
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrazone-based boron difluoride complexes as triplet photosensitizers for singlet oxygen generation†
Sven
van Vliet
,
Johannes G. H.
Hermens
 ,
Youxin
Fu
,
Lukas
Pfeifer‡
and
Ben L.
Feringa
*
,
Youxin
Fu
,
Lukas
Pfeifer‡
and
Ben L.
Feringa
*
Stratingh Institute for Chemistry, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, Groningen 9747 AG, The Netherlands
First published on 20th December 2022
Abstract
Due to the highly selective nature of singlet oxygen as an oxidant, it has received considerable interest in various areas of (organic) chemistry. Two green light activated hydrazone-based boron difluoride triplet photosensitizers possessing high quantum yields for 1O2 formation are reported. These photostable complexes are promising in applications in synthesis and catalysis.
Since the discovery of singlet oxygen in the early 1930's by Kautsky,1 it has received great interest and numerous applications have emerged in the fields of (drug)synthesis,2–6 photooxygenation,7–13 photodynamic therapy (PDT),14,15 bioimaging16 and in vivo oxygen sensing.17 Currently, it attracts renewed interest in the context of sustainable oxidations reactions using light and molecular oxygen.3,4,18,19 The most facile and convenient way of singlet oxygen generation is photosensitization, in which a sensitizer is electronically excited upon irradiation, populating its triplet state followed by energy transfer to ground state molecular oxygen (3O2) producing singlet oxygen (1O2).14 The triplet photosensitizer should fulfil certain criteria to achieve high photosensitizing efficiency: strong absorption in the visible or near-infrared region (NIR), high photostability, high triplet state interconversion probability and diminished phosphorescence.20 Although porphyrins,21 fullerene-based systems22,23 and transition metal complexes24 meet several of these criteria, their applicability is often constrained due to synthetic accessibility, high production costs, utilization of scarce metals, toxicity and processibility. To avoid such challenges, small molecule, metal-free organic photosensitizers represent a class of explicitly attractive alternatives. Organic dyes, such as Methylene Blue and Rose Bengal (RB),25 are commonly used photosensitizers for singlet oxygen generation, however their quantum yield for 1O2 generation might be limited.14 In this respect, organoboron complexes seem to be ideal candidates for employment as singlet oxygen photosensitizers due to their well-established and facile synthesis, the ability to absorb visible light and their tuneable and modular structure.26,27 Boron dipyrromethanes (BODIPYs) represent a privileged class of organoboron complexes which are frequently used for (biological) imaging applications28,29 due to their excellent photophysical and fluorescent properties.30 Moreover, it has been shown that certain BODIPYs are adequate triplet photosensitizers capable of singlet oxygen generation.31,32 Senge and co-workers have demonstrated that the formation of the triplet state and their lifetime can be strongly enhanced in donor–acceptor BODIPY dyads, in which a polyaromatic hydrocarbon or heteroaromatic arene donor moiety is connected via the meso position to the BODIPY acceptor (Fig. 1A).33,34 In these dyads, the charge transfer state, which is generated after photoinduced electron transfer, is directly converted into the lowest triplet state by spin–orbit charge transfer intersystem crossing. Recently, Durka et al. illustrated that rigid borafluorene complexes are behaving as singlet oxygen producing photosensitizers with quantum yields up to 78% in reference to methylene blue via a mechanism resembling the donor–acceptor BODIPY dyads (Fig. 1B).35
