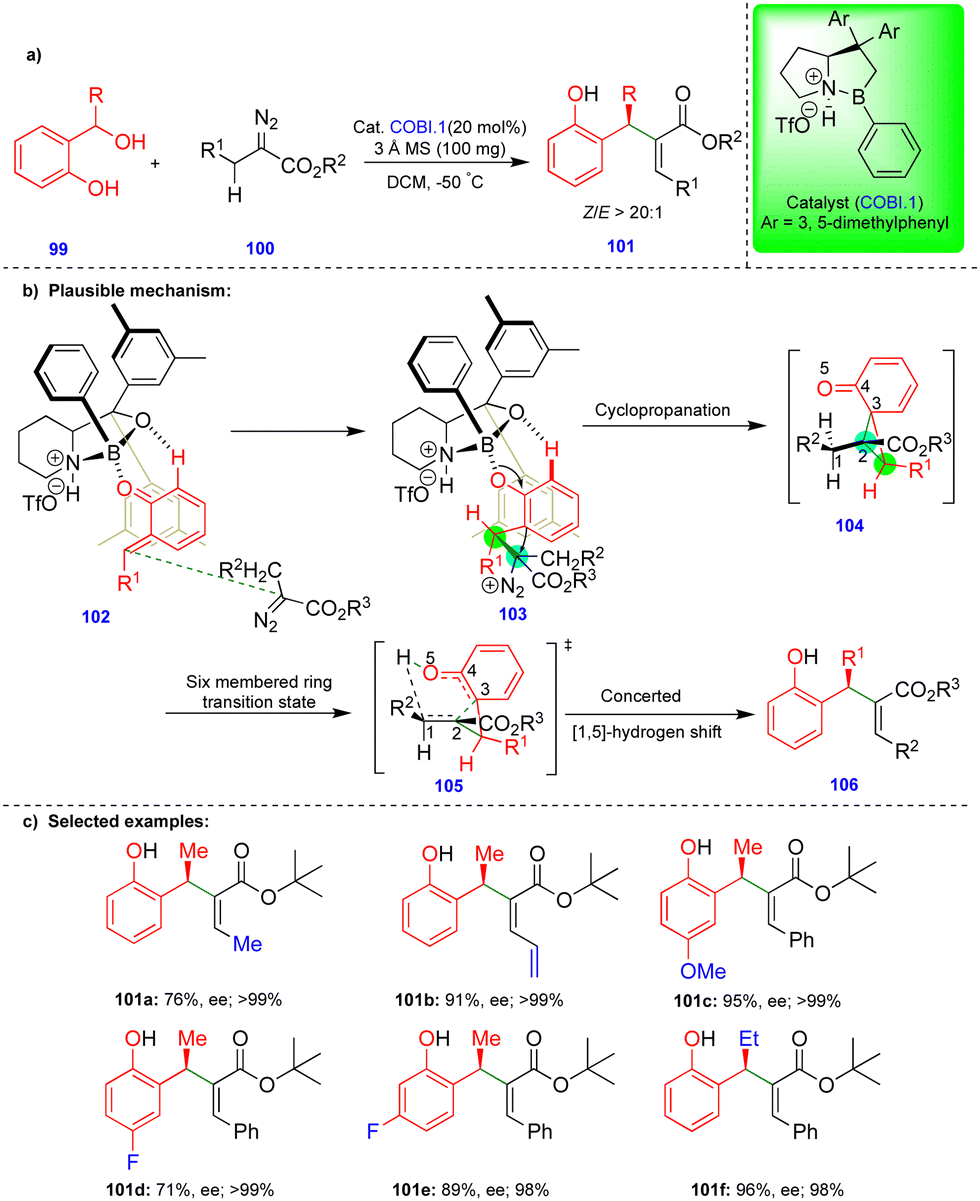 | ||
| Scheme 18 Enantioselective synthesis of Rauhut–Currier products. | ||
Recently, Lautens's group reported the rhodium and camphorsulfonic acid (CSA)-catalyzed dearomative cyclopropanation of 2-naphthol compounds via cyclopropene ring-opening, involving initial carbene insertion, followed by water elimination.32 The ring-opening of cyclopropene33107 derivatives in the presence of a rhodium catalyst (Rh2(OAc)4) delivered rhodium carbene compounds. Thus, they reacted with (1-hydroxyalkyl)-naphthalene-2-ol compounds 108 in a dearomative fashion to deliver a variety of vinyl cyclopropane compounds 109, in the presence of CSA and MgSO4, with good yields and moderate diastereoselectivities (Scheme 19c). Interestingly, when the authors switched the substrate to the tertiary-alcohol-containing (1-hydroxyalkyl)naphthalene-2-ol 114, the dihydrofuran derivative 115 was provided instead of the cyclopropanation product. Formation of the dihydrofuran could be explained via the in situ generation of an o-QM from tertiary-alcohol-containing (1-hydroxyalkyl) naphthalene-2-ol 114 in the presence of CSA, which then reacted with the corresponding rhodium carbene in a [4+1]-cycloannulation fashion to deliver the dihydrofuran 115 (Scheme 19d).
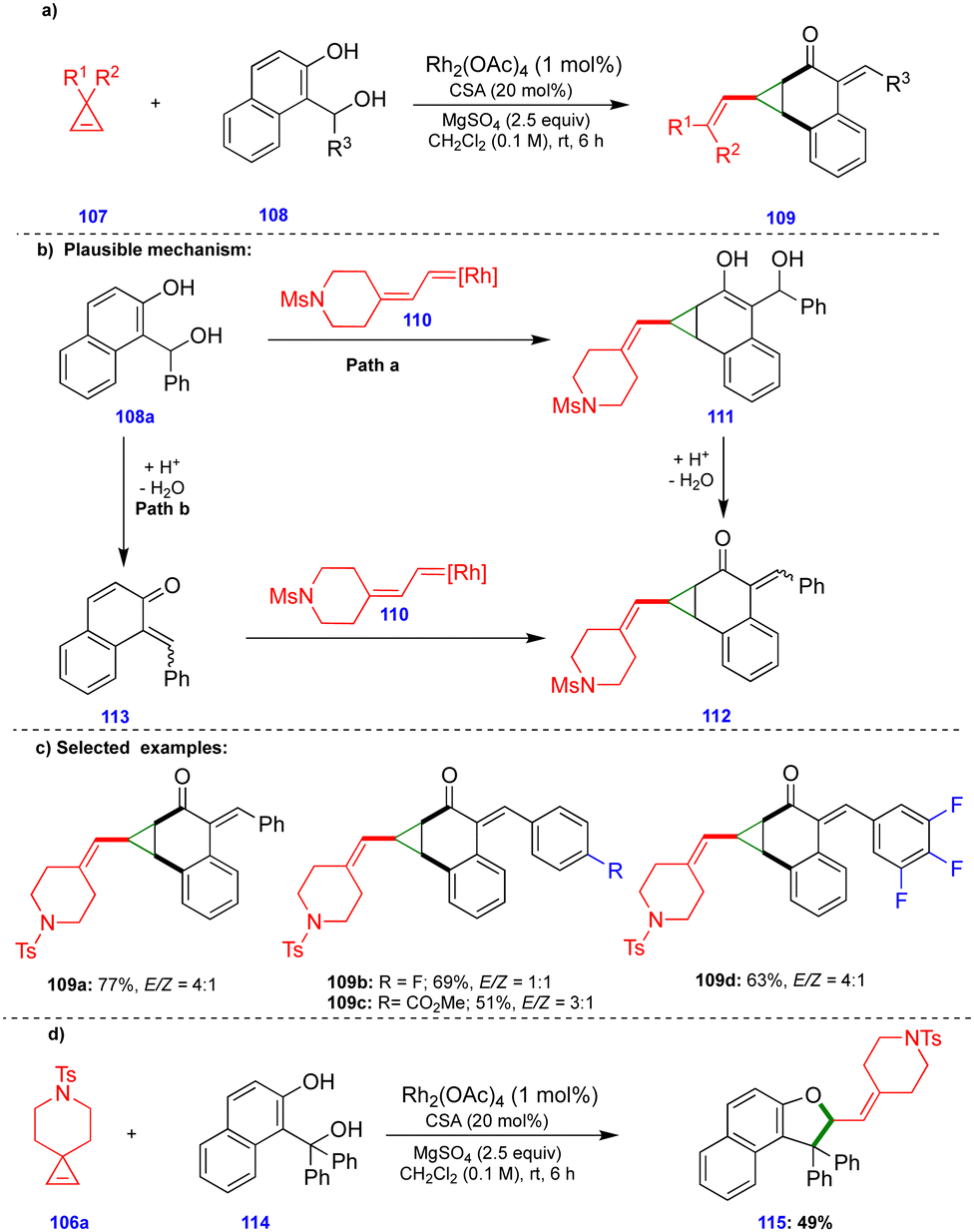 | ||
| Scheme 19 Dearomative cyclopropanation of naphthol compounds. | ||
Based on control experiments, the observed products, and DFT calculations, the product formation could be explained via two pathways. The in situ generated rhodium carbene 110 reacted with the naphthol derivative to deliver compound 111via Path a (Scheme 19b). Next, the CSA-catalyzed elimination of the benzylic hydroxy group delivered the product 112 through elimination of a water molecule. In Path b, the o-QM 113, which was