
Mixed oxides as multi-functional reaction media for chemical looping catalysis
Junchen
Liu
and
Fanxing
Li
 *
*
Chemical and Bimolecular Engineering, North Carolina State University, Raleigh, NC, USA. E-mail: fli5@ncsu.edu
First published on 1st December 2022
Abstract
Over the past two decades, chemical looping combustion (CLC) has been extensively investigated as a promising means to produce electric power while generating a concentrated carbon dioxide stream for sequestration. We note that the chemical looping strategy can be extended well outside of combustion-based carbon capture. In fact, application of the chemical looping strategy in areas beyond combustion can result in somewhat unexpected energy and carbon dioxide savings without producing a concentrated CO2 stream at all. Furthermore, it allows the looping-based technologies to tap into applications such as chemical production – a $4 trillion per year industrial sector with high energy and carbon intensities. The key resides in the design of effective oxygen carriers, also known as redox catalysts in the context of selective chemical conversion through chemical looping catalysis (CLCa). This contribution focuses on the design and applications of mixed oxides as multi-function reaction media in CLCa. Since typical mixed oxide oxygen carriers tend to be nonselective for hydrocarbon conversion, the first part of this article presents generalized design principles for surface modification of mixed oxides to improve their selectivity and catalytic activity. Applications of these redox catalysts in chemical looping – oxidative dehydrogenation (CL-ODH) of a variety of light alkanes and alkyl-benzenes are presented. This is followed with a discussion of computation assisted mixed oxide design based upon thermodynamic criteria. Finally, a few new directions for the chemical looping technologies are introduced.
1. Introduction
The chemical industry contributes to nearly 1 gigaton of direct CO2 emission each year.1 In addition, nearly 50% of the oil and natural gas input in the chemical sector is consumed as the feedstock, which can lead to secondary emissions in the downstream industrial and/or consumer sectors.1 Despite the projected strong growth, the chemical industry is expected to curb its emission within this decade according to the Net Zero by 2050 roadmap proposed by the International Energy Agency (IEA).2 In fact, several leading chemical and petrochemical companies have pledged more aggressive emission reduction targets over the recent years. Not surprisingly, CO2 emissions from state-of-the-art chemical production processes tie directly to the energy intensity of the manufacturing process as well as the annual production capacity. Fig. 1a summarizes the production capacity and emission levels from a few key commodity chemicals. Although the overall CO2 emissions can be reduced, to some extent, by switching to low carbon intensity fuels and increasing the renewable contribution to the electrical grid, such incremental improvements to the existing technologies are unlikely to attain the net zero target by 2050. Rather, transformative chemical production technologies that are fundamentally different from state-of-the-art approaches are required.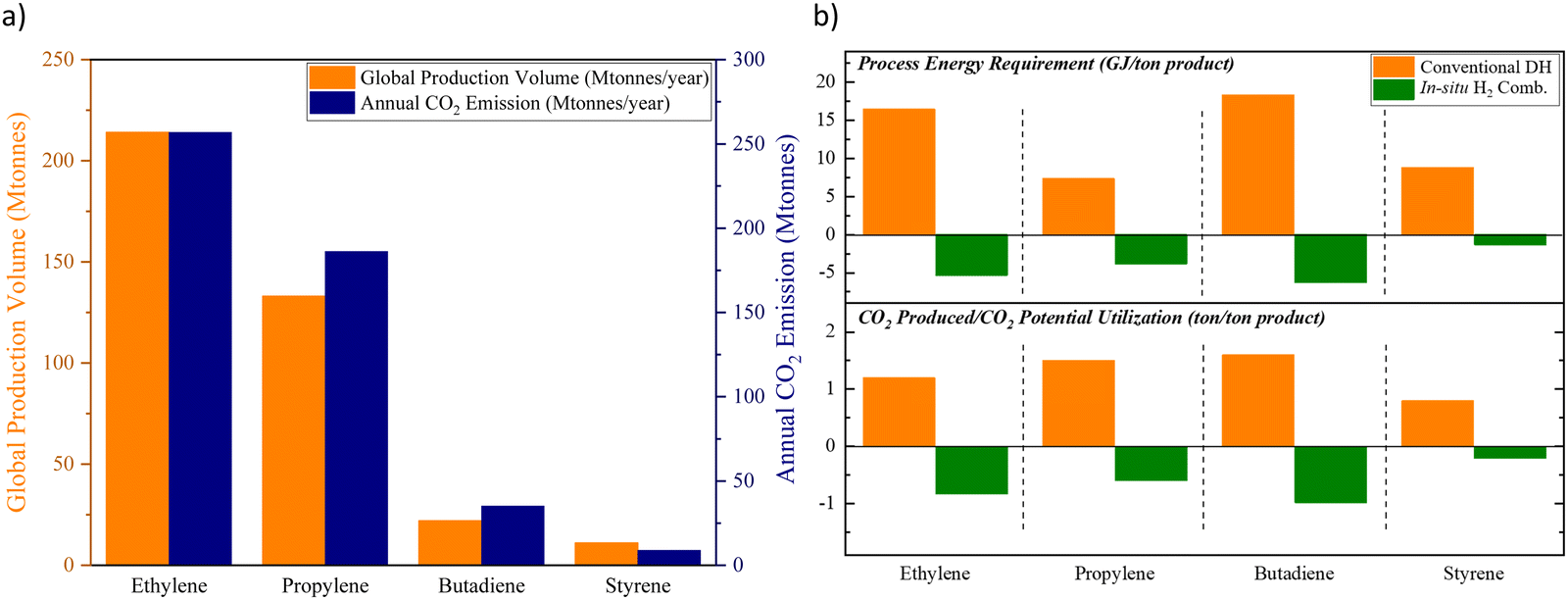 | ||
| Fig. 1 (a) The global annual production volumes and the associated CO2 emissions of ethylene, propylene, butadiene, and styrene;3–10 (b) top: comparison between the energy requirement of conventional dehydrogenation (DH) processes to that of the oxidative processes with in situ H2 combustion (assuming an ideal process with 100% hydrogen byproduct combustion); bottom: comparison between the CO2 emission from the existing processes to the CO2 utilization potential from the energy resulting from in situ H2 combustion (assuming an ideal scenario of utilizing the energy from H2 combustion for CO2 splitting to CO).3–10 | ||
At present, the unsaturated hydrocarbons (i.e. Ethylene,3 Propylene,4–8 Butadiene3,7 and Styrene9,10) listed in Fig. 1 are primarily produced by dehydrogenation or cracking processes, either thermally or in the presence of a heterogeneous catalyst. Common limitations for such processes include large heat requirement and equilibrium-limited single-pass yield due to the highly endothermic and endergonic nature of the reactions, shown in eqn (1). The limited product yield and selectivity in turn drive up the energy consumption and CO2 emissions associated with product separation.
| CxHy → CxHy−2n + nH2(n ≥ 1) ΔH > 0, ΔG > 0 (unless at very high temperatures) | (1) |
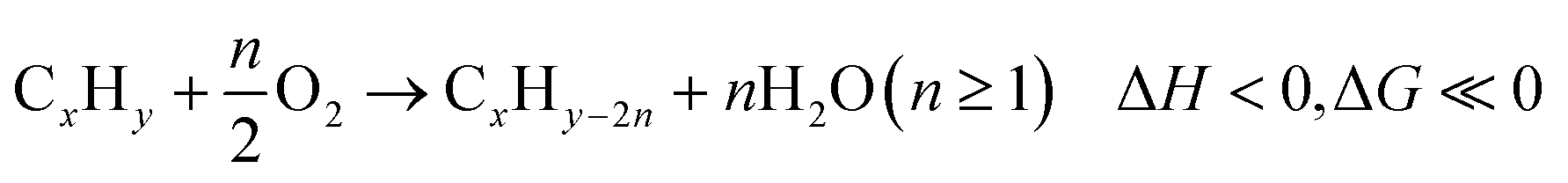 | (2) |
Resulting from three decades of research, a wealth of knowledge has been created for the chemical looping technology in terms of the oxygen carrier selection and performance, reactor design and operation, and the overall technological feasibility, primarily in the context of CO2 capture from fossil fuel combustion via chemical looping combustion (CLC).13–51 Shown in Fig. 2a, CLC technologies generally involves two cyclic steps: (1) the use of metal oxide(s) oxygen carrier as the oxidant to fully combust fossil fuels such as coal or methane; (2) the regeneration of the reduced metal oxide(s) in step 1 with air. We note that the core ideas of the CLC strategy reside in in situ oxygen separation from the air and indirect oxidation of carbonaceous fuels. Therefore, the use of chemical looping for efficient air separation (CLAS) represents a natural extension of CLC, as explored by many chemical looping researchers.52–94 Furthermore, marrying the chemical looping strategy with oxidative catalysis offers a unique opportunity to intensify the production of a few important commodity chemicals with substantially decreased energy consumption and CO2 emissions.95–136 Given that separation processes consume ∼60% of the total energy usage in chemical and petroleum industries and heterogeneous catalysts are responsible for >80% of all chemical products worldwide, chemical looping catalysis (CLCa) in this article, has the potential to facilitate process intensification throughout the chemical manufacturing sector by combining catalytic reactions with separations.120,137–142 The abovementioned chemical looping process types are summarized in Table 1. In the context of CLCa, the oxygen carriers are denoted as redox catalysts to capture their dual functionality.
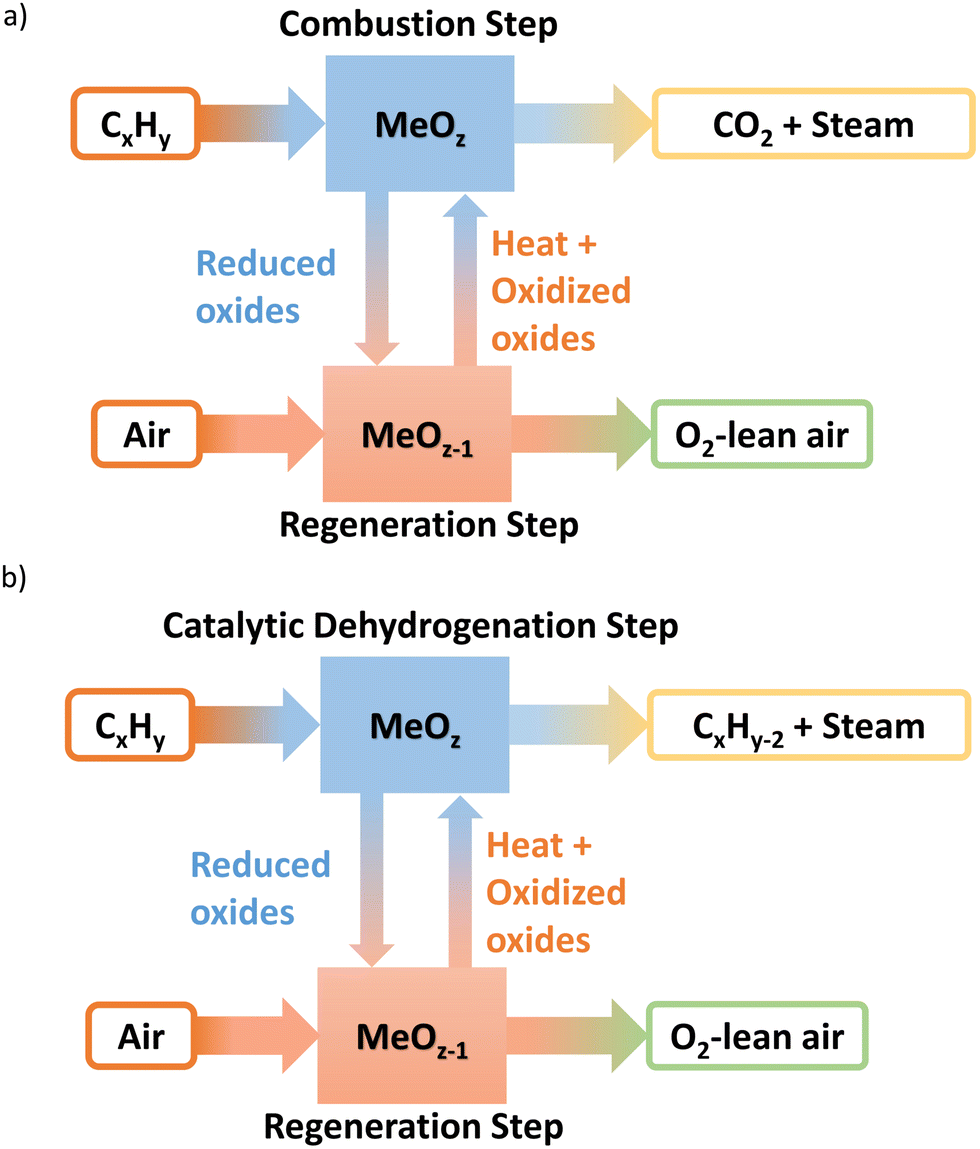 | ||
| Fig. 2 Schematic illustration of: (a) a generic CLC process: a metal oxide redox catalyst combusts fuel into CO2 in the oxidation step; the reduced metal oxide is reoxidized in the regeneration step with air; (b) a metal oxide redox catalyst oxidatively converts a light alkane into an olefin product in the oxidation step; the reduced metal oxide is reoxidized in the regeneration step with air. | ||
| Process type | Reactions |
|---|---|
| CLC | Fuels + MeOx → CO2 + H2O + MeOx−δ |
| Air + MeOx−δ → MeOx + N2 | |
| CLAS | MeOx → O2 + MeOx−δ |
| Air + MeOx−δ → MeOx + N2 | |
| CLCa | CxHy + MeOx → CxHy−2n + 2nH2O + MeOx−δ |
| Air + MeOx−δ → MeOx + N2 | |
Taking ethane ODH as an example, the redox catalyst particles first convert ethane into ethylene and water using its active lattice oxygen. After completing this ODH step, the oxygen-depleted redox catalyst is exposed to air (and/or steam), to replenish the lattice oxygen.143 Compared to conventional co-feed type ODH reactions, chemical looping process could partition the gaseous oxidant and the hydrocarbons, decreasing the selectivities towards unwanted COx and oxygenates. This CLCa process can be carried out either in circulating fluidized beds similar to a CFB combustor or parallel packed beds operated similar to the Houdry process.3,137,144 The advantages of the redox catalysts and CLCa compared to conventional, heterogeneous ODH catalysts include: (i) integration of catalytic reaction with air separation (a simpler and safer process); (ii) potential to achieve high selectivity (absence of gaseous oxygen inhibits side reactions); (iii) potential to tailor heat of reactions, by varying the metal oxide's redox properties, for improved heat management in the redox steps.137,145 Our recent studies indicated that up to 84% energy savings and emission reductions can be realized by CLCa.137Fig. 2 illustrates a generalized schematic for CLCa in the context of oxidative dehydrogenation of hydrocarbons.
As will be discussed in the following sections, the potential of chemical looping extends well beyond oxidation catalysis.
2. Redox catalyst design in CLCa for olefin production
Complete oxidation of carbonaceous fuels (to CO2 and H2O) is expected for CLC because this will maximize the heat release, increase the power generation efficiency, and produce a near “sequestration ready” CO2 stream.46 In contrast, CLCa needs to avoid full oxidation since the target products would be value-add fuels and chemicals such as H2, CO, or unsaturated organic molecules. With such expectations, typical CLC oxides alone would not be effective for CLCa because they tend to result in poor product selectivity. This is not at all surprising given that typical CLC oxygen carriers, particularly those composed of mixed oxides, are often specifically designed to have high equilibrium oxygen chemical potential (μO2), facile oxygen evolution kinetics, and (in many cases) highly basic surfaces.121,146–149 Therefore, the most critical aspect for CLCa resides in selectivity enhancement of redox-active metal oxides. Given that the (non-selective) reactions occur at the gas–oxide interfaces, surface modification of the oxides is almost always necessary in order to enhance the product selectivity.Addition of dopants and/or modulating the metal–oxygen bonding strength on the surface have been shown be effective to enhance the selectivity towards light olefins for a number of redox oxides and reactions.106,150 This section focuses on presenting a more “generalized strategy” for surface modification of mixed oxide in the context of oxidative dehydrogenation of hydrocarbons: instead of doping or impregnation of small amount of heteroatoms on the surface, we reasoned that complete or near-complete coverage of a non-selective oxide surface with a catalytically active layer would effectively suppress side reactions. This approach was first validated in our group for methane partial oxidation reactions,122,151,152 and was recently extended to a series of ODH reactions using various core and shell materials, as illustrated in Fig. 3.119–121,138,143,144,147,153–161 We categorize this generalized ODH strategy into three types: Type 1, shown in Fig. 3a, involves covering the non-selective mixed oxides with an “inert” molten salt layer to simply block the non-selective sites on the surface, thereby inhibiting nonselective oxidation. In this case, the redox catalyst primarily function as an oxygen carrying agent for selective hydrogen combustion (SHC); Type 2, as illustrated in Fig. 3b, utilizes an “active” molten salt as the surface layer. With the assistance of the oxygen species supplied from the mixed oxide, this active surface layer can accommodate and/or generate active oxidants, e.g. electrophilic oxygen species or halogen atoms, to initiate C–H bond activation at the gas-molten salt interface. The reactions likely proceed through a surface-initiated gas phase radical reaction pathway, with concurrent hydrogen oxidation; Type 3, shown in Fig. 3c, involves covering the oxide substrate with a solid catalytic shell to catalyze the dehydrogenation reactions, and the H2 byproduct is sequentially combusted by the oxygen supplied by the oxide core. The following sections further elaborate on these core–shell redox catalyst design strategies.