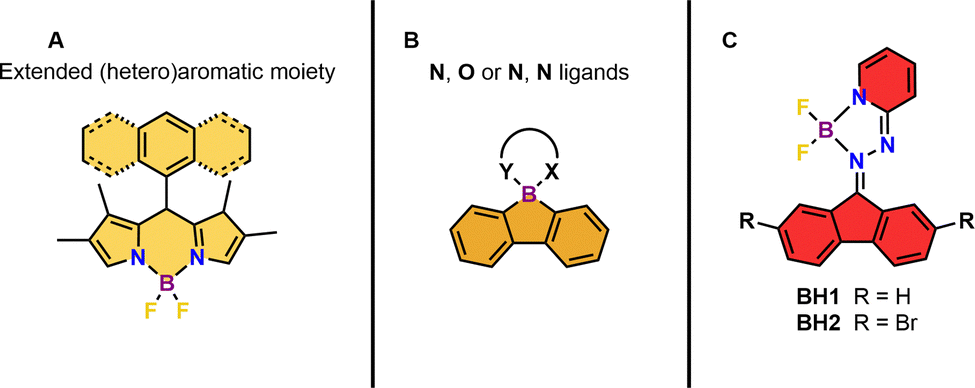 | ||
| Fig. 1 Earlier studies on organoboron-based triplet state photosensitizers (A) BODIPY-based sensitizers34 (B) borafluorene-based sensitizers.35 and (C) this work. | ||
Nevertheless, there are limitations regarding both systems in terms of multiple synthetic and purification steps, the introduction of polyaromatic and/or heteroaromatic functionalities at the meso position, the necessity of a second bidentate ligand, instability of the donor–acceptor dyad towards produced 1O2 and moderate singlet oxygen production in most cases.
Taking notice of the photosensitizing properties of BODIPY's and borafluorene complexes described above, we envisioned a hybrid photosensitizer in which a fluorene core is attached to a BODIPY-like upper half, creating a suitable ligand for boron complexation (Fig. 1C). In such hybrid systems, a fluorenone-based ketone is condensed with 2-pyridylhydrazine in order to obtain a bidentate ligand for the subsequent BF2 complexation. Herein, we report the design and short synthesis starting from cheap, readily available starting materials of hydrazone-based boron difluoride complexes acting as excellent triplet photosensitizers capable of producing singlet oxygen in high yields.
Starting from simple condensation reactions involving commercially available and inexpensive 2-hydrazinopyridine and 9-fluorenone or 2,7-dibromo-9-fluorenone, the bidentate hydrazone ligands were obtained in good yields after recrystallization. BF2 complexation was performed at elevated temperatures in toluene facilitated by Hünig's base in order to yield boron hydrazone complexes BH1 and BH2 in excellent yields (95% and 90%, respectively, Fig. 1C). Interestingly, these novel hydrazone-based organoboron compounds could be obtained in only two steps without the need of purification by column chromatography, pointing out the practical convenience of their synthesis.
Complex BH2 contains two bromide handles at the 2′ and 7′ position of the fluorene core for the purposes of (a) inducing a heavy atom effect for enhanced intersystem crossing due to increased spin–orbit coupling14 and (b) allowing further functionalization, through for example transition metal catalysed cross coupling reactions, of the whole system for the utilization in more complex, advanced materials, e.g. organic frameworks or polymers.
Single crystals suitable for X-ray diffraction of complex BH1 were grown by slow diffusion of hexane into a saturated CH2Cl2 solution (Fig. 2F). The boron coordination to the pyridyl nitrogen along with the hydrazino nitrogen forms a five-membered ring. The bond length between the pyridyl nitrogen and the boron centre is 1.546(2) Å, whereas the dative bond between the hydrazine nitrogen and the boron atom is slightly elongated 1.608(2) Å. In structurally related systems,36 no light-triggered B–N bond cleavage is observed when the bond length is 1.64 Å or shorter, making photoinduced B–N cleavage very unlikely in these complexes. Both complexes show excellent stability in the solid state as well in solution, such as CH2Cl2 and CH3CN, and appeared to be bench stable under aerobic conditions for at least three years.