generated from (1-hydroxyalkyl)naphthalene-2-ol 108a in the presence of an acid catalyst, reacted with rhodium carbene 110 to afford the cyclopropanation product 112 (Scheme 19b). The activation energy of Path a is more favorable by 5.6–6.3 kcal mol−1 compared with Path b, which suggests that the reaction proceeds through carbene insertion followed by an elimination sequence.
3.5 Other cycloannulations
Schneider's group continued to be interested in cooperative catalysis. Recently, they reported the stereoselective [3+3]-cycloannulation of in situ generated carbonyl ylides with indolyl-2-methides in the presence of rhodium and chiral phosphoric acid catalysts for the synthesis of oxa-bridged azepino[1,2-a]indole derivatives (Scheme 20).34 Carbonyl ylide 121 is generated from the carbonyl tethered α-diazoester 117 in the presence of a rhodium catalyst (Rh2(OAc)4) via the formation of rhodium carbene 120 (Scheme 20b), followed by trapping with a tethered carbonyl group. Indolyl-2-methide 119 derivatives are generated from indolyl-2-carbinol 116 in the presence of CPA.5, stabilized through hydrogen bonding (Scheme 20b). Thus, the two transient intermediates undergo [3+3]-cycloannulation to deliver the oxa-bridged azepino[1,2-a]indole derivatives 118.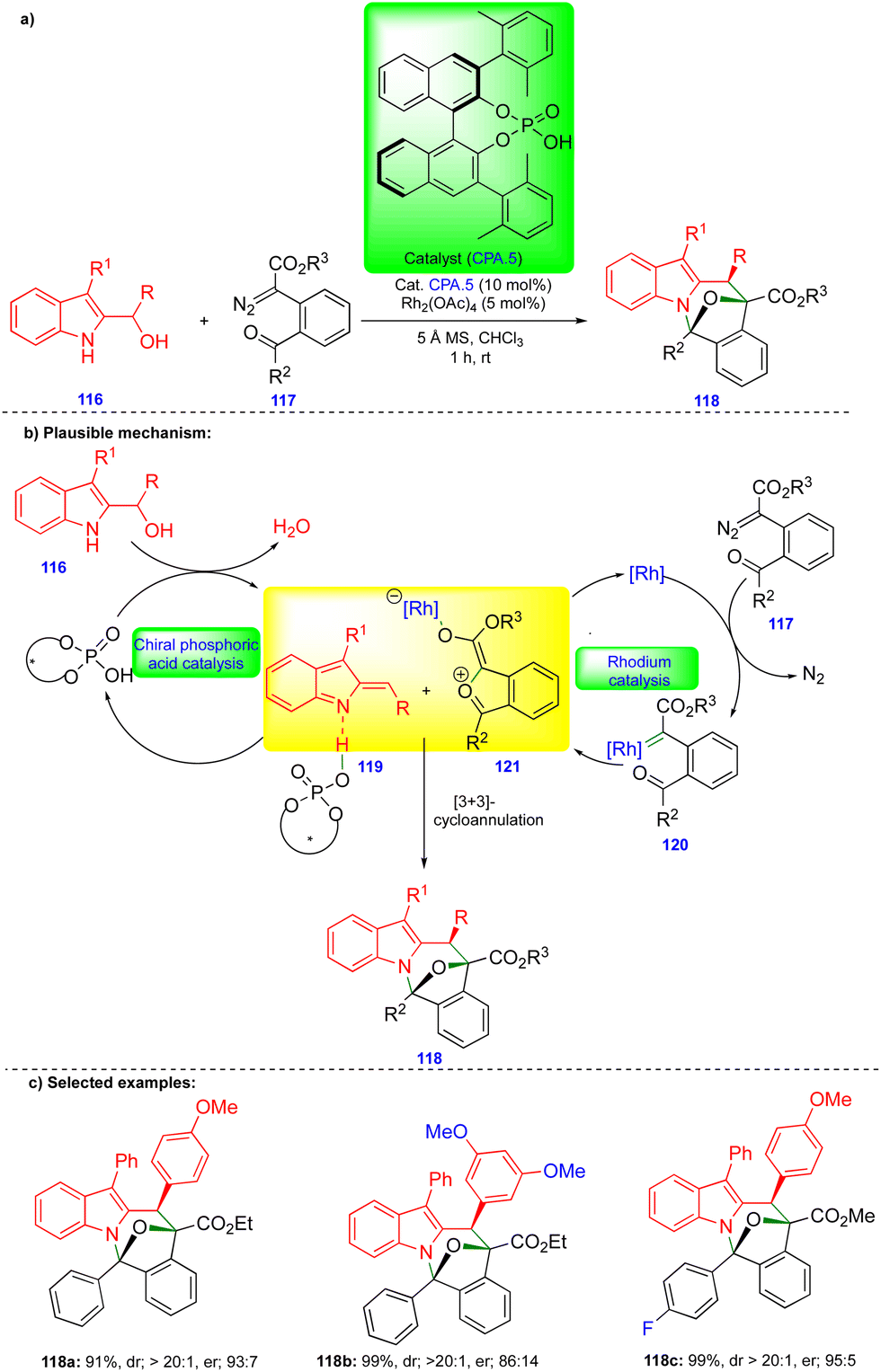 | ||
| Scheme 20 Stereoselective [3+3]-cycloannulation of carbonyl-ylides with indolyl-2-methides. | ||
The same authors showed the synthesis of a wide variety of oxa-bridged azepino[1,2-a]indole derivatives 118 (Scheme 20c) in both good yield and good to excellent stereoselectivity from the reaction between indolyl-2-carbinol 116 and α-diazoester 117via cooperative catalysis. However, the alkyl-substituted indolyl-2-carbinol substrate was unsuccessful under the optimized conditions due to the instability of indolyl-2-methides.