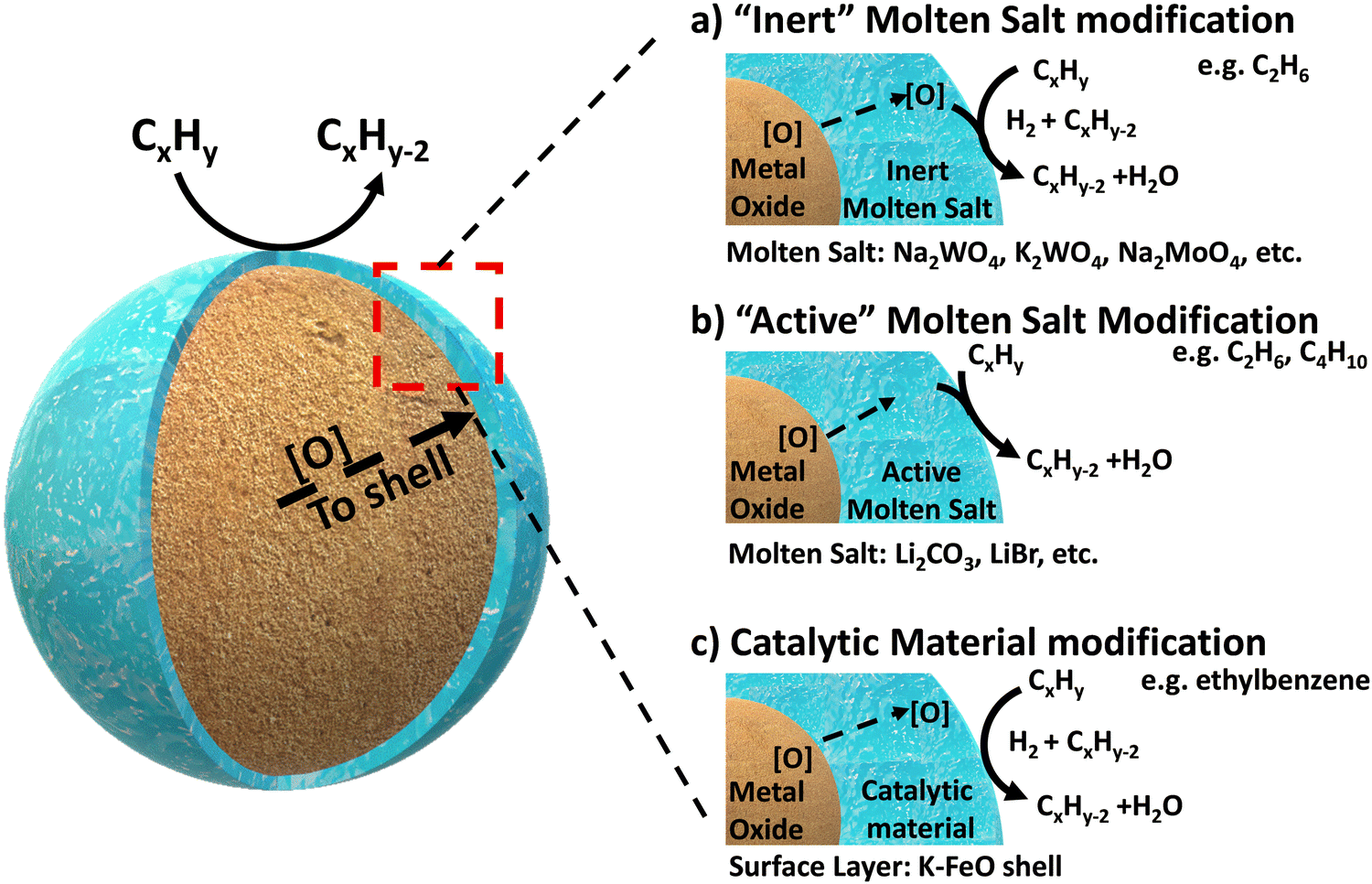 | ||
| Fig. 3 General core–shell design frameworks for mixed oxide@molten salt catalysts and mixed oxide@catalytic shell catalysts: (a) Type 1: “Inert” molten salt promoted mixed oxides; (b) Type 2: “Active” molten salt promoted mixed oxide; (c) Type 3: catalytic material promoted mixed oxides. [O] stands for the oxygen species donated from the oxide surface, which is responsible for the oxidation reaction. Details of the various oxygen species and their evolution are discussed in Section 2.1. | ||
2.1. Mixed oxide @ molten salt redox catalyst for light alkane conversion
Among these redox catalysts, Mg6MnO8@Na2WO4 was the most thoroughly studied.121,147 As shown in the Fig. 4a, co-impregnating Na and W at any ratios on Mg6MnO8 would lead to significant increase in olefin selectivity and yield.147 Of the various Na![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :W ratio investigated, the Mg6MnO8@Na2WO4 (Na:W = 2:1) redox catalyst exhibited one of the highest increase in C2H4 yield (∼45% increase, on an absolute scale) compared to the unpromoted Mg6MnO8 at 850 °C and 4500 h−1. The yield increase was primarily resulted from the substantially decreased COx selectivity (>80%). Mg6MnO8@Na2WO4 also exhibited well defined crystalline phases at room temperature (Mg6MnO8 and Na2WO4), with only a minor Na4Mg(WO4)3 phase (Fig. 4b). The core–shell structure of the catalyst was verified by low-energy ion scattering spectroscopy (LEIS) (Fig. 4c) and X-ray photoelectron spectroscopy (XPS) (Fig. 4d).121 Even after 10 cycles of sputtering, W and Na/Mg are still far more prevalent than Mn according to LEIS. It is noted that LEIS cannot accurately differentiate Na and Mg due to their similar atomic weights. XPS, which provides near surface elemental compositions (Fig. 4c), also confirmed the suppression of Mn by Na2WO4. In fact, impregnation of Na2WO4 onto many Mn-containing oxides has shown to create an oxide@Na2WO4 core shell structure. For instance, transmission electron microscope with energy dispersive x-ray spectroscopy (TEM-EDS) images published from our group by Hao et al. indicated a core shell structure of CaMn0.75Fe0.25O3(CMFO)@Na2WO4 (Fig. 4e)157 whereas LEIS and/or XPS data indicated surface enrichment of Na2WO4 on mixed (Fe/Mn)Ox,153 (Mn/Si)Ox,154 (Mn/Cu)Ox,118,164 and a number of perovskite oxides. The formation of core–shell structure is likely to be due to the relatively low melting point of Na2WO4 (698 °C)165 and low surface tension of the molten salt on typical oxide surfaces.
:W ratio investigated, the Mg6MnO8@Na2WO4 (Na:W = 2:1) redox catalyst exhibited one of the highest increase in C2H4 yield (∼45% increase, on an absolute scale) compared to the unpromoted Mg6MnO8 at 850 °C and 4500 h−1. The yield increase was primarily resulted from the substantially decreased COx selectivity (>80%). Mg6MnO8@Na2WO4 also exhibited well defined crystalline phases at room temperature (Mg6MnO8 and Na2WO4), with only a minor Na4Mg(WO4)3 phase (Fig. 4b). The core–shell structure of the catalyst was verified by low-energy ion scattering spectroscopy (LEIS) (Fig. 4c) and X-ray photoelectron spectroscopy (XPS) (Fig. 4d).121 Even after 10 cycles of sputtering, W and Na/Mg are still far more prevalent than Mn according to LEIS. It is noted that LEIS cannot accurately differentiate Na and Mg due to their similar atomic weights. XPS, which provides near surface elemental compositions (Fig. 4c), also confirmed the suppression of Mn by Na2WO4. In fact, impregnation of Na2WO4 onto many Mn-containing oxides has shown to create an oxide@Na2WO4 core shell structure. For instance, transmission electron microscope with energy dispersive x-ray spectroscopy (TEM-EDS) images published from our group by Hao et al. indicated a core shell structure of CaMn0.75Fe0.25O3(CMFO)@Na2WO4 (Fig. 4e)157 whereas LEIS and/or XPS data indicated surface enrichment of Na2WO4 on mixed (Fe/Mn)Ox,153 (Mn/Si)Ox,154 (Mn/Cu)Ox,118,164 and a number of perovskite oxides. The formation of core–shell structure is likely to be due to the relatively low melting point of Na2WO4 (698 °C)165 and low surface tension of the molten salt on typical oxide surfaces.
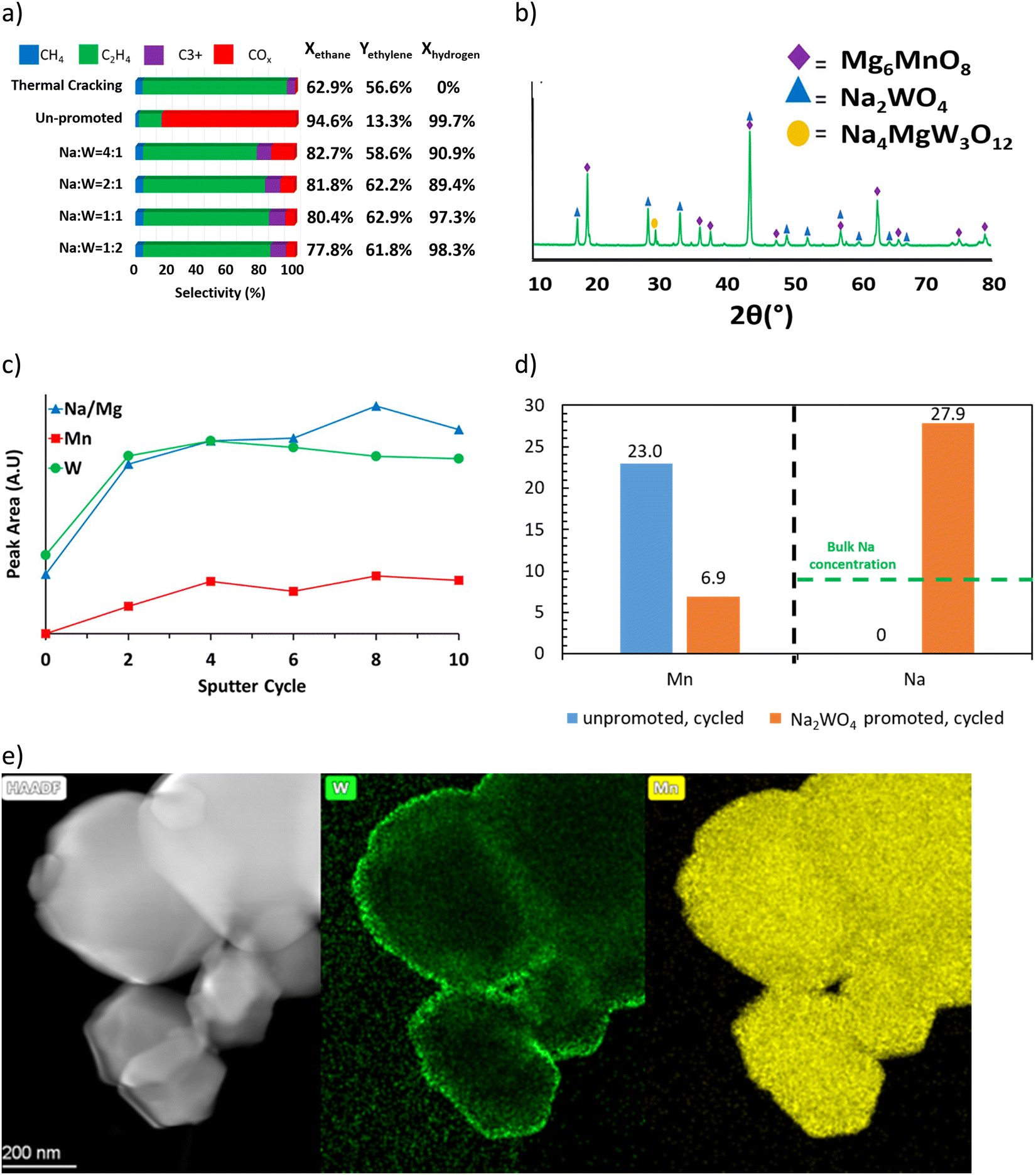 | ||
| Fig. 4 (a) Performance data of the CL-ODH with Na, W promoted Mg6MnO8 at 850 °C and GHSV = 4500 h−1;121 (b) X-ray diffraction characterization of Mg6MnO8@Na2WO4;121 (c) LEIS peak area over sputter cycles for Mg6MnO8@Na2WO4;121 (d) XPS near surface concentration of Na and Mn of the cycled sample;147 (e) TEM-EDS of CMFO@Na2WO4.157 | ||
Detailed mechanistic studies from our group were conducted by Yusuf et al. to understand the function and mechanism of the molten salt promoter for Type 1 redox catalyst.121,147 Mg6MnO8@Na2WO4 was used as the model catalyst. Methanol TPSR experiment (Fig. 5a) showed that the presence of Na2WO4 led to significantly decreased CO2 signal. This suggests that Na2WO4 would suppress the basic sites on the surface of Mg6MnO8.121 Mn 2p3/2 region scan from XPS (Fig. 5b) showed that the peaks corresponding to Mn4+(HBE) and Mn4+ at 644.1 eV and 641.7 eV were suppressed in Mg6MnO8@Na2WO4 as compared to the unpromoted Mg6MnO8.121 This corresponds well to the decrease in the basic sites on the surface. Oxygen donation/reincorporation dynamics were also probed with 18O/16O isotope exchange experiments, shown in Fig. 5c and Table 2.121 The presence of molten Na2WO4 inhibits oxygen exchange, particularly in terms of limiting the incorporation rates of dissociatively adsorbed oxygen into the Na2WO4 layer. Gas switching experiments between H2 and O2 further indicated that H2 solubility in Na2WO4 was negligible, thus indicating that the hydrogen combustion reaction would occur on the molten salt – gas interface. Therefore, it can be inferred that Na2WO4 decreases the COx formation by increasing the energy barrier for the release of the active lattice oxygen at the reaction interface. Electrochemical impedance spectroscopy (EIS) measurements confirmed that oxygen and electron can transport within the Na2WO4 molten layer to facilitate selective H2 combustion (Fig. 5d).158 It was further shown that Na2WO4 was redox active between W6+ and W5+ even though the bulk Na2WO4 phase was difficult to be fully reduced. As such, WO42−/WO3− redox pair would act as the intermediate to shuttle oxygen to and from the Na2WO4 core for selective hydrogen combustion following the following mechanism (Fig. 5e).
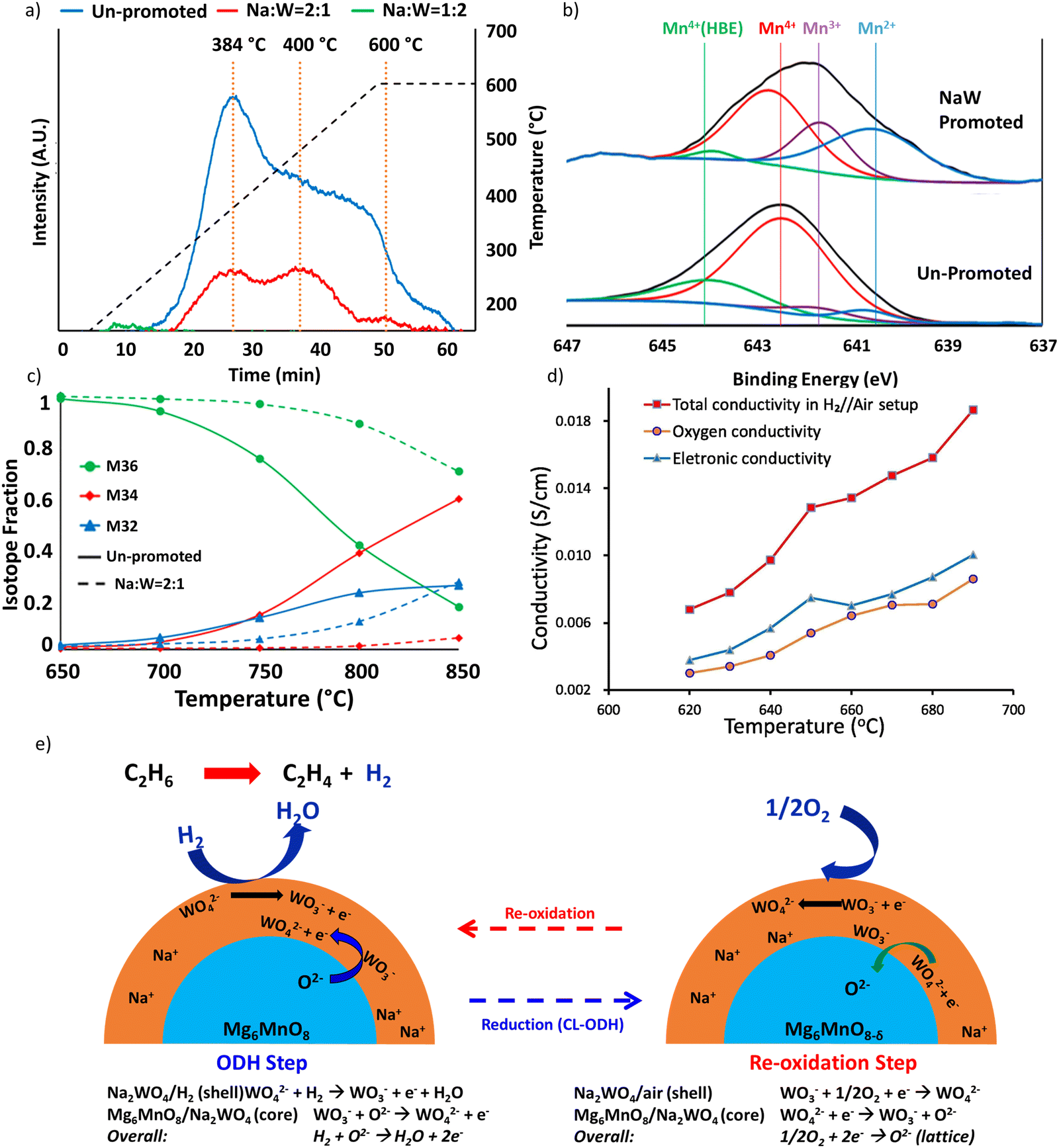 | ||
| Fig. 5 (a) CO2 signal Methanol TPSR of Mg6MnO8 and Mg6MnO8@Na2WO4;121 (b) XPS spectra of manganese 2p3/2 peaks of cycled redox catalysts;147 (c) 18O–16O isotope exchange experiment;121 (d) oxygen ion conductivity and electronic conductivity of Na2WO4;158(e) proposed mechanism of Mg6MnO8@Na2WO4.121 | ||
| Activation energy | Mg6MnO8 (kJ mol−1) | Mg6MnO8@Na2WO4 (kJ mol−1) (800∼850 °C) |
|---|---|---|
| Ro: overall oxygen exchange rate; Ra: dissociative oxygen adsorption rate; Ri: adsorbed oxygen incorporation rate. | ||
| E Ro | 163.83 | 202.58 |
| E Ra | 227.18 | 205.04 |
| E Ri | 123.82 | 174.68 |
The same strategy can be extended to many other Mn based oxides and molten salts.153,154,157,158,164 In a recent study, we demonstrated this strategy with Mg6MnO8 and (Cu/Mn)Ox as the redox core and W, V, Mo based alkali salts as the surface promoters.164 All the abovementioned molten salts were shown to be effective to decrease the COx selectivities from >90% (unpromoted oxides) to <15% (Fig. 6a and b) at 850 °C. C2H4 selectivities were maintained >80% while achieving up to ∼90% H2 conversion. As shown in Fig. 6a to ∼74% C2H4 yield at 80 vol% C2H6 feed and 850 °C can be achieved. Fig. 6a also demonstrated the effect of different molten promoters. The use of multiple alkali metal cations (e.g. Li and Na) and/or transition metals (e.g. Mo and W) for the salt anions were shown to improve the redox catalyst performance in several cases. Such combinative effect further increases the flexibility for designing the Type 1 catalyst.