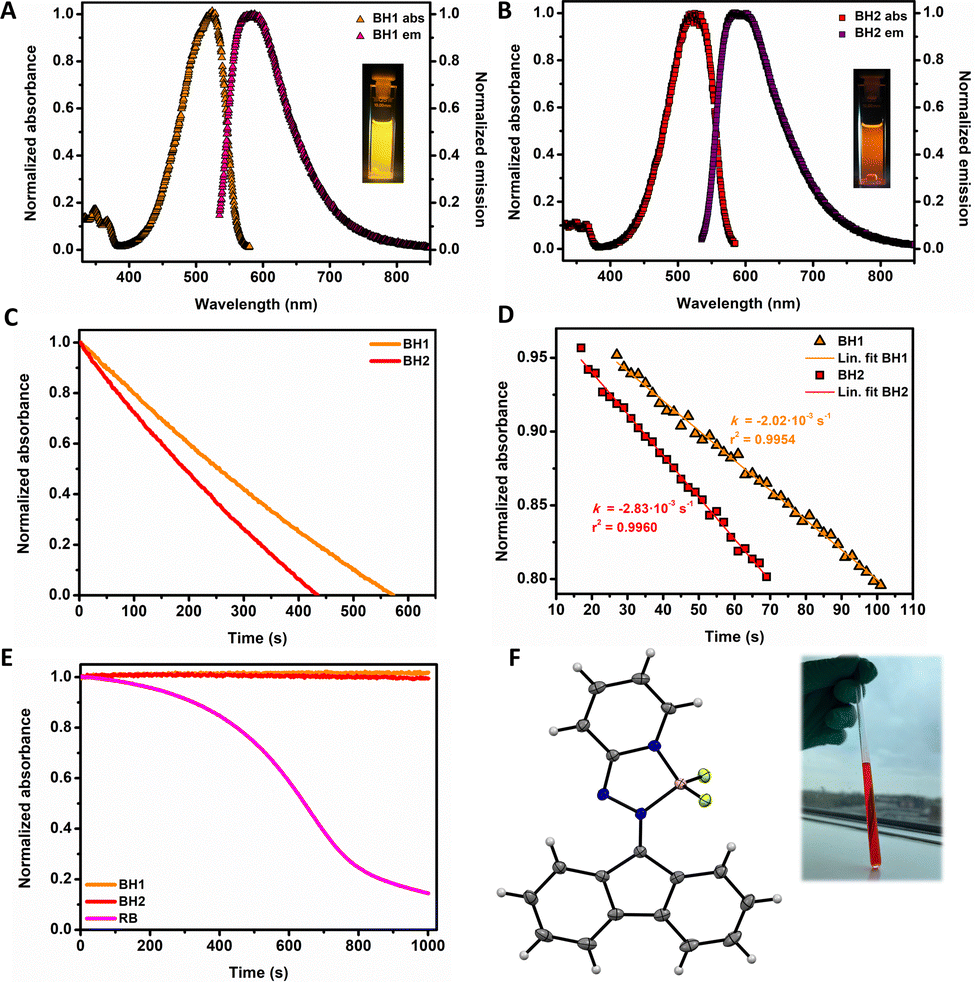 | ||
| Fig. 2 (A and B) Normalized UV-Vis and emission spectrum of BH1 and BH2 (CH2Cl2, 293 K) (C and D) Rate of singlet oxygen production of BH1 and BH2 monitored as function of decay of singlet oxygen scavenger followed by UV-Vis spectroscopy measured in CH3CN (293 K). (E) Decay of photosensitizers BH1, BH2 and RB (3 μM) over time (s) monitored at the main absorption bands in CH3CN at 293 K. (F) ORTEP image (ellipsoid at 50% probability) of complex BH1 Inset: BH1 in solution (CD2Cl2). | ||
We started our study of these new photosensitizers by comparing the elementary photophysical properties of both complexes in solution (Table 1, Fig. 2). Complexes BH1 and BH2 show sharp absorption maxima around 520 nm (λmax = 525 nm and 519 nm, resp.), whilst having the emission maxima around 580 nm (λmax = 588 nm and 584 nm, resp.), giving rise to Stokes shifts of approximately 2100 cm−1 (Table 1). BH2 exhibits a lower luminescence quantum yield in comparison to complex BH1, presumably due to advantageous heavy atom effect on the triplet state interconversion probability.24 As the onset of phosphorescence occurs at approximately 820 nm for both complexes (Fig. 2), the corresponding energy gaps between the ground state and triplet state are 146 kJ mole−1, indicating there is sufficient energy for sensitization of molecular oxygen to its singlet excited state (95 kJ mole−1).14
To investigate the efficiency of 1O2 generation of both complexes, the singlet oxygen sensitization was monitored by UV-Vis spectroscopy as a function of the decay of a singlet oxygen scavenger (followed at λ = 441 nm) upon green light irradiation (λirr = 535 nm) (Fig. 2). For this study, 1,3-diphenylisobenzofuran (DPBF) was used as a singlet oxygen scavenger as its absorption maximum (see ESI†) does not interfere with the absorption maxima of both organoboron complexes. Furthermore a stable oxidation product, 1,3-dibenzoylbenzene, is formed which does not absorb in the visible light region and does not further react under the conditions employed. Derived from the decay of scavenger, which is present in a 40-fold excess, essentially capturing every formed molecule of singlet oxygen, BH1 and BH2 show excellent quantum yields for singlet oxygen evolution (ΦΔ); both complexes BH1 and BH2 exhibit a high 1O2 quantum yield of 0.41(±0.02) and 0.46(±0.02), resp., (Table 1), comparable to RB (lit. value ΦΔ = 0.53).37