Anbarasan's group reported the synthesis of benzopyran compounds from in situ generated α-imino rhodium carbenes and o-QM intermediates via the rhodium-catalyzed denitrogenative transannulation of N-sulfonyl-1,2,3-triazoles with ortho-hydroxy benzhydryl alcohols.35N-Sulfonyl-1,2,3-triazole derivatives 122 are the precursors of α-diazoimines36via ring–chain isomerism, in the presence of a rhodium catalyst (Rh2(Oct)4), to form α-imino rhodium carbenes 125 (Scheme 21a). The nucleophilic attack of the carbonyl group of o-QM 126 onto the electrophilic α-imino rhodium carbene 125 then generates the carbonyl ylide 127 (Scheme 21a). A 1,3-rhodium shift to 128 followed by 6π-electrocylization and subsequent deprotonation and isomerization leads to the corresponding benzopyran derivative 124 (Scheme 21a), and the proposed mechanism is well-supported by control experiments. Electronically diverse starting materials reacted well and produced a broad spectrum of benzopyran compounds via denitrogenative transannulation (Scheme 21b). Interestingly, the one-pot synthesis of benzopyran was obtained using phenylacetylene, tosyl azide, and ortho-hydroxy benzhydryl alcohol in the single-step integration of copper and rhodium catalysts via a two-step, one-pot protocol.
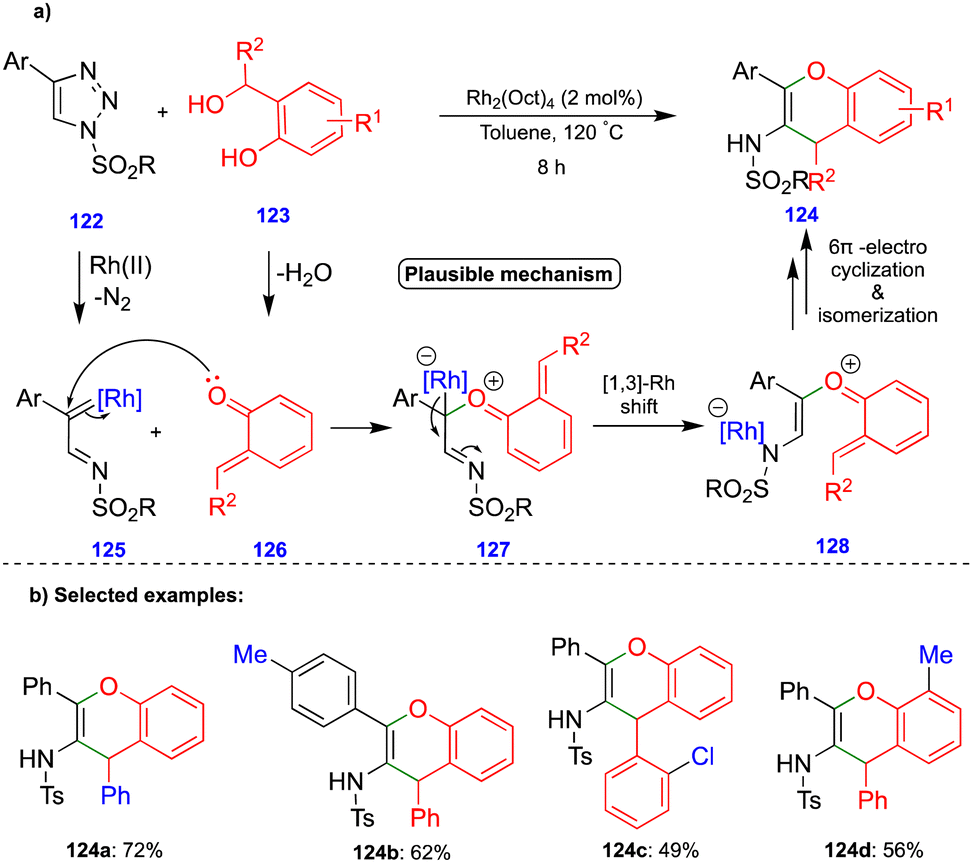 | ||
| Scheme 21 Rhodium-catalyzed transannulation of N-sulfonyl-1,2,3-triazoles with o-QMs. | ||
3.6 1,6-Conjugate additions of diazo compounds with p-QMs
Mohanan's group reported the synthesis of 1,2-diaryl alkenylphosphonates via a two-step protocol using a Seyferth–Gilbert reagent (SGR), such as dimethyl diazomethylphosphonate, and p-QMs (Scheme 22).37 The first step involves generation of the diazomethylphosphonate (DAMP) anion 133 from SGR 130, and subsequent 1,6-conjugate addition an electrophilic p-QM 129 leads to the intermediate 134, the protonation of which delivers the diarylmethylated diazophosphonate 131 (Scheme 22b). Rhodium carbenes are generated from the diarylmethylated diazophosphonate 131 and a rhodium catalyst (Rh2(OAc)4) upon the elimination of nitrogen (Scheme 22b). Then, 1,2-aryl migration occurs, leading to the corresponding product 132via formation of the phenonium ion intermediate 136 (Scheme 22b). 1,2-Aryl group migration is observed only with hydroxy-bearing aryl groups compared with another aryl moieties due to the aryl group's high nucleophilicity.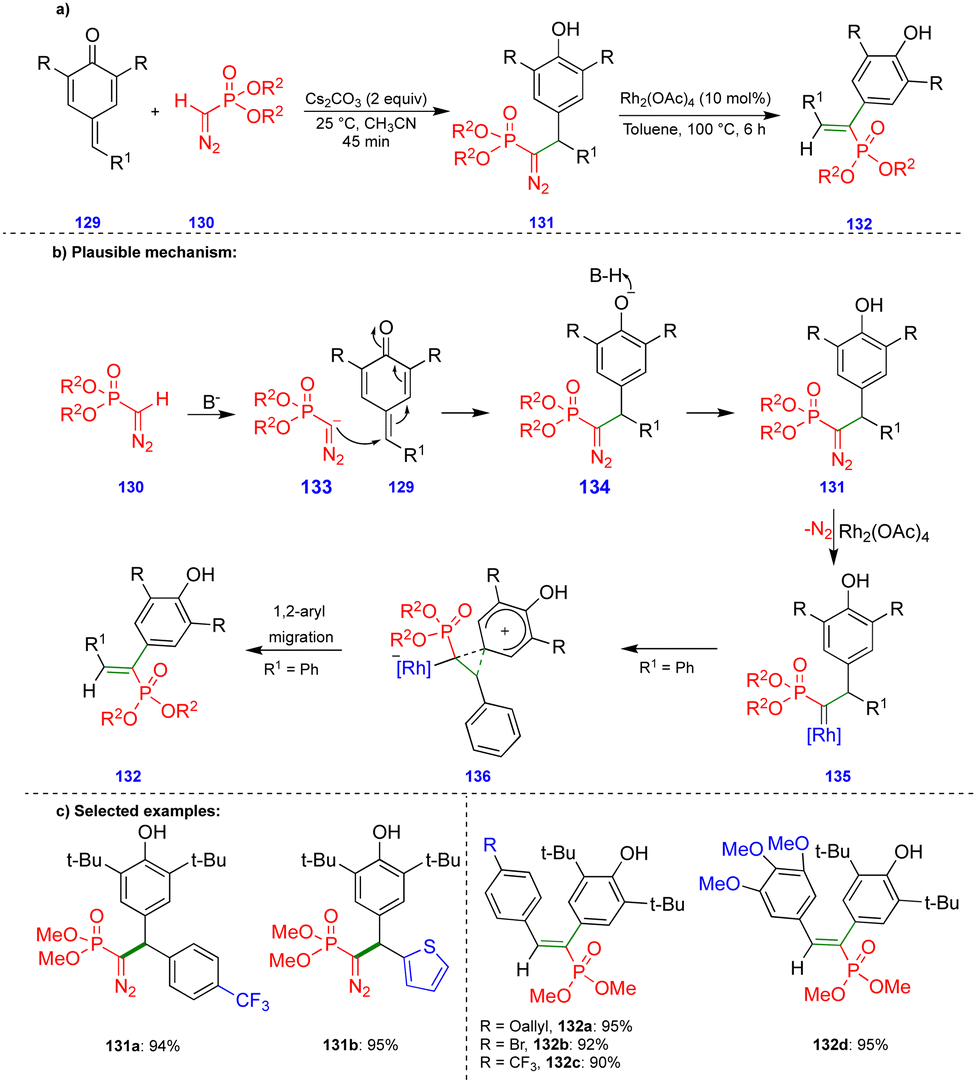 | ||