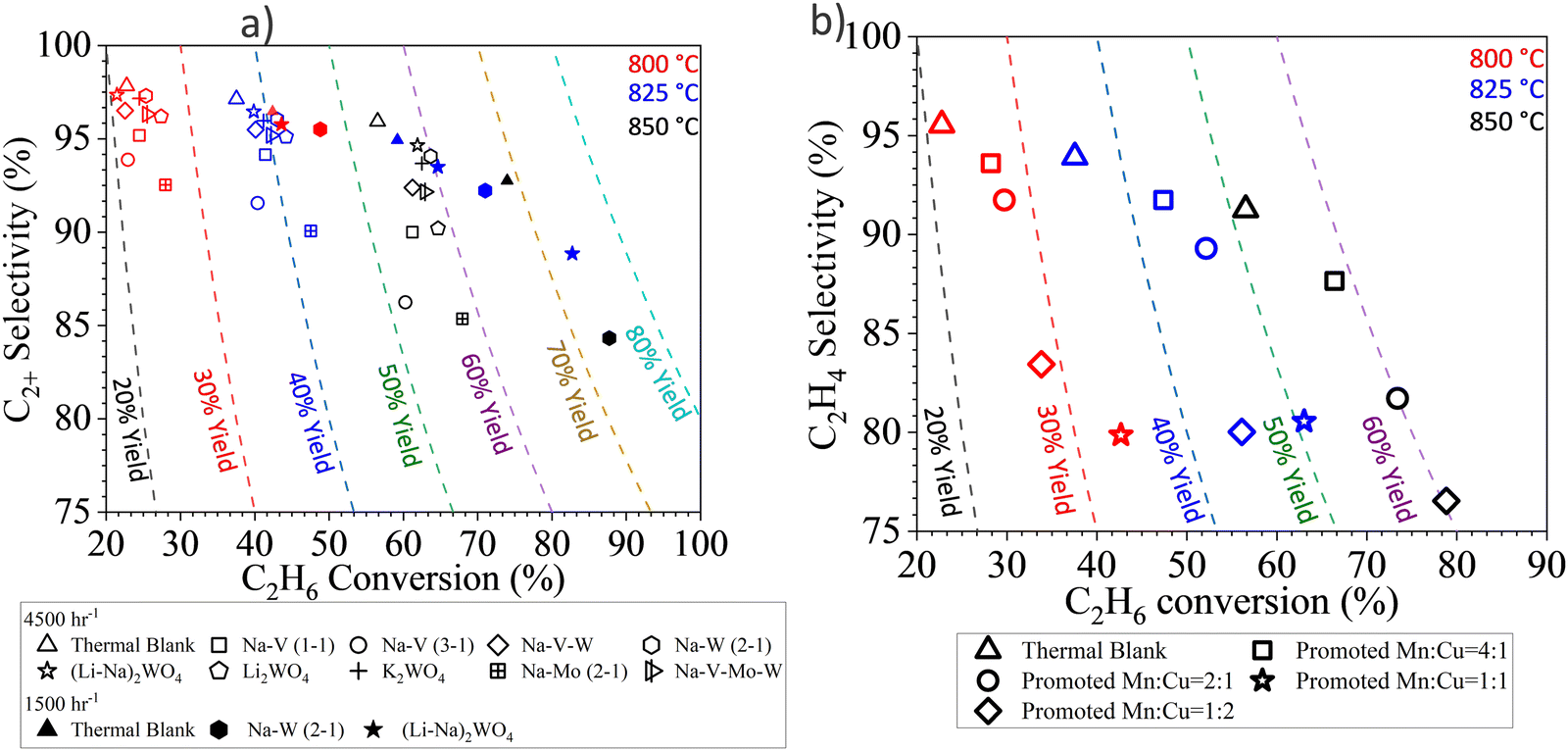 | ||
| Fig. 6 (a) Performance data of the CL-ODH with Li, Na, K and W, V, Mo molten salt shell and Mg6MnO8 core at 850 °C at 4500 h−1.164 (b) Performance data of the CL-ODH with Na2WO4 molten salt shell and Cu/MnOx core at 850 °C at 4500 h−1.164 | ||
Catalyst stability was also demonstrated through a long-term study using a more mechanically robust prototype redox catalyst designed based on the aforementioned principles.129 As shown in Fig. 7a, stable product selectivity and ethane conversion were maintained, throughout 1400 redox cycles, in a bubbling fluidized bed reactor operated at 845 °C with space velocities varying between 2000 and 3250 h−1. The pressure drop across the fluidized bed was also quite stable, indicating proper fluidization without significant attrition and particle entrainment.129 The demonstrated higher conversion, along with in situ H2 oxidation, leads to a lower energy requirement and potential for near an order of magnitude CO2 emission reduction (Fig. 7b).
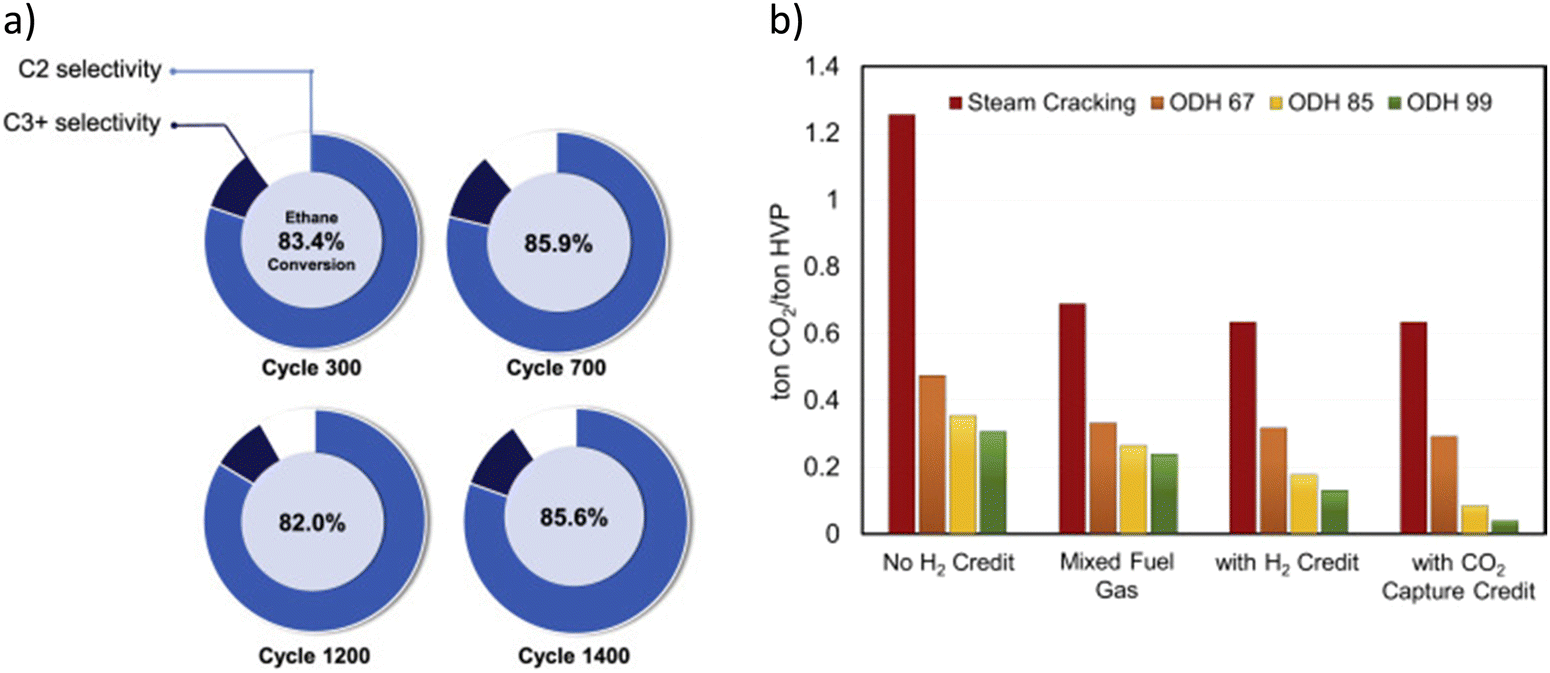 | ||
| Fig. 7 (a) C2H6 conversion and selectivity profile of the prototype redox catalyst over 1400 cycles at 845 °C, 15–30 mol% ethane and GHSV = 2000–3250 h−1; (b) CO2 emissions of steam cracking and ODH processes under different scenarios for ODH 67/85/99, the numerical value indicates the ethane conversion value in percentage.129 | ||
While Type 1 redox catalysts were most frequently used in the context of ethane thermal cracking, they can also be applied for oxidative cracking of naphtha. As shown in Fig. 8a and b, the promotion of Na2WO4 on BaFe6Al6O19, CaMnO3, or La0.8Sr0.2FeO3 decreases the COx selectivity and increases the olefin yields from the cracking of naphtha model compounds such as n-hexane and cyclohexane.158–160
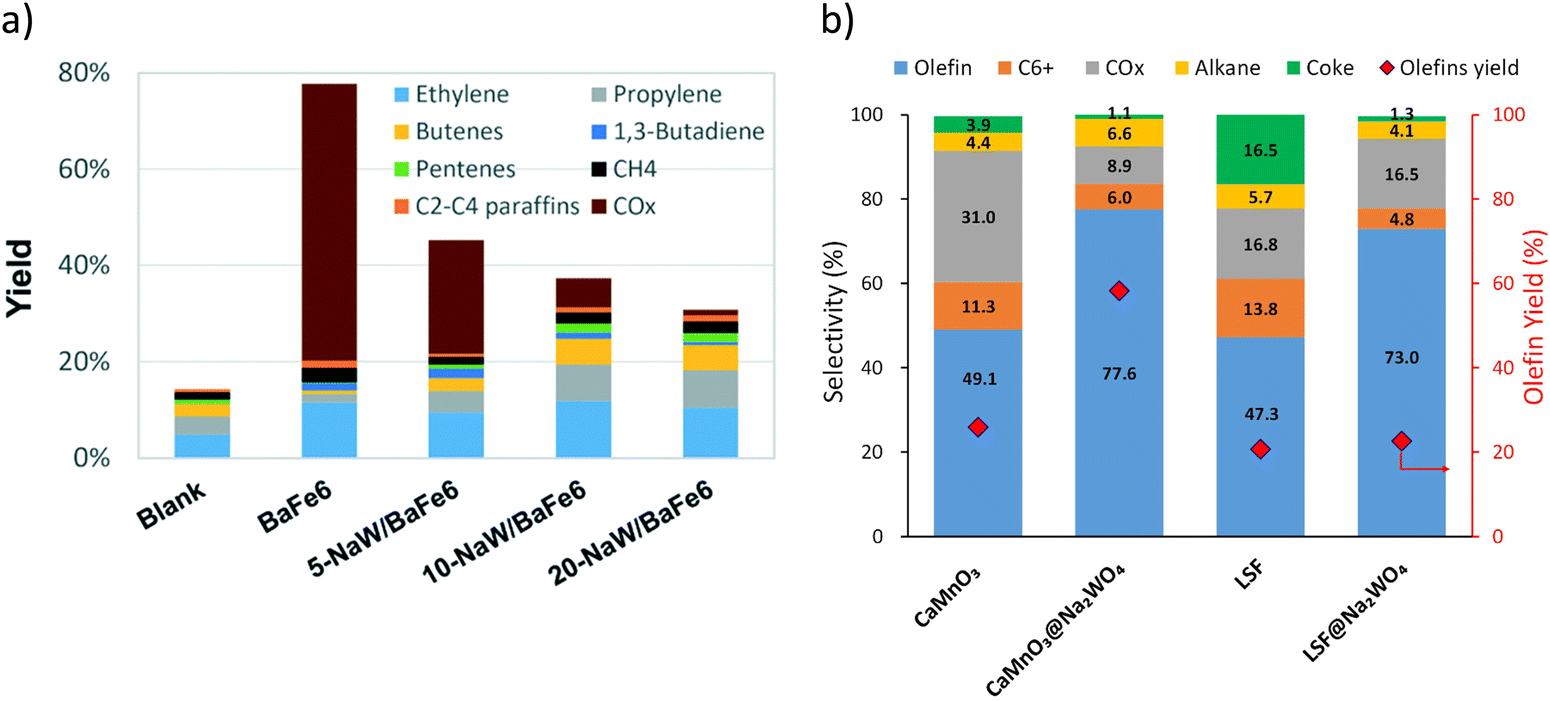 | ||
| Fig. 8 (a) Products yields of BaFe6Al6O19 after promotion with different amounts of Na2WO4 in n-hexane cracking. Reaction condition: T = 700 °C; GHSV = 9000 h−1 cyclohexane concentration ≈13 mol%160 (b) selectivity profile of cyclohexane cracking with CaMnO3@Na2WO4 and La0.8Sr0.2FeO3@Na2WO4T = 750 °C; GHSV = 5400 h−1; cyclohexane concentration ≈7 mol%.158 | ||
A similar strategy has also been recently adopted by a few other researchers in the context of CL-ODH and methane oxydehydroaromatizations.96,97,118,166 In all cases, higher H2 combustion selectivity were observed after promoting the oxide surface with the “inert” molten salt. This further demonstrates the feasibility of the Type 1 catalyst architecture. In fact, the selective hydrogen combustion function of Type 1 redox catalyst would work well when combined with other DH or cracking catalysts. This will be further detailed in Section 2.3.
To sum up, the design of Type 1 redox catalyst mainly aims to increase the selectivity towards hydrogen combustion, reducing the unwanted deep oxidation products such as COx. We apply an “inert” layer of molten salt, which is largely inactive towards C–H bond activation, while still being able to selectively oxidize hydrogen using the active lattice oxygen from the transition metal oxides. The term inert mainly refers to the inhibition of C–H bond activation since the molten salt promoters do actively participate in oxygen and electron transport as well as the hydrogen combustion reaction.