To assess the stability of both photosensitizers, oxygen saturated solutions of the complexes in CH2Cl2 and CH3CN were irradiated for a prolonged period of time in the absence of the singlet oxygen scavenger (Fig. S5 and S6, ESI†). To our delight, both complexes show exceptional stability in CH3CN over time compared to the commercially available and widely used photosensitizer RB (Fig. 2E), thereby indicating these complexes are highly stable towards 1O2 mediated self-sensitized photochemical oxidation, a common challenge in the field of singlet oxygen photosensitization. In CH2Cl2, dibromo complex BH2 showed to be slightly more stable over time compared to non-functionalized complex BH1.
Prompted by these encouraging results, the complexes were employed as photocatalysts for green light-induced 1O2-mediated oxidation reactions of selected organic model substrates, namely 1,3-diphenylisobenzofuran (DPBF), 2-furoic acid (2-FA) and triphenylphosphine (TPP) (Fig. 3). Photocatalytic oxidations were conducted with 5 mol% of photosensitizer and irradiation performed at their absorption maxima (λirr = 535 nm) in CH2Cl2 or CH3CN (0.1 M), two commonly used solvents in organic synthesis. For both complexes, all 1O2-mediated reactions reached full conversion within a reasonable and practical period of time; the maximum reaction time showed to be 5 h. The starting point was the photooxidation of DPBF, initially used as 1O2 scavenger, going via a [4+2] cycloaddition of singlet oxygen in order to generate the endo-peroxide which quickly decomposes at ambient temperatures.38 In CH3CN, both photosensitizers BH1 and BH2 quantitatively oxidize DBPF within 2 h (Fig. 3A). The oxidation of 2-FA proceeds via a similar mechanism, a [4+2] cycloaddition, as DPBF, to produce the endo-peroxide, which in this case subsequently undergoes a decarboxylation reaction to yield 5-hydroxyfuran-2(5H)-one as photooxidation product (Fig. 3B).39 Both photocatalysts quantitatively oxidized the 2-FA substrate, however BH2 showed to be a more efficient catalyst as full conversion was reached within 2 h instead of reaching full conversion after 5 h for BH1. TPP, like other triaryl phosphines, is slowly oxidized in air under ambient conditions. It is known that the oxidation process for triaryl phosphines is greatly enhanced in the presence of 1O2.40
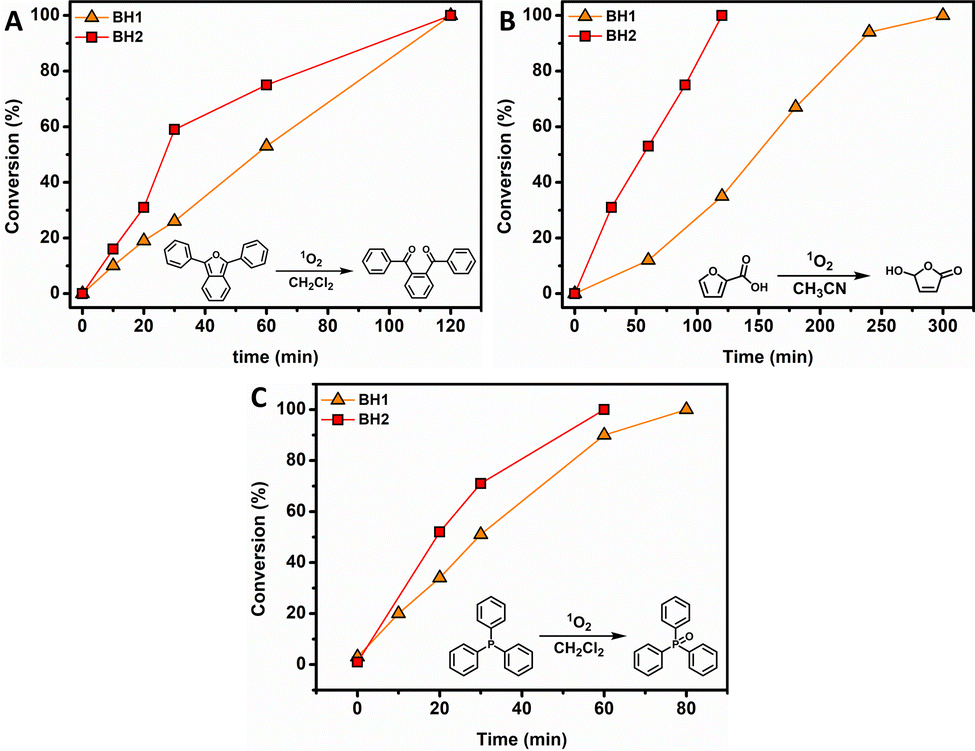 | ||
| Fig. 3 Kinetic plots of 1O2 mediated oxidation reactions involving model substrates 1,3-diphenylisobenzofuran (A), 2-furoic acid (B) and triphenylphosphine (C). | ||
The oxidation of TPP, used as model substrate, in the presence of 1O2 showed to be completed within 1 h for photocatalyst BH2, whereas BH1 exhibited a slightly longer reaction time (80 min, Fig. 3C). Control experiments show no product formation in the absence of BH1 and BH2, confirming their photosensitization properties.
In line with the efficiency of 1O2 formation, complex BH2 outperformed complex BH1 in all model oxidations. The findings above prove that these kind of easily accessible hydrazone-based BF2 complexes serve as excellent photocatalysts, especially the bromo-functionalized complex BH2, opening up pathways for their use in (organic) synthesis and photocatalysis.
The photooxidation of 2-FA using BH2 was performed on a preparative 1 g scale using only 0.5 mol% of photosensitizer and provided us quantitative amounts of 5-hydroxyfuran-2(5H)-one (hydroxybutenolide), an emerging biobased acrylate alternative precursor.3
To conclude, we have shown a pair of promising triplet state photosensitizers consisting of hydrazone-based boron difluoride complexes. The complexes have demonstrated to possess excellent (photo)physical properties, such as green light absorption, high singlet oxygen evolution quantum yields (up to 46%) and desirable stability towards self-sensitized photooxidation in solution. The photooxidation experiments clearly indicate these hydrazone-based organoboron triplet state sensitizers behave as practical photocatalysts, as various model substrates were oxidized within only a few hours. In combination with the commercial availability of the inexpensive starting materials, their short synthesis route, the absence of tedious purification steps and the opportunity for further functionalization, it is evident that these kind of boron difluoride photosensitizers serve as attractive candidates for exploration in the areas of (heterogeneous) photooxygenation and sustainable synthesis.
B. L. F. designed the study. S. V. V. and B. L. F. prepared the manuscript with input from J. G. H. H. S. V. V. synthesized the investigated compounds and performed characterization. J. G. H. H. performed the quantum yield and photooxygenation experiments. Y. F. measured the luminescence. L. P. performed the X-ray elucidation and analysed the data. All authors reviewed the manuscript. B. L. F. acquired funding.
S. V. V. gratefully acknowledges J. L. Sneep for performing HRMS experiments. Financial support is provided by the Netherlands Ministry of Education, Culture and Science (Gravitation Programme to B. L. F.), the Ministry of Economic Affairs and Climate (Advanced Research Center for Chemical Building Blocks, ARC CBBC, which is cofounded and cofinanced by the Netherlands Organization for Scientific Research, NWO contract 736.000.000 to B. L. F.) and the European Commission (MSCA-IF No. 793082 to L. P.).
Conflicts of interest
The authors declare there to be no conflicts of interest.Notes and references
- H. Kautsky, Ber. Dtsch. Chem. Ges., 1931, 64, 2677–2683 CrossRef
.
- A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116(17), 9994–10034 CrossRef CAS PubMed
- J. G. H. Hermens, T. Freese, K. J. Van den Berg, R. Van Gemert and B. L. Feringa, Sci. Adv., 2020, 6, eabe0026 CrossRef CAS PubMed
- J. G. H. Hermens, A. Jensma and B. L. Feringa, Angew. Chem., Int. Ed., 2022, 61, e202112618 CrossRef CAS PubMed
- B. L. Feringa, Tetrahedron Lett., 1981, 22, 1443–1446 CrossRef CAS
- L. Melters, O. J. Gelling and B. L. Feringa, Tetrahedron Lett., 1990, 31, 7201–7204 CrossRef
-
C. S. Foote and E. L. Clennan, Properties and Reactions of Singlet Dioxygen, in Active Oxygen in Chemistry, ed. C. S. Foote, J. S. Valentine, A. Greenberg and J. F. Liebman, Springer, Dordrecht, Netherlands, 1995, pp. 105–140 Search PubMed
-
B. König, in Chemical Photocatalysis, ed. B. B. König, De Gruyter, Boston, Berlin, 2013, pp. 45–61 Search PubMed
- F. Lévesque and P. H. Seeberger, Org. Lett., 2011, 13, 5008–5011 CrossRef PubMed
- P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181–3209 RSC
- C. Sambiagio and T. Noël, Trends Chem., 2020, 2, 92–106 CrossRef CAS
- B. L. Feringa, Recl. Trav. Chim. Pays-Bas, 1987, 106, 469–488 CrossRef CAS
- B. L. Feringa and R. J. Butselaar, Tetrahedron Lett., 1981, 23, 1941–1942 CrossRef
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233, 351–371 CrossRef
- I. Pibiri, S. Buscemi, A. P. Piccionello and A. Pace, ChemPhotoChem, 2019, 2, 535–547 CrossRef
- J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795–2838 CrossRef CAS
- J. A. Spencer, F. Ferraro, E. Roussakis, A. Klein, J. Wu, J. M. Runnels, W. Zaher, L. J. Mortensen, C. Alt, R. Turcotte, R. Yusuf, D. Côté, S. A. Vinogradov, D. T. Scadden and C. P. Lin, Nature, 2014, 208, 269–273 CrossRef PubMed
- C. A. Clark, D. S. Lee, S. J. Pickering, M. Poliakoff and M. W. George, Org. Process Res. Dev., 2016, 20, 1792–1798 CrossRef CAS
- D. S. Lee, Z. Amara, C. A. Clark, Z. Xu, B. Kakimpa, H. P. Morvan, S. J. Pickering, M. Poliakoff and M. W. George, Org. Process Res. Dev., 2017, 21, 1042–1050 CrossRef CAS PubMed
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC
- L. Cheng, H. Bai, J. F. Xu, S. Wang and X. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 13950–13957 CrossRef PubMed
- A. Blacha-Grzechnik, M. Krzywiecki, R. Motyka and M. Czichy, J. Phys. Chem. C, 2019, 123, 25915–25924 CrossRef CAS
- D. M. Guldi and M. Prato, Acc. Chem. Res., 2000, 33, 695–703 CrossRef CAS PubMed
- L. K. McKenzie, H. E. Bryant and J. A. Weinstein, Coord. Chem. Rev., 2019, 379, 2–29 CrossRef CAS
- P. Murasecco-Suardi, E. Gassmann, A. M. Braun and E. Oliveros, Helv. Chim. Acta, 1987, 70, 1760–1773 CrossRef CAS
- A. Haque, R. A. Al-Balushi, P. R. Raithby and M. S. Khan, Molecules, 2020, 25, 2645 CrossRef PubMed
- A. N. Bismillah and I. Aprahamian, Chem. Soc. Rev., 2021, 50, 5631–5649 RSC
- T. Kowada, H. Maedab and K. Kikuchi, Chem. Soc. Rev., 2015, 44, 4953–4972 RSC
- Y. Niab and J. Wu, Org. Biomol. Chem., 2014, 12, 3774–3791 RSC
- L. J. Patalag, J. Hoche, R. Mitric, D. B. Werz and B. L. Feringa, Angew. Chem., Int. Ed., 2022, 61, e202116834 CrossRef
- A. Turksoy, D. Yildiz and E. U. Akkaya, Coord. Chem. Rev., 2019, 379, 47–64 CrossRef
- K. Chen, Y. Dong, X. Zhao, M. Imran, G. Tang, J. Zhao and Q. Li, Front. Chem., 2019, 7, 821 CrossRef CAS
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, H. Savoie, K. J. Flanagan, C. Sy, E. Sitte, M. Telitchko, F. Laquai, R. W. Boyle and M. O. Senge, J. Am. Chem. Soc., 2017, 139, 6282–6285 CrossRef CAS
- M. A. Filatov, Org. Biomol. Chem., 2020, 18, 10–27 RSC
- P. H. Marek-Urban, M. Urban, M. Wiklińska, K. Paplińska, K. Woźniak, A. Blacha-Grzechnik and K. Durka, J. Org. Chem., 2021, 86, 12714–12722 CrossRef CAS PubMed
- Y. Yang, R. P. Hughes and I. Aprahamian, J. Am. Chem. Soc., 2012, 134, 15221–15224 CrossRef CAS PubMed
- W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1993, 22, 113–262 CrossRef
- J. A. Howard and G. D. Mendenhall, Can. J. Chem., 1975, 53, 2199–2201 CrossRef CAS
- J. A. Navio, J. F. Mota, M. A. Pradera Adrian and M. Garcia Gomez, J. Photochem. Photobiol., A, 1990, 52, 91–95 CrossRef CAS
- R. Gao, D. G. Ho, T. Dong, D. Khuu, N. Franco, O. Sezer and M. Selke, Org. Lett., 2001, 3, 3719–3722 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2209988. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc05336e |
| ‡ Current address: EPFL, CH G1 614 (Bâtiment CH), Station 6, CH-1015, Lausanne, Switzerland. |
| This journal is © The Royal Society of Chemistry 2023 |