| Scheme 22 1,6-Conjugate addition reaction of diazomethylphosphonate compounds with p-QMs. | ||
Excellent yields of the diarylmethylated diazophosphonate derivatives 131 were synthesized (Scheme 22c) from SGR and electronically unbiased p-QMs through Cs2CO3-mediated 1,6-conjugate addition. Interestingly, when treated with the rhodium-catalyst, the synthesized diarylmethylated diazophosphonates produced unusual 1,2-aryl migrated products such as the trans-compound of 1,2-diaryl alkenylphosphonate derivative 132 in excellent yield.
Similar reactivity was observed by Namboothiri's group in the reaction of p-QMs with the Bestmann–Ohira reagent38 (BOR) for the synthesis of alkenylphosphonates (Scheme 23).39 Initially, the BOR reagent 137 underwent deacylation in the presence of ethanolic KOH to generate the anionic intermediate 140, and the subsequent 1,6-conjugate addition of 141 followed by protonation delivered the 1,6-addition product of diazophosphonate 142 (Scheme 23b). Alkenylphosphonate derivatives 139 were obtained from diazophosphonates 142 in the presence of Rh2(OAc)4via the formation of a spiro-cyclopropane intermediate. E-Alkenylphosphonate compounds 139 were predominantly synthesized with good to excellent yields (Scheme 23c) via 1,2-aryl migration of the phenonium ion intermediate in the presence of a rhodium-catalyst.
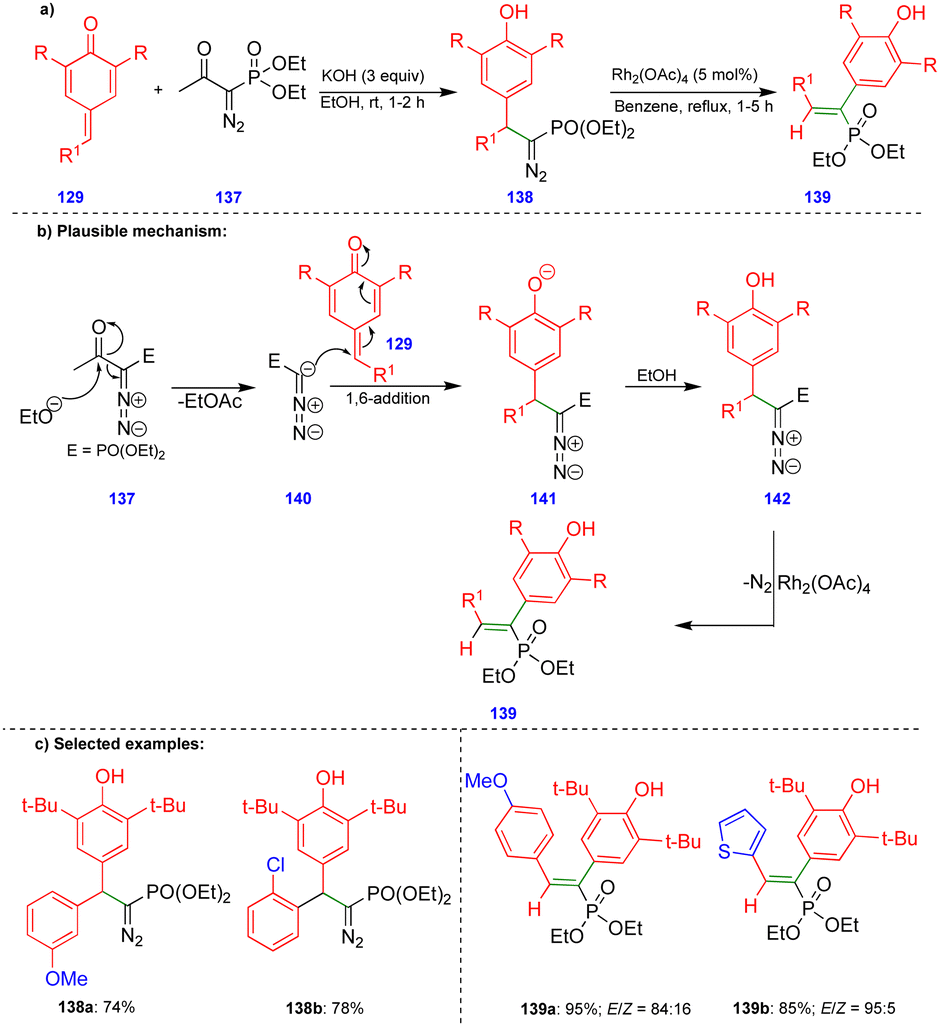 | ||
| Scheme 23 Deacylative conjugate addition reaction of diazo-phosphonate with p-QMs. | ||
Later, the same group reported the single-step synthesis of tri- and tetrasubstituted alkenes from α-diazo-β-ketosulfone and α-diazo-β-carbonyl compounds with p-QMs (Schemes 24 and 25).40 A wide variety of tri- and tetrasubstituted alkenes 144 and 152 were synthesized through the initial 1,6-conjugate addition of α-diazo-β-ketosulfone 143 and α-diazo-β-dicarbonyl 151 compounds to p-QMs. Subsequent protonation and the elimination of nitrogen delivered the corresponding alkenes with good to excellent yields and selectivity in the absence of a rhodium-catalyst (Schemes 24 and 25).