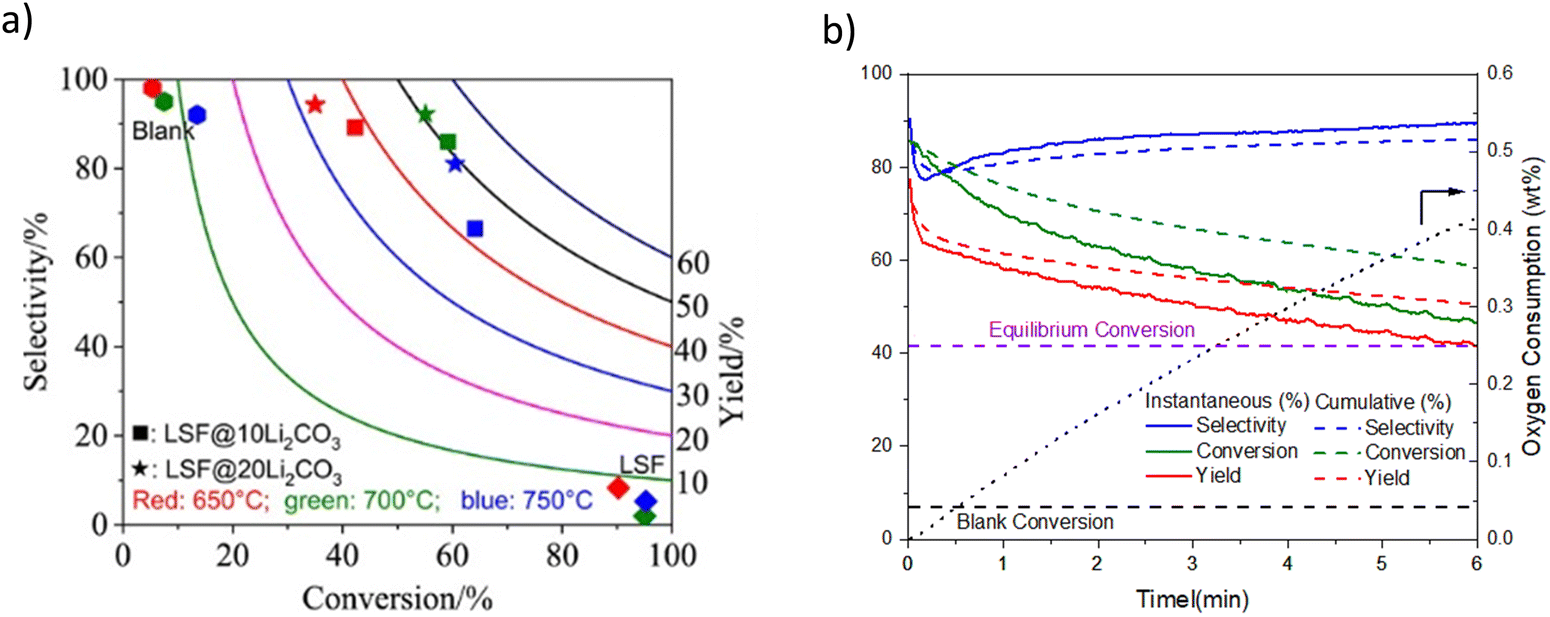 | ||
| Fig. 9 (a) Ethylene selectivity and conversion profiles for LSF@10 wt% (or 20 wt%) Li2CO3via CL-ODH of ethane (our work) compared with literature results with various catalysts;167 (b) ethane conversion profile vs. time-on-stream for: LSF@Li2CO3, thermal cracking (denoted as blank) and equilibrium conversion at 700 °C and 480 h−1. | ||
in situ XRD was performed to study LSF@10 wt% Li2CO3 (Fig. 10a) and LSF (Fig. 10b).167 The peak analysis indicates that the perovskite (LSF) phase largely remained stable in LSF@10 wt% Li2CO3 during the ODH step. In contrast, a significant portion of unpromoted LSF decomposed to La2O3, (La/Sr)2FeO4 and Fe phases. TGA studies showed that the oxygen capacity of LSF@10 wt% Li2CO3 was significantly lower than pure LSF (0.5 wt% vs. 12 wt%). These observations indicate that Li2CO3 inhibits the oxygen release from LSF and limits the reduction of Fe cation in the perovskite structure. Coupled with Mössbauer spectroscopy results, it was determined that for LSF@10 wt% Li2CO3, Fe4+ in LSF was reduced to Fe3+, whereas in unpromoted LSF, a notable fraction of Fe4+/Fe3+ were deeply reduced to form metallic Fe. The role of Li2CO3 was further characterized by electrochemical impedance spectroscopy (EIS, Fig. 10c). The results indicate that, at near melting point, Li2CO3 has increased oxygen ion conductivity but the electronic conductivity stayed near zero. This suggests that Li2CO3 could transport ionic oxygen species, but not electron. Therefore, such oxygen species must be in the oxidized form. Among all possible species, peroxide ion (O22−) is the most likely after excluding other potential species (molecular O2, O2−, O2−, and C2O42−) based on O2-TPD, 13C-NMR, and literature study.169 This was also supported by the ethane and O2 co-feed experiment with and without co-feeding CO2 (Fig. 10d). It was shown that the cofeed of CO2 would decrease the activity for ethylene formation, indicating that the peroxide species is likely suppressed by CO2 (2O22− + 2CO2 → 2CO32− + O2). Density functional theory (DFT) calculation also indicates that the formation of the peroxide from the Fe4+ to Fe3+ is thermodynamically favorable (Fig. 10e). Based on these findings, the following reaction steps were proposed for the ODH reaction: (1) peroxide formation at the LSF/Li2CO3 interface through Fe4+ → Fe3+ transition; (2) dissolution and transport of the O22− by Li2CO3; (3) H abstraction from C2H6 facilitated by O22− at the molten-salt/gas interface. The subsequent reactions are likely to proceed through a surface initiated homogenous reaction pathway by releasing ethyl radicals into the gas phase.
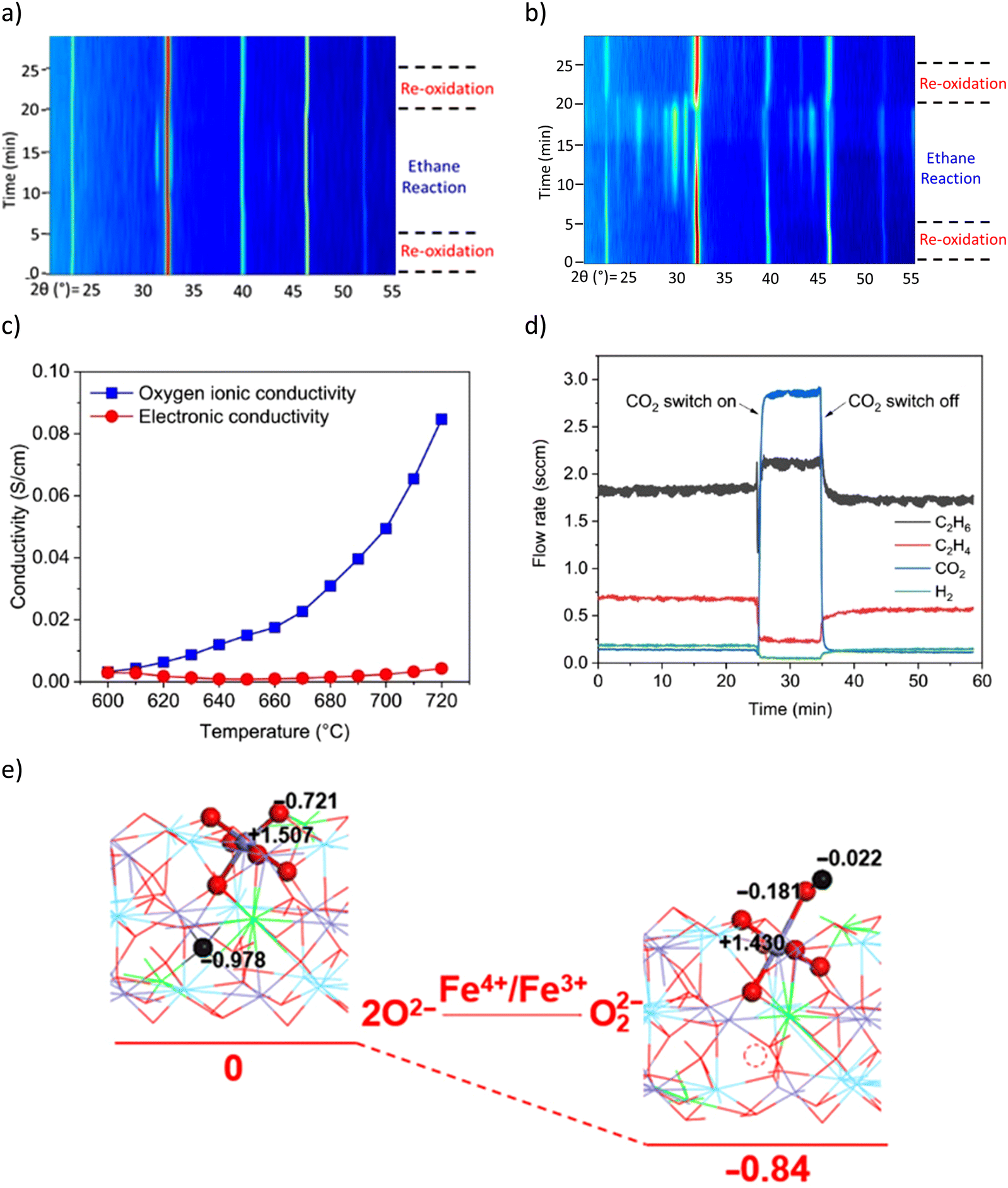 | ||
| Fig. 10 (a) In situ XRD of LSF@10 wt% Li2CO3 under CL-ODH cycles at 700 °C, the LSF phase remained stable, with a small amount of (La/Sr)2FeO4 formed during the ODH step; (b) in situ XRD of unpromoted LSF under CL-ODH cycles at 700 °C. Decomposition of the LSF phase to La2O3, (La/Sr)2FeO4, and Fe occurred during the ODH step; (c) electrochemical impedance spectroscopy (EIS) of Li2CO3; (d) ethane and O2 co-feed experiment on pure Li2CO3 with and without CO2 co-feed at 730 °C, 4000 h−1; (e) DFT calculation of peroxide formation with Fe4+ to Fe3+ transition.167 | ||
As one would expect, for different reactions and conditions, the required active species may change. For example, butane ODH to 1,3 butadiene conversion needs to be carried out at temperatures far below Li2CO3's melting point (∼710 °C). As such, LSF@10 wt% Li2CO3 would not be effective. As can be seen from Fig. 11, LSF@10 wt% Li2CO3 does not have a high selectivity towards 1,3-butadiene.168 For this reaction, LSF@AX (A = alkali, X = halogen) redox catalysts showed significant butadiene yields whereas the blank experiment indicated minimal thermal cracking.155 The unmodified LSF exhibited 71% butane conversion, but with ∼93% selectivity towards COx products. In contrast, LSF@20 wt% LiBr showed 56.2% 1,3-butadiene selectivity with 75.6% n-butane conversion.168
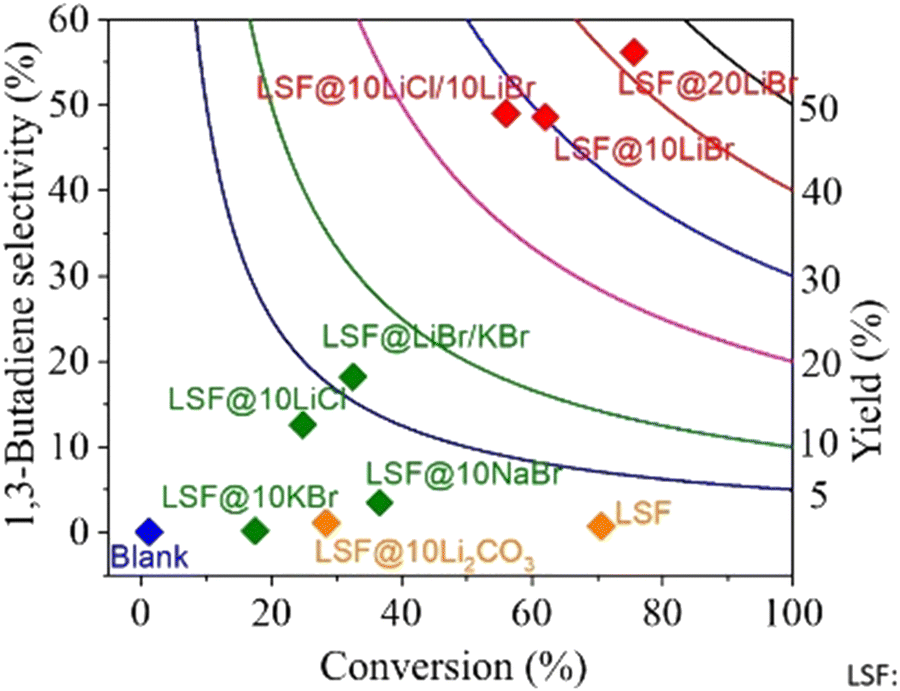 | ||
| Fig. 11 1,3-Butadiene selectivity and conversion profiles for LSF@10 wt% LiBr via CL-ODH of n-butane (our work) compared with literature results with various catalysts at GHSV = 4500 h−1 and 500 °C.168 | ||
Compared to LiBr, other molten and active halogenated salt promoters such as NaBr, KBr and/or LiCl were less effective. This indicates that Br possibly act in conjunction with Li to facilitate selective dehydrogenation of butane. The role and interaction of Li and Br species were probed by ab initio molecular dynamics (AIMD) shown in Fig. 12a, the LiBr is likely oxidized by Li2O2 to form atomic Br, which then diffuse to the surface and facilitate H abstraction from butane at the gas/molten salt interface. DFT calculation shown in Fig. 12b indicates that the Br assisted C–H activation have energy barriers of 91.6 kJ mol−1 (butane to butyl radical) and 34.7 kJ mol−1 (1-butene to butenyl radical), a significant decrease compared to the C–H bond dissociation energy (∼400 kJ mol−1). In addition, the desorption energies of the radicals are both ∼30 kJ mol−1; it is therefore more likely that the radicals would be converted to the dehydrogenated counterpart on the surface of the molten salt layer. Additional gas-phase Reaction Mechanism Generator and Chemkin-Pro results indicate the 2-butene, instead of 1,3-butadiene, would be the primary product via gas phase radical reactions in the presence of butyl radical. The absence of 2-butene rejects the surface initiated radical reaction pathway. As such, the as-formed atomic Br would be primarily responsible for all the dehydrogenation steps. Based on these findings, the following reaction steps were proposed: (1) the as-formed peroxide on LSF oxidizes Li2O to Li2O2; (2) Li2O2 oxidizes LiBr to atomic Br; (3) Atomic Br then abstracts H from C–H bond from n-butane and 1-butene, sequentially forming 1,3-butadiene and HBr; (4) HBr then react with Li2O to form LiBr to complete the reaction cycle.
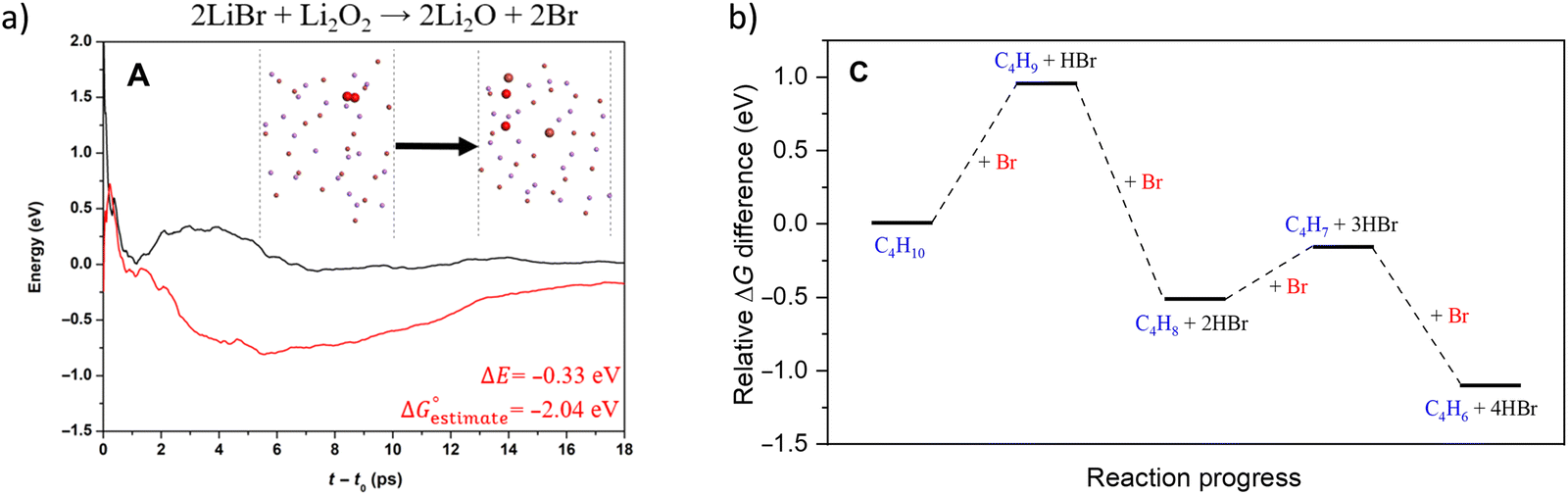 | ||
| Fig. 12 (a) AIMD calculation of reaction pathway of 2LiBr + Li2O2 → 2Li2O + 2Br; (b) AIMD calculation of reaction pathway of n-butane reacting with atomic Br-containing molten LiBr.168 | ||
Generally speaking, a Type 2 redox catalyst functions through: (1) inhibiting CO2 formation by covering the mixed oxide with the promoter, this aspect is similar to Type 1 redox catalysts;167,168 (2) selectively catalyzing ODH reactions through the formation of active species from molten salt and oxide core interactions. It is also worth noting that different promoters in Type 2 redox catalysts can lead to different reaction pathways and mechanisms (Table 3). Selection of the oxide substrate is also important in terms compatibility with the promoter and ability to form desirable active oxygen species such as peroxide ions.
| C2H6 ODH with LSF@Li2CO3 | C4H10 ODH with LSF@LiBr |
|---|---|
| At the LSF/molten Li2CO3 interface: | At the LSF-molten LiBr interface: |
| 2Fe4+ + 2O2− → 2Fe3+ + O22− | O22− + 2Li2O → O2− + Li2O2 |
| Li2O2 + 2LiBr → 2Li2O + 2Br | |
| At the gas-molten Li2CO3 interface: | At gas-molten LiBr interface: |
| Overall reaction: |
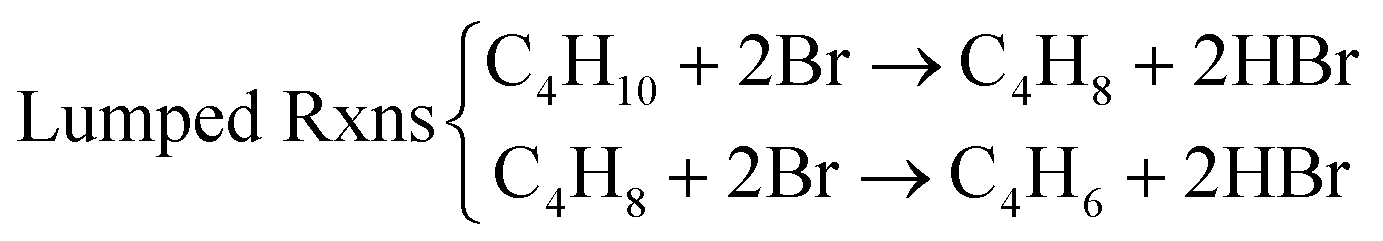
|
| C2H6 + O22− → C2H4 + O2− + H2O | Li2O + 2HBr → 2LiBr + H2O |
2.2. Type 3: mixed oxide @ catalytic phase redox catalyst for hydrocarbon conversion
Type 3 redox catalysts are based on a design concept like Type 2 but differs in that a solid catalytic phase is used and a radical formation step is not likely to be involved. For instance, a well-studied K–Fe–O catalyst is highly active towards ethylbenzene DH to styrene.170,171 By combining the K–Fe–O's catalytic function with transitional metals oxides’ SHC capability, equilibrium limitation can be circumvented, intensifying the overall process.A notable example is the multi-functional (Ca/Mn)1−xO@KFeO2 redox catalyst developed from our group, which showed high activity towards ethylbenzene CL-ODH.161 As shown in Fig. 13a, up to 91% single-pass styrene yield can be achieved. This represents a 72% yield increase, on a relative basis, compared to commercial dehydrogenation. As can be seen in Fig. 13a, there is an unselective region at the beginning of the ODH reaction step, during which the ST selectivity was ∼54%. The selectivity of ST would increase to ∼95% as more ethylbenzene (EB) is injected. This unselective region can be avoided by limiting the air injection during the regeneration step, as shown in Fig. 13b to maintain a high selectivity (∼94.2%). Higher EB partial pressures closer to industrial conditions has also been tested at a lab scale (Fig. 13c), and the styrene yield surpassed the equilibrium limitation. In addition, the stability of this redox catalyst was tested for 100 cycles (Fig. 13d), albeit at a relatively low EB partial pressure.
 | ||
| Fig. 13 Performance of redox-ODH of ethylbenzene with (a) fully reoxidized and (b) partially reoxidized redox catalyst (EB partial pressure = ∼0.01 atm, temperature = 600 °C). (c) Redox-ODH performance comparing to DH equilibrium conversions in the range of 0.01–0.1 atm ethylbenzene feed partial pressure (balance Ar) using partially reoxidized redox catalyst. (d) Long-term cycle and product distributions in redox-ODH using fully reoxidized redox catalyst.161 | ||
Fig. 13a and b signify that there were dynamic changes in the redox catalyst. in situ XRD (Fig. 14a) and ex situ XPS (Fig. 14b and c) were used to characterize the changes in the shell and the core at different stages of the redox ODH reactions.161in situ XRD indicated that the unselective region shown in Fig. 13a was resulted from the solid-state reactions between the core and shell phases, forming a nonselective K0.296Mn0.926O2 phase while decreasing the surface concentration of potassium. As the injection of EB continues, this nonselective phase disappeared and the catalytic layer KFeO2 appeared throughout the rest of the EB injection step. Meanwhile, there was a continuous lattice oxygen release from the CaO–Mn2+/3+O solid solution, which was responsible for selective hydrogen combustion. From Fig. 14b and c, the Mn 2p peaks shifted to lower binder energy level when in contact with EB (in both reduced and deeply reduced region), while the Fe 2p peaks only shifted after prolonged contact with the EB (deeply reduced region). Both results suggest that Fe3+ oxidation state, which is key to high DH activity, is reserved by sacrificing Mn3+/4+ in the operating regime.
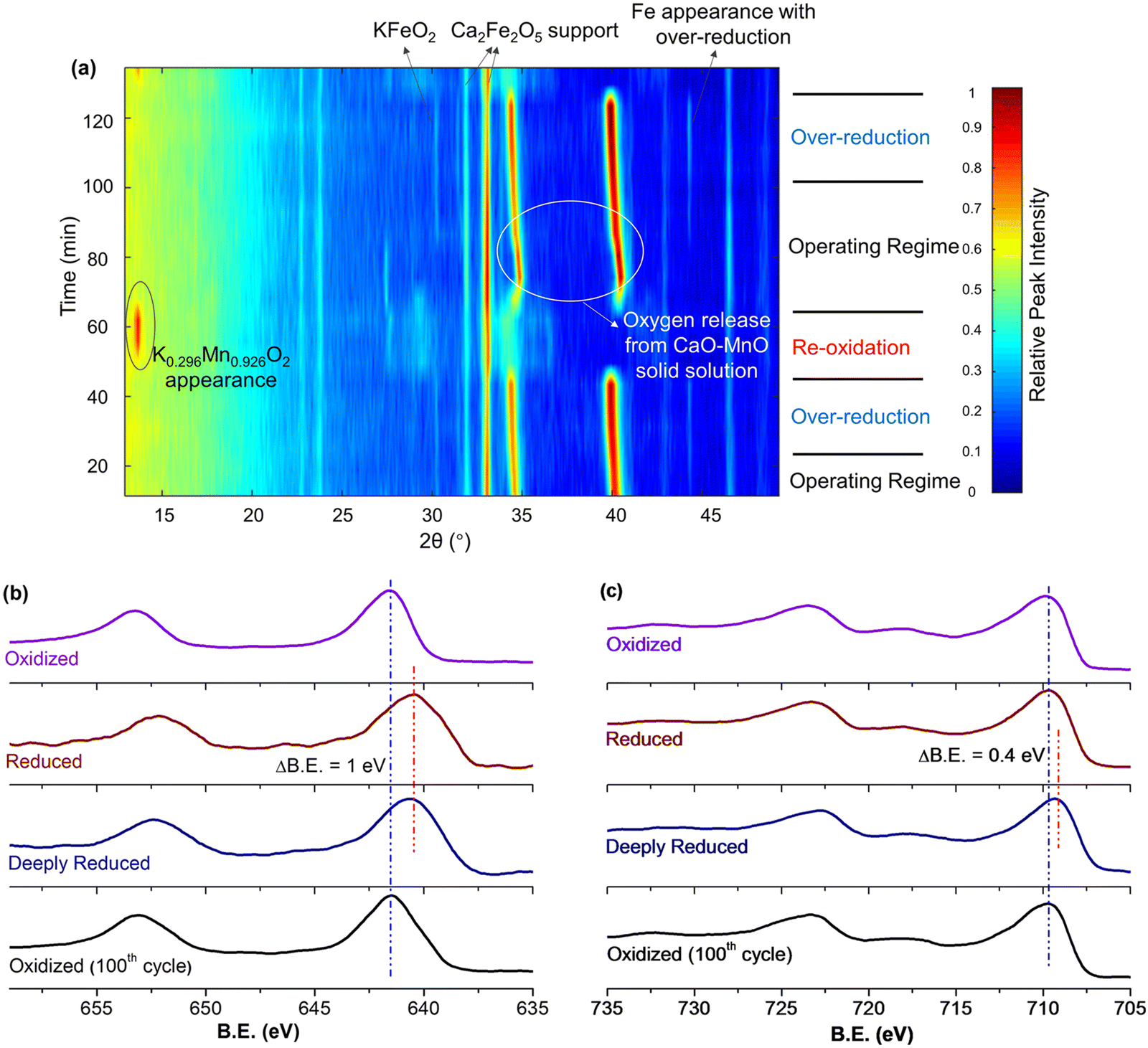 | ||
| Fig. 14 (a) In situ XRD under cyclic ethylbenzene ODH and air reoxidation steps at 600 °C; (b) XPS of Mn 2p and (c) XPS of Fe 2p for redox catalysts at different reaction stages.161 | ||
The reaction mechanism was further studied by isotope experiments in our group. Similar to conventional catalytic DH, during the dehydrogenation reaction, ethylbenzene on the CLCa catalyst undergoes a two-step H abstraction on the KFeO2 surface.170–172 The KIE value (Fig. 15a) measured in the isotope experiment confirmed that H abstraction was the rate limiting step. This was further supported by the DFT results (Fig. 15b-1), where the α-H abstraction step was shown to be rate limiting. DFT results also indicated that the presence of water would facilitate the proton transfer in the water formation step on the KFeO2 surface, leading to more than 4-fold decrease in activation energy (Fig. 15b-2). This was verified by dehydrogenation experiments on KFeO2 both with and without cofeeding steam.
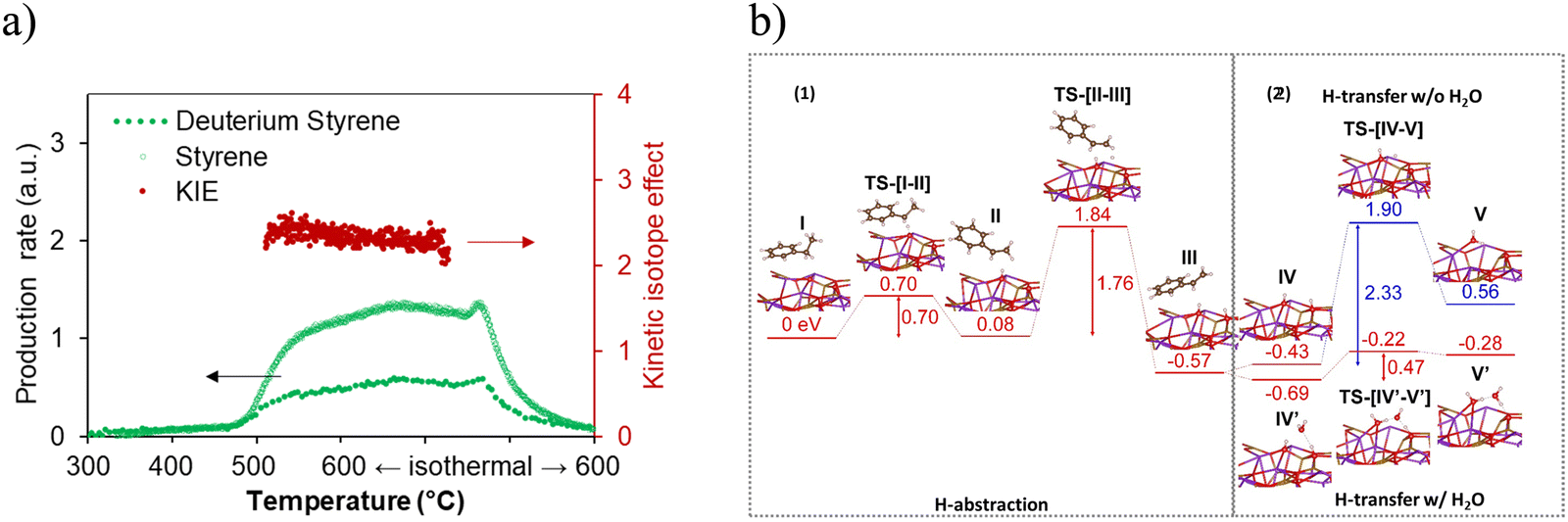 | ||
| Fig. 15 (a) Kinetic isotope effect study with both temperature-programmed reduction and isothermal reduction of the (Ca/Mn)1−xO@KFeO2 using C8H10 and C8D10161; (b) computed energy potential profiles of (1) α and β H abstraction and (2) proton transfer and water formation with or without water assistance.161 | ||
Similar to Type 2 redox catalysts, design of Type 3 needs to consider the compatibility of the core and shell materials. The dynamic changes observed during the redox reactions signify the potential complexity of CLCa when compared to conventional heterogeneous catalysis systems.161
2.3. Synergistic effects between heterogeneous and redox catalysts
As briefly mentioned in Section 2.1, the abovementioned redox catalyst design, particularly Type 1, has the potential to function in concert with conventional heterogeneous catalysts. We recently demonstrated this approach towards catalytic cracking of cyclohexane by integrating cation exchanged ZSM-5 with a Type 1 redox catalyst, i.e. perovskite oxide@Na2WO4.157 The primary function of the redox catalyst was to selectively oxidize hydrogen and to enhance the naphtha conversion. It is interesting to note that a significant increase in ethylene (C2=) and propylene (C3=) selectivity and C3=/C2= ratio was observed by mixing the ZSM-5 based catalyst with the Type 1 redox catalyst (Fig. 16a and b). The presence of the perovskite oxide@Na2WO4 did not deactivate the zeolite catalyst. Rather, it changed the acidic properties of the zeolite by creating additional strong Brønsted acid sites, which is likely to be responsible for the enhanced olefin selectivity and yield. A similar concept was also shown to be effective for n-hexane conversion: Ba-exchanged ZSM5 mixed with CaMnO3@Na2WO4 could exhibit up to 67% single-pass olefin yield from n-hexane with tunable propylene to ethylene ratio, with <5% CO2 yield.173 We note that in both cases, the nature of the interactions between ZSM-5 and the redox catalyst particles, as well as the fundamental reasons for the change in product selectivity have yet to be understood.157 This represents an interesting area for in-depth studies. Synergistic utilization of heterogeneous catalysts with redox-active oxides can also significantly broaden the applicability for CLCa.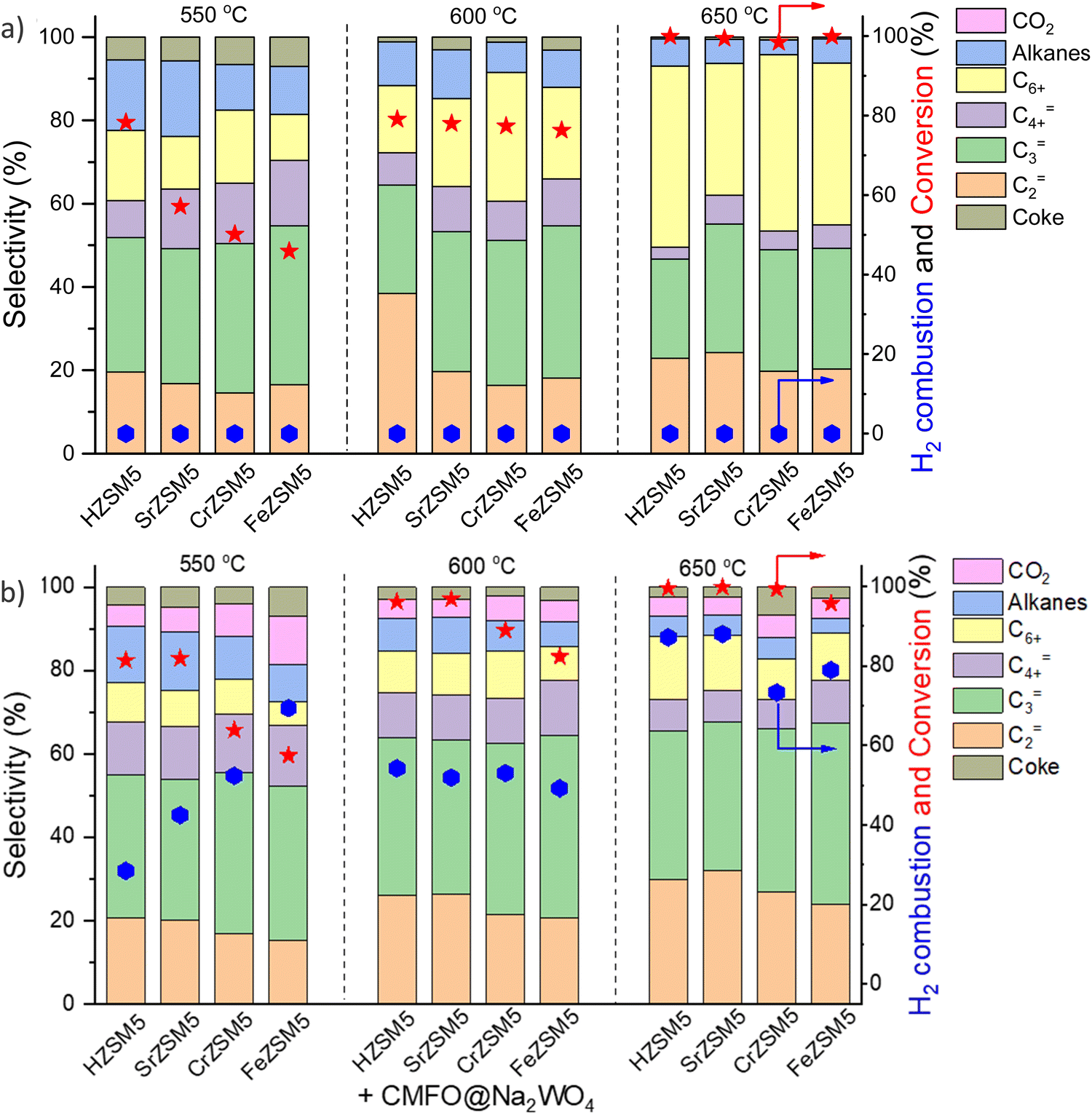 | ||
| Fig. 16 Cyclohexane conversion and product distributions over (a) HZSM5 and different metal cation (Sr, Ca, Cr or Fe) exchanged ZSM-5; (b) composite composed of ZSM-5s and CaMn0.75Fe0.25O3−δ@Na2WO4 redox catalyst.157 | ||
When integrating CLCa with a heterogeneous catalyst, an important aspect to consider is whether the heterogeneous catalyst would function well under significant oxygen partial pressure swings. In addition, steam generated from hydrogen combustion by the CLCa redox catalyst can have a negative impact on the active sites of the catalyst and can lead to side reactions (e.g. steam reforming). For example, MoCx, active for dehydroaromatization (DHA) of methane,174–181 can react with steam produced from SHC to form MoOx, which would deactivate the catalyst. Therefore, care must be taken in selecting robust and compatible hybrid catalyst systems involving both chemical looping and heterogeneous catalysts.
2.4. Computational assisted oxide selection
Although the previous section focused on surface promoters for the redox-active oxides, the selection of the oxide also represents a crucial and enabling step to CL-ODH. In fact, a sizable amount of chemical looping research focused on discoveries and the selections of oxide-based oxygen carriers. To date, over 2000 research articles have covered different aspects of oxygen carrier compositions, performance, and underlying mechanism. Despite the extensive research and development efforts, a rationalized strategy for the effective design and selection of oxygen carriers is still lacking. In fact, the high complexity of redox reactions, which extends beyond the interactions of gaseous molecules with poorly defined oxide surfaces, makes it near impossible for ab initio design of redox-oxides. On the other hand, the emergence of oxygen carriers composed of redox-active mixed oxides, e.g. perovskite oxides, has greatly expanded the design space for chemical looping material selection, making oxygen carrier optimization an even more daunting task.We note that while it is impractical to expect an ab initio oxygen carrier selection model to comprehensively consider all aspects of oxygen carrier design, some relatively simple selection criteria can nevertheless be adopted, particularly when capitalizing on modern computational and data science tools, to greatly reduce the experimental efforts in oxygen carrier development. Take CLCa of light alkanes as an example, the light olefin yields tie directly with the equilibrium oxygen partial pressure (PO2,eq) or equivalently oxygen chemical potential (μO2,eq) of the redox oxide from a thermodynamic standpoint (Fig. 17a). In fact, the thermodynamic selection rule of the oxides in CLCa of alkane ODH largely overlaps with that of chemical looping combustion and chemical looping air separation (CLAS). This is not at all surprising given that a primary function of the oxygen carrier in CLCa is in situ air separation, besides acting as a heterogeneous catalyst. Given that the thermodynamic requirement represents a prerequisite for oxygen carrier selection, we recently used such a criterion to select oxides with a general formulation of SrxA1−xFeyB1−yO3−δ (A = Ca, K, Y, Ba, La, Sm; B = Ti, Ni, Mn, Mg, Cu, Co) for applications such as CLAS and chemical looping dry reforming of methane. Fig. 17b illustrates the generalized simulation and experimental validation strategies as well as representative results.182 Despite adopting simplified assumptions to facilitate density functional theory (DFT) based high throughput computations, the simulation results were shown to be quite effective in predicting various perovskite oxides with interesting properties, that could be adopted in the both CLAS or CLCa.182 These redox active oxides materials have excellent potential to be adopted for CLCa. We also note that the proposed model is not without limitations. For instance, the model was based exclusively on thermodynamic criteria without considering kinetic effects. To ensure manageable computational intensity, we assumed random distribution of the dopants and did not consider potential defect clustering. The U parameters selected to correct the strong on-site coulomb interaction of the d-electrons may need to be further tuned for the perovskite oxides. Potential formation of alternative phases and structures besides the base SrFeO3 structure were not considered. These represents potential areas of improvements for future modelling efforts. To date, only a few other groups have also use high-throughput method for materials screening.183,184 Fan et al. screened over 1500 M1–M2–O/N pairs and 170 M1–M2–Nrich/Npoor pairs to identify optimized bicationic materials for CL-ammonia synthesis from different route.183In the context of CLC of methane, Singstock et al. screened over 1300 redox pairs (i.e. ABOx/AOy + B, A(SO4)x/ASx) and predicted 152 pair to demonstrate >99% methane conversion with limited byproducts.184
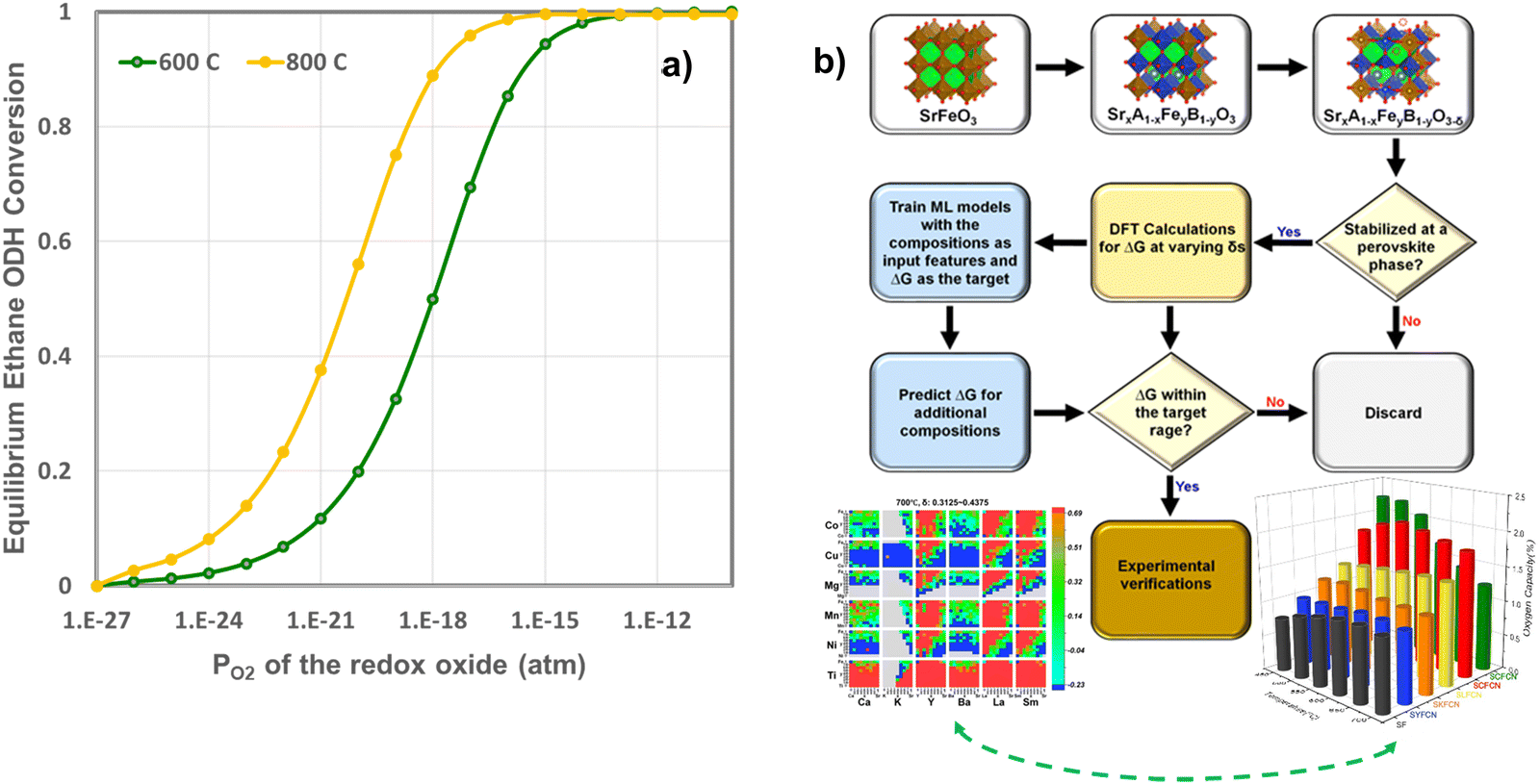 | ||
| Fig. 17 (a) Relationship between the equilibrium ethane conversion in CL-ODH and the equilibrium PO2 of the redox oxide at 600 and 800 °C (only the ODH reaction is considered in this calculation without considering the dehydrogenation reaction); (b) a general strategy for computationally guided oxide selection and the key results related to CLAS material selection published by Wang et al.182 | ||
3. Summary and outlook
The pressing need for decarbonizing the modern society calls for transformative technologies to intensify state-of-the-art manufacturing processes. The chemical looping strategy, with an inherent capability for chemical transformation combined with feedstock and/or product separations, offers unique opportunities to significantly simply the chemical manufacturing processes, improve energy efficiency, and reduce CO2 emissions.This review covered a generalized redox catalyst design strategy, involving a redox-active mixed metal oxide core and a surface promoter shell, for the CL-ODH of hydrocarbons to olefins. An interesting aspect of this design strategy is the ability to independently tune the properties of the oxide core and the promoter shell provided that the two materials are compatible under the operating conditions. For example, for ethane conversion with Type 1 catalyst, we recently predicted that C2+ yield could achieve 78.2–86.7% based on the existing data from molten salt promoted Mg6MnO8 and Cu/MnOx. These analyses suggest that further increase in single-pass olefin yield is highly possible by optimizing the core and the shell compositions and ratios. For Type 2 redox catalysts, the heat of reactions of ethane CL-ODH can be tuned to be mildly exothermic in both the ethane ODH and the re-oxidation steps, allowing more efficient heat management. On the other hand, the selection of the core would also affect the activity and selectivity of the redox catalysts. In the case of LSF, Fe4+ in the perovskite structure was proposed to be responsible for the formation of the peroxide in molten phase. As an example, further adjusting the La:Sr ratio could tune the propensity to form peroxide and therefore optimize the ODH performance. Type 3 redox catalysts, and the potential to integrate CLCa redox catalyst with a heterogeneous catalyst in tandem, also offer exciting opportunities.
From a practical standpoint, molten salt promoted oxides may not be easily fluidizable. However, it is possible to adjust the surface promoter compositions to ensure good fluidization properties and long-term stability.129 Molten salt promoted redox catalysts have also been operated in packed bed reactors in our recent studies, showing satisfactory long-term stability up to 1000 hours.119,120 Other issues to consider for future studies and practical applications include the potential vaporization of molten salts at high temperatures and reactor material selection to minimize potential corrosion. A recent long term study on Li2CO3 promoted LSF showed signs of slight increase in COx selectivity.119 This may be attributed to the loss of Li2CO3. Therefore, replenishing of Li2CO3 either ex situ or during the reactor operation may be considered for future studies.
The application of chemical looping concept is not limited to olefin productions. Table 4 summarizes recent publications related to CLCa. An extensively investigated topics is methane to syngas/H2 conversion.95,109–117,123,125–127,130,136,185,193–208 A few other studies explored a phase transition sorbent concept where a single mixed oxide particle combines the function of a redox oxide (for oxygen separation), a carbonate sorbent (for CO2 separation), and a catalyst (to catalyze chemical reactions).95,123,130,197,198,209–211 For instance, a recent study adopted the chemical looping strategy to selectively oxidize ammonia with excellent NO selectivity for nitric acid production.192 A few other studies explored the feasibility of using nitrides/imides/hydrides131–134,189–191,212–214 as intermediates for ammonia synthesis and using alkali molten salt as the medium for combined CO2 capture and utilization.215 Engineering the chemical potential of redox oxides has also shown to be able to circumvent reaction equilibrium limitations in conventional processes while maintaining high selectivity, making it possible to achieve previously unattainable yields for water gas shift reactions.126,194
| Target product | Carrier | Operating condition | Key performance result | Ref. |
|---|---|---|---|---|
| Syngas/CO | Fe2O3@SBA-15 | 750–935 °C, 17.8–37.5 mL per h per mgFe2O3 | ∼100% CO selectivity | 108 |
| La0.5Ce0.5FeO3 | 850 °C, flow rate: 80 mL min−1 | X methane: 82% | 185 | |
| 0.8 g catalyst | S Syngas: 93% | |||
| Reduction: 10 mol% CH4/N2 | ||||
| Oxidation: 10 mol% CO2/N2 | ||||
| BaFe1−xSnxO3−δ | 900 °C, 0.1 g catalyst | >99% syngas selectivity | 114 | |
| Reduction: 5% CH4/He (20 mL min−1) | Y syngas = 19.2 mmol g−1 | |||
| Oxidation: 5% O2/He (20 mL min−1) | ||||
| MgO supported Ca2Fe2O5 | 1000 °C, 2 g catalyst 1500 h−1 | X methane > 99% | 117 | |
| Reduction: 10% CH4/He (25 mL min−1) | S Syngas: 98.1%, H2/CO ∼2 | |||
| Oxidation: 40% CO2/He (25 mL min−1) | ||||
| Ni-doped Fe2O3/Al2O3 | 900 °C, 2 g catalyst 1500 h−1 | X methane > 95% | 186 | |
| Reduction: 20% CH4 (75 mL min−1) | S Syngas: ∼96%, H2/CO ∼2.3 | |||
| Oxidation: 20% O2 (75 mL min−1) | ||||
| Ethylene (via CL-OCM) | (Li,W)–Mg6MnO8 | 850 °C, 2400 h−1 | X methane: 50% | 106 |
| Reduction: pure% CH4 (200 mL min−1) | C2+ Yield: 28.6% | |||
| Ethylene (via CL-ODH) | 0.2Ce/SrFeO3 | 700∼750 °C 3000∼6000 h−1 | Ethylene Yield: 23.8% | 100 |
| Reduction: ∼12.5 mol % C2H6 | X CO2 69.4% | |||
| Sr0.8Ca0.2FeO3−δ@Na2CO3 + MoVNbTeOx | 500 °C, 600 h−1 GHSV | S ethylene: 91% | 96 | |
| Reduction: 6 mol% C2H6, balance N2 | X ethane: 42% | |||
| Ce-incorporated FeTiOx | 600 °C, reduction: 20 vol% C2H6/N2, 30 mL min−1 | S ethylene: 84.1% | 187 | |
| Oxidation 20 vol% CO2/N2, 30 mL min−1 | X ethane: 14% | |||
| Propylene (via CL-ODH) | Mo–V–O (V/Mo = 6)188 | 500 °C, 2500 h−1 GHSV | X propane: 36% | 188 |
| Reduction: pure% CH4 (200 mL min−1) | S propylene: 89% | |||
| Ammonia | Li–Pd/Li2NH | 300 °C and 1 bar | r NH3 = 693 μmolNH3 g−1 h−1 | 189 |
| 100% N2 or H2 at 30 mL min−1 | ||||
| Si modified γ-Al2O3 ZrO2 supported | 1 bar | X AlN = 60.4% | 190 | |
| N-sorption: 1200 °C 100 mL min−1 N2 | ||||
| NH3-desorption: 1200 °C 80 vol% H2O 500 mL min−1 | ||||
| 20 wt%TiN/MgO | Nitridation: 1 atm, 808 nm laser at 30 W cm−2 (∼550 °C) | r NH3 = 1.67 μmolNH3 g−1 h−1 | 191 | |
| NO (NH3 oxidation) | V2O5 | 300–650 °C | NO selectivity: 99.8% | 192 |
| Reduction: 5 mol % NH3/Ar, 30 mL min−1 | NH3 conversion: 97% | |||
| Oxidation: 30 mol % O2/Ar, 30 mL min−1 |
Most of the processes summarized above harness the oxidizing potential (e.g. SHC) of the metal oxides at high temperatures during the half cycle for metal oxide reduction. The reduced metal oxides are regenerated in an O2, CO2, or steam rich environment, allowing integrated air separation, CO2-splitting, or hydrogen production. It can be fully anticipated that well-designed metal oxides, after reduction, could be used for producing complex, value-added chemicals via hydrogenation and/or carbonylation during the regeneration half cycle. This offers the potential to produce two different value-added products in a two-step redox process or even multi-step chemical loops. Therefore, many new and highly exciting areas of fundamental research and practical applications can be anticipated through the unique combination of chemical looping with heterogeneous catalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. National Science Foundation (Award No. CBET-2116724 and CBET-1923468), US Department of Energy (Award No. FE0031869, EE0008809, FE0031918), and the Kenan Institute for Engineering, Technology and Science at NC State University.References
- International Energy Agency, Chemicals, https://www.iea.org/fuels-and-technologies/chemicals, accessed July 23rd, 2022.
- I. E. Agency, Net Zero by 2050, https://www.iea.org/reports/net-zero-by-2050.
- T. Ren, M. Patel and K. Blok, Energy, 2006, 31, 425–451 CrossRef CAS.
- H. A. Maddah, Am. Acad. Sci. Res. J. Eng., Technol. Sci., 2018, 45, 49–63 Search PubMed.
- A. Agarwal, D. Sengupta and M. El-Halwagi, ACS Sustainable Chem. Eng., 2018, 6, 2407–2421 CrossRef CAS.
- H. Kim, B. Lee, D. Lim, C. Choe and H. Lim, Green Chem., 2021, 23, 7635–7645 RSC.
- R. C. Müller, A. Schiessl, R. Volk and F. Schultmann, J. Cleaner Prod., 2021, 317, 128086 CrossRef.
- S. Yuanyuan, W. Di, Y. Junjie and Z. Liang, Acta Pet. Sin., Pet. Process. Sect., 2020, 36, 1361–1369 Search PubMed.
- A. C. Dimian and C. S. Bildea, Ind. Eng. Chem. Res., 2019, 58, 4890–4905 CrossRef CAS.
- W. L. Luyben, Ind. Eng. Chem. Res., 2010, 50, 1231–1246 CrossRef.
- G. Rochelle, E. Chen, S. Freeman, D. Van Wagener, Q. Xu and A. Voice, Chem. Eng. J., 2011, 171, 725–733 CrossRef CAS.
- E. Krzystowczyk, V. Haribal, J. Dou and F. Li, ACS Sustainable Chem. Eng., 2021, 9, 12185–12195 CrossRef CAS.
- J. Bao, Z. Li and N. Cai, Ind. Eng. Chem. Res., 2013, 52, 6119–6128 CrossRef CAS.
