
A self-delivery photodynamic sensitizer for enhanced DNA damage by PARP inhibition†
Ren-Jiang
Kong
a,
Xin-Yu
Li
a,
Jia-Qi
Huang
a,
Xiang
Zhou
a,
Fu-An
Deng
b,
Yan-Mei
Li
a,
Ling-Shan
Liu
a,
Shi-Ying
Li
 b and
Hong
Cheng
*a
b and
Hong
Cheng
*a
aSchool of Biomedical Engineering & Guangdong Provincial Key Laboratory of Construction and Detection in Tissue Engineering, Southern Medical University, Guangzhou 510515, P. R. China. E-mail: chengh@smu.edu.cn
bSchool of Pharmaceutical Sciences, Guangzhou Medical University, Guangzhou 511436, P. R. China
First published on 4th November 2022
Abstract
Tumor cells activate DNA repair pathways to combat the oxidative damage induced by reactive oxygen species (ROS), contributing to their resistance to photodynamic therapy (PDT). Herein, a self-delivery photodynamic sensitizer is developed to enhance oxidative damage by blocking the DNA repair pathway through poly(ADP-ribose) polymerase (PARP) inhibition. Specifically, the photodynamic sensitizer (CeOla) is constructed based on the self-assembly of the photosensitizer chlorine e6 (Ce6) and the PARP inhibitor olaparib (Ola). Of note is that carrier free CeOla has a high drug content and favorable water stability, which could be effectively internalized by tumor cells for robust PDT upon light irradiation. Moreover, CeOla could inhibit the activation of PARP, promote the upregulation of γ-H2AX and reduce the expression of Rad51, thereby blocking the DNA repair pathway to sensitize tumor cells for PDT. As a consequence, the self-delivery CeOla greatly promotes the tumor cell apoptosis and shows a high antitumor performance with low side effects. It serves as a novel platform for the development of self-delivery nanomedicine to overcome oxidative resistance in tumor treatment.
Introduction
Photodynamic therapy (PDT) is a powerful strategy for surficial tumor elimination, which possesses great advantages of non-invasiveness and on-demand controllability.1–4 Mechanically, appropriate light excited photosensitizers can transfer protons to the nearby O2 to produce reactive oxygen species (ROS), resulting in oxidative damage of DNA, proteins and unsaturated lipids.5–7 Particularly, ROS-induced DNA damage, including DNA strand breaks (DSBs) and base disruptions, could directly cause functional inactivation and growth arrest of tumor cells.8–10 However, tumor cells would immediately initiate a sophisticated cellular repair machinery to combat DNA damage, which is recognized as a major obstacle to hinder DNA damage-associated therapeutic modalities.11–13 Of note is that poly(ADP-ribosyl)ation is a DNA-damage dependent post-translational modifier for protecting injured proliferating cells from death. Among which, poly(ADP-ribose) polymerase (PARP) family proteins, especially PARP1 and PARP2, are the sole catalytic enzymes that respond fast to initiate DNA repair.14–16 Some recent works found that PARP inhibitors were able to sensitize tumor cells and amplify the antitumor effect of DNA damaging agents.17–21 Among which, olaparib as an oral PARP inhibitor has been approved for the treatment of patients with recurrent ovarian cancer and a BRCA mutation. Moreover, it also shows promising activity in patients with metastatic breast or prostate cancer. However, investigations about PARP inhibition and PDT have been rarely reported, and an integrated system could be developed based on a photosensitizer and PARP inhibitor for enhanced DNA damage.In the past few decades, drug delivery systems (DDSs) have met with huge successes in improving the solubility and selectivity and reducing the blood/renal clearance and adverse side effects of drugs.22–24 Very recently, self-delivery nanomedicine (SDN) was developed by the self-assembly of therapeutic agents in the absence of non-therapeutic carriers, which attracted great attention for high-performance tumor therapy. Compared with traditional DDSs, SDN could not only improve the physicochemical and pharmacological behaviors of drugs, but also avoid the carrier-induced toxicity and immunogenicity.25–28 More importantly, the simplified drug composition and synthetic procedure of SDN provided a repeatable and scalable strategy to prepare nanomedicine with extremely high drug loading contents. Unfortunately, the self-assembly performance is rarely satisfactory with the synergistic mechanism due to the inherent structure of drugs.29–31 In this case, the development of photosensitizer and PARP inhibitor based SDN becomes deeply rewarding but really challenging.
In view of this, we developed a self-delivery photodynamic sensitizer (designated as CeOla) for PDT amplified DNA damage by PARP inhibition. As illustrated in Scheme 1, the photosensitizer chlorine e6 (Ce6) and the PARP inhibitor olaparib (Ola) could self-assemble into stable nanosheets of CeOla through intermolecular hydrophobic interactions. Importantly, carrier free CeOla possessed a high drug loading rate (nearly 100%), good dispersibility and uniform nanosize distribution. Furthermore, CeOla improved the cellular uptake behavior for effective drug delivery, which was beneficial for exerting synergistic activities. Under light irradiation, large amounts of 1O2 could be produced to destroy DNA and promote the upregulation of γ-H2AX. Meanwhile, CeOla could also successively inhibit the DNA-damage response and immediately activate PARP1, which would block the DNA repair pathway and sensitize the PDT effect. Both in vitro and in vivo investigations demonstrated that CeOla exhibited a high antitumor performance with no obvious adverse effects. This work provides an insight into the development of SDN and also paves a new way to enhance the PDT efficiency by inhibiting the cell repair machinery.
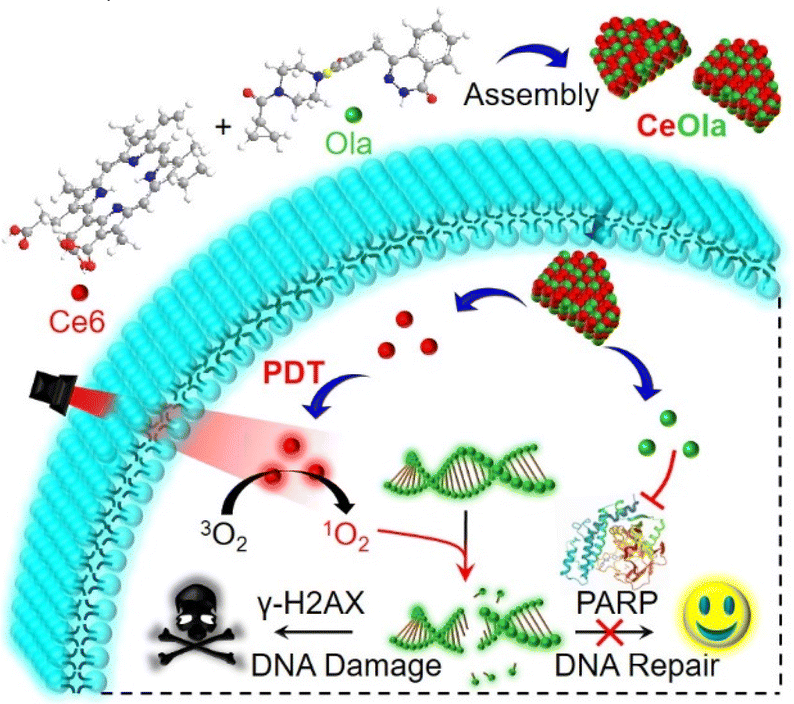 | ||
| Scheme 1 Schematic illustration of CeOla for enhanced DNA damage by PARP inhibition. Ce6 and Ola could self-assemble into the nanosheet of CeOla without any excipients, which increased the drug uptake by tumor cells. Under light irradiation, the photodynamic therapy (PDT) of CeOla produced large amounts of ROS to induce DNA damage and increase γ-H2AX accumulation. The concomitant delivery of Ola would inhibit the activation of PARP1, which would reduce the DNA repair and enhance the oxidative damage for enhanced PDT. | ||
Results and discussion
Preparation and characterization
A self-delivery photodynamic sensitizer was prepared by the self-assembly of Ce6 and olaparib (Ola) through non-covalent interactions. In order to obtain the optimum nanomedicine, the self-assembly behaviors of Ce6 and Ola at different feeding ratios were investigated. As shown in Fig. 1A, the morphologies of the harvested formulations were observed by transmission electron microscopy (TEM). When the dosage ratio of Ce6 and Ola was 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, there was difficulty in forming nanoparticles. After adjusting the ratio to 2:1, Ce6 and Ola seemed to assemble into uniform nanoparticles. However, at lower feed ratios of 1:1 and 1:2, the obtained nanoparticles had wide particle size distributions (Fig. S1†) and non-uniform morphologies (Fig. 1A), and could easily aggregate in water. By contrast, the nanomedicine assembled at the 2:1 ratio exhibited a narrow size distribution and the zeta potential was confirmed to be negative (Fig. 1B). The hemolytic test indicated that the nanomedicine had a low hemolysis rate (less than 5%) in vitro (Fig. S2†), suggesting good biosafety. Moreover, it was found that the mean size and PDI values had no significant difference in 60 h (Fig. 1C). Of special note is that the size and PDI of the nanomedicine also exhibited a relatively small fluctuation in a week in PBS and cell culture medium (Fig. S3†). This favorable stability of the self-delivery nanomedicine suggested its potential for biomedical applications, and it was thus chosen for following studies.
:1, there was difficulty in forming nanoparticles. After adjusting the ratio to 2:1, Ce6 and Ola seemed to assemble into uniform nanoparticles. However, at lower feed ratios of 1:1 and 1:2, the obtained nanoparticles had wide particle size distributions (Fig. S1†) and non-uniform morphologies (Fig. 1A), and could easily aggregate in water. By contrast, the nanomedicine assembled at the 2:1 ratio exhibited a narrow size distribution and the zeta potential was confirmed to be negative (Fig. 1B). The hemolytic test indicated that the nanomedicine had a low hemolysis rate (less than 5%) in vitro (Fig. S2†), suggesting good biosafety. Moreover, it was found that the mean size and PDI values had no significant difference in 60 h (Fig. 1C). Of special note is that the size and PDI of the nanomedicine also exhibited a relatively small fluctuation in a week in PBS and cell culture medium (Fig. S3†). This favorable stability of the self-delivery nanomedicine suggested its potential for biomedical applications, and it was thus chosen for following studies.
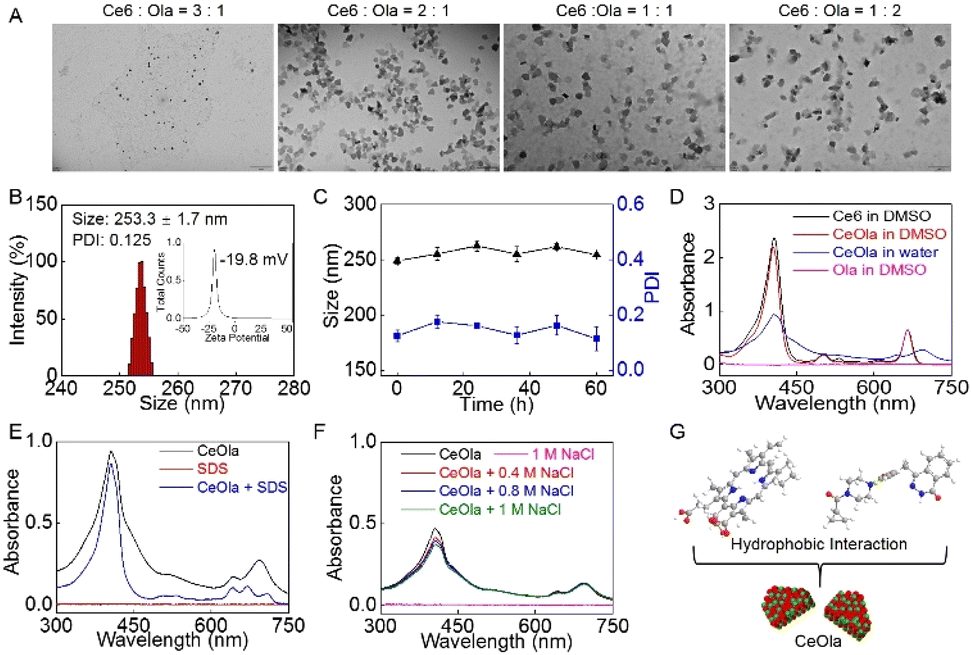 | ||
| Fig. 1 The self-assembly of Ce6 and Ola to prepare CeOla. (A) TEM observations of various formulations obtained at the feed ratio of 3:1, 2:1, 1:1 or 1:2. Scale bar: 200 nm. (B) The hydrodynamic size distribution and zeta potential (inserted) of CeOla. (C) Particle size and PDI variations of CeOla in 60 h. (D) UV-vis spectra of Ce6, CeOla and Ola in water or DMSO. (E) UV-vis spectrum of CeOla in the absence or presence of SDS. (F) UV-vis spectra of CeOla treated with different concentrations of NaCl. UV-vis spectra of SDS and NaCl were used as controls. (G) The proposed mechanism of Ce6 and Ola to self-assemble into CeOla through hydrophobic interactions. | ||
In order to explore the self-assembly mechanism of the nanomedicine, the ultraviolet-visible (UV-Vis) absorption spectrum of CeOla was recorded under different conditions. First, CeOla was dissolved in water or DMSO to record the UV-Vis spectrum (Fig. 1D). Compared with free Ce6, CeOla had a wider and red-shifted Soret band, proving the existence of non-covalent forces in the self-assembly of Ce6 and Ola. But CeOla in DMSO presented a similar absorption spectrum to Ce6, which implied the destruction of the assembled nano-structure by DMSO. Furthermore, CeOla was also treated with hydrophobic sodium dodecyl sulfate (SDS, 0.2% w/v). As displayed in Fig. 1E, the addition of SDS obviously altered the characteristic absorption of CeOla, demonstrating that hydrophobic interactions should be responsible for the assembly of the nanomedicine. Specifically, the hydrophobic interaction between the nonpolar groups of Ce6 and Ola molecules could promote their aggregation and assembly in water solution. Nevertheless, sodium chloride (NaCl) had a very limited impact on the absorption of CeOla even at a high concentration (Fig. 1F), indicating that there was negligible electrostatic interaction between Ce6 and Ola and NaCl could not destroy their self-assembly. Based on the above, it was inferred that the hydrophobic interaction might be the main driving force behind the self-assembly of CeOla (Fig. 1G). Besides, since the self-delivery nanomedicine had no additional carriers, CeOla exhibited a rather high drug loading efficiency. The standard curves of Ce6 and Ola were obtained by high performance liquid chromatography (HPLC) and UV-Vis spectroscopy (Fig. S4†). The contents of Ce6 and Ola in the nanomedicine were calculated to be nearly 59.4% and 40.6%, respectively. Meanwhile, the encapsulation efficiencies of Ce6 and Ola were 63.3% and 43.5% respectively. In an acidic tumor environment, CeOla could be disassembled to release the active drugs for tumor treatment (Fig. S5†).
Cell internalization of CeOla for ROS production and PARP inhibition
The cellular uptake behavior of CeOla was investigated by using Ce6 as the control. Briefly, after incubation with different concentrations of Ce6 or CeOla, 4T1 cells were analyzed by CLSM and flow cytometry to detect the intracellular fluorescence. As shown in Fig. 2A, 4T1 cells treated with CeOla had much stronger red fluorescence than those treated with Ce6, which illustrated an improved drug delivery efficiency of CeOla. Quantitative outcomes of flow cytometry also led us to arrive at the same conclusion (Fig. 2B). Regardless of the administration dosages, the uptake of CeOla was several times more than that of Ce6, which might be mainly ascribed to the enhanced solubility of the drugs after their assembly into the nanomedicine. Moreover, the cellular uptake gradually increased with the increased drug concentration, suggesting a dose-dependent cell internalization. In short, the findings highlighted the superiority of this self-assembled nanomedicine in improving the drug delivery efficiency.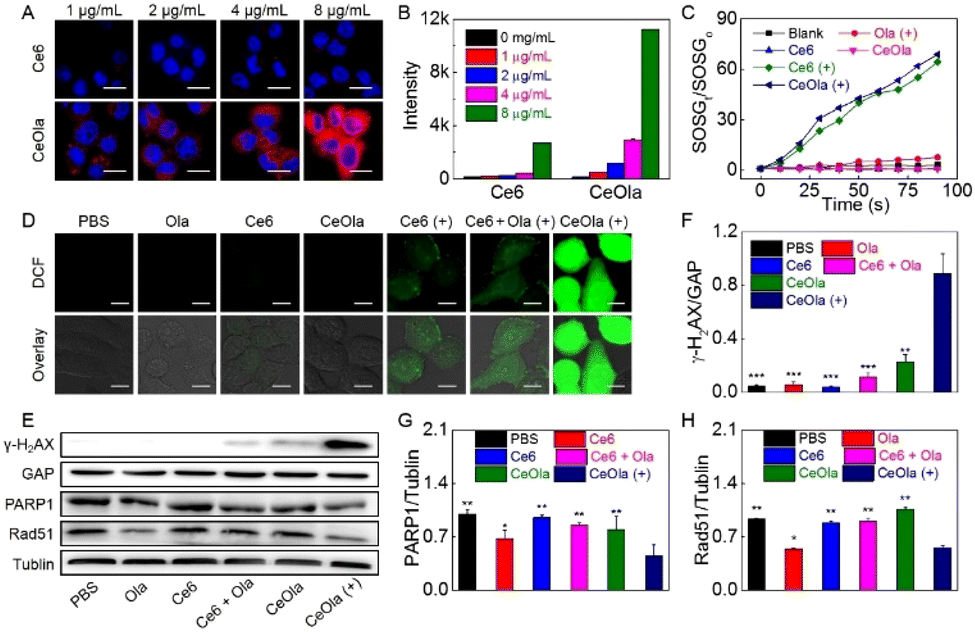 | ||
| Fig. 2 Cellular uptake and ROS production of CeOla to enhance DNA damage by PARP inhibition. (A) CLSM images and (B) flow cytometry analysis of 4T1 cells after treatment with gradient concentrations of Ce6 and CeOla. Scale bar: 25 μm. (C) 1O2 production of Ola, Ce6, and CeOla in the presence or absence of light. (D) CLSM images of ROS generation of 4T1 cells after treatment with Ce6, Ola, Ce6 + Ola or CeOla in the presence or absence of light. Scale bar: 20 μm. (E) Western blot analysis of γ-H2AX, PARP1 and Rad51 expressions on 4T1 cells after treatment with Ola, Ce6, Ce6 + Ola or CeOla in the presence or absence of light. The relative expression analysis of (F) γ-H2AX, (G) PARP1 and (H) Rad51 in 4T1 cells after treatment with Ola, Ce6, Ce6 + Ola or CeOla in the presence or absence of light. *P < 0.05, **P < 0.01 and ***P < 0.005 were tested via a Student's t-test. | ||
The in vitro ROS generation of CeOla was assessed using a singlet oxygen fluorescence probe (SOSG) (Fig. 2C). In the absence of light or photosensitizers, there were no obvious trends of increasing SOSG fluorescence, indicating negligible singlet oxygen generation. In contrast, both Ce6 and CeOla after irradiation led to a continuous fluorescence enhancement of SOSG over time, demonstrating high ROS yields. Besides, self-assembled CeOla exhibited a comparable photodynamic performance to Ce6. Furthermore, DCFH-DA was used as a probe to test the production of ROS in cells. DCFH-DA could be oxidized by ROS to show green fluorescence. As shown in Fig. 2D, without light irradiation, almost no green fluorescence was observed in 4T1 cells after various treatments due to the failure of initiating the photodynamic reaction process. Conversely, once exposed to excitation light, 4T1 cells treated with CeOla could generate large amounts of ROS with a strong green fluorescence. However, Ce6 was observed to generate very little ROS in the cells, regardless of its good photodynamic properties. The main reason for this might be the limited uptake of Ce6 by 4T1 cells. These results demonstrated that the improved drug delivery efficiency of CeOla contributed to the improvement of the PDT efficiency.
To assess the ability of CeOla to inhibit DNA repair, the expression of γ-H2AX was first assessed by western blotting analysis (Fig. 2E). γ-H2AX is a biomarker of DNA double bond breakage, which can clearly reflect the degree of DNA damage. The quantitative results showed that the expression of γ-H2AX in the CeOla with light group was significantly higher than that in other control groups (Fig. 2F), indicating serious DNA damage in the cells owing to the robust PDT. Besides, PARP1 is the most important member of the PARP family, which carries out more than 90% of the functions of the PARP family in cells and is a key factor in DNA damage repair.32,33 Disruption of PARP could suppress the expression of RAD51, which plays an important role in repairing double-strand breaks in the biological genetic material DNA through homologous recombination (HR). In this work, CeOla also showed an inhibitory effect on PARP1 and Rad51 expressions (Fig. 2G and H), further confirming that CeOla could affect the repair of DNA damage to enhance the PDT efficacy.
Oxidative damage enhanced PDT
Encouraged by the favorable photodynamic properties and DNA repair capacity, the antitumor effect of CeOla was then investigated in vitro. Above all, the cytotoxicities of free Ce6, Ola and CeOla on 4T1 cells were detected by the MTT assay. Apparently, Ce6, Ola and CeOla all had very weak cytotoxicity in darkness because the cell viability was more than 90% even with a higher drug concentration (Fig. 3A). However, after laser irradiation, Ce6 showed enhanced cytotoxicity due to the ROS generation (Fig. 3B). Even then, the PDT of Ce6 had a poor therapeutic effect on account of its limited cell internalization. Importantly, CeOla upon light irradiation inhibited the proliferation of tumor cells efficiently, suggesting a much stronger antitumor capacity than free Ce6. In addition, cell apoptosis analysis was also performed by flow cytometry (Fig. 3C). In the absence of light, the percentages of normal cells in all groups were greater than 92%, indicating a low dark toxicity. Under light conditions, the apoptotic proportion of cells in the Ce6, Ce6 + Ola and CeOla groups increased successively (Fig. 3D). Among which, Ce6 + Ola caused more cell apoptosis than Ce6, demonstrating that Ola facilitated the antitumor effect of PDT because Ola mediated PARP inhibition reduced DNA damage repair and increased the lethality of ROS to tumor cells. What's more, CeOla after light irradiation induced more apoptosis of tumor cells relative to Ce6 + Ola. One of the main reasons should be the increased drug delivery efficiency of agents after assembly into nanomedicine. Additionally, the co-delivery of Ce6 and Ola could improve their varied pharmacokinetics profiles, which enhanced the synergistic effect of PDT and DNA repair inhibition. Besides, the tumor cells after various treatments were used for live/dead cell staining. Calcein AM could easily penetrate into intact living cells and be hydrolyzed by esterase to produce a strong fluorescence. PI could only penetrate into the destroyed cells and bind to the DNA to show red fluorescence. As revealed in Fig. 3E, the strongest red fluorescence and weakest green fluorescence were observed in the cells treated with CeOla upon light irradiation, suggesting an overwhelming superiority in tumor inhibition over other groups. Based on the above, it could be concluded that the Ce6 and Ola based self-delivery nanomedicine destructed the ROS defensive system and amplified the DNA damage for highly efficient PDT by PARP inhibition.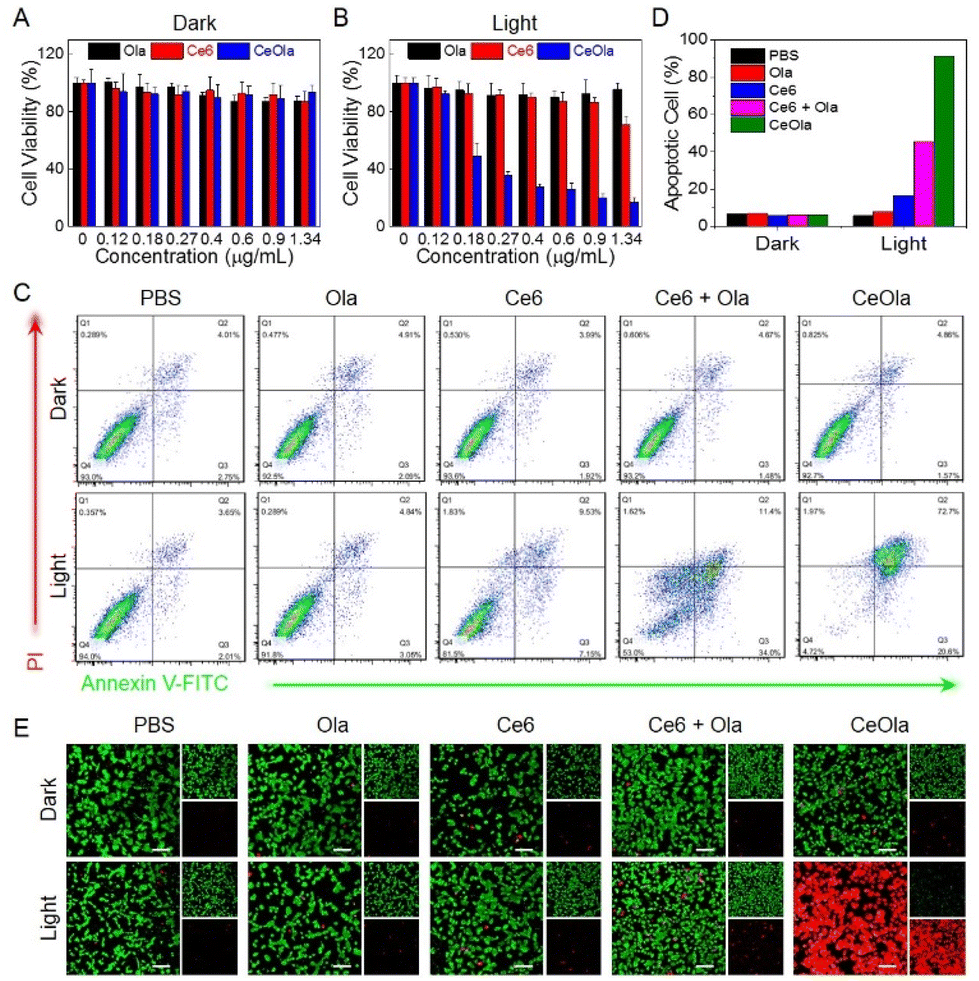 | ||
| Fig. 3 Antitumor effect of CeOla in vitro. The MTT assay for detecting the viability of 4T1 cells after treatment with Ola, Ce6 or CeOla (A) in the absence of light or (B) in the presence of light. (C) Flow cytometry analysis and (D) corresponding apoptotic proportions of 4T1 cells after treatment with Ola, Ce6 or CeOla in the absence or presence of light. (E) Live/dead cell staining analysis of 4T1 cells after treatment with Ola, Ce6 or CeOla in the absence or presence of light. Scale bar: 100 μm. | ||
In vivo antitumor study
It has been reported that nano-sized particles are more likely to accumulate at tumor sites through an enhanced penetration and retention (EPR) effect for targeted therapy.34 As shown in Fig. 4A, the in vivo biodistribution of CeOla was investigated prior to conducting the antitumor study. Under light conditions, 0.5 h after the drug was intravenously injected, a large range of fluorescence accumulation appeared in the mouse with unceasing blood circulation. Subsequently, preferential fluorescence accumulation was found in the tumor tissue, which confirmed the EPR-mediated passive targeting of the nanomedicine. After that, the overall fluorescence intensity gradually decreased as the drug was metabolized. 12 h later, the main organs and tumor tissues were taken for ex vivo imaging. It could be observed that in addition to tumor tissue, liver as one of the main metabolic organs also exhibited much fluorescence aggregation. Even then, CeOla exhibited a considerable accumulation on tumor tissues compared to other normal tissues. Good tumor accumulation and retention of drugs were beneficial for efficient tumor therapy.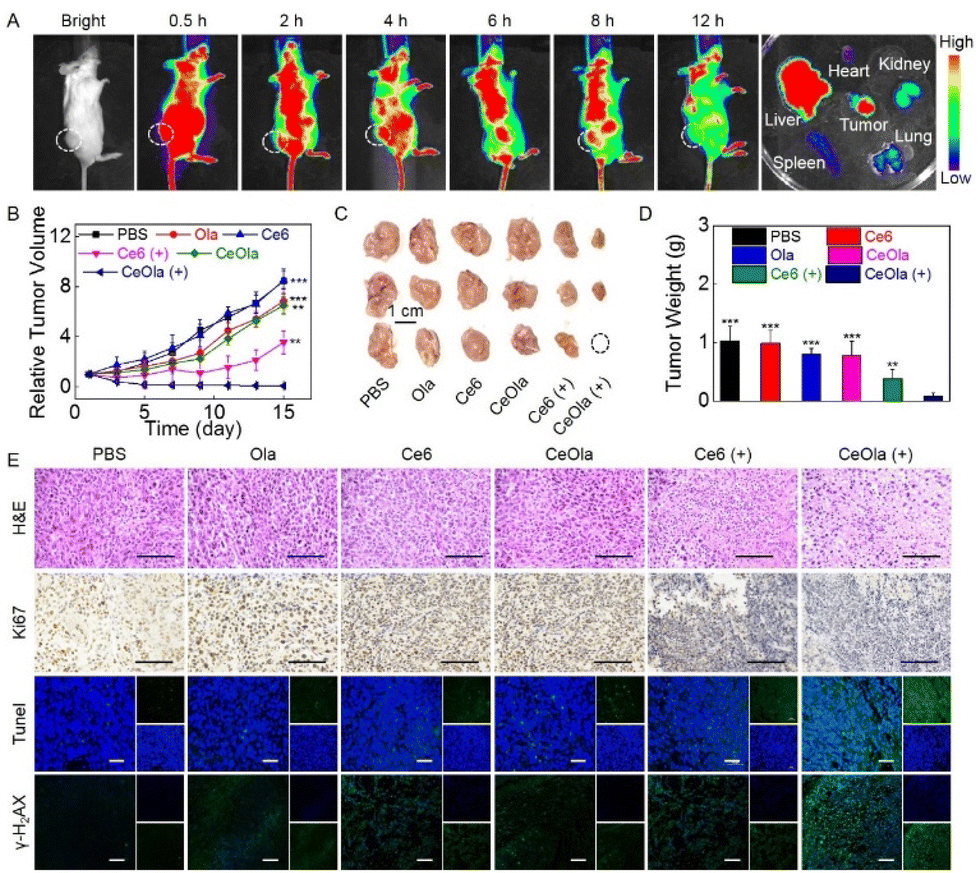 | ||
| Fig. 4 In vivo antitumor study of CeOla. (A) Biodistribution of CeOla in 4T1 tumor-bearing mice after intravenous injection. (B) Relative tumor volume variations of 4T1 tumors after treatment with Ola, Ce6, and CeOla in the presence or absence of light. (C) The represented tumor tissues and (D) the mean tumor weight of the mice after treatment with Ola, Ce6, or CeOla in the presence or absence of light on the 15th day. (E) H&E, Ki67, TUNEL and γ-H2AX immunofuorescence staining of tumor tissues after treatment with Ola, Ce6, and CeOla in the presence or absence of light. Scale bar: 100 μm. **P < 0.01 and ***P < 0.005 were tested via a Student's t-test. | ||
Next, antitumor studies were conducted on 4T1 tumor-bearing mice. After intravenous administration, the tumor volume was recorded every other day to monitor the tumor growth. As shown in Fig. 4B, without any treatments, tumors of the mice in the PBS group grew very fast. Due to the low dark toxicity, Ce6 did not inhibit the tumor growth at all. Notably, CeOla could enhance DNA oxidative damage by inhibiting PARP mediated DNA repair. Ce6 induced PDT exhibited a robust antitumor effect initially, which significantly reduced the growth rate of tumors. Even then, the tumors were found to regrow rapidly at a later stage. To our delight, CeOla with light could maximally inhibit tumor growth, which can be attributed to the improved drug delivery and enhanced PDT efficiency. Specifically, self-assembled CeOla had increased tumor accumulation and improved cellular uptake behaviors. More importantly, the internalized CeOla could inhibit the activity of PARP to hinder the repair of damaged DNA in tumor cells, thereby enhancing the PDT efficacy in tumor suppression. At the end of the treatment, tumors were collected for imaging (Fig. 4C) and weighing (Fig. 4D). The results were consistent with previous findings that CeOla with light irradiation had the best antitumor efficiency and significantly outperformed other groups. Furthermore, H&E and immunofluorescence staining were performed on tumor tissues (Fig. 4E). Compared with other groups, CeOla with light led to the greatest damage to tumor tissue. Moreover, tumor cells in the CeOla with light group exhibited the weakest Ki67 signal and the strongest TUNEL signal, indicating that it could maximally inhibit cell proliferation and promote cell apoptosis. Of special note is that the PDT of CeOla was confirmed to effectively increase the expression ofγ-H2AX, which implied serious DNA destruction. It was deduced that Ola prevented the DNA damage repair causing an amplified oxidative damage by the PDT of Ce6.
Biosafety evaluation
To evaluate the biosafety of CeOla, the body weight of the mice was measured during the treatment. As shown in Fig. 5A, the body weight steadily increased, suggesting a low systemic toxicity of therapeutic agents. In addition, the blood samples were extracted from the mice for blood biochemical and blood routine tests to compare the levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN) and uric acid (UA) in different groups (Fig. 5B). There was no significant abnormity found in the CeOla with light group, meaning that the damage to liver and kidney function was basically negligible. Moreover, the primary hematological indexes also remained in normal ranges (Fig. 5C), which indicated a good blood compatibility of therapeutic agents. Besides, the major organs of the mice were histologically examined by H&E staining (Fig. 5D). The results showed that the heart, liver, spleen, lung and kidney had no obvious morphological abnormalities. Although intravenously injected CeOla could aggregate in normal tissues, it had a less influence because of its biological compatibility and low dark toxicity. Taken together, the photodynamic sensitizer CeOla significantly increased the DNA damage of tumor cells by PARP inhibition and induced few side effects in vivo.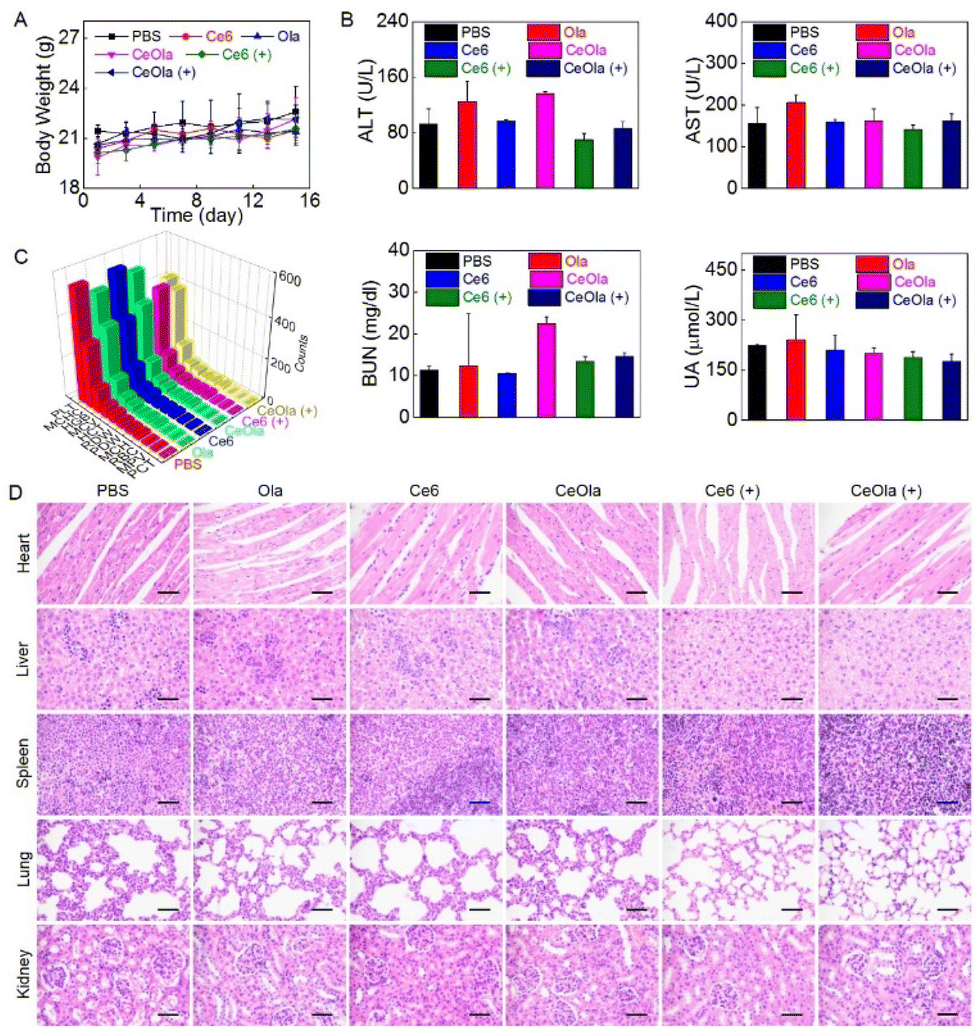 | ||
| Fig. 5 The biosafety evaluation of PhotoSyn. (A) The body weight changes of the mice after treatment with Ola, Ce6, and CeOla in the presence or absence of light. (B) ALT, AST, BUN and UA levels in the serum, (C) blood routine examination and (D) H&E staining analysis of the main organs of the mice at the end of the treatment on the 15th day. Scale bar: 50 μm. | ||
Conclusions
In summary, we successfully prepared CeOla, a self-assembled photodynamic sensitizer based on Ce6 and Ola, to enhance oxidative damage for highly efficient tumor inhibition. By virtue of the hydrophobic interaction, Ce6 and Ola at an appropriate feed ratio could self-assemble into CeOla with a nanosheet structure, which possessed a narrow size distribution and favorable dispersion. In addition, CeOla had extremely high drug loading rates and good water-phase stability. Compared with free Ce6, CeOla showed much better cellular uptake behavior and photodynamic performance. More importantly, CeOla could prevent DNA damage repair by inhibiting the activity of PARP, thereby hindering the ROS defense system and sensitizing tumor cells to ROS. Consequently, the antitumor effect of PDT was significantly improved. In vivo investigations revealed that the intravenously injected CeOla preferred to accumulate at tumor tissue and promoted cell apoptosis by amplifying the DNA damage, leading to an effective tumor suppression. This design of CeOla improved the PDT efficacy by sensitizing tumor cells to ROS, but not increasing the ROS production, which would reduce the side effect caused by superfluous ROS.Experimental
Materials and instruments
Olaparib (Ola) was obtained from Aladdin® (Shanghai, China). Chlorin e6 (Ce6) was purchased from Frontiersci (Shanghai, China). An Annexin V-FITC/PI apoptosis detection kit, 2,7-dichloroflorescein diacetate (DCFH-DA), and methylthiazolyldiphenyl-tetrazolium bromide (MTT) were provided by Beyotime Biotechnology Co., Ltd (Shanghai, China). Sodium dodecyl sulfate (SDS) and sodium chloride (NaCl) were purchased from Aladdin. Cell culture related reagents such as Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), trypsin, antibiotics and Dulbecco's phosphate buffered saline (PBS) were obtained from Gibco Invitrogen Corp. Chromatographic grade acetonitrile and methyl alcohol were purchased from Oceanpak. The instruments used in this work included a dynamic light scattering system (DLS, Brookhaven, NanoBrook 90plus PALS), UV-vis spectrophotometer (Thermo Scientific, Evolution 300), fluorescence spectrophotometer (Thermo Scientific, Lumina), small animal imaging system (Bruker, FX Pro), microplate reader (BioTek, SYNERGY H1), confocal laser scanning microscope (CLSM, Carl Zeiss, LSM880), inverted microscope (Nikon, Eclipse Ni-U), high performance liquid chromatography system (HPLC, SHIMADZU, SPD M20A), and transmission electron microscope (TEM, JEOL, JEM 1400plus).Preparation and characterization
CeOla was self-assembled from Ce6 (10 mg mL−1) and Ola (10 mg mL−1). Frist of all, Ce6 and Ola were dissolved in DMSO. In order to obtain CeOla, 60 μL of Ce6 and 22.5 μL of Ola solution were mixed and stirred for 2 h. After that, the above mixed solution was added to 2 mL of water under ultrasonication for 30 min and then stirred for 8 h. Subsequently, the solution was subjected to dialysis (MWCO 1000 Da) and centrifugation (3000 rpm for 10 min) to remove the remaining free Ce6, Ola and the aggregated Ce6. Other formulations with different mass ratios of Ce6 to Ola were prepared in the same way. The contents of Ola and Ce6 were measured by HPLC and UV-vis spectroscopy, respectively. The drug loading efficiency (DLE) and entrapment efficiency (EE) were calculated as follows: DLE = mass of the drug loaded in CeOla/mass of CeOla × 100%, EE = mass of the drug loaded in CeOla/mass of the drug fed initially × 100%. To investigate the drug release behavior, 3 mL of CeOla (0.297 mg mL−1) was placed in a dialysis tube (3500 Da) for dialysis against PBS (15 mL) at pH 5.5. At the predetermined time, 1 mL of PBS was taken out for fluorescence analysis while isopyknic fresh PBS was added.Cell culture
Murine mammary carcinoma (4T1) cells were obtained from the American Type Culture Collection (ATCC) and they were cultured in the DMEM containing 10% FBS and 1% penicillin–streptomycin at 37 °C.In vitro singlet oxygen (1O2) detection
1O2 generation was evaluated by fluorescence spectroscopy using SOSG as the sensor. In brief, 1 μL of SOSG (5 μM) was mixed with 1 mL of CeOla (3.34 μg mL−1), Ce6 (2.0 μg mL−1) or Ola (1.34 μg mL−1). The light groups were treated with a 638 nm laser (0.38 W). After irradiation for 10 s, the fluorescence intensity at 530 nm (excitation wavelength: 488 nm) was detected with a fluorescence spectrophotometer, and this was repeated 10 times. The dark groups were detected by a similar method but without light irradiation. Besides, the cellular ROS production was measured by CLSM using DCFH-DA as the probe. 4T1 cells were seeded and treated with CeOla (16.67 μg mL−1), Ce6 (10 μg mL−1) or Ola (6.67 μg mL−1) for 6 h. After washing the cells 3 times with PBS, DCFH-DA was added and incubated for 30 min. And then the cells of light groups were irradiated with an LED (638 nm, 1.8 J cm−2, 29.8 mW cm−2) for 2 min. Finally, the fluorescence of DCF in cells was observed by CLSM (excitation wavelength: 488 nm).Cellular uptake experiment
The cellular uptake behavior was investigated by flow cytometry and CLSM. For flow cytometry analysis, 4T1 cells were seeded and cultured for 24 h. Then, the cells were incubated with gradient concentrations of CeOla or Ce6 for 8 h. After washing with PBS, the cells were collected by 0.25% trypsin digestion and the intracellular fluorescence intensity was analyzed by flow cytometry (excitation wavelength: 570 nm). For CLSM observation, 4T1 cells were similarly treated with gradient concentrations of CeOla or Ce6 for 8 h. Then, the cells were washed with PBS three times. Then, the fluorescence of 4T1 cells was detected by CLSM to analyze the cellular uptake (excitation wavelength: 633 nm).MTT assay
MTT assay was used to measure the cytotoxicity of different drugs. First, 4T1 cells were seeded in a 96-well plate, which were treated with gradient concentrations of CeOla, Ce6, Ola or a mixture of Ce6 and Ola. After incubation for 12 h, the cells in light groups were irradiated with an LED (638 nm, 1.8 J cm−2, 29.8 mW cm−2) for 2 min, and the cells in other groups were incubated in darkness. 2 h later, 20 μL of MTT (5 mg mL−1) was added and incubated for another 4 h. Finally, the culture medium was replaced by 150 μL of DMSO and the well plate was shaken slowly for 15 min on a shaker to fully dissolve the crystal violet. The OD value at 570 nm was detected with a microplate reader.Live/dead cell staining analysis
4T1 cells were seeded and cultured for 24 h. Then, the cells were treated with CeOla (16.67 μg mL−1), Ce6 (10 μg mL−1) or Ola (6.67 μg mL−1) for 8 h. After that, the cells were irradiated with an LED panel (638 nm, 1.8 J cm−2, 29.8 mW cm−2) for 1 min or cultured in darkness. After incubation at 37 °C for another 2 h, calcein-AM (1 μL) and PI (1.5 μL) were added for staining for 30 min. Finally, the intracellular fluorescence was observed by CLSM.Cell apoptosis detection
4T1 cells were seeded in 6-well plates and cultured for 24 h. Then, the cells were incubated with CeOla (16.67 μg mL−1), Ce6 (10 μg mL−1) or Ola (6.67 μg mL−1). After 8 h, 4T1 cells in light groups were irradiated with an LED (638 nm, 1.8 J cm−2, 29.8 mW cm−2) for 3 min, while the cells in dark groups did not receive any treatment. After incubation at 37 °C for 2 h, the cells were digested with EDTA-free trypsin and the collected cells were washed twice with PBS. After staining with V-FITC and PI for 20 min, the cellular fluorescence was analyzed by flow cytometry.In vivo fluorescence imaging
In vivo research studies were conducted based on the guidelines of the Institutional Animal Care and Use Committee (IACUC) of Animal Experiment Center of Guangzhou Medical University (Guangzhou, China) and the Regulations for the Administration of Affairs Concerning Experimental Animals. A solid tumor model was established by subcutaneous injection of 4T1 cells in the right hind leg region of female BALB/c mice (6–8 weeks old). The mice were fed for one week to allow the cells to form solid tumor tissue. Subsequently, the 4T1 tumor-bearing mouse was intravenously injected with CeOla (3.74 mg kg−1). In vivo fluorescence imaging was carried out at the predetermined time. 12 h later, the mice were sacrificed, and the major organs (heart, liver, spleen, lungs, kidneys) as well as tumor tissues were obtained for ex vivo fluorescence imaging (excitation wavelength: 630 nm, emission wavelength: 700 nm).Hemolysis test
The whole blood from BALB/c mice was placed in a pre-heparinized tube and centrifuged at 3500 rpm for 5 min. The upper layer of plasma was discarded. It was then washed repeatedly with saline until the upper layer was colorless. The resulting red blood cells (RBC) were diluted with saline into 2% RBC suspension. Subsequently, different concentrations of CeOla and physiological saline were mixed with 2% RBC suspension and co-cultured for 4 h at 37 °C. All samples were centrifuged at 1500 rpm for 10 min to evaluate the hemolysis. The RBC in water and saline without adding CeOla were respectively used as the positive and negative controls. The OD value was measured at 577 nm. The hemolysis rate (%) was calculated as [(OD sample − OD negative control)/(OD positive control − OD negative control)] × 100%.In vivo antitumor study
A 4T1 tumor-bearing mouse model was constructed as described above. Subsequently, the tumor-bearing mice were randomly divided into 6 groups. At the first and eighth days, CeOla (3.63 mg kg−1), Ce6 (2.44 mg kg−1) and Ola (1.19 mg kg−1) were respectively injected into the mice via the tail vein. 6 h after each administration, laser light (0.68 W) was applied to the tumor site for 10 min. Throughout the treatment process, the tumor volume and body weight of the mice were measured every two days. The mice were sacrificed on the fifteenth day to collect the tumors and main organs (heart, liver, spleen, lungs, kidneys) for H&E staining. Meanwhile, the tumors were also collected for immunofluorescence staining of Ki67 and TUNEL. Moreover, the blood samples from the BALB/c mice were also obtained for blood biochemistry and blood routine analysis.Western blot analysis
4T1 cells were seeded in 6-well plates and cultured for 24 h. CeOla (16.67 μg mL−1), Ce6 (10 μg mL−1) and Ola (6.67 μg mL−1) were respectively added to the culture and incubated for 8 h. Subsequently, 4T1 cells of light groups were irradiated with a LED (638 nm, 1.8 J cm−2, 29.8 mW cm−2) for 2 min, while the cells in dark groups did not receive any treatment. After incubation for another 4 h, 4T1 cells were washed with PBS three times and 100 μL RIPA cell lysis buffer was added. The cells were lysed on ice for 30 min. Then, the lysis solution was centrifuged and the supernatant was collected for quantitative analysis using a BCA protein assay kit. 20 μg of the protein sample was separated by electrophoresis using SDS-PAGE gel and then transferred to a nitrocellulose membrane. After washing with TBST, the membrane was blocked for 1 h using TBST with 5% skimmed milk. Then the membrane was washed with TBST again and cultured with the rabbit anti-tublin, rabbit anti-Rad51, rabbit anti-PARP1, rabbit anti-γ-H2AX, or the rabbit anti-GAPDH primary antibody at 4 °C overnight. After that, the membrane was further treated with the HRP conjugated goat anti-rabbit secondary antibody for 2 h at RT. Finally, the western lightning ECL substrate was loaded on the membrane under dark conditions and the fluorescence was monitored by enhanced chemiluminescence exposure. The protein expressions were evaluated by analyzing the gray levels.Γ-H2AX immunofluorescence
4T1 tumor-bearing mice were randomly divided into 6 groups, and CeOla (3.63 mg kg−1), Ce6 (2.44 mg kg−1) or Ola (1.19 mg kg−1) was injected respectively via the tail vein. 6 h later, the tumor site was irradiated with laser light for 10 min (0.68 W). After another 12 h, the mice were sacrificed to collect the tumors for immunofluorescence staining of γ-H2AX.Statistical analysis
Statistical analysis was performed by a Student's t-test. P-Values <0.05 were considered to be statistically significant differences.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Guangdong Basic and Applied Basic Research Foundation (2022B1515020095, 2021A1515010418, 2021B1515020043), the National Natural Science Foundation of China (52073140), the National Key R&D Program of China (2021YFD1800600) and the Open Research Foundation of State Key Laboratory of Respiratory Diseases (SKLRD-OP-202204).References
- S. Gai, G. Yang, P. Yang, F. He, J. Lin, D. Jin and B. Xing, Nano Today, 2018, 19, 146–187 CrossRef CAS.
- S. Li, Q. Zou, Y. Li, C. Yuan, R. Xing and X. Yan, J. Am. Chem. Soc., 2018, 140, 10794–10802 CrossRef CAS PubMed.
- J. Liu, W. Bu and J. Shi, Chem. Rev., 2017, 117, 6160–6224 CrossRef CAS PubMed.
- X. Li, S. Lee and J. D. Huang Yoon, Angew. Chem., Int. Ed., 2018, 31, 9885–9890 CrossRef PubMed.
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef PubMed.
- C. Chen, C. Wu, J. Yu, X. Zhu, Y. Wu, J. Liu and Y. Zhang, Coord. Chem. Rev., 2022, 461, 214495 CrossRef CAS.
- J. J. Hu, Q. Lei and X. Zhang, Prog. Mater. Sci., 2020, 114, 100685 CrossRef CAS.
- T. D. Halazonetis, V. G. Gorgoulis and J. Bartek, Science, 2008, 319, 1352–1355 CrossRef CAS.
- D. Hou, Z. Liu, X. Xu, Q. Liu, X. Zhang, B. Kong, J. Wei, Y. Gong and C. Shao, Redox Biol., 2018, 17, 99–111 CrossRef CAS PubMed.
- U. S. Srinivas, B. W. Q. Tan, B. A. Vellayappan and A. D. Jeyasekharan, Redox Biol., 2019, 25, 101084 CrossRef CAS PubMed.
- X. Y. Li, F. A. Deng, R. R. Zheng, L. S. Liu, Y. B. Liu, R. J. Kong, A. L. Chen, X. Y. Yu, S. Y. Li and H. Cheng, Small, 2021, 17, 2102470 CrossRef CAS.
- J. J. Hu, Y. Chen, Z. H. Li, S. Y. Peng, Y. X. Sun and X. Z. Zhang, Nano Lett., 2019, 19, 5568 CrossRef CAS PubMed.
- G. Bridge, S. Rashid and S. Martin, Cancers, 2014, 6, 1597 CrossRef PubMed.
- F. Dantzer, V. Schreiber, C. Niedergang, C. Trucco, E. Flatter, G. D. L. Rubia, J. Oliver, V. Rolli, J. M. Murcia and G. Murcia, Biochimie, 1999, 81, 69–75 CrossRef CAS PubMed.
- F. R. Sallmann, M. D. Vodenicharov, Z. Wang and G. G. Poirier, J. Biol. Chem., 2000, 275, 15504–15511 CrossRef CAS PubMed.
- Y. Mao, X. Huang, Z. Shuang, G. Lin, J. Wang, F. Duan, J. Chen and S. Li, Cancer Med., 2018, 7, 1285–1296 CrossRef CAS PubMed.
- H. E. Bryant, N. Schultz, H. D. Thomas, K. M. Parker, D. Flower, E. Lopez, S. Kyle, M. Meuth, N. J. Curtin and T. Helleday, Nature, 2007, 447, 346–346 CrossRef CAS.
- J. Murai, S. N. Huang, B. B. Das, A. Renaud, Y. Zhang, J. H. Doroshow, J. Ji, S. Takeda and Y. Pommier, Cancer Res., 2012, 72, 5588–5599 CrossRef CAS.
- H. Farmer, N. McCabe, C. J. Lord, A. N. J. Tutt, D. A. Johnson, T. B. Richardson, M. Santarosa, K. D. Dillon, I. Hickson, C. Knights, N. M. B. Martin, S. P. Jackson, G. C. M. Smith and A. Ashworth, Nature, 2005, 434, 917–921 CrossRef CAS PubMed.
- P. Fong, D. S. Boss, T. A. Yap, A. Tutt, P. Wu, M. M. Roelvink, P. Mortimer, H. Swaisland, A. Lau, M. J. O'Connor, A. Ashworth, J. Carmichael, S. B. Kaye, J. H. M. Schellens and J. S. Bono, N. Engl. J. Med., 2009, 361, 123–134 CrossRef CAS PubMed.
- S. Y. Zanden, J. J. Luimstra, J. Neefjes and J. B. Ovaa, Trends Immunol., 2020, 41, 493–511 CrossRef PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- J. Liang and B. Liu, Bioeng. Transl. Med., 2016, 1, 239–251 CrossRef CAS PubMed.
- Q. Hu, Q. Chen and Z. Gu, Biomaterials, 2018, 178, 546–558 CrossRef CAS PubMed.
- L. Huang, S. Zhao, F. Fang, T. Xu, M. Lan and J. Zhang, Biomaterials, 2021, 268, 120557 CrossRef CAS.
- L. P. Zhao, R. R. Zheng, R. J. Kong, C. Y. Huang, X. N. Rao, N. Yang, A. L. Chen, X. Y. Yu, H. Cheng and S. Y. Li, ACS Nano, 2022, 16, 1182 CrossRef CAS PubMed.
- C. Y. Ang, S. Y. Tan, C. The, J. M. Lee, M. F. E. Wong, Q. Qu, L. Q. Poh, M. Li, Y. Zhang, V. Korzh and Y. Zhao, Small, 2017, 13, 1602379 CrossRef.
- X. Y. Chen, R. R. Zheng, L. P. Zhao, R. J. Kong, N. Yang, Y. B. Liu, A. L. Chen, C. Wang, H. Cheng and S. Y. Li, Chem. Eng. J., 2022, 435, 134783 CrossRef CAS.
- L. Liu and X. Zhang, Prog. Mater. Sci., 2022, 125, 100919 CrossRef CAS.
- L. P. Zhao, R. R. Zheng, H. Q. Chen, L. S. Liu, X. Y. Zhao, H. H. Liu, X. Z. Qiu, X. Y. Yu, H. Cheng and S. Y. Li, Nano Lett., 2020, 20, 2062 CrossRef CAS PubMed.
- S. Y. Qin, A. Zhang, S. Cheng, L. Rong and X. Zhang, Biomaterials, 2017, 112, 234–247 CrossRef CAS.
- J. A. Ledermann, Ann. Oncol., 2016, 27, i40–i44 CrossRef PubMed.
- M. Langelier, A. A. Riccio and J. M. Pascal, Nucleic Acids Res., 2014, 42, 7762–7775 CrossRef CAS.
- J. Fang, W. Islam and H. Maeda, Adv. Drug Delivery Rev., 2020, 157, 142–160 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm01320g |
| This journal is © The Royal Society of Chemistry 2023 |