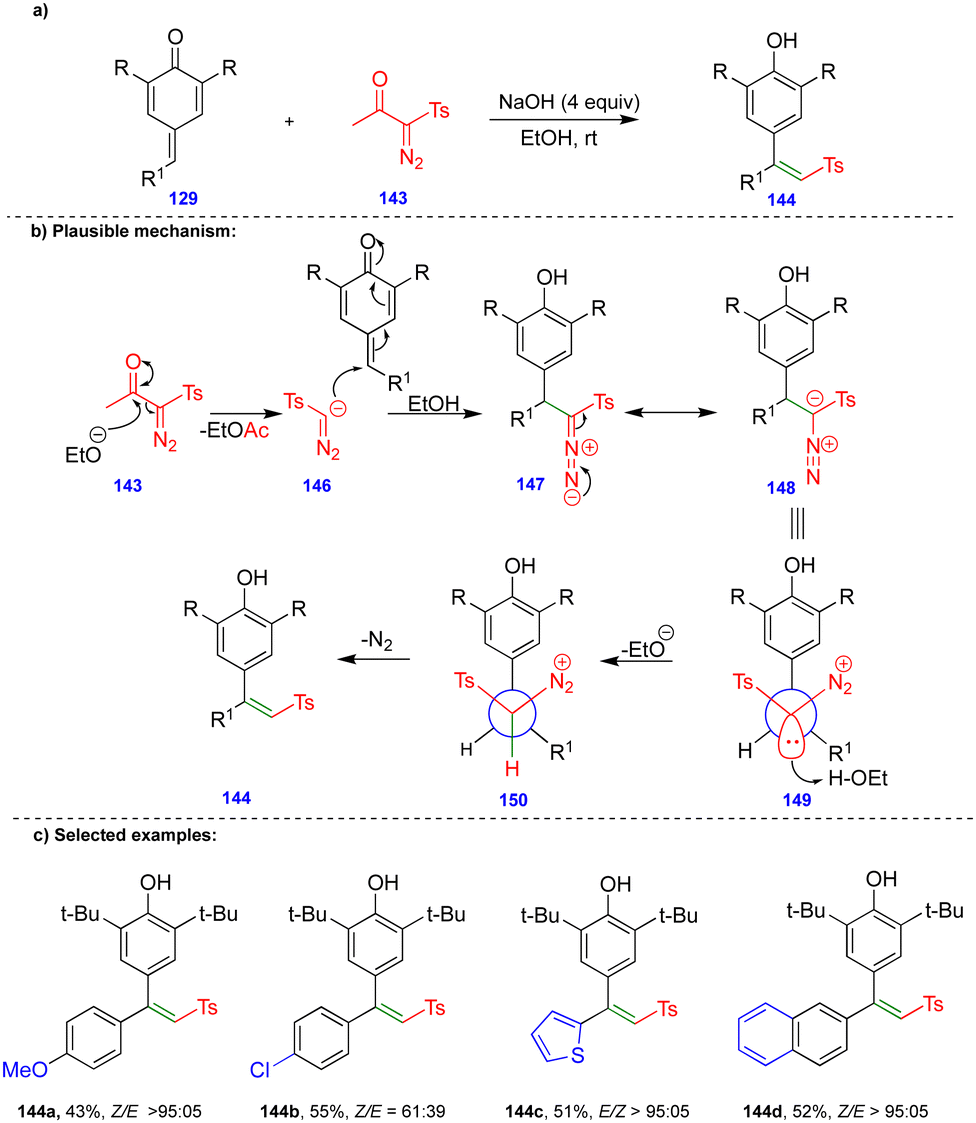 | ||
| Scheme 24 Synthesis of alkenyl phosphonates from p-QMs with Bestmann–Ohira reagent. | ||
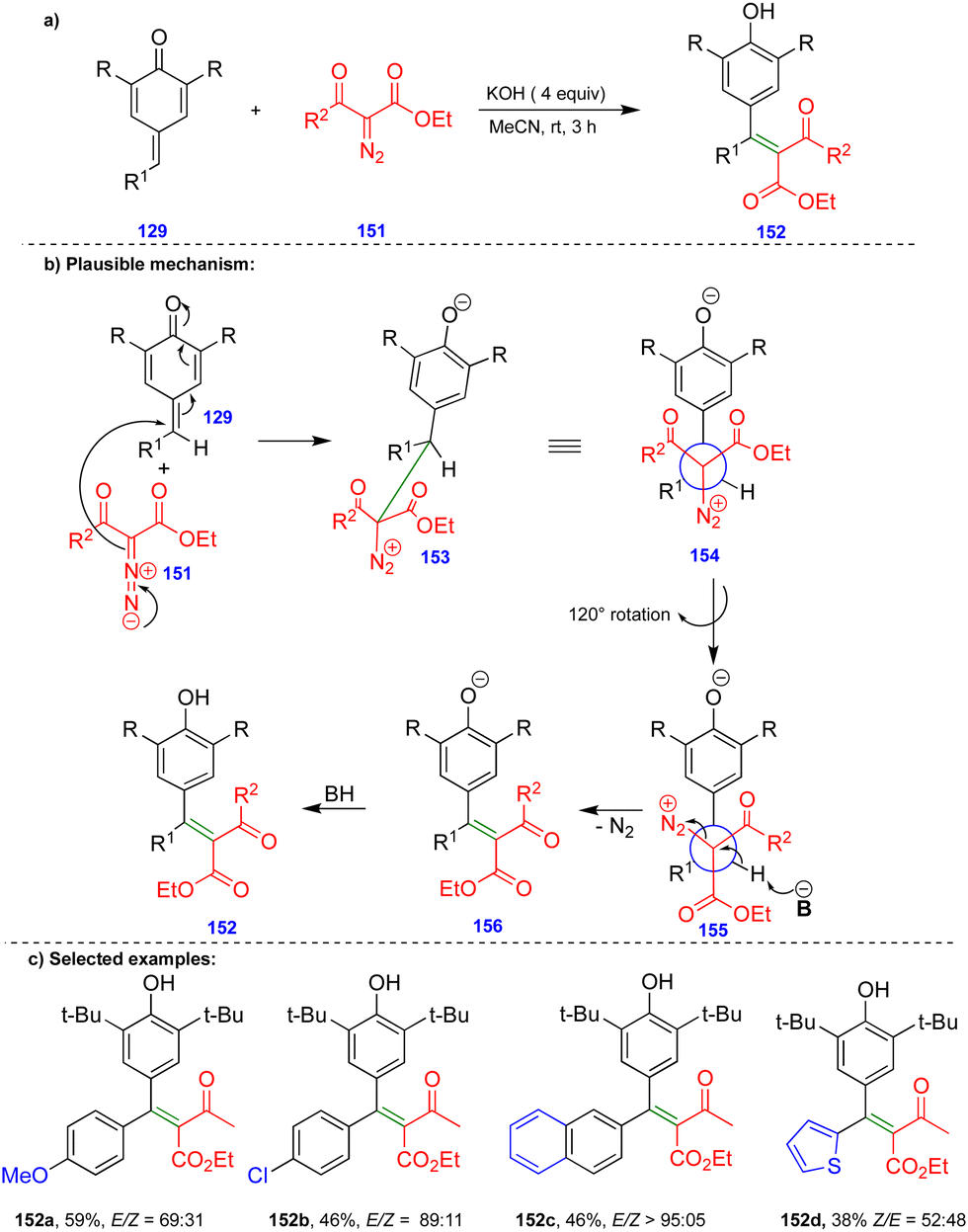 | ||
| Scheme 25 Synthesis of tri- and tetrasubstituted alkenes. | ||
The group of Huang and Peng recently reported the asymmetric 1,6-conjugate addition of an SGR with p-QMs to access chiral diarylmethylated diazomethylphosphonates (Scheme 26).41 Chiral diarylmethylated diazomethylphosphonate derivatives 158 can be obtained from the reaction between SGR 157 and p-QM 129 in the presence of a chiral phase-transfer catalyst (CPTC.1), which is derived from the cinchona alkaloid. Bulky substituents on the SGR 157 are necessary for better yields and enantioselectivities. Various p-QMs 129 reacted very well, giving good yields and enantioselectivities. However, in case of p-QMs substituted with electron-withdrawing groups, reduced yields and enantioselectivities were observed (Scheme 26b). The authors explained the product formation based on the experimental results, and they proposed through the transition state model 159 (Scheme 26b) that the p-QMs are activated via hydrogen bonding of the hydroxy group of CPTC.1, which shows an attraction with the oxygen of the diazomethylphosphonate, perhaps due to their close proximity. The nucleophilic carbon of the α-diazomethylphosphonate attacks the δ-carbon of the p-QM via a Re-face attack, and subsequent protonation leads to the corresponding product. Furthermore, the synthesized chiral diarylmethylated diazomethylphosphonates were utilized to access chiral dihydrocinnoline phosphonate and chiral α-amino phosphonate.