- L. Xu, R. Edland, Z. Li, H. Leion, D. Zhao and N. Cai, Energy Fuels, 2014, 28, 7085–7092 CrossRef CAS.
- H. Gu, L. Shen, J. Xiao, S. Zhang and T. Song, Energy Fuels, 2010, 25, 446–455 CrossRef.
- A. M. Kierzkowska and C. R. Müller, ChemPlusChem, 2013, 78, 92–100 CrossRef CAS.
- Z. T. Yaqub, B. O. Oboirien, M. Hedberg and H. Leion, Chem. Eng. Technol., 2021, 44, 1075–1083 CrossRef CAS.
- D. Yilmaz, B. M. Steenari and H. Leion, ACS Omega, 2021, 6, 16649–16660 CrossRef CAS.
- L. Liu, Z. Li, L. Wang, Z. Zhao, Y. Li and N. Cai, Ind. Eng. Chem. Res., 2019, 59, 7238–7246 CrossRef.
- J. Yan, H. Ge, S. Jiang, H. Gu, T. Song, Q. Guo and L. Shen, Energy Fuels, 2019, 33, 2153–2165 CrossRef CAS.
- Y. Li, H. Wang, W. Li, Z. Li and N. Cai, Energy Fuels, 2018, 33, 449–459 CrossRef.
- T. Pröll and A. Lyngfelt, Energy Fuels, 2022, 36, 9502–9512 CrossRef.
- D. Mei, A. Lyngfelt, H. Leion, C. Linderholm and T. Mattisson, Energy Fuels, 2022, 36, 9470–9484 CrossRef CAS.
- S. Bhavsar, M. Najera, A. More and G. Veser, Reactor and Process Design in Sustainable Energy Technology, 2014, pp. 233–280 DOI:10.1016/b978-0-444-59566-9.00007-7.
- I. Gogolev, C. Linderholm, D. Gall, M. Schmitz, T. Mattisson, J. B. C. Pettersson and A. Lyngfelt, Int. J. Greenhouse Gas Control, 2019, 88, 371–382 CrossRef CAS.
- H. Chen, M. Cheng, L. Liu, Y. Li, Z. Li and N. Cai, Int. J. Greenhouse Gas Control, 2020, 93, 102889 CrossRef CAS.
- T. Song, E.-U. Hartge, S. Heinrich, L. Shen and J. Werther, Int. J. Greenhouse Gas Control, 2018, 70, 22–31 CrossRef CAS.
- D. C. Ozcan, A. Macchi, D. Y. Lu, A. M. Kierzkowska, H. Ahn, C. R. Müller and S. Brandani, Int. J. Greenhouse Gas Control, 2015, 43, 198–212 CrossRef CAS.
- A. Abad, P. Gayán, L. F. de Diego, F. García-Labiano and J. Adánez, Proc. Combust. Inst., 2019, 37, 4361–4369 CrossRef CAS.
- L. Liu, Z. Li, Z. Li, Y. Larring and N. Cai, Chem. Eng. J., 2021, 417, 128054 CrossRef CAS.
- G. Deng, K. Li, Z. Gu, X. Zhu, Y. Wei, X. Cheng and H. Wang, Chem. Eng. J., 2018, 341, 588–600 CrossRef CAS.
- H. Ge, L. Shen, H. Gu and S. Jiang, Chem. Eng. J., 2015, 262, 1065–1076 CrossRef CAS.
- L. Liu, Z. Li, S. Wu, D. Li and N. Cai, Fuel Process. Technol., 2021, 213, 106711 CrossRef CAS.
- S. Lin, Z. Gu, X. Zhu, Y. Wei, Y. Long, K. Yang, F. He, H. Wang and K. Li, Energy, 2020, 197, 117202 CrossRef CAS.
- S. Zhang, H. Gu, J. Zhao, L. Shen and L. Wang, Energy, 2019, 186, 115893 CrossRef CAS.
- M. Rydén and A. Lyngfelt, Int. J. Hydrogen Energy, 2006, 31, 1271–1283 CrossRef.
- Z. Gu, L. Zhang, C. Lu, S. Qing and K. Li, Appl. Energy, 2020, 277, 115590 CrossRef CAS.
- G. Deng, K. Li, G. Zhang, Z. Gu, X. Zhu, Y. Wei and H. Wang, Appl. Energy, 2019, 253, 113534 CrossRef CAS.
- J. Bao, Z. Li and N. Cai, Appl. Energy, 2014, 115, 549–558 CrossRef CAS.
- I. Gogolev, T. Pikkarainen, J. Kauppinen, C. Linderholm, B.-M. Steenari and A. Lyngfelt, Fuel, 2021, 297, 120743 CrossRef CAS.
- J. Wang and H. Zhao, Fuel, 2016, 165, 235–243 CrossRef CAS.
- H. Zhao and J. Wang, Combust. Flame, 2018, 191, 9–18 CrossRef CAS.
- Z. Xu, H. Zhao, Y. Wei and C. Zheng, Combust. Flame, 2015, 162, 3030–3045 CrossRef CAS.
- J. Bao, Z. Li, H. Sun and N. Cai, Combust. Flame, 2013, 160, 808–817 CrossRef CAS.
- X. Zhao, H. Zhou, V. S. Sikarwar, M. Zhao, A.-H. A. Park, P. S. Fennell, L. Shen and L.-S. Fan, Energy Environ. Sci., 2017, 10, 1885–1910 RSC.
- L.-S. Fan, L. Zeng, W. Wang and S. Luo, Energy Environ. Sci., 2012, 5, 7254–7280 RSC.
- V. Purnomo, D. Mei, A. H. Soleimanisalim, T. Mattisson and H. Leion, Energy Fuels, 2022, 36, 9768–9779 CrossRef CAS PubMed.
- I. Sampron, L. F. de Diego, F. Garcia-Labiano, M. T. Izquierdo, A. Abad and J. Adanez, Bioresour. Technol., 2020, 316, 123908 CrossRef CAS PubMed.
- D. Wang, A. Joshi and L.-S. Fan, Powder Technol., 2022, 409, 117814 CrossRef CAS.
- J. Dai and K. J. Whitty, Fuel, 2020, 263, 116780 CrossRef CAS.
- S. Sundqvist, T. Mattisson, H. Leion and A. Lyngfelt, Fuel, 2018, 232, 693–703 CrossRef CAS.
- J. Dou, E. Krzystowczyk, X. Wang, T. Robbins, L. Ma, X. Liu and F. Li, ChemSusChem, 2020, 13, 385–393 CrossRef CAS PubMed.
- E. Krzystowczyk, X. Wang, J. Dou, V. Haribal and F. Li, Phys. Chem. Chem. Phys., 2020, 22, 8924–8932 RSC.
- Q. Zheng, M. Lail, S. Zhou and C. C. Chung, ChemSusChem, 2019, 12, 2598–2604 CrossRef CAS PubMed.
- J. Vieten, B. Bulfin, P. Huck, M. Horton, D. Guban, L. Zhu, Y. Lu, K. A. Persson, M. Roeb and C. Sattler, Energy Environ. Sci., 2019, 12, 1369–1384 RSC.
- Z. Sarshar and S. Kaliaguine, Ind. Eng. Chem. Res., 2013, 52, 6946–6955 CrossRef CAS.
- K. Shah, B. Moghtaderi and T. Wall, Energy Fuels, 2012, 26, 2038–2045 CrossRef CAS.
- J. Vieten, B. Bulfin, M. Senholdt, M. Roeb, C. Sattler and M. Schmücker, Solid State Ionics, 2017, 308, 149–155 CrossRef CAS.
- L. Hou, Q. Yu, K. Wang, T. Wang, F. Yang and S. Zhang, J. Therm. Anal. Calorim., 2018, 137, 317–325 CrossRef.
- J. Vieten, B. Bulfin, F. Call, M. Lange, M. Schmücker, A. Francke, M. Roeb and C. Sattler, J. Mater. Chem. A, 2016, 4, 13652–13659 RSC.
- D. K. Khosla, S. K. Gupta and D. N. Saraf, Fuel Process. Technol., 2007, 88, 51–63 CrossRef CAS.
- B. Bulfin, J. Vieten, S. Richter, J. M. Naik, G. R. Patzke, M. Roeb, C. Sattler and A. Steinfeld, Phys. Chem. Chem. Phys., 2020, 22, 2466–2474 RSC.
- I. S. Metcalfe, B. Ray, C. Dejoie, W. Hu, C. de Leeuwe, C. Dueso, F. R. Garcia-Garcia, C. M. Mak, E. I. Papaioannou, C. R. Thompson and J. S. O. Evans, Nat. Chem., 2019, 11, 638–643 CrossRef CAS PubMed.
- G. Luongo, F. Donat and C. R. Muller, Phys. Chem. Chem. Phys., 2020, 22, 9272–9282 RSC.
- V. Sereda, A. Sednev, D. Tsvetkov and A. Zuev, J. Mater. Res., 2019, 34, 3288–3295 CrossRef CAS.
- B. Bulfin, J. Lapp, S. Richter, D. Gubàn, J. Vieten, S. Brendelberger, M. Roeb and C. Sattler, Chem. Eng. Sci., 2019, 203, 68–75 CrossRef CAS.
- C.-C. Cormos, Energy, 2020, 191, 116579 CrossRef CAS.
- K. Shah, B. Moghtaderi, J. Zanganeh and T. Wall, Fuel, 2013, 107, 356–370 CrossRef CAS.
- C. Tian, Q. Fu, Z. Ding, Z. Han and D. Zhang, Sep. Purif. Technol., 2017, 189, 54–65 CrossRef CAS.
- A. Moran and O. Talu, Ind. Eng. Chem. Res., 2018, 57, 11981–11987 CrossRef CAS.
- E. Marek, W. Hu, M. Gaultois, C. P. Grey and S. A. Scott, Appl. Energy, 2018, 223, 369–382 CrossRef CAS.
- B. Sankararao and S. K. Gupta, Ind. Eng. Chem. Res., 2007, 46, 3751–3765 CrossRef CAS.
- L. Jiang, L. T. Biegler and V. G. Fox, AIChE J., 2003, 49, 1140–1157 CrossRef CAS.
- A. V. Nikonov, K. A. Kuterbekov, K. Z. Bekmyrza and N. B. Pavzderin, Eur. J. Phys. Funct. Mater., 2018, 2, 274–292 CrossRef.
- Y. Hao, C.-K. Yang and S. M. Haile, Chem. Mater., 2014, 26, 6073–6082 CrossRef CAS.
- D. de Ligny and P. Richet, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 3013–3022 CrossRef CAS PubMed.
- H.-C. Wu and Y. S. Lin, Ind. Eng. Chem. Res., 2017, 56, 6057–6064 CrossRef CAS.
- N. Miura, H. Ikeda and A. Tsuchida, Ind. Eng. Chem. Res., 2016, 55, 3091–3096 CrossRef CAS.
- M. Xu, H.-C. Wu, Y. S. Lin and S. Deng, Chem. Eng. J., 2018, 354, 62–74 CrossRef CAS.
- H. Ikeda, S. Nikata, E. Hirakawa, A. Tsuchida and N. Miura, Chem. Eng. Sci., 2016, 147, 166–172 CrossRef CAS.
- Q. Ji, L. Bi, J. Zhang, H. Cao and X. S. Zhao, Energy Environ. Sci., 2020, 13, 1408–1428 RSC.
- Q. Yin, J. Kniep and Y. S. Lin, Chem. Eng. Sci., 2008, 63, 5870–5875 CrossRef CAS.
- Y. Wang, B. Hu, Z. Zhu, H. J. M. Bouwmeester and C. Xia, J. Mater. Chem. A, 2014, 2, 136–143 RSC.
- R. H. Görke, E. J. Marek, F. Donat and S. A. Scott, Int. J. Greenhouse Gas Control, 2020, 94, 102891 CrossRef.
- F. Donat, W. Hu, S. A. Scott and J. S. Dennis, Ind. Eng. Chem. Res., 2015, 54, 6713–6723 CrossRef CAS.
- J. Dou, E. Krzystowczyk, A. Mishra, X. Liu and F. Li, ACS Sustainable Chem. Eng., 2018, 6, 15528–15540 CrossRef CAS.
- J. Dou, E. Krzystowczyk, X. Wang, A. R. Richard, T. Robbins and F. Li, JPhys Energy, 2020, 2, 025007 CrossRef CAS.
- C. Tagliaferri, R. Görke, S. Scott, J. Dennis and P. Lettieri, Chem. Eng. Res. Des., 2018, 131, 686–698 CrossRef CAS.
- C. Y. Lau, M. T. Dunstan, W. Hu, C. P. Grey and S. A. Scott, Energy Environ. Sci., 2017, 10, 818–831 RSC.
- S. Wang, P. A. W. van der Heide, C. Chavez, A. J. Jacobson and S. B. Adler, Solid State Ionics, 2003, 156, 201–208 CrossRef CAS.
- J. Yoo, A. Verma, S. Wang and A. J. Jacobson, J. Electrochem. Soc., 2005, 152(3), A497 CrossRef CAS.
- L. Hou, Q. Yu, T. Wang, K. Wang, Q. Qin and Z. Qi, Korean J. Chem. Eng., 2018, 35, 626–636 CrossRef CAS.
- B. Bulfin, L. Hoffmann, L. de Oliveira, N. Knoblauch, F. Call, M. Roeb, C. Sattler and M. Schmucker, Phys. Chem. Chem. Phys., 2016, 18, 23147–23154 RSC.
- H. Song, K. Shah, E. Doroodchi, T. Wall and B. Moghtaderi, Energy Fuels, 2013, 28, 173–182 CrossRef.
- A. N. Antzaras, E. Heracleous and A. A. Lemonidou, Fuel Process. Technol., 2020, 208, 106513 CrossRef CAS.
- G. Luongo, F. Donat, A. H. Bork, E. Willinger, A. Landuyt and C. R. Müller, Adv. Energy Mater., 2022, 12, 2200405 CrossRef CAS.
- W. Ding, K. Zhao, S. Jiang, Z. Zhao, Y. Cao and F. He, Appl. Catal., A, 2021, 609, 117910 CrossRef CAS.
- X. Huang, Z. Yang, J. Qiu, B. Tang, C. Qin, Y. Yan and J. Ran, Fuel, 2022, 327, 125210 CrossRef CAS.
- Y. Tian, P. R. Westmoreland and F. Li, Catal. Today, 2022 DOI:10.1016/j.cattod.2022.04.026.
- X. Tian, C. Zheng and H. Zhao, Appl. Catal., B, 2022, 303, 120894 CrossRef CAS.
- W. Sun, G. Zhao, Y. Gao, J. Si, Y. Liu and Y. Lu, Appl. Catal., B, 2022, 304, 120948 CrossRef CAS.
- S. Parishan, P. Littlewood, A. Arinchtein, V. Fleischer and R. Schomäcker, Catal. Today, 2018, 311, 40–47 CrossRef CAS.
- J. Huang, K. Zhao, S. Jiang, S. Kang, Y. Lin, Z. Huang, A. Zheng and Z. Zhao, Fuel Process. Technol., 2022, 235, 107352 CrossRef CAS.
- V. Fleischer, U. Simon, S. Parishan, M. G. Colmenares, O. Görke, A. Gurlo, W. Riedel, L. Thum, J. Schmidt, T. Risse, K.-P. Dinse and R. Schomäcker, J. Catal., 2018, 360, 102–117 CrossRef CAS.
- S. Jiang, W. Ding, K. Zhao, Z. Huang, G. Wei, Y. Feng, Y. Lv and F. He, Fuel, 2021, 299, 120932 CrossRef CAS.
- D. S. Baser, Z. Cheng, J. A. Fan and L.-S. Fan, ACS Sustainable Chem. Eng., 2021, 9, 2651–2660 CrossRef CAS.
- Z. Cheng, D. S. Baser, S. G. Nadgouda, L. Qin, J. A. Fan and L.-S. Fan, ACS Energy Lett., 2018, 3, 1730–1736 CrossRef CAS.
- Y. Liu, L. Qin, Z. Cheng, J. W. Goetze, F. Kong, J. A. Fan and L.-S. Fan, Nat. Commun., 2019, 10, 5503 CrossRef CAS PubMed.
- M. Tian, C. Wang, Y. Han and X. Wang, ChemCatChem, 2021, 13, 1615–1637 CrossRef CAS.
- V. Shah, Z. Cheng, P. Mohapatra and L.-S. Fan, React. Chem. Eng., 2021, 6, 1928–1939 RSC.
- Y. Kang, M. Tian, C. Huang, J. Lin, B. Hou, X. Pan, L. Li, A. I. Rykov, J. Wang and X. Wang, ACS Catal., 2019, 9, 8373–8382 CrossRef CAS.
- L. Zhang, W. Xu, J. Wu, Y. Hu, C. Huang, Y. Zhu, M. Tian, Y. Kang, X. Pan, Y. Su, J. Wang and X. Wang, ACS Catal., 2020, 10, 9420–9430 CrossRef CAS.
- F. Donat, A. Kierzkowska and C. R. Müller, Energy Fuels, 2022, 36, 9780–9784 CrossRef CAS.
- L. Zhang, Y. Hu, W. Xu, C. Huang, Y. Su, M. Tian, Y. Zhu, H. Gong and X. Wang, Energy Fuels, 2020, 34, 6991–6998 CrossRef CAS.
- Y. Kang, Y. Han, M. Tian, C. Huang, C. Wang, J. Lin, B. Hou, Y. Su, L. Li, J. Wang and X. Wang, Appl. Catal., B, 2020, 278, 119305 CrossRef CAS.
- A. More, C. J. Hansen and G. Veser, Catal. Today, 2017, 298, 21–32 CrossRef CAS.
- V. Shah, Z. Cheng, D. S. Baser, J. A. Fan and L.-S. Fan, Appl. Energy, 2021, 282, 116111 CrossRef CAS.
- T. Wang, Y. Gao, Y. Liu, M. Song, J. Liu and Q. Guo, Fuel, 2021, 303, 121286 CrossRef CAS.
- L. Brody, L. Neal, J. Liu and F. Li, Energy Fuels, 2022, 36, 9736–9744 CrossRef CAS.
- L. Brody, L. Neal, V. Haribal and F. Li, Chem. Eng. J., 2021, 417, 128886 CrossRef CAS.
- S. Yusuf, L. Neal, Z. Bao, Z. Wu and F. Li, ACS Catal., 2019, 9, 3174–3186 CrossRef CAS.
- L. Neal, A. Shafiefarhood and F. Li, Appl. Energy, 2015, 157, 391–398 CrossRef CAS.
- Y. Ni, C. Wang, Y. Chen, X. Cai, B. Dou, H. Chen, Y. Xu, B. Jiang and K. Wang, Appl. Therm. Eng., 2017, 124, 454–465 CrossRef CAS.
- J. Y. Kim, N. Ellis, C. J. Lim and J. R. Grace, Fuel, 2020, 271, 117665 CrossRef CAS.
- Y. Han, M. Tian, C. Wang, Y. Kang, L. Kang, Y. Su, C. Huang, T. Zong, J. Lin, B. Hou, X. Pan and X. Wang, ACS Sustainable Chem. Eng., 2021, 9, 17276–17288 CrossRef CAS.
- C. de Leeuwe, W. Hu, J. Evans, M. von Stosch and I. S. Metcalfe, Chem. Eng. J., 2021, 423, 130174 CrossRef CAS.
- A. Hafizi, M. R. Rahimpour and M. Heravi, Int. J. Hydrogen Energy, 2019, 44, 17863–17877 CrossRef CAS.
- O. Condori, L. F. de Diego, F. Garcia-Labiano, M. T. Izquierdo, A. Abad and J. Adanez, Energy Fuels, 2021, 35, 17182–17196 CrossRef CAS PubMed.
- L. M. Neal, V. P. Haribal and F. Li, iScience, 2019, 19, 894–904 CrossRef CAS PubMed.
- R. Y. Chein and W. H. Hsu, Renewable Energy, 2020, 153, 117–129 CrossRef CAS.
- Q. Lai, T. Cai, S. C. E. Tsang, X. Chen, R. Ye, Z. Xu, M. D. Argyle, D. Ding, Y. Chen, J. Wang, A. G. Russell, Y. Wu, J. Liu and M. Fan, Sci. Bull., 2022, 67(20), 2124–2138 CrossRef CAS.
- R. J. Lee Pereira, P. A. Argyris and V. Spallina, Appl. Energy, 2020, 280, 115874 CrossRef CAS.
- M. M. Sarafraz and F. C. Christo, Energy Convers. Manage., 2021, 229, 113735 CrossRef CAS.
- W. Gao, J. Guo, P. Wang, Q. Wang, F. Chang, Q. Pei, W. Zhang, L. Liu and P. Chen, Nat. Energy, 2018, 3, 1067–1075 CrossRef CAS.
- X. Tian, C. Zheng, F. Li and H. Zhao, ACS Sustainable Chem. Eng., 2021, 9, 8002–8011 CrossRef CAS.
- M. S. Sukma, Y. Zheng, P. Hodgson and S. A. Scott, Energy Fuels, 2022, 36, 9410–9422 CrossRef CAS PubMed.
- V. P. Haribal, L. M. Neal and F. Li, Energy, 2017, 119, 1024–1035 CrossRef CAS.
- X. Zhu, Q. Imtiaz, F. Donat, C. R. Müller and F. Li, Energy Environ. Sci., 2020, 13, 772–804 RSC.
- S. Bhavsar and G. Veser, RSC Adv., 2014, 4, 47254–47267 RSC.
- Y. Zhang, F. Kong, A. Tong and L.-S. Fan, Ind. Eng. Chem. Res., 2020, 59, 5877–5890 CrossRef CAS.
- D. Li, R. Xu, X. Li, Z. Li, X. Zhu and K. Li, Energy Fuels, 2020, 34, 5381–5413 CrossRef CAS.
- S. Bhavsar, M. Najera, R. Solunke and G. Veser, Catal. Today, 2014, 228, 96–105 CrossRef CAS.
- L. M. Neal, S. Yusuf, J. A. Sofranko and F. Li, Energy Technol., 2016, 4, 1200–1208 CrossRef CAS.
- R. B. Dudek and F. Li, Fuel Process. Technol., 2021, 218, 106827 CrossRef CAS.
- V. P. Haribal, Y. Chen, L. Neal and F. X. Li, Engineering, 2018, 4, 714–721 CrossRef CAS.
- M. Badlani and I. E. Wachs, Catal. Lett., 2001, 75, 137–149 CrossRef CAS.
- S. Yusuf, L. M. Neal and F. Li, ACS Catal., 2017, 7, 5163–5173 CrossRef CAS.
- L. Zeng, Z. Cheng, J. A. Fan, L.-S. Fan and J. Gong, Nat. Rev. Chem., 2018, 2, 349–364 CrossRef CAS.
- A. Joshi, V. Shah, P. Mohapatra, S. Kumar, R. K. Joshi, M. Kathe, L. Qin, A. Tong and L.-S. Fan, Adv. Appl. Energy, 2021, 3, 100044 CrossRef.
- S. Chen, C. Pei, X. Chang, Z. J. Zhao, R. Mu, Y. Xu and J. Gong, Angew. Chem. Int. Ed., 2020, 59, 22072–22079 CrossRef CAS PubMed.
- L. M. Neal, A. Shafiefarhood and F. Li, ACS Catal., 2014, 4, 3560–3569 CrossRef CAS.
- A. Shafiefarhood, J. C. Hamill, L. M. Neal and F. Li, Phys. Chem. Chem. Phys., 2015, 17, 31297–31307 RSC.
- S. Yusuf, V. Haribal, D. Jackson, L. Neal and F. Li, Appl. Catal., B, 2019, 257, 117885 CrossRef CAS.
- S. Yusuf, L. Neal, V. Haribal, M. Baldwin, H. H. Lamb and F. Li, Appl. Catal., B, 2018, 232, 77–85 CrossRef CAS.
- Y. Gao, X. Wang, N. Corolla, T. Eldred, A. Bose, W. Gao and F. Li, Sci. Adv., 2022, 8, eabo7343 CrossRef CAS PubMed.
- Y. Gao, X. Wang, J. Liu, C. Huang, K. Zhao, Z. Zhao, X. Wang and F. Li, Sci. Adv., 2020, 6, eaaz9339 CrossRef CAS PubMed.
- F. Hao, Y. Gao, J. Liu, R. Dudek, L. Neal, S. Wang, P. Liu and F. Li, Chem. Eng. J., 2021, 409, 128192 CrossRef CAS.
- F. Hao, Y. Gao, L. Neal, R. B. Dudek, W. Li, C. Chung, B. Guan, P. Liu, X. Liu and F. Li, J. Catal., 2020, 385, 213–223 CrossRef CAS.
- R. B. Dudek, X. Tian, M. Blivin, L. M. Neal, H. Zhao and F. Li, Appl. Catal., B, 2019, 246, 30–40 CrossRef CAS.
- X. Tian, R. B. Dudek, Y. Gao, H. Zhao and F. Li, Catal. Sci. Technol., 2019, 9, 2211–2220 RSC.
- X. Zhu, Y. Gao, X. Wang, V. Haribal, J. Liu, L. M. Neal, Z. Bao, Z. Wu, H. Wang and F. Li, Nat. Commun., 2021, 12, 1329 CrossRef CAS PubMed.
- R. B. Dudek, X. Tian, M. Blivin, L. M. Neal, H. B. Zhao and F. X. Li, Appl. Catal., B, 2019, 246, 30–40 CrossRef CAS.
- S. Yusuf, L. M. Neal, V. P. Haribal, M. Baldwin, H. H. Lamb and F. Li, Appl. Catal., B, 2018, 232, 77–85 CrossRef CAS.
- J. Liu, S. Yusuf, D. Jackson, W. Martin, D. Chacko, K. Vogt-Lowell, L. Neal and F. Li, Appl. Catal., A, 2022, 646, 118869 CrossRef CAS.
- P. H. Bottelberghs, E. Everts and G. H. J. Broers, Mater. Res. Bull., 1976, 11, 263–267 CrossRef CAS.
- X. Ji, Y. Liu, J. Liu and J. Zhang, Appl. Catal., B, 2022, 307, 121194 CrossRef CAS.
- Y. Gao, X. Wang, J. Liu, C. Huang, K. Zhao, Z. Zhao, X. Wang and F. Li, Sci. Adv., 2020, 6(17), eaaz9339 CrossRef CAS PubMed.
- Y. Gao, X. Wang, N. Corolla, T. Eldred, A. Bose, W. Gao and F. Li, Sci. Adv., 2022, 8(30), eabo7343 CrossRef CAS PubMed.
- M. Cassir, G. Moutiers and J. Devynck, J. Electrochem. Soc., 2019, 140, 3114–3123 CrossRef.
- M. Muhler, J. Schütze, M. Wesemann, T. Rayment, A. Dent, R. Schlögl and G. Ertl, J. Catal., 1990, 126, 339–360 CrossRef CAS.
- T. Hirano, Appl. Catal., 1986, 26, 65–79 CrossRef CAS.
- O. Shekhah, W. Ranke and R. Schlögl, J. Catal., 2004, 225, 56–68 CrossRef CAS.
- Y. Gao, S. Wang, F. Hao, Z. Dai and F. Li, ACS Sustainable Chem. Eng., 2020, 8, 14268–14273 CrossRef CAS.
- N. K. Razdan and A. Bhan, J. Catal., 2020, 389, 667–676 CrossRef CAS.
- L. Y. Chen, L. W. Lin, Z. S. Xu, X. S. Li and T. Zhang, J. Catal., 1995, 157, 190–200 CrossRef CAS.
- M. Rahman, A. Sridhar and S. J. Khatib, Appl. Catal., A, 2018, 558, 67–80 CrossRef CAS.
- P. Mériaudeau, L. V. Tiep, V. T. T. Ha, C. Naccache and G. Szabo, J. Mol. Catal. A: Chem., 1999, 144, 469–471 CrossRef.
- D. Wang, J. H. Lunsford and M. P. Rosynek, Top. Catal., 1996, 3, 289–297 CrossRef CAS.
- R. W. Borry, Y. H. Kim, A. Huffsmith, J. A. Reimer and E. Iglesia, J. Phys. Chem. B, 1999, 103, 5787–5796 CrossRef CAS.
- Y. Xu, W. Liu, S.-T. Wong, L. Wang and X. Guo, Catal. Lett., 1996, 40, 207–214 CrossRef CAS.
- L. Wang, L. Tao, M. Xie, G. Xu, J. Huang and Y. Xu, Catal. Lett., 1993, 21, 35–41 CrossRef CAS.
- X. Wang, Y. Gao, E. Krzystowczyk, S. Iftikhar, J. Dou, R. Cai, H. Wang, C. Ruan, S. Ye and F. Li, Energy Environ. Sci., 2022, 15, 1512–1528 RSC.
- J. Fan, W. Li, S. Li and J. Yang, Adv. Sci., 2022, 9, e2202811 CrossRef PubMed.
- N. R. Singstock, C. J. Bartel, A. M. Holder and C. B. Musgrave, Adv. Energy Mater., 2020, 10(27), 2000685 CrossRef CAS.
- X. Zhang, C. Pei, X. Chang, S. Chen, R. Liu, Z. J. Zhao, R. Mu and J. Gong, J. Am. Chem. Soc., 2020, 142, 11540–11549 CrossRef CAS PubMed.
- D. Kang, H. S. Lim, M. Lee and J. W. Lee, Appl. Energy, 2018, 211, 174–186 CrossRef CAS.
- M. Hye Jeong, D. Hyung Lee, J. Won Moon, J. Sun, J. Soon Choi, D. Sik Hong, C.-H. Chung and J. Wook Bae, Chem. Eng. J., 2022, 433, 134621 CrossRef CAS.
- S. Chen, L. Zeng, R. Mu, C. Xiong, Z.-J. Zhao, C. Zhao, C. Pei, L. Peng, J. Luo, L.-S. Fan and J. Gong, J. Am. Chem. Soc., 2019, 141, 18653–18657 CrossRef CAS PubMed.
- H. Yan, W. Gao, Q. Wang, Y. Guan, S. Feng, H. Wu, Q. Guo, H. Cao, J. Guo and P. Chen, J. Phys. Chem. C, 2021, 125, 6716–6722 CrossRef CAS.
- C. Xiong, Y. Wu, M. Feng, J. Fang, D. Liu, L. Shen, M. D. Argyle, K. A. M. Gasem and M. Fan, Appl. Energy, 2022, 323, 119519 CrossRef CAS.
- D. F. Swearer, N. R. Knowles, H. O. Everitt and N. J. Halas, ACS Energy Lett., 2019, 4, 1505–1512 CrossRef CAS.
- C. Ruan, X. Wang, C. Wang, L. Zheng, L. Li, J. Lin, X. Liu, F. Li and X. Wang, Nat. Commun., 2022, 13, 718 CrossRef CAS PubMed.
- Q. Yang, M. Yan, L. Zhang, X. Xia, Y. Zhu, C. Zhang, B. Zhao, X. Ma and X. Wang, Energy Convers. Manage., 2021, 231, 113845 CrossRef CAS.
- I. S. Metcalfe, B. Ray, C. Dejoie, W. Hu, C. de Leeuwe, C. Dueso, F. R. Garcia-Garcia, C. M. Mak, E. I. Papaioannou, C. R. Thompson and J. S. O. Evans, Nat. Chem., 2019, 11, 638–643 CrossRef CAS PubMed.
- K. Kousi, D. Neagu, L. Bekris, E. Calì, G. Kerherve, E. I. Papaioannou, D. J. Payne and I. S. Metcalfe, J. Mater. Chem. A, 2020, 8, 12406–12417 RSC.
- R. Liu, C. Pei, X. Zhang, S. Chen, H. Li, L. Zeng, R. Mu and J. Gong, Chin. J. Catal., 2020, 41, 1140–1151 CrossRef CAS.
- Y. Q. Xu, B. W. Lu, C. Luo, J. Chen, Z. W. Zhang and L. Q. Zhang, Chem. Eng. J., 2021, 406, 126903 CrossRef CAS.
- C. Dang, Z. Li, J. Long, W. Yang and W. Cai, Fuel, 2022, 324, 124468 CrossRef CAS.
- L. C. Buelens, V. V. Galvita, H. Poelman, C. Detavernier and G. B. Marin, Science, 2016, 354, 449–452 CrossRef CAS PubMed.
- T. Xu, X. Wang, B. Xiao, H. Zhao and W. Liu, Fuel Process. Technol., 2022, 228, 107169 CrossRef CAS.
- V. P. Haribal, F. He, A. Mishra and F. Li, ChemSusChem, 2017, 10, 3402–3408 CrossRef CAS PubMed.
- A. Mishra, A. Shafiefarhood, J. Dou and F. Li, Catal. Today, 2020, 350, 149–155 CrossRef CAS.
- S. Iftikhar, W. Martin, Y. Gao, X. Yu, I. Wang, Z. Wu and F. Li, Catal. Today, 2022 DOI:10.1016/j.cattod.2022.07.022.
- H. S. Lim, M. Kim, Y. Kim, H. S. Kim, D. Kang, M. Lee, A. Jo and J. W. Lee, ACS Appl. Energy Mater., 2022, 5, 8437–8442 CrossRef CAS.
- M. Lee, H. S. Lim, Y. Kim and J. W. Lee, Energy Convers. Manage., 2020, 207, 112507 CrossRef CAS.
- V. Singh, L. C. Buelens, H. Poelman, M. Saeys, G. B. Marin and V. V. Galvita, J. CO2 Util., 2022, 61, 102014 CrossRef CAS.
- B. Jin, N. V. Srinath, H. Poelman, C. Detavernier, Z. Liang, G. B. Marin and V. V. Galvita, AIChE J., 2022, 68(9), e17779 CrossRef CAS.
- A. Longo, S. A. Theofanidis, H. Poelman, D. Banerjee, G. B. Marin and V. V. Galvita, ChemCatChem, 2022, 14(5) DOI:10.1002/cctc.202101627.
- H. Gu, Y. Gao, S. Iftikhar and F. Li, J. Mater. Chem. A, 2022, 10, 3077–3085 RSC.
- L. Brody, R. Cai, A. Thornton, J. Liu, H. Yu and F. Li, ACS Sustainable Chem. Eng., 2022, 10(19), 6434–6445 CrossRef CAS.
- R. Chang, X. Wu, O. Cheung and W. Liu, J. Mater. Chem. A, 2022, 10, 1682–1705 RSC.
- J. Hua, K. Wang, Q. Wang and R. Peng, J. Therm. Anal. Calorim., 2021, 146, 673–680 CrossRef CAS.
- S. W. Brown, B. Robinson, Y. Wang, C. Wildfire and J. Hu, J. Mater. Chem. A, 2022, 10, 15497–15507 RSC.
- R. J. L. Pereira, W. Hu and I. S. Metcalfe, Energy Fuels, 2022, 36, 9757–9767 CrossRef CAS PubMed.
- J. Liu, Y. Gao, X. Wang and F. Li, Cell Rep. Phys. Sci., 2021, 2, 100503 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |