
A chemical probe for proteomic analysis and visualization of intracellular localization of lysine-succinylated proteins†
Keitaro
Umezawa
 *,
Hiroki
Tsumoto
,
Kyojiro
Kawakami
and
Yuri
Miura
*
*,
Hiroki
Tsumoto
,
Kyojiro
Kawakami
and
Yuri
Miura
*
Research Team for Mechanism of Aging, Tokyo Metropolitan Institute of Gerontology, 35-2 Sakae-cho, Itabashi-ku, Tokyo, Japan. E-mail: umezawa@tmig.or.jp; miura@center.tmig.or.jp
First published on 5th December 2022
Abstract
Protein acylation is a vital post-translational modification that regulates various protein functions. In particular, protein succinylation has attracted significant attention because of its potential relationship with various biological events and diseases. In this report, we establish a new method for the comprehensive detection and analysis of potentially succinylated proteins using a chemical tagging technology. The newly synthesized alkyne-containing succinyl substrate successfully labeled lysine residues of proteins through intracellular metabolic labeling independent of other acylation pathways such as protein malonylation. Furthermore, reporter molecules such as biotin moieties and fluorescent dyes were conjugated to alkyne-tagged succinylated proteins via Click reactions, permitting enrichment for proteomic analysis and fluorescence imaging of the labeled proteins. We successfully analyzed and identified numerous potential succinylated proteins associated with various biological processes using gel electrophoresis, proteomic and bioinformatic analyses, and their visualization in cells.
Introduction
Lysine acylation of proteins is widely known to play a crucial role in regulating protein activities such as changes in the chromatin structure, gene expression, enzyme activity, and protein–protein interactions, as represented by lysine acetylation of proteins.1 Currently, more than ten different structures of lysine acylation have been discovered in mammals (e.g., malonylation,2 succinylation,3 glutarylation,4 crotonylation,5 propionylation/butyrylation,6 2-hydroxy(iso)butyrylation,7,8 lactylation9), and numerous proteins are also highly acylated in microorganisms such as gut microbiota.10 Hence, thorough elucidation of the diverse mechanisms associated with functional control of proteins derived from structural differences of acylation and their correlation with human health is of major interest.Among them, anionic acylations, such as malonylation, succinylation, and glutarylation, have attracted attention as important post-translational modifications that are highly conserved from bacteria to mammals3,11,12 and potentially reflect the state of cellular metabolism because the corresponding malonyl-/succinyl-/glutaryl-CoAs (precursors of lysine acylation) are also key compounds related to energy metabolism.13,14 Taking lysine succinylation as an example, abnormalities in the succinylation level have been shown to disrupt various biological functions (mitochondrial metabolic imbalance,15,16 respiratory chain inhibition and associated redox imbalance,17,18 increased oncogenesis because of induced apoptosis resistance,19 impaired cardiac function,18,20 effects on inflammatory responses,21 infections,22,23 and Alzheimer's disease24). Thus, comprehensive and precise measurement of the acylation levels of proteins in living organisms should clarify various disease mechanisms and enable early diagnosis of these diseases, including those mentioned above. Therefore, further innovations in proteomic analysis technologies are required to achieve detailed protein acylation measurements.
Standard powerful techniques for detecting or enriching post-translationally modified proteins typically use antibodies that recognize targeted modifications. However, methods using antibodies have several problems, for example, non-specific binding and heterogeneity of antibody quality. These shortcomings have been overcome by developing new analytical methods that combine metabolic labeling and Click chemistry for analyzing post-translational modifications.25,26 Incubation/administration of artificial metabolites containing tag units such as alkynes and azides with/to cells or animals enables selective incorporation of these tags into target proteins by metabolic labeling. In addition, various reporter molecules such as fluorescent dyes and biotin can be orthogonally labeled on the tag sites via Click chemistry (copper(I)-catalyzed alkyne–azide cycloaddition, CuAAC reaction), making it possible to realize selective enrichment and visualization of metabolically tagged proteins. Despite these advantages, only a few substrates have been developed to metabolically tag acylated proteins,27–30 and no substrates are available for lysine succinylation. Therefore, we aimed to develop a new substrate that enables comprehensive analysis of succinylated proteins and visualization of their intracellular distribution, which will contribute to unveiling the previously unknown phenomenon of protein succinylation.
Results and discussion
We designed a new substrate, alkynylated succinate acetoxymethyl (AM) ester (SucAM-yne), suitable for Click ligation and metabolic labeling of protein succinylation (Scheme 1a). SucAM-yne has an alkyne unit as a tag for Click ligation and acetoxymethyl moieties for cell permeability, and these esters are easily hydrolyzed back to carboxylic acids (Suc-yne) by intracellular esterases (Schemes 1a and S1†). The substrate was synthesized successfully according to Scheme 1b and used for the following experiments. | ||
| Scheme 1 (a) Metabolic labeling strategy for the visualization and identification of lysine-succinylated proteins using SucAM-yne. Details of possible structures are also mentioned in Scheme S1.† (b) Synthetic scheme for SucAM-yne. Reaction and conditions: (a) propargyl bromide, NaOMe, MeOH, 0 °C to reflux, overnight. (b) i) Methyl bromoacetate, NaH, THF, 0 °C to reflux, 2 h; (ii) MeOH and then H2O. (c) i) 5 N NaOH, MeCN, r.t., 2 h; (ii) conc. HClaq.; (iii) bromomethyl acetate, N,N-diisopropylethylamine, MeCN, r.t., overnight. | ||
Initially, an evaluation of the proof-of-concept was performed. In brief, SucAM-yne was incubated with cultured HEK293T cells for 1 h, and after washing with phosphate-buffered saline, the cells were lysed with NP-40 buffer to extract the proteins. The lysate was then subjected to a Click reaction with a fluorescent dye (Cy5-azide), allowing specific conjugation of Cy5 to alkyne-incorporated proteins. Control samples were generated by performing the same procedure without SucAM-yne or Cy5-azide. These proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), followed by fluorescence scanning of the resulting gels. Only samples treated with SucAM-yne and Cy5-azide showed intense ladder-like fluorescence bands in the gel (Fig. 1a, lane 4), indicating that Suc-yne was successfully conjugated to many proteins in living cells. Furthermore, we found that the signal patterns of Cy5 (tagged proteins) and SYPRO Ruby (total proteins) differed in that lane. Non-specific labeling of intracellular proteins independent of metabolic pathways by Suc-yne should give an estimated labeling rate approximately proportional to the total amount of proteins, thereby resulting in similar signal patterns between Cy5 and SYPRO Ruby. Because this was not observed, the result implies that Suc-yne conjugated to proteins based on particular metabolic pathways in vivo rather than a non-specific reaction. To support this hypothesis, we confirmed that different Cy5 signal patterns in gels were observed between the sample treated with SucAM-yne or MalAM-yne28 (an alkyne-containing substrate for protein malonylation), as shown in Fig. 1b and S1.† In contrast, the total protein expression level and pattern were essentially identical upon treatment with each substrate (Fig. S1†). Detailed analysis by two-dimensional PAGE (2D-PAGE) revealed apparent differences in patterns on the 2D gels (Fig. 1c and S2†) with protein spots in green (Cy3) and red (Cy5) corresponding to Mal-yne- and Suc-yne-conjugated proteins, respectively. Malonylation and succinylation of proteins have been reported previously to occur through different metabolic modification mechanisms,13 and these previous observations might be correlated with the abovementioned results. The amount of metabolically labeled proteins depended on the concentration of SucAM-yne (Fig. S3,† left panel), and no significant toxicity was observed over the concentration range examined (Fig. S3, right panel, and S4†). The experimental conditions were further tested, revealing the optimal concentration of Cy5-azide as 20–100 μM and a metabolic labeling time of one hour (Fig. S5 and S6†).
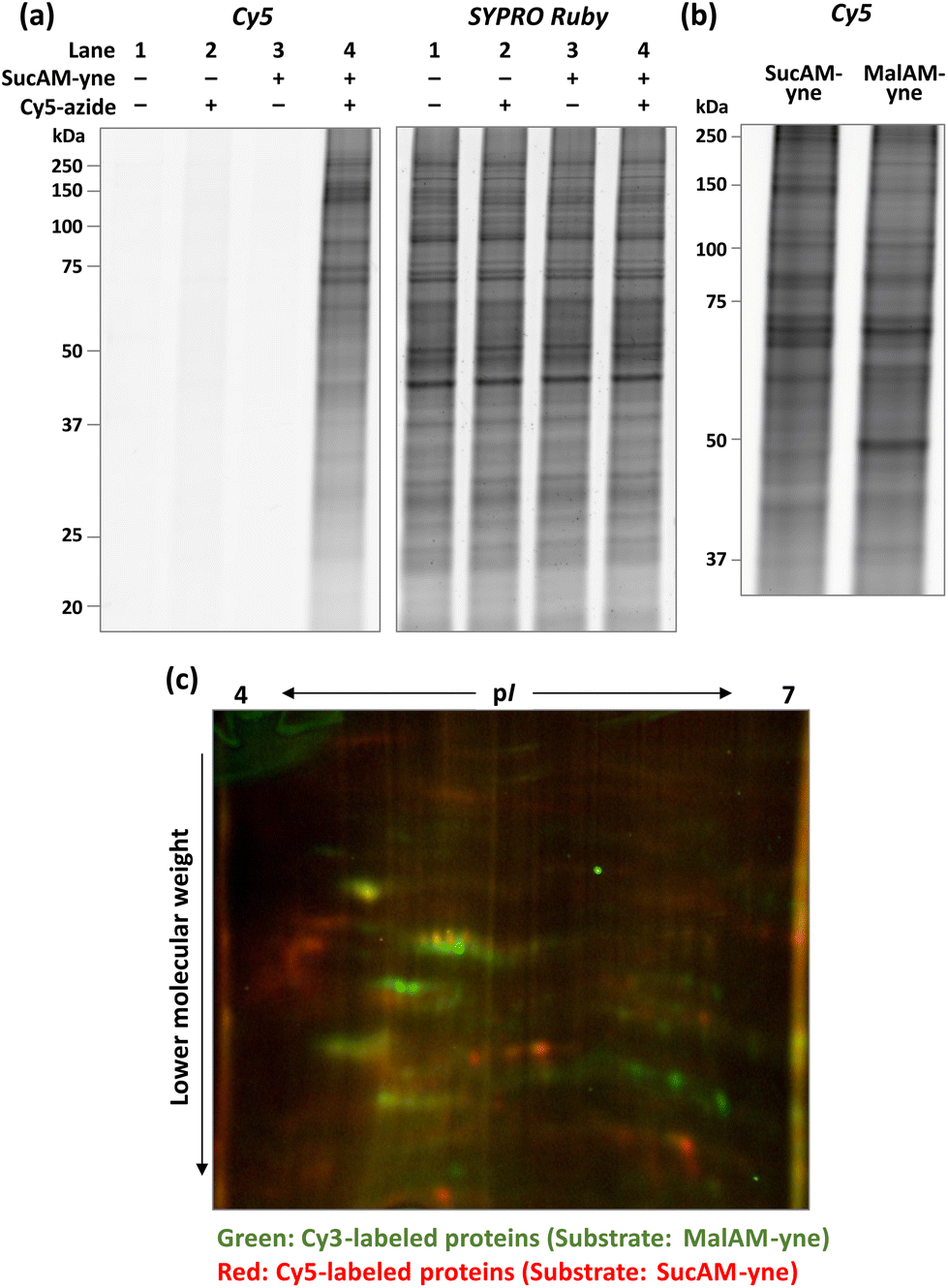 | ||
| Fig. 1 (a) Confirmation of the metabolic labeling of proteins in HEK293T cells with SucAM-yne. Lysates of HEK293T cells incubated with/without SucAM-yne (500 μM, 1 h) were reacted with/without Cy5-azide and analyzed by fluorescence scanning. Visualization of total proteins was undertaken by staining with SYPRO Ruby. (b) Comparison of labeled proteins using different substrates (SucAM-yne, left; MalAM-yne, right) analyzed by SDS-PAGE. (c) Differences in Mal-yne-conjugated and Suc-yne-conjugated proteins labeled with Cy3-azide (green) and Cy5-azide (red), respectively, were analyzed using a fully automated two-dimensional chip gel electrophoresis system (Auto2D). | ||
Encouraged by these results, we next attempted to enrich succinylated proteins using SucAM-yne and Dde biotin-azide. Succinylated proteins labeled with biotin-azide were subject to specific enrichment by avidin (NeutrAvidin) beads for proteomic analyses. A biotin-azide substrate containing a chemically cleavable linker (Dde biotin-azide), which is cleaved by adding hydrazine, was used for the efficient and mild elution of labeled proteins from avidin beads (Scheme 2). The lysates of SucAM-yne-applied HEK293T cells were reacted with Dde biotin-azide, and the degree of biotinylation of the proteins was evaluated by immunoblotting analysis. As shown in Fig. S7,† intense chemiluminescence signals were observed in the lane of the SucAM-yne/Dde biotin-azide-treated sample (lane 4). In contrast, the signals disappeared upon treatment with hydrazine (lane 6). These results indicate that succinylated proteins were biotinylated by reaction with Dde biotin-azide and that the linker was cleaved by adding hydrazine. Next, selective enrichment of biotinylated proteins with NeutrAvidin beads was investigated (protein-based enrichment). In brief, cell lysates that reacted with Dde biotin-azide were incubated with NeutrAvidin beads, and the beads were washed with appropriate solvents to remove non-specifically bound proteins. After hydrazine treatment to elute captured proteins, the eluate was analyzed by gel electrophoresis. As shown in Fig. 2a, a large number of protein bands were detected in the Suc-yne-supplemented cell lysate (lane 4), whereas only a few protein bands were observed in the DMSO-supplemented cell lysate (lane 3). Thus, selective enrichment of succinylated proteins was achieved as expected.
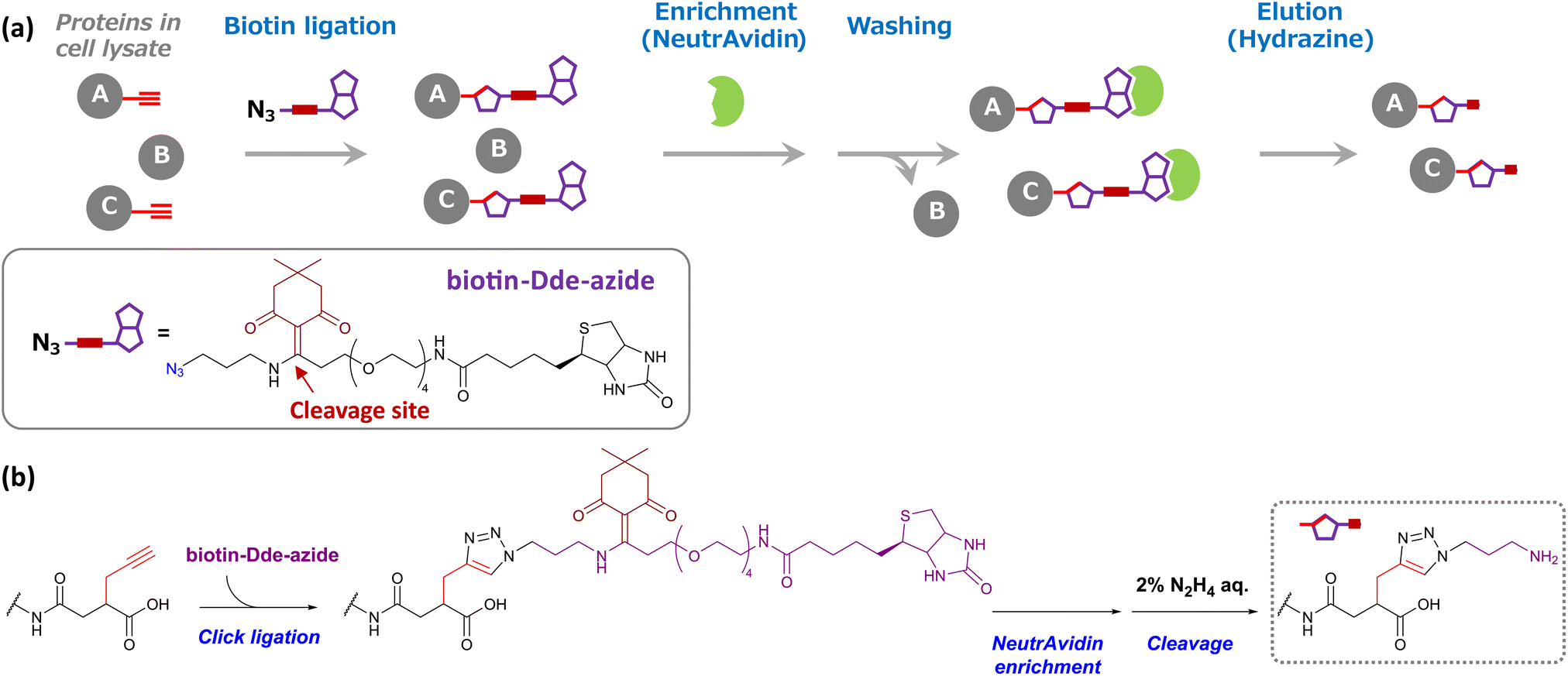 | ||
| Scheme 2 (a) Schematic model of succinylated protein enrichment using Dde biotin-azide and NeutrAvidin agarose gel. Cell lysates, including Suc-yne-labeled proteins (A and C) upon addition of SucAM-yne to living cells, were conjugated with Dde biotin-azide via Click chemistry. These biotinylated proteins were captured by NeutrAvidin beads (green), whereas non-biotinylated proteins (B) were not captured and removed by the washing step. NeutrAvidin-bound proteins were eluted by the addition of hydrazine. (b) Changes in the chemical structure of lysine during the enrichment process. | ||
 | ||
| Fig. 2 Proteomic analysis of lysine-succinylated proteins. (a) Confirmation of the enrichment of succinylated proteins stained with SYPRO Ruby (10% acrylamide gel) by fluorescence scanning. The percentage value given in parentheses for the input lanes represents the amount of loaded protein relative to the amount of protein used for enrichment. (b) Venn diagram of identified enriched proteins. (c) Immunoblotting analysis of enriched proteins, which were identified by LC-MS/MS. IB represents the primary antibodies used. The percentage value in parentheses for the input lanes represents the amount of loaded protein relative to the amount of protein used for enrichment. (d) MS/MS spectrum for identifying K88 modification on EMD (emerin). b and y ions indicate peptide backbone fragment ions containing the N and C termini, respectively. ++ indicates the divalent ion. | ||
The enriched proteins then underwent proteomic analysis. The eluted samples from NeutrAvidin beads were dried in vacuo, followed by reductive alkylation with dithiothreitol (DTT) and iodoacetamide (IAA), trypsin digestion, and desalting. The resulting peptides were analyzed by nano liquid chromatography-tandem mass spectrometry (nanoLC-MS/MS), and protein identification was carried out using the PEAKS XPro software. As a result, 1802 or 156 proteins were identified in the sample of SucAM-yne-treated or DMSO-treated HEK293T cells, respectively (Fig. 2b). By extracting proteins present in only the SucAM-yne-treated sample, 1669 potentially succinylated proteins were identified. Recording independent experiments confirmed experimental reproducibility (Fig. S8†), and these duplicates were merged to obtain total identified proteins. As a result, 2438 potentially succinylated proteins were identified (Table S1†). Some of the identified proteins were also cross-checked by immunoblotting analysis using antibodies against known succinylated proteins (fumarate hydratase, FH; succinate dehydrogenase, SDHA)12 and succinylated protein candidates identified in this research (cathepsin B, CTSB; sirtuin-3, SIRT3) (Fig. 2c and S9†).
Evaluating the conjugation of the synthesized substrate to lysine residues of proteins was carried out by nanoLC-MS/MS analysis. According to the protein-based enrichment strategy, intact proteins were first enriched and the collected proteins were digested and used for proteome analysis. The digests contained not only succinylated peptides but also much larger amounts of unmodified peptides derived from the enriched proteins, which generally interferes with the accurate detection and identification of succinylated peptides. Therefore, we performed enrichment with NeutrAvidin beads using pre-digested samples of SucAM-yne-applied cell lysates (peptide-based enrichment). This approach enhanced the analytical sensitivity of the lysine-modified peptides by eliminating non-modified peptides. As illustrated in Fig. 2d and S10,† we validated particular peptide sequences containing the target molecular structure at the ε-amino group of lysine residues. Thus, direct evidence of lysine modification by the synthetic substrate was obtained. The identification data are summarized in Table S2,† and many of the identified proteins were found to be among those shown in Fig. S8d.†
Gene ontology (GO) analysis and pathway analysis of the identified proteins revealed that protein succinylation is associated with various biological events. For example, the identified proteins were localized in many organelles, especially in the nucleus and cytoplasm (Fig. 3a). Additionally, KEGG pathway analysis (Fig. 3b and c and Table S3†) revealed that mechanisms related to quality control of proteins (ribosomal events, protein spliceosomes, proteasomes, processing in the endoplasmic reticulum, endocytosis), metabolic regulation related to energy production (citrate cycle, oxidative phosphorylation, fatty acid metabolism, glycolytic system) and intracellular metabolic regulations (carbon metabolism, cysteine and methionine metabolism, pyruvate metabolism, pentose phosphate pathway) correlated with protein succinylation. Some of these observations support previous reports (e.g., ribosomal event in bacteria,31,32 citrate cycle,15–18,20 oxidative phospolyration17,18,20), whereas others include several previously unreported events (e.g. protein spliceosomes, proteasomes, processing in the endoplasmic reticulum, cysteine and methionine metabolism). Interestingly, a different pathway analysis (KeyMolnet) revealed an overall high detection of signaling pathways (Table S4†).
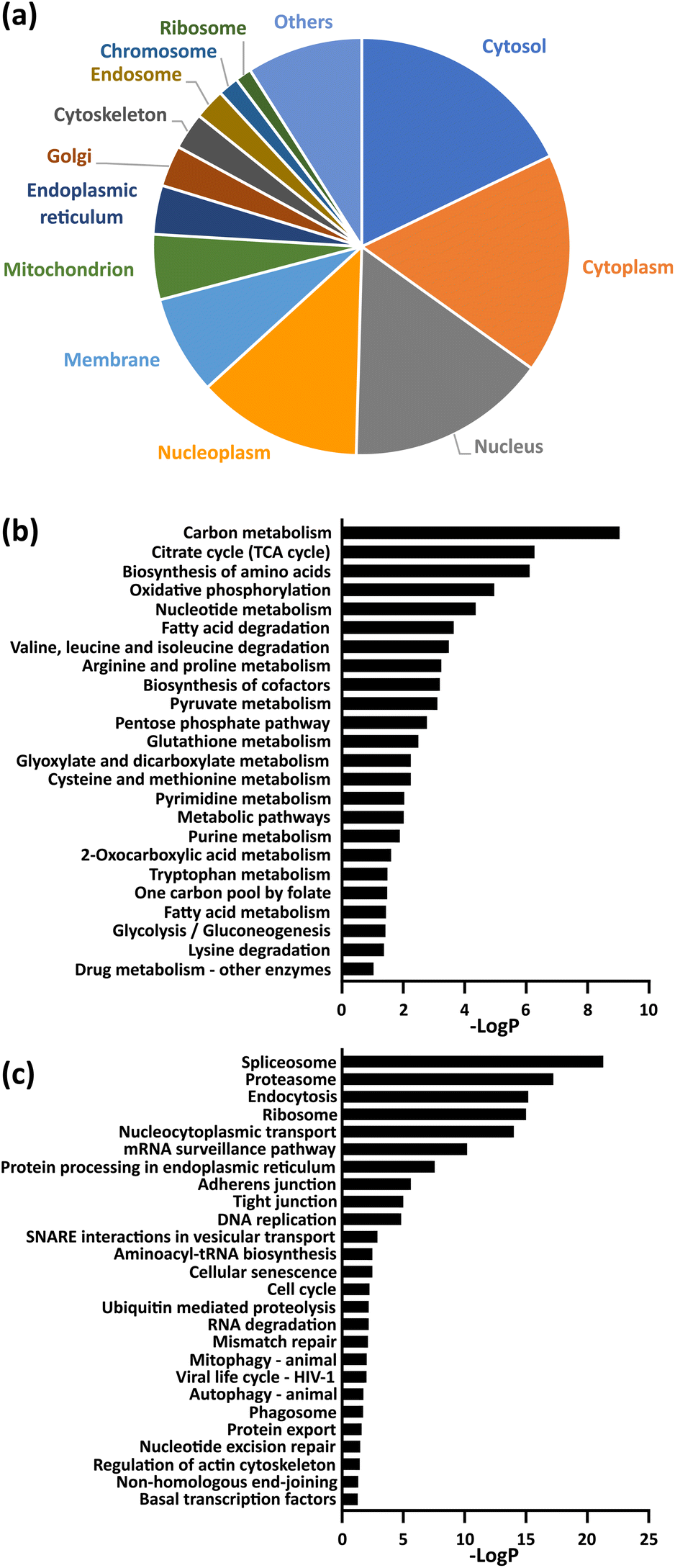 | ||
Fig. 3 (a) Cellular compartment distribution of enriched proteins by GO analysis. (b and c) KEGG pathway analysis of enriched proteins. The entire data are compiled in Table S3,† and the term corresponding to metabolism (b) and genetic information or cellular processing (c) were extracted in each graph. −Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P (logarithm of P-value) indicates the statistical significance of the term. P (logarithm of P-value) indicates the statistical significance of the term. | ||
Fluorescence visualization of succinylated proteins in cells was attempted. SucAM-yne-treated or DMSO-treated HEK293T and HeLa cells were washed, followed by fixation, permeabilization, and Click ligation of Cy3-azide. Intense signals were observed in the fluorescence images, especially in the cytoplasm and nucleus (Fig. S11 and S12†), which is consistent with the cellular component analysis presented in Fig. 3a. In addition, the distribution of succinylated proteins in many compartments was observed by confocal fluorescence imaging (Fig. 4, S13 and S14†). Therefore, our developed method achieved fluorescence imaging of succinylated proteins in cells for the first time.
 | ||
| Fig. 4 Confocal images of succinylated proteins labeled with Cy3-azide in HeLa cells. After incubation with SucAM-yne (500 μM, 1 h), the cells were fixed and permeabilized, followed by Click ligation with Cy3-azide. Scale bar: 25 μm. ROI1 and ROI2 correspond to Fig. S14.† | ||
Finally, we evaluated whether endogenous desuccinylation can be observed using the synthetic substrate. Because sirtuin-5 (SIRT5) is known as a major desuccinylation enzyme candidate,33 we examined the effects of SIRT5-knockdown on the modification levels of Suc-yne using RNA interference of SIRT5. However, only a minor difference in the modification level of Suc-yne was observed between SIRT5-knockdown and control cells (Fig. S15†). Similar results have been reported previously using artificial alkyne-containing tags for lysine acylation in SIRT5-knockdown experiments (for 3-hydroxyl-3-methylglutarylation)29 and SIRT5-inhibiting experiments (for malonylation).28 These common issues of using artificial substrates may arise from limited substrate recognition capacity by deacylase, SIRT5, or a contribution by other deacylation mechanisms (presumably including unknown mechanisms).34 As in this example, the complete mechanism(s) of general protein acylation and deacylation, as well as the enzymes involved in these processes, remains unresolved. Thus, the results in this study should be interpreted with a careful understanding of such background and limitations, and in some cases with a combination of multiple analytical approaches such as immunoaffinity-based analysis.
Nevertheless, even considering these factors, some significant results and findings have been obtained by using this method. For example, the results of the difference in the labeled proteins using SucAM-yne and MalAM-yne (Fig. 1b and c) represent the first step toward a new comprehensive analysis of each type of protein acylation (integrated protein acylome analysis). In addition, the large numbers of identified proteins (2438 proteins) will advance exploratory studies of succinylated protein candidates. Furthermore, the visualization of intracellular localization of succinylated proteins contributes to developing next-generation comprehensive studies that combine cell imaging and proteome analysis. Therefore, for these reasons, we believe that the strengths of the newly developed method and its potential for proteomics have been demonstrated in this study.
Finally, we point out the advantage of our methods over other approaches using antibodies. Immunoaffinity-based enrichment of acylated proteins is widely known as a powerful strategy because of its high affinity for targets. The standard protocol recommends the use of peptide samples for enrichment, and previous reports referred to in the Introduction basically followed this protocol. Therefore, this method can be specialized for LC-MS/MS analysis, and its strengths clearly emerge in the detailed identification of succinylation sites in peptides. The general problem of nonspecific binding/recognition of antibodies might be responsible for limiting the types of samples applicable for measurement. However, the enriched peptides cannot be used for other analyses such as gel-based assays. In contrast, our protocol is applicable for enriching peptides and intact proteins, and fluorescence labeling of tagged proteins is feasible by choosing azide-containing fluorophores instead of biotins. This flexibility enabled a wide range of applications, not only in MS/MS-based proteomic analyses but also in various biochemical approaches including convenient gel-based assays and cellular fluorescence imaging. Consequently, we believe that our developed analytical method complements available conventional immunoaffinity-based methods because the strengths of each method compensate for the shortcomings of the other. Therefore, we anticipate that combining these methods will greatly advance the comprehensive analysis of protein succinylation.
Conclusions
We have developed an alkyne-tagged substrate to detect lysine succinylated proteins by intracellular metabolic labeling. The uptake of the synthetic alkyne-tagged substrate by cells was rapid and covalently bound to the ε-amino moiety of lysine residues in specific proteins. The alkyne unit in the succinylated proteins reacted with azide-containing compounds such as fluorescent dyes and biotin via Click chemistry, whose structures were further confirmed by LC-MS/MS analysis. Biotinylation of succinylated proteins facilitated specific enrichment of these proteins with avidin beads and subsequent LC-MS/MS proteomic analysis, which resulted in the identification of 2438 potentially succinylated proteins. Moreover, the conjugation of fluorophores to succinylated proteins enabled their visualization by cellular imaging and other biochemical approaches. The intracellular localization of succinylated proteins was achieved for the first time. In combination with cellular imaging data and pathway analysis of the identified proteins, we determined that succinylated proteins are delocalized in living cells and closely related to various events that regulate cellular activities. Thus, the strengths of our developed method exist in its flexibility and diversity of applicable analytical techniques. In conclusion, this new and powerful method should support exploratory studies of succinylated proteins and provide an approach to performing comprehensive observation of protein succinylation. In the future, through further verification of the issues mentioned and optimization of analytical conditions, this new analytical tool should contribute to discovering unknown biological phenomena related to protein succinylation.Experimental
Preparation of cells
HEK293T or HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Fujifilm Wako Pure Chemical) supplemented with 10% fetal bovine serum (FBS; Invitrogen) and 50 U mL−1 penicillin–50 μg mL−1 streptomycin (PS, Invitrogen) at 37 °C in a 5%/95% CO2/air incubator.Metabolic labeling and cell lysis
The cells were seeded on a 100 mm dish or 12-well dish and cultured in DMEM (including 10% FBS and 1% PS) overnight so that cell confluency reached ca. ∼90%, and then treated with either DMSO or SucAM-yne (stock solution in DMSO) at concentrations of 0–1000 μM (0.5% DMSO) in DMEM and incubated for 1 hour at 37 °C in a 5%/95% CO2/air incubator. The cells were washed with cold PBS (×2), and then ice-cold lysis buffer (50 mM HEPES, 1% NP-40, 150 mM NaCl, 2 mM MgCl2, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride (PMSF), EDTA-free protease inhibitor cocktail (Roche), ca. 10–20 U benzonate (Sigma)) was applied to the dish and incubated for 10 minutes at 4 °C. The lysed cells were scraped and collected in 2 mL tubes, followed by rotation for a further 20 minutes at 4 °C. After centrifugation (13000g, 30 minutes, 4 °C), the supernatant was collected into new tubes. The total protein concentration was determined using the Pierce™ 660 nm protein assay reagent (Thermo Fisher Scientific).
Ligation of Cy3-azide or Cy5-azide via Click chemistry35
A stock aqueous solution of Cy3-azide (Sigma, 400 μM) or Cy5-azide (Sigma, 400 μM), CuSO4 (100 mM), tris(3-hydroxypropyltriazolylmethyl)amine (THPTA, 100 mM), and sodium ascorbate (100 mM) were prepared, and CuSO4 and THPTA solution were premixed at 1/1 ratio (v/v) to form a Cu(II)/THPTA complex. Cell lysate (1–2 μg μL−1 final), Cy3-azide or Cy5-azide (100 μM final, unless otherwise noted) and CuSO4/THPTA premix (1 mM/1 mM final, respectively) were dissolved in 100 mM sodium phosphate buffer (pH 7.0), followed by addition of sodium ascorbate (2 mM final) to initiate the reaction and incubated for 1.5 hours at room temperature. Four volumes of acetone were added to the reaction mixture and stored at −20 °C for at least 2 hours, and the resulting precipitates were collected by centrifugation (13000g, 10 minutes, room temperature). The precipitates were dissolved in 1× Laemmli buffer (Bio-rad) and heated at 95 °C for 10 minutes, and then sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) was undertaken using Any kD™ Mini-PROTEAN® TGX™ Precast Protein Gels (Bio-rad) unless otherwise noted. Fixation of the proteins on a gel was performed using 50% MeOH and 7% AcOH solution (30 minutes ×2), followed by staining with SYPRO™ Ruby Protein Gel Stain (Thermo Fisher Scientific) according to the manufacturer's instructions. The stained gel was scanned on a Typhoon 9500 laser scanner (GE Healthcare), and total proteins, Cy3-labeled proteins and Cy5-labeled proteins on a gel were detected at excitation/emission wavelengths of 450/610 nm, 550/570 nm and 649/670 nm, respectively.
Two-dimensional gel electrophoresis (2D-PAGE)
For the comparison of labeled proteins with different substrates, MalAM-yne was used as a comparison target, which was synthesized according to the previous report.28 A schematic diagram of 2D-PAGE analysis is shown in Fig. S2a.† Metabolic labeling of MalAM-yne or SucAM-yne was performed independently, and subsequent cell lysis and Click ligation (Cy3-azide for the MalAM-yne-applied sample, Cy5-azide for SucAM-yne-applied sample, respectively) were performed according to the above-described procedure. After the acetone precipitation step, the precipitated proteins were dissolved in isoelectric focusing buffer (IEF buffer; 8 M urea, 2 M thiourea, 4% CHAPS, 50 mM DTT, and 0.2% Pharmalyte 3–10 in distilled water). Cy3-labeled and Cy5-labeled samples were mixed equivalently (50 μg proteins each; total proteins: 100 μg) and 2D PAGE was performed using a fully automated two-dimensional chip gel electrophoresis system (Auto2D®) (Sharp Manufacturing Systems Co. Osaka, Japan), with an Auto2D® IEF Chip pH 4–7 (Sharp) and an Auto2D® PAGE Chip (7.5% acrylamide gel, Sharp) in accordance with the manufacturer's instructions. Equilibration buffer (500 mM Tris-HCl (pH 6.6), 50 mM DTT, 4% SDS, 12.5% glycerol, 0.005% bromophenol blue) and rehydration solution (8 M urea, 2 M thiourea, 4% CHAPS, 50 mM DTT, and 0.2% Pharmalyte pH 3–10 in distilled water) were also used. The running conditions were programmed as follows: sample application by the rehydration method, 35 minutes; isoelectric focusing (IEF) as first-dimension separation, 50 minutes; equilibration, 5 minutes; and SDS-PAGE as second-dimension separation, 38 minutes. IEF was performed at 200 V constantly for 5 minutes, gradually increased to 1000 V for 5 minutes, constantly at 1000 V for 5 minutes, gradually increased to 7000 V for 15 minutes, and constantly at 7000 V for 20 minutes. SDS-PAGE was performed constantly at 2 W for 38 minutes. The temperature was controlled at 20 °C for sample application, IEF and equilibration, and at 4 °C for SDS-PAGE.Ligation of Dde biotin-azide via Click chemistry
In order to achieve specific enrichment of succinylated proteins, Dde biotin-azide (Sigma-Aldrich or Click Chemistry Tools) was used as a biotinylation reagent. Cell lysate (1–2 μg μL−1 final), Dde biotin-azide (100 μM final) and CuSO4/THPTA premix (1 mM/1 mM final, respectively) were dissolved in 100 mM sodium phosphate buffer (pH 7.0), followed by the addition of sodium ascorbate (2 mM final) to initiate the reaction and incubated for 1.5–2 hours at room temperature. Four volumes of acetone were added to the reaction mixture and stored at −20 °C for at least 2 hours, and the resulting precipitates were collected by centrifugation (13000g, 10 minutes, room temperature). The precipitated proteins were dissolved in RIPA buffer (Upstate Biotechnology Inc.), followed by vortexing and sonication. The supernatant was collected in a new tube by centrifugation (15000g, 10 minutes, room temperature) and the total protein concentration was determined by the Pierce™ 660 nm protein assay reagent. These samples were used for the following experiments.
Confirmation of biotinylation of sucinylated proteins by immunoblot analysis
The above-mentioned biotinylated samples were divided into two (one with 2% hydrazine, the other without) and incubated at room temperature for 30 minutes. The reaction solution was purified using an Amicon Ultra 3K (Cytiva) and the resulting concentrated solution was dried in vacuo. 1× Laemmli buffer was added to these samples and heated at 95 °C for 10 minutes, followed by SDS-PAGE and immunoblotting as described in the ESI† for the confirmation of biotinylation of succinylated proteins.Enrichment of succinylated proteins in lysate using NeutrAvidin beads (protein-based enrichment)
The biotinylated sample (ca. 400–800 μg) was diluted with PBS (0.1–0.25 μg μL−1 protein, final) in the presence of Pierce® NeutrAvidin® Agarose HC (Thermo Fisher Scientific, 10 μL per 200–400 μg proteins), and the suspension was gently mixed by an end-to-end rotator for 2 hours at room temperature. The suspension was poured into a filter-embedded empty column (Pierce® Spin Columns, Thermo Fisher Scientific) and the supernatant was removed by spinning down. The collected beads on a filter were washed with 6 M urea, 1% SDS in PBS (200 μL ×4), PBS (200 μL, ×3), distilled water (200 μL ×3), and the washing solvents were removed by centrifugation. The washed beads were suspended in 2% hydrazine aqueous solution (containing 0.1% SDS (for SDS-PAGE and immunoblotting) or 0.1% RapiGest SF (Waters, for LC-MS/MS analysis)) and stirred for 30 minutes at room temperature. The supernatant was collected in a new tube by centrifugation, and the solvents were evaporated in vacuo. For the samples for SDS-PAGE, 1× Laemmli buffer was added to the sample tubes and heated at 95 °C for 10 minutes, followed by SDS-PAGE, SYPRO Ruby staining and gel scanning as described above. For the samples for immunoblotting analysis, the details are described in the ESI.† For the samples for LC-MS/MS analysis, the details are described below.Tryptic digestion of proteins for LC-MS/MS analysis
The enriched samples were dissolved in 50 mM ammonium bicarbonate buffer containing 0.1% RapiGest SF (pH 8.1, 100 μL), and dithiothreitol (DTT) was added to the solution (5 mM, final) and heated at 60 °C for 30 minutes. After cooling to room temperature, iodoacetamide (IAA) was added to the solution (10 mM, final) and incubated at room temperature for 30 minutes in the dark. Trypsin Gold, Mass Spectrometry Grade (Promega), was added to the reaction mixture (0.5 μg enzyme) and incubated at 37 °C overnight (ca. 16 hours). The solution was acidified with a TFA aliquot (pH < 3) and further incubated at 37 °C for 30 minutes. After centrifugation (15000g, 10 minutes, room temperature), the solution was desalted by homemade StageTip embedded Empore™ SDB-XC disks (GL Science) according to the previously published literature.36 The eluate was dried in vacuo and dissolved in 2% MeCN and 0.1% formic acid in distilled water. LC-MS/MS analyses were performed on an Ultimate 3000 RSLCnano system (Thermo Fisher Scientific) coupled to a Q Exactive hybrid quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific) equipped with a nanoelectrospray ionization source. LC-MS/MS measurements were performed in triplicate, and the results of each measurement were merged and analyzed to identify the proteins (Fig. 2b). Furthermore, the experiments were perfomed in duplicate (Fig. S8†), and the results were merged to obtain total identified proteins (Table S1†). 20 μg of proteins prior to enrichment was used per measurement. The detailed analytical conditions are described in the ESI.†
Enrichment of succinylated peptides in lysate using NeutrAvidin beads (peptide-based enrichment)
Cell lysates (total protein: ca. 2000 μg) were diluted with 50 mM ammonium bicarbonate buffer (pH 8.1, 1 mL; final protein concentration: ca. 1 μg μL−1) including 0.5% SDS, and DTT was added to the solution (20 mM, final) and heated at 65 °C for 30 minutes. After cooling to room temperature, IAA was added to the solution (50 mM, final) and incubated at room temperature for 30 minutes in the dark. Four volumes of acetone were added to the reaction mixture and stored at −20 °C for at least 2 hours, and the resulting precipitates were collected by centrifugation (13000g, 10 minutes, room temperature). The precipitated proteins were dissolved in a small aliquot of 100 mM ammonium bicarbonate buffer including 6 M urea (final protein concentration: ca. 4–8 μg μL−1), followed by vortexing and sonication until the precipitates were dissolved. 100 mM ammonium bicarbonate buffer was added to the solution until the concentration of urea becomes below 1 M, and then Trypsin Gold, Mass Spectrometry Grade (Promega), was added to the reaction mixture (total proteins/trypsin = 100/1, w/w) and incubated at 37 °C overnight (ca. 16 hours). Dde biotin-azide (100 μM final) and CuSO4/THPTA premix (1 mM/1 mM final, respectively) were added to the digested solution, followed by the addition of sodium ascorbate (2 mM final) to initiate the reaction and incubated for 2 hours at room temperature. Unreacted Dde biotin-azide was removed by strong cation exchange (SCX) purification using a Monospin SCX column (GL Science) according to the manufacturer's procedure. In brief, the samples were diluted with loading buffer (10 mM KH2PO4 in 25% MeCN, pH 2.8), and then subjected to a MonoSpin™ SCX column. After washing the filter with the loading buffer 3 times, the peptides on an SCX filter were eluted with a loading buffer containing 1 M KCl, and the eluate was evaporated in vacuo. The resulting samples were dissolved in PBS including 2% MeCN (0.5 μg μL−1, final) in the presence of Pierce® NeutrAvidin® Agarose HC (10 μL per 500 μg peptides), and the suspension was gently mixed by an end-to-end rotator for 2 hours at room temperature. The suspension was poured into a filter-embedded empty column (Pierce® Spin Columns) and the supernatant was removed by spinning down. The collected beads on a filter were washed with 5% acetonitrile in PBS (200 μL ×4), PBS (200 μL, ×2), and distilled water (200 μL ×3), and the washing solvents were removed by centrifugation. The washed beads were suspended in 2% hydrazine aqueous solution and stirred for 30 minutes at room temperature. The supernatant was collected in a new tube by centrifugation, and the solvents were evaporated in vacuo. The sample was dissolved in 2% MeCN and 0.1% TFA in distilled water, and desalted using homemade StageTip embedded Empore™ SDB-XC disks (GL Science). The eluate was dried in vacuo and dissolved in 2% MeCN and 0.1% formic acid in distilled water for LC-MS/MS analysis. 500 μg of proteins prior to enrichment was used per measurement. The experiments were undertaken in duplicate, and the results were merged and analyzed to identify the succinylated peptides (Table S2†).
Fluorescence imaging
Cells grown on a 35 mm glass-bottom dish coated with poly-L-lysine (Matsunami Glass) were metabolically labeled with DMSO or SucAM-yne (500 μM final), fixed with 4% paraformaldehyde solution (Fujifilm Wako Pure Chemical), permeabilized with 0.5% Triton X-100 and then reacted with Cy3-azide (20 μM final) in the presence of a CuSO4/THPTA mixture (1 mM/1 mM final) and sodium ascorbate (2 mM final) for 1 hour at room temperature. The cells were washed with PBS-T (0.1% Tween-20 in PBS, ×3) and incubated with DAPI in PBS for 5 minutes. After washing with distilled water (×3), the dishes were filled with PBS and subjected to fluorescence microscopes. Epi-fluorescence (FL) and bright-field (BF) imaging were undertaken with a BZ-X800 all-in-one fluorescence microscope (Keyence) instrument equipped with BZ-X filters (OP-87762 for DAPI; OP-87764 for Cy3, respectively) and an objective lens (BZ-PA40). Confocal fluorescence imaging was performed with a TCS SP8 (Leica) instrument equipped with a diode (405 nm), an argon laser (561 nm), and an objective lens (HC PL APO CS2 93x/1.3; Leica). The microscope was controlled using LAS AF software (Leica).Author contributions
K. U. and Y. M. designed the research. K. U. performed experiments. K. U., H. T. and K. K. analyzed the data. All authors discussed the results and co-wrote the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was supported in part by MEXT/JSPS KAKENHI grant number 19K11685 (to K. U.) and a grant from the Koyanagi Foundation (to K. U.). The authors thank Dr Yasuteru Urano (The University of Tokyo), Dr Mako Kamiya (Tokyo Institute of Technology), and Drs Jun Toyohara and Tetsuro Tago (Tokyo Metropolitan Institute of Gerontology) for their support with the organic synthesis of the substrates, and Dr Tamao Endo (Tokyo Metropolitan Institute of Gerontology) for helpful discussions and advice. We thank Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.References
- I. Ali, R. J. Conrad, E. Verdin and M. Ott, Chem. Rev., 2018, 118, 1216–1252 CrossRef PubMed.
- C. Peng, Z. Lu, Z. Xie, Z. Cheng, Y. Chen, M. Tan, H. Luo, Y. Zhang, W. He, K. Yang, B. M. M. Zwaans, D. Tishkoff, L. Ho, D. Lombard, T.-C. He, J. Dai, E. Verdin, Y. Ye and Y. Zhao, Mol. Cell. Proteomics, 2011, 10, M111.012658 CrossRef PubMed.
- Z. Zhang, M. Tan, Z. Xie, L. Dai, Y. Chen and Y. Zhao, Nat. Chem. Biol., 2010, 7, 58–63 CrossRef.
- M. Tan, C. Peng, K. A. Anderson, P. Chhoy, Z. Xie, L. Dai, J. Park, Y. Chen, H. Huang, Y. Zhang, J. Ro, G. R. Wagner, M. F. Green, A. S. Madsen, J. Schmiesing, B. S. Peterson, G. Xu, O. R. Ilkayeva, M. J. Muehlbauer, T. Braulke, C. Mühlhausen, D. S. Backos, C. A. Olsen, P. J. McGuire, S. D. Pletcher, D. B. Lombard, M. D. Hirschey and Y. Zhao, Cell Metab., 2014, 19, 605–617 CrossRef CAS PubMed.
- M. Tan, H. Luo, S. Lee, F. Jin, J. S. Yang, E. Montellier, T. Buchou, Z. Cheng, S. Rousseaux, N. Rajagopal, Z. Lu, Z. Ye, Q. Zhu, J. Wysocka, Y. Ye, S. Khochbin, B. Ren and Y. Zhao, Cell, 2011, 146, 1016–1028 CrossRef CAS.
- Y. Chen, R. Sprung, Y. Tang, H. Ball, B. Sangras, S. C. Kim, J. R. Falck, J. Peng, W. Gu and Y. Zhao, Mol. Cell. Proteomics, 2007, 6, 812–819 CrossRef CAS.
- L. Dai, C. Peng, E. Montellier, Z. Lu, Y. Chen, H. Ishii, A. Debernardi, T. Buchou, S. Rousseaux, F. Jin, B. R. Sabari, Z. Deng, C. D. Allis, B. Ren, S. Khochbin and Y. Zhao, Nat. Chem. Biol., 2014, 10, 365–370 CrossRef CAS PubMed.
- Z. Xie, D. Zhang, D. Chung, Z. Tang, H. Huang, L. Dai, S. Qi, J. Li, G. Colak, Y. Chen, C. Xia, C. Peng, H. Ruan, M. Kirkey, D. Wang, L. M. Jensen, Oh K. Kwon, S. Lee, S. D. Pletcher, M. Tan, D. B. Lombard, K. P. White, H. Zhao, J. Li, R. G. Roeder, X. Yang and Y. Zhao, Mol. Cell, 2016, 62, 194–206 CrossRef CAS.
- D. Zhang, Z. Tang, H. Huang, G. Zhou, C. Cui, Y. Weng, W. Liu, S. Kim, S. Lee, M. Perez-Neut, J. Ding, D. Czyz, R. Hu, Z. Ye, M. He, Y. G. Zheng, H. A. Shuman, L. Dai, B. Ren, R. G. Roeder, L. Becker and Y. Zhao, Nature, 2019, 574, 575–580 CrossRef CAS.
- X. Zhang, Z. Ning, J. Mayne, Y. Yang, S. A. Deeke, K. Walker, C. L. Farnsworth, M. P. Stokes, J.-F. Couture, D. Mack, A. Stintzi and D. Figeys, Nat. Commun., 2020, 11, 4120 CrossRef CAS.
- Z. Xie, J. Dai, L. Dai, M. Tan, Z. Cheng, Y. Wu, J. D. Boeke and Y. Zhao, Mol. Cell. Proteomics, 2012, 11, 100–107 CrossRef CAS.
- B. T. Weinert, C. Schölz, S. A. Wagner, V. Iesmantavicius, D. Su, J. A. Daniel and C. Choudhary, Cell Rep., 2013, 4, 842–851 CrossRef CAS.
- M. D. Hirschey and Y. Zhao, Mol. Cell. Proteomics, 2015, 14, 2308–2315 CrossRef CAS.
- S. Trefely, C. D. Lovell, N. W. Snyder and K. E. Wellen, Mol. Metab., 2020, 100941 CrossRef.
- J. Park, Y. Chen, D. X. Tishkoff, C. Peng, M. Tan, L. Dai, Z. Xie, Y. Zhang, B. M. M. Zwaans, M. E. Skinner, D. B. Lombard and Y. Zhao, Mol. Cell, 2013, 50, 919–930 CrossRef.
- M. J. Rardin, W. He, Y. Nishida, J. C. Newman, C. Carrico, S. R. Danielson, A. Guo, P. Gut, A. K. Sahu, B. Li, R. Uppala, M. Fitch, T. Riiff, L. Zhu, J. Zhou, D. Mulhern, R. D. Stevens, O. R. Ilkayeva, C. B. Newgard, M. P. Jacobson, M. Hellerstein, E. S. Goetzman, B. W. Gibson and E. Verdin, Cell Metab., 2013, 18, 920–933 CrossRef.
- Y. Zhang, S. S. Bharathi, M. J. Rardin, J. Lu, K. V. Maringer, S. Sims-Lucas, E. V. Prochownik, B. W. Gibson and E. S. Goetzman, J. Biol. Chem., 2017, 292, 10239–10249 CrossRef PubMed.
- L. Zhou, F. Wang, R. Sun, X. Chen, M. Zhang, Q. Xu, Y. Wang, S. Wang, Y. Xiong, K. L. Guan, P. Yang, H. Yu and D. Ye, EMBO Rep., 2016, 17, 811–822 CrossRef PubMed.
- F. Li, X. He, D. Ye, Y. Lin, H. Yu, C. Yao, L. Huang, J. Zhang, F. Wang, S. Xu, X. Wu, L. Liu, C. Yang, J. Shi, X. He, J. Liu, Y. Qu, F. Guo, J. Zhao, W. Xu and S. Zhao, Mol. Cell, 2015, 60, 661–675 CrossRef PubMed.
- S. Sadhukhan, X. Liu, D. Ryu, O. D. Nelson, J. A. Stupinski, Z. Li, W. Chen, S. Zhang, R. S. Weiss, J. W. Locasale, J. Auwerx and H. Lin, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4320–4325 CrossRef PubMed.
- F. Wang, K. Wang, W. Xu, S. Zhao, D. Ye, Y. Wang, Y. Xu, L. Zhou, Y. Chu, C. Zhang, X. Qin, P. Yang and H. Yu, Cell Rep., 2017, 19, 2331–2344 CrossRef.
- M. Yang, Y. Wang, Y. Chen, Z. Cheng, J. Gu, J. Deng, L. Bi, C. Chen, R. Mo, X. Wang and F. Ge, Mol. Cell. Proteomics, 2015, 14, 796–811 CrossRef.
- C. Gaviard, I. Broutin, P. Cosette, E. Dé, T. Jouenne and J. Hardouin, J. Proteome Res., 2018, 17, 2449–2459 CrossRef PubMed.
- D. M. Acosta, C. Mancinelli, C. Bracken and D. Eliezer, J. Biol. Chem., 2022, 298, 101442 CrossRef PubMed.
- E. Thinon and H. C. Hang, Biochem. Soc. Trans., 2015, 43, 253–261 CrossRef.
- C. G. Parker and M. R. Pratt, Cell, 2020, 180, 605–632 CrossRef.
- Y.-Y. Yang, J. M. Ascano and H. C. Hang, J. Am. Chem. Soc., 2010, 132, 3640–3641 CrossRef PubMed.
- X. Bao, Q. Zhao, T. Yang, Y. M. E. Fung and X. D. Li, Angew. Chem., Int. Ed., 2013, 52, 4883–4886 CrossRef.
- X. Bao, Y. Xiong, X. Li and X. D. Li, Chem. Sci., 2018, 9, 7797–7801 RSC.
- Y. Sun, Y. Chen and T. Peng, Chem. Sci., 2022, 13, 6019–6027 RSC.
- J. Pan, R. Chen, C. Li, W. Li and Z. Ye, J. Proteome Res., 2015, 14, 4309–4318 CrossRef PubMed.
- Y. Yang, H. Zhang, Z. Guo, S. Zou, F. Long, J. Wu, P. Li, G.-P. Zhao and W. Zhao, Mol. Cell. Proteomics, 2021, 20, 100148 CrossRef.
- J. Du, Y. Zhou, X. Su, J. J. Yu, S. Khan, H. Jiang, J. Kim, J. Woo, J. H. Kim, B. H. Choi, B. He, W. Chen, S. Zhang, R. A. Cerione, J. Auwerx, Q. Hao and H. Lin, Science, 2011, 334, 806–809 CrossRef PubMed.
- K. A. Anderson, F. K. Huynh, K. Fisher-Wellman, J. D. Stuart, B. S. Peterson, J. D. Douros, G. R. Wagner, J. W. Thompson, A. S. Madsen, M. F. Green, R. M. Sivley, O. R. Ilkayeva, R. D. Stevens, D. S. Backos, J. A. Capra, C. A. Olsen, J. E. Campbell, D. M. Muoio, P. A. Grimsrud and M. D. Hirschey, Cell Metab., 2017, 25, 838–855 CrossRef PubMed.
- S. I. Presolski, V. P. Hong and M. G. Finn, Curr. Protoc. Chem. Biol., 2011, 3, 153–162 CrossRef.
- J. Rappsilber, M. Mann and Y. Ishihama, Nat. Protoc., 2007, 2, 1896–1906 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental procedures, and supplementary figures and tables. See DOI: https://doi.org/10.1039/d2an01370c |
| This journal is © The Royal Society of Chemistry 2023 |