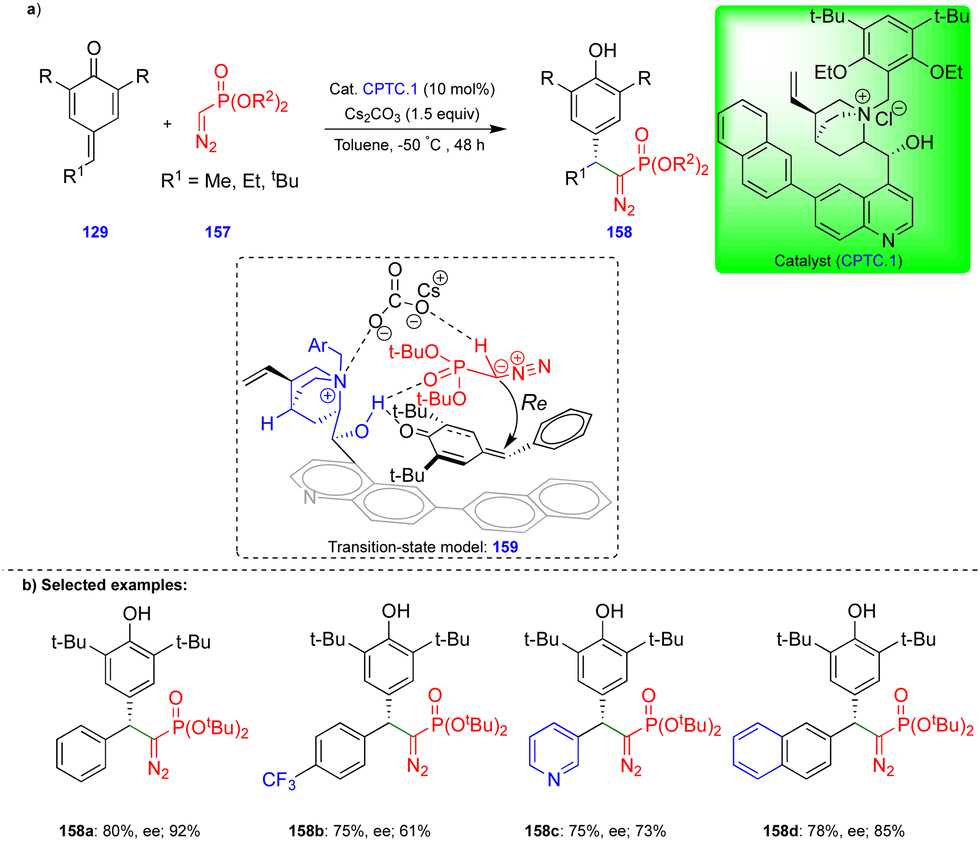 | ||
| Scheme 26 Enantioselective synthesis of diarylmethylated diazomethylphosphonates. | ||
4. Conclusions
In this feature article, we discussed the reactivity of QMs with carbenes generated from α-diazocarbonyl derivatives and their related compounds to access enantioenriched synthetic intermediates and oxygen-containing and fused(spiro)-heterocyclic compounds. In particular, o-QMs that possess an exo-methylene moiety with a reactive carbonyl group are generated either in situ or ex-situ from ortho-hydroxy benzhydryl alcohols. Thus, they undergo a variety of cycloannulation reactions and other transformations, e.g., [4+3], [4+1], [2+1], etc., with carbenes derived from α-diazocarbonyl compounds, N-tosylhydrazones, difluorocarbenes, cyclopropenes, and N-sulfonyl-1,2,3-triazoles in the presence of various rhodium or palladium catalysts in combination with chiral phosphoric acids and chiral Lewis acids. Furthermore, the reactivity of o-QMs was also studied in the presence of Brønsted-acids, Lewis acids, visible light, and additives. Similarly, p-QMs showed widespread reactivity with various diazo precursors for the synthesis of valuable synthetic intermediates and heterocyclic compounds in the presence of chiral phosphoric acids, phase transfer catalysts, and base mediated transformations. Interestingly, aza-o-QMs and indolyl-2-methides also underwent cycloannulation, giving nitrogen-containing heterocyclic compounds.So far, the chemistry of o-QMs with carbenes has mainly been studied for achiral compounds, apart from a few examples. Still, one needs to use an expensive rhodium catalyst and primarily study the intermolecular or intramolecular version for the initial formation of an oxonium ylide or carbonyl ylide followed by cyclization. The chemistry of QMs not been studied extensively with carbenes, and there is much room for expanding this chemistry by utilizing the multi-component approach for synthesizing enantioenriched heterocyclic compounds. We are hopeful that this article will direct future discoveries on the reactivity of carbenes with o-QMs, p-QMs, aza-o-QMs, and indolyl-2-methides without the use of any expensive catalysts or chiral ligands. The reactivity of QMs with carbenes will find applications in total synthesis, natural product synthesis, and the synthesis of biologically relevant molecules.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the DST-Inspire IFA20-CH-348 and IIT Roorkee FIG-100936 for financial support. H. S. and M. J. sincerely thank IIT Roorkee for their MHRD fellowship.Notes and references
- (a) A. A. Larsen, Nature, 1969, 224, 25–27 CrossRef CAS; (b) D. Hamels, P. M. Dansette, E. A. Hillard, S. Top, A. Vessieres, P. Herson, G. Jaouen and D. Mansuy, Angew. Chem., Int. Ed., 2009, 48, 9124–9126 CrossRef CAS; (c) G. B. Messiano, T. da Silva, I. R. Nascimento and L. M. X. Lopes, Phytochemistry, 2009, 70, 590–596 CrossRef CAS; (d) R. Dehn, Y. Katsuyama, A. Weber, K. Gerth, R. Jansen, H. Steinmetz, G. Hçfle, R. Müller and A. Kirschning, Angew. Chem., Int. Ed., 2011, 50, 3882–3887 CrossRef CAS; (e) C. Sridar, J. D’Agostino and P. F. Hollenberg, Drug Circ. Drug Metab. Dispos., 2012, 40, 2280–2288 CrossRef CAS PubMed; (f) C. G. S. Lima, F. P. Pauli, D. C. S. Costa, A. S. de Souza, L. S. M. Forezi, V. F. Ferreira and F. de Carvalho da Silva, Eur. J. Org. Chem., 2020, 2650–2692 CrossRef CAS.
- S. E. Rokita, Quinone Methides, Wiley, New York, 2009 Search PubMed.
- (a) B. Yang and S. Gao, Chem. Soc. Rev., 2018, 47, 7926–7953 RSC; (b) S. Kulkarni, Asian J. Res. Chem., 2018, 11, 72–74 CrossRef; (c) Y. Lu, Y. Liu, J. Zhou, D. Li and W. Gao, Med. Res. Rev., 2021, 41, 1022–1060 CrossRef CAS PubMed; (d) T. N. Purdy, B. S. Moore and A. L. Lukowski, J. Nat. Prod., 2022, 85, 688–701 CrossRef CAS PubMed.
- (a) K. Fries and K. I. Liebigs, Ann. Chem., 1907, 353, 335–356 CrossRef CAS; (b) W. J. Bai, J. G. David, Z. G. Feng, M. G. Weaver, K. L. Wu and T. R. R. Pettus, Acc. Chem. Res., 2014, 47, 3655–3664 CrossRef CAS PubMed.
- (a) R. Rodriguez, R. M. Adlington, J. E. Moses, A. Cowley and J. E. Baldwin, Org. Lett., 2004, 6, 3617–3619 CrossRef CAS PubMed; (b) K. Wojciechowski and K. Dolatowska, Tetrahedron, 2005, 61, 8419–8422 CrossRef CAS; (c) A. F. Barrero, J. F. Quílez del Moral, M. Mar Herrador, P. Arteaga, M. Cortés, J. Benites and A. Rosellón, Tetrahedron, 2006, 62, 6012–6017 CrossRef CAS; (d) H. Sugimoto, S. Nakamura and T. Ohwada, J. Org. Chem., 2007, 72, 10088–10095 CrossRef CAS PubMed; (e) C. D. Bray, Org. Biomol. Chem., 2008, 6, 2815–2819 RSC; (f) J. T. Spence and J. H. George, Org. Lett., 2013, 15, 3891–3893 CrossRef CAS.
- (a) A. K. Shaikh, A. J. A. Cobb and G. Varvounis, Org. Lett., 2012, 14, 584–587 CrossRef CAS; (b) A. E. Mattson and K. A. Scheidt, J. Am. Chem. Soc., 2007, 129, 4508–4509 CrossRef CAS PubMed; (c) J. Izquierdo, A. Orue and K. A. Scheidt, J. Am. Chem. Soc., 2013, 135, 10634–10637 CrossRef CAS; (d) Y. Zhu, L. Zhang and S. Luo, J. Am. Chem. Soc., 2016, 138, 3978–3981 CrossRef CAS PubMed; (e) J. Xu, S. Yuan, J. Peng, M. Miao, Z. Chen and H. Ren, Org. Biomol. Chem., 2017, 15, 7513–7517 RSC; (f) G. Q. Xu, Z. T. Feng, J. T. Xu, Z. Y. Wang, Y. Qin and P. F. Xu, Chem. – Eur. J., 2018, 24, 13778–13782 CrossRef CAS; (g) T. B. Hua, F. Chao, L. Wang, C. Y. Yan, C. Xiao, Q. Q. Yang and W. J. Xiao, Adv. Synth. Catal., 2020, 362, 2615–2619 CrossRef CAS.
- (a) Y. Chiang, A. J. Kresge and Y. Zhu, J. Am. Chem. Soc., 2001, 123, 8089–8094 CrossRef CAS PubMed; (b) M. Flegel, M. Lukeman, L. Huck and P. Wan, J. Am. Chem. Soc., 2004, 126, 7890–7897 CrossRef CAS; (c) Y. Chen and M. G. Steinmetz, J. Org. Chem., 2006, 71, 6053–6060 CrossRef CAS; (d) S. Arumugam and V. V. Popik, J. Org. Chem., 2010, 75, 7338–7346 CrossRef CAS; (e) F. Zhou, Y. Cheng, X. P. Liu, J. R. Chen and W. J. Xiao, Chem. Commun., 2019, 55, 3117–3120 RSC.
- (a) D. Liao, H. Li and X. Lei, Org. Lett., 2012, 14, 18–21 CrossRef CAS PubMed; (b) B. Wu, X. Gao, Z. Yan, M. W. Chen and Y. G. Zhou, Org. Lett., 2015, 17, 6134–6137 CrossRef CAS; (c) K. Gebauer, F. Reuss, M. Spanka and C. Schneider, Org. Lett., 2017, 19, 4588–4591 CrossRef CAS PubMed.
- (a) P. Batsomboon, W. Phakhodee, S. Ruchirawat and P. Ploypradith, J. Org. Chem., 2009, 74, 4009–4012 CrossRef CAS PubMed; (b) A. L. Lawrence, R. M. Adlington, J. E. Baldwin, V. Lee, J. A. Kershaw and A. L. Thompson, Org. Lett., 2010, 12, 1676–1679 CrossRef CAS; (c) J. H. George, J. E. Baldwin and R. M. Adlington, Org. Lett., 2010, 12, 2394–2397 CrossRef CAS; (d) D. Wilcke, E. Herdtweck and T. Bach, Synlett, 2011, 1235–1238 CAS; (e) O. El-Sepelgy, S. Haseloff, S. K. Alamsetti and C. Schneider, Angew. Chem., Int. Ed., 2014, 53, 7923–7927 CrossRef CAS PubMed; (f) C.-C. Hsiao, H.-H. Liao and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 13258–13263 CrossRef CAS.
- (a) J. Wang, X. Pan, J. Liu, L. Zhao, Y. Zhi, K. Zhao and L. Hu, Org. Lett., 2018, 20, 5995–5998 CrossRef CAS PubMed; (b) Y. J. Fan, L. Zhou and S. Li, Org. Chem. Front., 2018, 5, 1820–1824 RSC.
- B. Koutek, L. Pavlickova and M. Soucek, Synth. Commun., 1976, 6, 305–308 CrossRef CAS.
- D. Richter, N. Hampel, T. Singer, A. R. Ofial and H. Mayr, Eur. J. Org. Chem., 2009, 3203–3211 CrossRef CAS.
- (a) R. W. Van De Water and T. R. R. Pettus, Tetrahedron, 2002, 58, 5367–5405 CrossRef CAS; (b) Z. Wang and J. Sun, Synthesis, 2015, 3629–3644 Search PubMed; (c) L. Caruana, M. Fochi and L. Bernardi, Molecules, 2015, 20, 11733–11764 CrossRef CAS PubMed; (d) A. A. Jaworski and K. A. Scheid, J. Org. Chem., 2016, 81, 10145–10153 CrossRef CAS PubMed; (e) C. D.-T. Nielsen, H. Abas and A. C. Spivey,, Synthesis, 2018, 4008–4018 CAS; (f) Y. H. Ma, X. Y. He, Q. Q. Yang, A. Boucherif and J. Xuan, Asian J. Org. Chem., 2021, 10, 1233–1250 CrossRef CAS; (g) D. Caroline and S. Christoph, Synthesis, 2022, 3125–3141 Search PubMed; (h) K. Ali, P. Mishra, A. Kumar, D. N. Reddy, S. Chowdhury and G. Panda, Chem. Commun., 2022, 58, 6160–6175 RSC; (i) M. T. M. Martins, F. R. F. Dias, R. S. M. de Moreas, M. F. V. da Silva, K. R. Lucio, K. D’Oliveira Goes, P. A. do Nascimento, A. S. S. da Silva, V. F. Ferreira and A. C. Cunha, Chem. Rec., 2022, 22, e202100251, DOI:10.1002/tcr.202100251; (j) H. H. Liao, S. Minoza, S. C. Lee and M. Rueping, Chem. – Eur. J., 2022, 28 DOI:10.1002/chem.202201112.
- M. M. Toteva and J. P. Richard, J. Am. Chem. Soc., 2000, 122, 11073–11083 CrossRef CAS.
- J. Y. Wang, W. J. Hao, S. J. Tu and B. Jiang, Org. Chem. Front., 2020, 7, 1743–1778 RSC.
- (a) A. Parra and M. Tortosa, ChemCatChem, 2015, 7, 1524–1526 CrossRef CAS; (b) W. Li, X. Xu, P. Zhang and P. Li, Chem. – Asian J., 2018, 13, 2350–2359 CrossRef CAS PubMed.
- S. K. Alamsetti, M. Spanka and C. Schneider, Angew. Chem., Int. Ed., 2016, 55, 2392–2396 CrossRef CAS PubMed.
- Selected examples (a) A. Padwa, Acc. Chem. Res., 1991, 24, 22–28 CrossRef CAS; (b) A. Padwa and S. F. Hornbuckle, Chem. Rev., 1991, 91, 263–309 CrossRef CAS; (c) W. Hu, X. Xu, J. Zhou, W. J. Liu, H. Huang, J. Hu, L. Yang and L. Z. Gong, J. Am. Chem. Soc., 2008, 130, 7782–7783 CrossRef CAS PubMed; (d) J. Jiang, H. D. Xu, J. B. Xi, B. Y. Ren, F. P. Lv, X. Guo, L. Q. Jiang, Z. Y. Zhang and W. H. Hu, J. Am. Chem. Soc., 2011, 133, 8428–8431 CrossRef CAS PubMed; (e) H. Qiu, M. Li, L. Q. Jiang, F. P. Lv, L. Zan, C. W. Zhai, M. P. Doyle and W. H. Hu, Nat. Chem., 2012, 4, 733–738 CrossRef CAS PubMed; (f) Y. Qian, C. Jing, S. Liu and W. Hu, Chem. Commun., 2013, 49, 2700–2702 RSC; (g) L. Ren, X. L. Lian and L. Z. Gong, Chem. – Eur. J., 2013, 19, 3315–3318 CrossRef CAS PubMed.
- K. X. Rodriguez, J. D. Vail and B. L. Ashfeld, Org. Lett., 2016, 18, 4514–4517 CrossRef CAS PubMed.
- A. Suneja and C. Schneider, Org. Lett., 2018, 20, 7576–7580 CrossRef CAS PubMed.
- G. Xu, S. Tang, Y. Shao and J. Sun, Chem. Commun., 2019, 55, 9096–9099 RSC.
- Y. H. Ma, F. X. Meng, R. N. Wang, Y. X. Fan, Q. Q. Su and J. Y. Du, Synthesis, 2022, 499–505 CrossRef CAS.
- Z. D. Tucker, H. M. Hill, A. L. Smith and B. L. Ashfeld, Org. Lett., 2020, 22, 6605–6609 CrossRef CAS PubMed.
- S. Zhou, B. Cai, C. Hu, X. Cheng, L. Li and J. Xuan, Chin. Chem. Lett., 2021, 32, 2577–2581 CrossRef CAS.
- Y. C. Wu, B. D. Cui, Y. Long, W. Y. Han, N. W. Wan, W. C. Yuan and Y. Z. Chen, Adv. Synth. Catal., 2021, 363, 1702–1713 CrossRef CAS.
- Y. Jia, Y. Yuan, J. Huang, Z. X. Jiang and Z. Yang, Org. Lett., 2021, 23, 2670–2675 CrossRef PubMed.
- R. P. Pandit, S. T. Kim and D. H. Ryu, Angew. Chem., Int. Ed., 2019, 58, 13427–13432 CrossRef CAS PubMed.
- H. Lam, Z. Qureshi, M. Wegmann and M. Lautens, Angew. Chem., Int. Ed., 2018, 57, 16185–16189 CrossRef CAS PubMed.
- A. Suneja, H. J. Loui and C. Schneider, Angew. Chem., Int. Ed., 2020, 59, 5536–5540 CrossRef CAS PubMed.
- B. Huang, Y. Shen, Z. Mao, Y. Liu and S. Cui, Org. Lett., 2016, 18, 4888–4891 CrossRef CAS PubMed.
- S. T. Kim, R. P. Pandit, J. Yun and D. H. Ryu, Org. Lett., 2021, 23, 213–217 CrossRef CAS PubMed.
- R. J. Baker, J. Ching, T. R. Hou, I. Franzoni and M. Lautens, Angew. Chem., Int. Ed., 2022, 61, e202116171, DOI:10.1002/anie.202116171.
- All the references are there in R. Vicente, Chem. Rev., 2021, 121, 162–226 CrossRef CAS PubMed.
- H. J. Loui, A. Suneja and C. Schneider, Org. Lett., 2021, 23, 2578–2583 CrossRef PubMed.
- D. Yadagiri, M. Chaitanya, A. C. S. Reddy and P. Anbarasan, Org. Lett., 2018, 20, 3762–3765 CrossRef CAS PubMed.
- All the references therein (a) B. Chattopadhyay and V. Gevorgyan, Angew. Chem., Int. Ed., 2012, 51, 862–872 CrossRef CAS PubMed; (b) H. M. L. Davies and J. S. Alford, Chem. Soc. Rev., 2014, 43, 5151–5162 RSC; (c) Y. Jiang, R. Sun, X. Y. Tang and M. Shi, Chem. – Eur. J., 2016, 22, 17910–17924 CrossRef CAS PubMed; (d) D. Yadagiri, M. Rivas and V. Gevorgyan, J. Org. Chem., 2020, 85, 11030–11046 CrossRef CAS PubMed; (e) D. Yadagiri and P. Anbarasan, Chem. Rec., 2021, 21, 3872–3883 CrossRef CAS PubMed; (f) M. Akter, K. Rupa and P. Anbarasan, Chem. Rev., 2022, 122, 13108–13205 CrossRef CAS PubMed.
- A. K. Gupta, S. Ahamad, N. K. Vaishanv, R. Kant and K. Mohanan, Org. Biomol. Chem., 2018, 16, 4623–4627 RSC.
- M. F. Jamali, N. K. Vaishnav and K. Mohanan, Chem. Rec., 2020, 20, 1394–1408 CrossRef CAS PubMed.
- S. Pati and I. N. N. Namboothiri, Tetrahedron, 2019, 75, 2246–2253 CrossRef CAS.
- S. Pati, S. Rayi and I. N. N. Namboothiri, ACS Org. Inorg. Au, 2021, 1, 51–59 CrossRef CAS.
- Y. Chen, R. Yu, M. Wang, Y. Huang and Y. Peng, Adv. Synth. Catal., 2021, 363, 4856–4861 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |