
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxygen transfer reactivity mediated by nickel perfluoroalkyl complexes using molecular oxygen as a terminal oxidant†
Shubham
Deolka
 a,
R.
Govindarajan
a,
Eugene
Khaskin
a,
Serhii
Vasylevskyi
a,
Janet
Bahri
a,
Robert R.
Fayzullin
b,
Michael C.
Roy
a and
Julia R.
Khusnutdinova
*a
a,
R.
Govindarajan
a,
Eugene
Khaskin
a,
Serhii
Vasylevskyi
a,
Janet
Bahri
a,
Robert R.
Fayzullin
b,
Michael C.
Roy
a and
Julia R.
Khusnutdinova
*a
aCoordination Chemistry and Catalysis Unit Okinawa Institute of Science and Technology Graduate University, 1919-1 Tancha, Onna-son, 904-0495, Okinawa, Japan. E-mail: juliak@oist.jp
bArbuzov Institute of Organic and Physical Chemistry, FRC Kazan Scientific Center, Russian Academy of Sciences, 8 Arbuzov Street, Kazan, 420088, Russian Federation
First published on 6th June 2023
Abstract
Nickel perfluoroethyl and perfluoropropyl complexes supported by naphthyridine-type ligands show drastically different aerobic reactivity from their trifluoromethyl analogs resulting in facile oxygen transfer to perfluoroalkyl groups or oxygenation of external organic substrates (phosphines, sulfides, alkenes and alcohols) using O2 or air as a terminal oxidant. Such mild aerobic oxygenation occurs through the formation of spectroscopically detected transient high-valent NiIII and structurally characterized mixed-valent NiII–NiIV intermediates and radical intermediates, resembling O2 activation reported for some Pd dialkyl complexes. This reactivity is in contrast with the aerobic oxidation of naphthyridine-based Ni(CF3)2 complexes resulting in the formation of a stable NiIII product, which is attributed to the effect of greater steric congestion imposed by longer perfluoroalkyl chains.
Introduction
Oxidation of organic substrates using molecular oxygen as a terminal oxidant is an important process in many biological processes1 and in industrial aerobic oxidation catalysis where oxygen or air is considered as an inexpensive, “green” oxidant that produces no hazardous waste products.2 Precious metal catalysts have been extensively used in enabling aerobic oxidation of hydrocarbon substrates or stoichiometric O2 activation, typically based on Pt,3 Pd,4 Ru,5 Rh,6 Ir,7etc. At the same time, metalloenzymes capable of aerobic oxygenation typically contain more abundant first row transition metals, which led to the development of bioinspired model complexes capable of O2 activation, most commonly based on Cu,8 Fe,9 Mn,10 and Co.11 Although Ni is known to be present at the active site of several O2-reactive enzymes,12 examples of synthetic “nickel oxygenase” reactivity in which Ni complexes assist in the transfer of O-atoms from O2 to a substrate remain scarce.13 While Ni complex examples in oxidation catalysis using strong oxidants, such as peroxides, NaOCl, PhIO, or meta-chloroperbenzoic acid, are well known,2a,14 their utilization in aerobic oxidation is often prohibited by the stability of NiII towards aerobic oxidation. At the same time, several recent examples of the generation of highly reactive Ni superoxide or peroxo-species indicate that such species can participate in O-transfer reactivity.15 Only a few examples of direct aerobic oxidation of NiII to produce high valent Ni species in +3 or +4 oxidation states are known, often enabled by the use of multidentate macrocyclic,16 scorpionate,17 and redox non-innocent ligands,15d with only one example featuring a simple organometallic tris(norbornyl)Ni complex that produced NiIV at low temperature under O2.18 More recently, we reported facile aerobic oxidation of a bis(trifluoromethyl)NiII complex supported by simple bidentate naphthyridine or methyl-substituted naphthyridine ligands to form stable bis(trifluoromethyl)NiIII complexes capable of NiIII–CF3 bond homolysis and C(sp2)–H bond radical trifluoromethylation (Scheme 1a).19 Later we found that longer fluoroalkyl chains can be transferred to organic substrates as well using naphthyridine-free Ni catalyst.20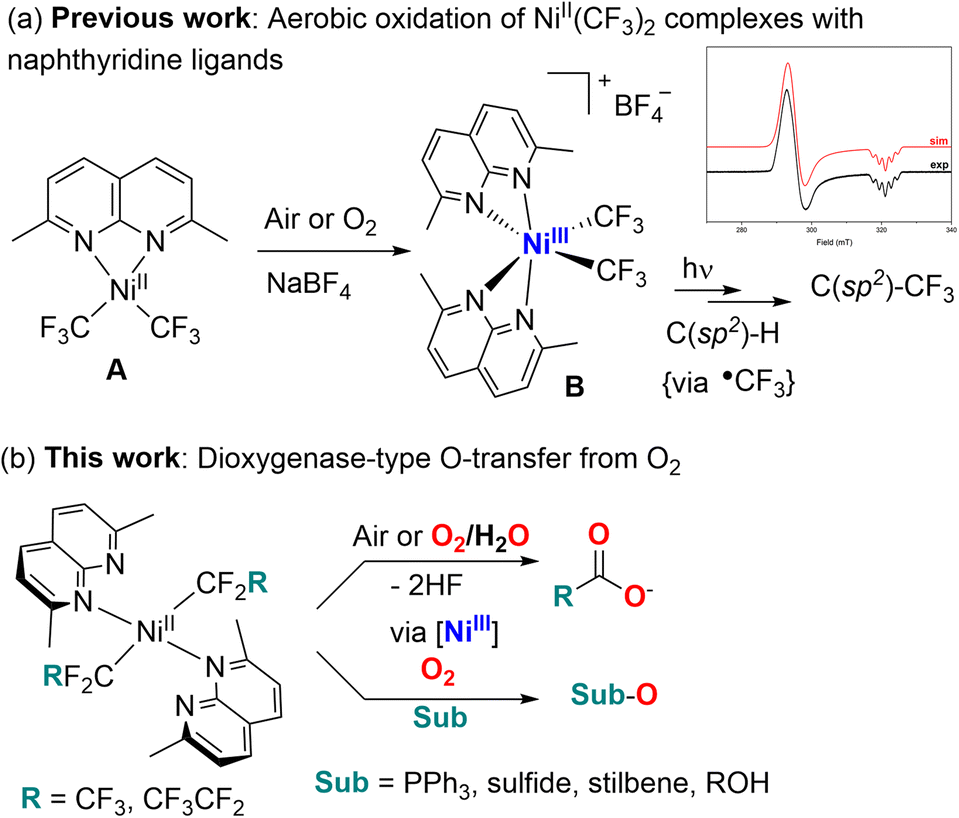 | ||
| Scheme 1 Aerobic oxidation and reactivity of bis(trifluoromethyl) (a) and longer chain bis(perfluoroalkyl) complexes (b). | ||
In this work, we report that naphthyridine-supported NiII complexes containing longer-chain pentafluoroethyl and heptafluoropropyl ligands show distinctly different reactivity with O2, leading to the formation of perfluorocarboxylates as main products formed via the formal removal of both α-fluorine atoms and O-atom transfer from O2 (Scheme 1b). In the presence of organic substrates, facile oxygen-atom transfer reactivity was induced, leading to aerobic oxidation of stilbenes to epoxides and benzaldehyde (via C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond cleavage), aerobic oxidation of alcohols, and oxygenation of phosphines and sulfides in a stoichiometric manner. These NiII complexes present a unique mode of O2 activation by perfluoroalkyl transition metal complexes that is unobserved in commonly studied trifluoromethyl analogs, and that leads to facile O-atom transfer reactivity and the formation of transient high valent Ni intermediates.
C bond cleavage), aerobic oxidation of alcohols, and oxygenation of phosphines and sulfides in a stoichiometric manner. These NiII complexes present a unique mode of O2 activation by perfluoroalkyl transition metal complexes that is unobserved in commonly studied trifluoromethyl analogs, and that leads to facile O-atom transfer reactivity and the formation of transient high valent Ni intermediates.
Results and discussion
Synthesis of longer chain NiII bis (perfluoroalkyl) complexes
First, complexation of 1,8-naphthyridine (L1) with (MeCN)2NiII(C2F5)2 (ref. 21) precursor proceeds smoothly at room temperature (RT) in acetonitrile solution to give complex 1, isolated in 58% yield and characterized by NMR, FT-IR, and UV/vis spectroscopy and electrospray ionization high-resolution mass spectrometry (ESI-HRMS) (Scheme 2). Single crystal X-ray diffraction (SC-XRD) study reveals that complex 1 contains one equivalent of L1 coordinated to a Ni atom in a bidentate fashion with a Ni atom featuring a distorted square planar geometry, with geometry indices for four-coordinate structures τ4 = 0.20 and τ4′ = 0.19 (the values for ideal square planar and ideal tetrahedral geometries are 0 and 1, respectively),22 with trans-N–Ni–C bond angles of 166.08(9)° and 165.69(7)° (Fig. 1). Such distortion from square planarity is likely due to steric repulsion between ortho-H atoms of naphthyridine and fluorine atoms of the C2F5 group. Similarly, Vicic and co-workers reported that (4,4′-bis-tert-butyl-bipyridine)Ni(RF)2 (RF = C2F5 or CF3) complexes feature a distorted square planar geometry around Ni, which was attributed to interactions between fluorine atoms and ortho-H atoms of the ligand.23 Among (4,4′-bis-tert-butyl-bipyridine)Ni(RF)2 series, a significantly greater degree of distortion was observed in a C2F5-analog compared to a trifluoromethyl complex implying that the introduction of a longer pentafluoroethyl chain imposes a notably greater steric hindrance compared to CF3, while (bpy)NiMe2 was essentially square planar.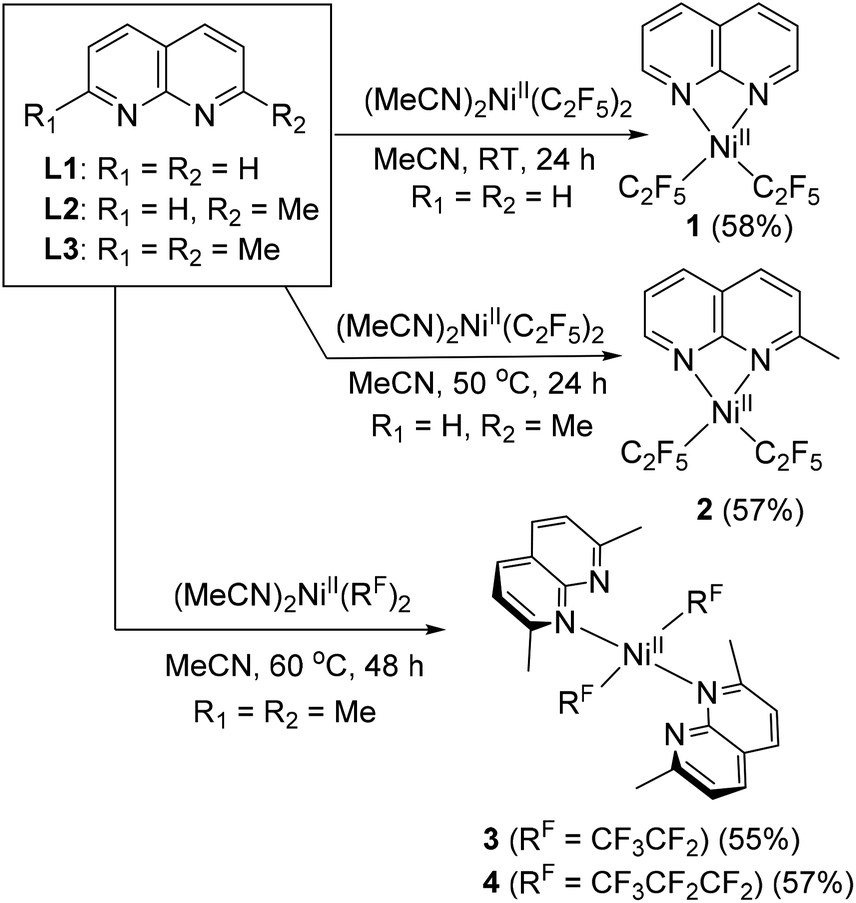 | ||
| Scheme 2 Synthesis of NiII pentafluoroethyl and heptafluoropropyl complexes supported by naphthyridine ligand (isolated yields are shown in parentheses). | ||
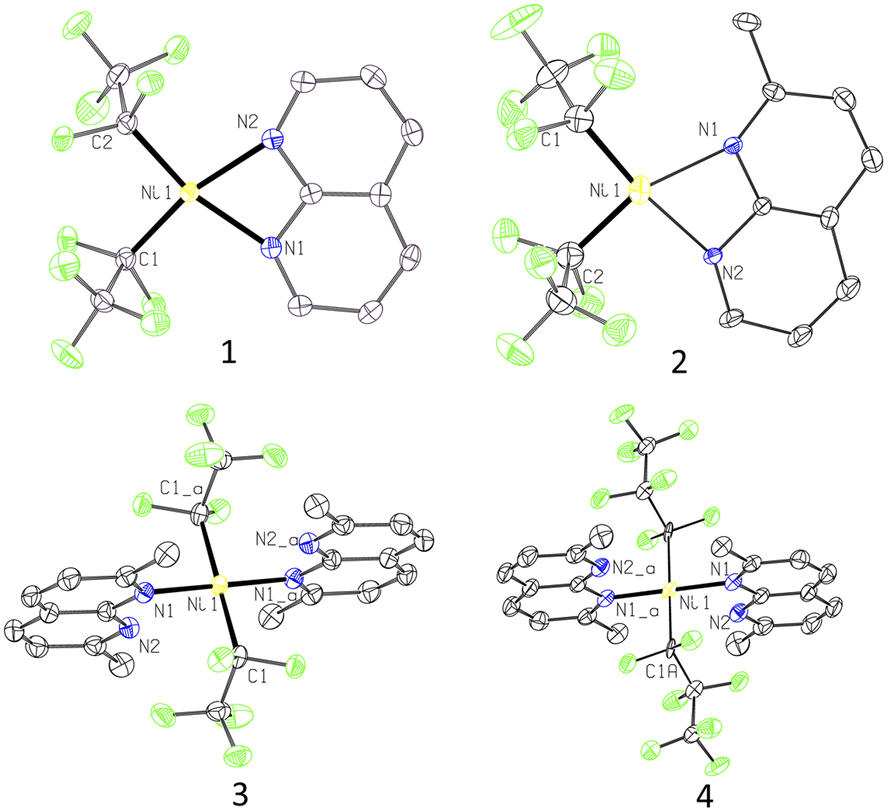 | ||
| Fig. 1 ORTEP of 1, 2, 3, and 4 at the 60% probability level. The disorder for 2 and 4 as well as hydrogen atoms are omitted for clarity. | ||
Interestingly, no complexation was observed at RT when more sterically hindered 2-methyl-1,8-naphthyridine (L2) was used, while further heating at 50 °C for 24 h afforded the yellow complex 2 with similar distorted square planar geometry and κ2-coordinated L2.
Treatment of (MeCN)2NiII(C2F5)2 with the even more sterically hindered 2,7-dimethyl-1,8-naphthyridine L3 at 60 °C resulted in the isolation of complex 3. Surprisingly, SC-XRD reveals that two equivalents of κ1-bound L3 coordinate to the nearly ideally square planar (τ4 = τ4′ = 0.00) Ni atom. The complex features two C2F5 groups in mutually trans-positions, with trans-N–Ni–N and C–Ni–C bond angles of 180°. This is strikingly different from the previously reported highly distorted square planar 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex (κ2-L3)NiII(CF3)2 with mutually cis-CF3 groups (τ4 = τ4′ = 0.23; C–Ni–N angle of 163.59(12)°). Such a configuration is uncommon for NiII dialkyl or bis(perfluoroalkyl) complexes due to the destabilizing presence of two strong trans-influencing ligands in trans-positions to each other (the trans-influencing properties of perfluoroalkyl group are expected to be similar to those of alkyl ligands).23,24 In complex 3, this unusual coordination mode is adopted to avoid steric congestion between two ortho-Me groups of L3 and the bulkier C2F5 ligands, as supported by computational analysis (see Scheme S8 and Fig. S134 in the ESI†). Additional stabilization is achieved through the formation of an ideal square planar geometry preferred by a low spin NiII complex.
:1 complex (κ2-L3)NiII(CF3)2 with mutually cis-CF3 groups (τ4 = τ4′ = 0.23; C–Ni–N angle of 163.59(12)°). Such a configuration is uncommon for NiII dialkyl or bis(perfluoroalkyl) complexes due to the destabilizing presence of two strong trans-influencing ligands in trans-positions to each other (the trans-influencing properties of perfluoroalkyl group are expected to be similar to those of alkyl ligands).23,24 In complex 3, this unusual coordination mode is adopted to avoid steric congestion between two ortho-Me groups of L3 and the bulkier C2F5 ligands, as supported by computational analysis (see Scheme S8 and Fig. S134 in the ESI†). Additional stabilization is achieved through the formation of an ideal square planar geometry preferred by a low spin NiII complex.
To examine if the same structural features will be observed in the complexes containing even longer perfluoroalkyl chains, we treated an analogous heptafluoropropyl (MeCN)2NiII(C3F7)2 (ref. 20) precursor with L3 at 60 °C for 48 h, which led to the formation of complex 4. SC-XRD reveals a similar, nearly ideal square planar structure with two trans-C3F7 groups and trans-N–Ni–N and C–Ni–C angles of 180°. Interestingly, both complexes 3 and 4 show well-resolved 1H NMR spectra at RT in CD2Cl2 solution exhibiting only two L3 ligand multiplets and one Me group, which could be due to the dynamic coordination of L3.
The Ni–C bonds in complex 3 (1.992(2) Å) are notably longer than in 1 (1.8904(19) and 1.899(2) Å), which is attributed to a stronger trans-influence between two C2F5 groups at 180°. Furthermore, Ni–C distances in longer-chain heptafluoropropyl complex 4 (2.021(6) Å) are slightly elongated compared to 3.
Aerobic reactivity of complexes 2–4
We have previously reported that bis(trifluoromethyl) complexes (L′)NiII(CF3)3 (L′ = L1, L2, and L3) undergo facile aerobic oxidation in the presence of air or O2 and NaBF4 at RT to produce isolable NiIII complexes, [(L′)NiIII(CF3)2](BF4) (Scheme 1a). In the case of longer-chain perfluoroalkyl analogs, we anticipated that different geometry around the Ni center and elongated Ni–C distances due to greater steric congestion could lead to a different reactivity in aerobic oxidation.Since the attempted aerobic oxidation of complex 1 resulted in the release of a free ligand, we investigated the aerobic reactivity of complexes 2–4. When solutions of 2–4 were exposed to O2, no stable NiIII product could be obtained, although the formation of a transient NiIII species was detected spectroscopically at low temperature (vide infra). Surprisingly, when complexes 2–4 were exposed to air at RT for 2 h in m-xylene solution containing an additive of 5 equiv. of water, crystallization under ambient conditions afforded single crystals of trifluoroacetate complexes 2a–3a and pentafluoropropionate complex 4a isolated in 38–48% yields and analyzed by SC-XRD, ESI-HRMS, FT-IR, and UV/vis spectroscopy (Scheme 3 and Fig. 2). These complexes feature a pseudooctahedral geometry around a Ni coordinated to two (in 2a and 4a) or one (in 3a) substituted naphthyridine ligand. Complexes 2a and 3a also contain coordinated water as terminal or bridging ligands, respectively. The Ni–O bond lengths in 2a and 3a, 2.0553(14) Å and 2.0758(10) Å, are consistent with typical Ni–O bond length in NiII aqua complexes,25 while significantly shortened Ni distance (ca. 1.86–1.89 Å) is expected from NiIII-hydroxo-bridged compounds.26 The hydrogen atoms were placed onto coordinated water molecules and were freely refined, their position were determined from Fourier difference electron density maps (Fig. S130–S131).† The perfluorocarboxylate ligands in 2a–4a retain the same number of carbon atoms, indicating that the carboxylate group is not formed via CO2 insertion into the Ni–C bond but rather via a formal removal of two F-atoms from the Ni-bound difluoromethylene fragment.
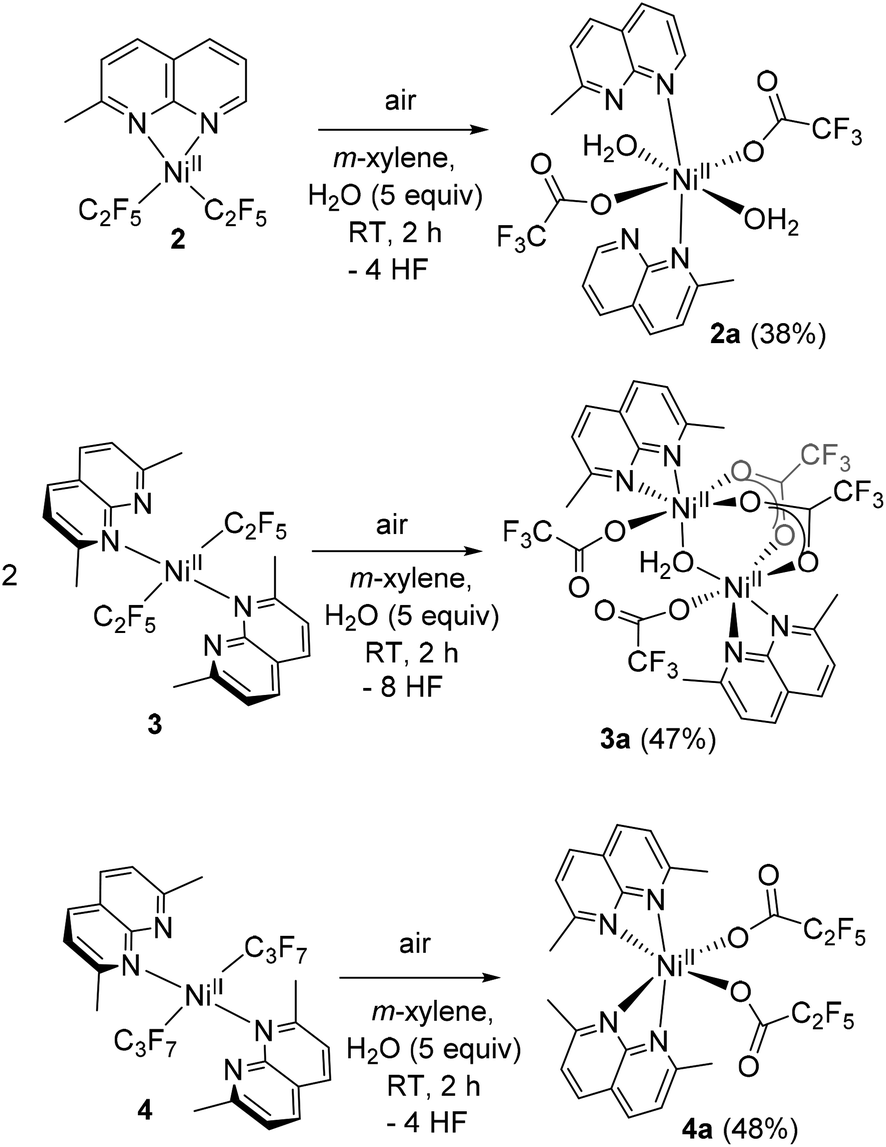 | ||
| Scheme 3 Isolation of perfluorocarboxylate complexes by aerobic oxidation of 2-4 (isolated yields are shown in parentheses). | ||
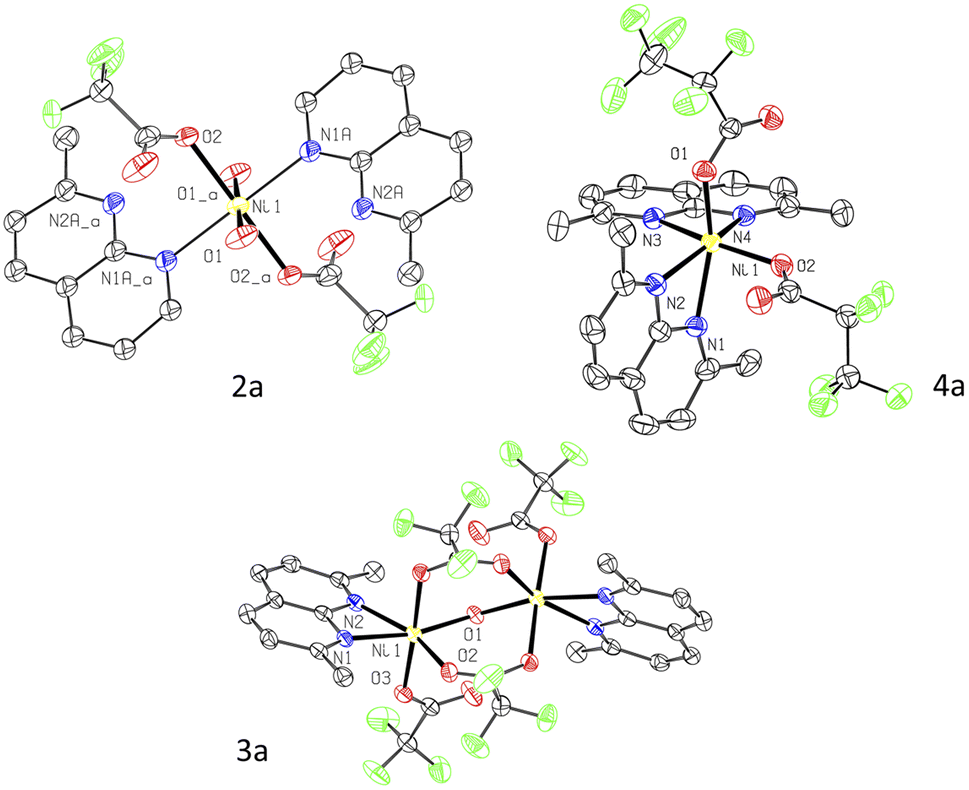 | ||
| Fig. 2 ORTEP of 2a, 3a, and 4a at the 60% probability level. | ||
The formation of perfluorocarboxylate products implies that free fluoride should be released into solution. This was confirmed using a fluoride paper test (see ESI†). In addition, the formation of an unstable fluoride-bridged dinickel complex was confirmed by SC-XRD (vide infra).
To quantify the amount of perfluorocarboxylate formed during aerobic oxidation, we performed aerobic oxidation in a protic coordinating solvent, methanol, which could promote trifluoroacetate dissociation. The yield of diamagnetic fluorine-containing products was then determined by 19F NMR integration against the internal standard, α,α,α-trifluorotoluene, and reported relative to the total number of available perfluoroalkyl groups (i.e., two per Ni). Exposure of a MeOH-d4 solution of 3 to ambient air at RT for 24 h produced a mixture of trifluoroacetate (70%) and pentafluoroethane (16%). Similarly, aerobic oxidation of 4 produced pentafluoropropionate (30%) and heptafluoropropane (38%). No oxidation was observed in MeOH solution of 3 under N2, or in MeCN solution in the presence of 100 equiv. of water, and only starting material could be detected, showing that complex 3 is stable in protic solvents in the absence of oxygen. When the oxidation of 3 was performed under dry O2 in methanol, the yield of trifluoroacetate increased to 76% (average for 2 trials), while no detectable amount of pentafluoroethane was observed.
To confirm the origin of O-atoms, we performed a series of experiments using 18O-isotopically labeled reagents and analyzed the products by ESI-HRMS. First, two sets of peaks were detected in the control reaction mixture obtained by oxidation of 3 with unlabeled O2 corresponding to cationic complexes of Ni with trifluoroacetate containing one or two L3 ligands per Ni: [(L3)Ni(CF3COO)]+ (m/z 329.0037, calc. 329.0042) and [(L3)2Ni(CF3COO)]+ (m/z 487.0880, calc. 487.0886) (Scheme 4a and Fig. S56†). The formation of both 2:1 and 1:1 complexes could be due to the labile nature of NiII complexes in solution and multiple modes of L3 coordination. Next, when the oxidation of 3 was performed using 18O2, the isotopic composition of these two peaks changes showing that the main product contained a doubly labeled trifluoroacetate, evident from the presence of the peaks corresponding to [(L3)Ni(CF3C18O2)]+ (m/z 333.0121, calc. 333.0127) and [(L3)2Ni(CF3C18O2)]+ (m/z 491.0954, calc. 491.0971). The minor peaks were assigned to the analogous adduct with a singly labeled CF3C16O18O− (m/z 331.0079 and 489.0909), and the non-labeled product was present in only small amounts. The ratio of the peaks corresponding to adducts with CF3C18O2−, CF3C18O16O−, and CF3C16O2− was 100:36:16 after 24 hours (Scheme 4b and Fig. S58†). Similarly, when the oxidation of heptafluoropropyl complex 4 was performed under the same conditions, the major peaks were assigned to the doubly labeled adduct as a major product, while singly and unlabeled isotopomers were present in smaller amounts (the C2F5C18O2−:C2F5C18O16O−:C2F5C16O2− ratio of 100:40:20 after 24 h) (Scheme 4c and Fig. S62†).
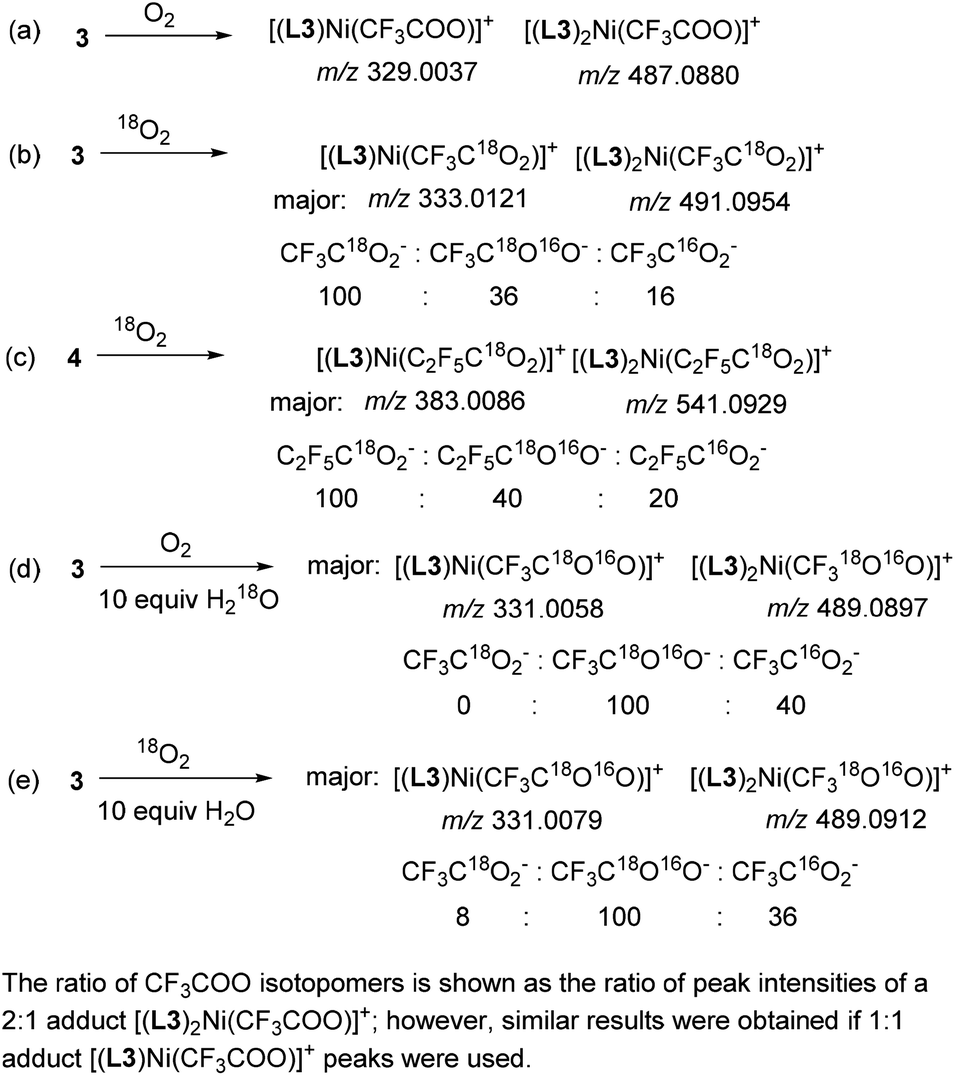 | ||
| Scheme 4 Reactivity of 3 and 4 with labeled O2 and water. | ||
The presence of a singly labeled product could result either from isotopomeric O2 impurities or more likely from adventitious water present in a solvent. To confirm this idea, we performed oxidation of 3 with O2 in the presence of 10 equiv. of H218O, which resulted in the formation of a singly labeled CF3C18O16O− adduct as a major product (Scheme 4d), while the analogous reaction under 18O2 with 10 equiv. of unlabeled water produced singly labeled adduct and doubly labeled adduct in 100:8 ratio (Scheme 4e). These results suggest that when excess water is introduced into a reaction mixture, it serves as a source of one of the O-atoms, while in the absence of added water, second oxygen atoms originate from O2 or O2-derived products (e.g., peroxides or hydroxide).
Intrigued by this dioxygenase-type reactivity, we examined if oxygen transfer from O2 could be directed to organic substrates in the presence of a suitable O-atom acceptor. Stirring solution of PPh3 in the presence of 1 equiv. of 3 under an O2 atmosphere at RT for 3–4 h afforded triphenylphosphine oxide with complete conversion (Scheme 5a). The PPh3 oxidation in the presence of 3 and 18O2 results in the formation of 18O-labeled Ph3P18O as a major product (90%) with only ca. 10% of unlabeled Ph3PO present as confirmed by HRMS and GC-MS (Scheme 5b). One equivalent of 3 relative to PPh3 was not required and using 0.5 equiv. 3 relative to PPh3 resulted in ca. 95% conversion to PPh3O after longer reaction time (20 h). Only a trace Ph3PO product was observed during oxidation of PPh3 with O2 under analogous conditions in the absence of 3.
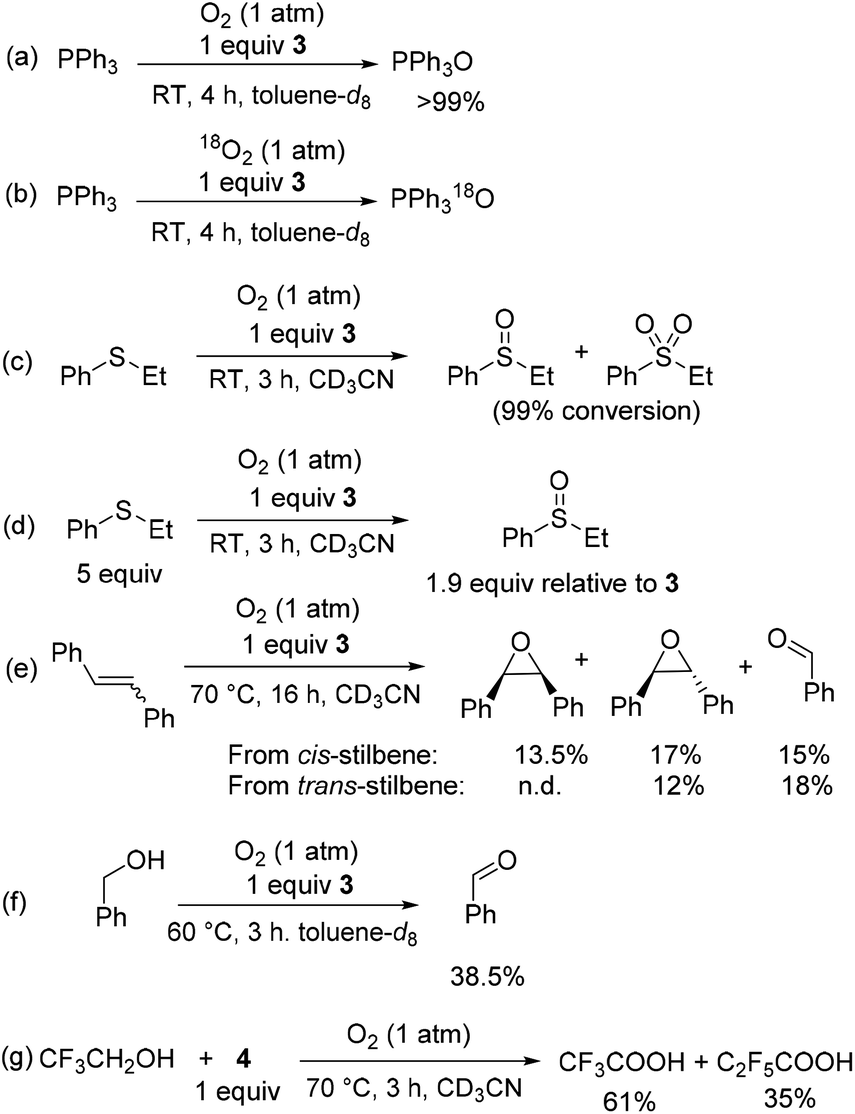 | ||
| Scheme 5 Substrate oxidation in the presence of O2 and complexes 3 or 4 under stoichiometric condition. Average yields are reported for 2–4 experiments. | ||
Oxidation of phenyl ethyl sulfide in the presence of only 1 equiv. of 3 under O2 atmosphere at RT produced a mixture of sulfoxide and sulfone at 99% conversion (Scheme 5c), whereas the analogous reaction with excess sulfide (5 equiv. relative to 3) lead to a cleaner reaction that gave 1.9 equiv. of sulfoxide relative to 3 (Scheme 5d).
Heating a solution of cis-stilbene and 1 equiv. of 3 under O2 at 70 °C for 16 h produced a mixture of cis-stilbene oxide (13.5%), trans-stilbene oxide (17%), and benzaldehyde (15%) along with unreacted starting material (Scheme 5e). Oxidation of trans-stilbene under O2 in the presence of 3 produced trans-epoxide and benzaldehyde as main products in 12% and 18% average yields, respectively (Scheme 5e).
Oxidation of benzyl alcohol in the presence of 1 equiv. of 3 at 60 °C produced benzaldehyde in 38.5% yield after 3 h (Scheme 5f).
In the case of 2,2,2-trifluoroethanol oxidation, the unstable aldehyde intermediate was not detected and only the product of full oxidation, trifluoroacetic acid, was produced in 61% yield when 2,2,2-trifluoroethanol was heated under 1 atm of O2 in the presence of complex 4 along with perfluoropropionate (35%) formed by C3F7 ligand oxidation (Scheme 5g).
During oxidation of PPh3 or trans-stilbene in the presence of 3 and O2, trifluoroacetate was detected only in minor amounts, 9–15%, showing that C2F5 groups might not be fully reacted when competitive oxygen acceptor was present. Although catalytic turnover has not yet been achieved, this might indicate the potential for substoichiometric use of nickel precursor in aerobic oxidation.
Overall, this reactivity shows that in the presence of suitable O-atom acceptors, activation of molecular oxygen by Ni perfluoroalkyl complexes resulted in oxidation or oxygenation of a number of substrates including phosphines, sulfides, alcohols, and alkenes.
Mechanistic investigation
To gain insight into the nature of possible high-valent Ni intermediates of O2 activation with 3, a solution of 3 in 2-methyltetrahydrofuran (2-MeTHF) was exposed to O2 for 2 min and the resulting purple solution was frozen at 91 K and analyzed by EPR spectroscopy. An anisotropic signal was detected characterized by g values of 2.228, 2.206, and 2.023 showing splitting to a single N-atom (AN = 16.3 G) (Fig. 3), which eventually disappears after warming up to RT at long reaction time. Significant g anisotropy and high g average values (gave = 2.152) are consistent with a NiIII species similar to the previously reported complex B which shows splitting from two N-atoms (Scheme 1a; AN = 17.9 G) (Scheme 1a, top right corner).19 This implies that while complex B containing two cis-CF3 groups is stabilized by two κ2-L3 ligands, similar κ2-coordination of two L3 ligands cannot be achieved upon oxidation of 3 and an exogenous ligand may be present. These differences could be attributed to a greater steric congestion around the Ni center in 3. A similar signal was obtained upon exposure of complex 4 to O2, showing superhyperfine splitting from only one N-atom (Fig. S105†). | ||
| Fig. 3 Experimental X-band EPR spectra of the product of oxidation of 3 with O2 (black line: MeTHF glass, 91 K, 9.082 GHz) and simulated EPR spectrum (red line). Simulation parameters: gx = 2.228, gy = 2.206, gz = 2.023 (AN = 16.3 G). | ||
Such lack of stabilization by bis-ligation to two cisL3 ligands could be imposed by steric congestion imposed by two ortho-Me groups of L3 and bulky C2F5 ligands: for example, computational analysis shows that significant degree of steric congestion is present in a hypothetical cis-(L3)2NiII(C2F5)2 already in +2 oxidation state leading to significantly distorted structure and its destabilization compared to trans-configuration (see Scheme S8 and Fig. S134†).
When the aerobic oxidation of 3 was performed in the presence of 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a radical trap for O-derived radicals, no DMPO-OH adducts were observed suggesting that Fenton-type chemistry (e.g. via Ni-mediated OH˙ radical formation by decomposition of O2-derived H2O2) is unlikely to occur under these conditions, and the inner-sphere O2 activation via superoxide formation may be involved in the initial oxidation to NiIII (see Fig. S102†).27
Considering that trans-arrangement of two perfluoroalkyl groups will likely result in their greater reactivity in Ni–C bond homolysis, especially in NiIII species, we then tested for the presence of the C2F5˙ radical using the (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) radical trap. When 3 was exposed to O2 for 24 h at RT in the presence of TEMPO in CD3OD, C6D6, or CD3CN solutions, the TEMPO-C2F5 adduct formed in 5–12% yields, while no TEMPO-C2F5 was detected in the absence of O2, showing that aerobic oxidation is required for radical formation.28
Based on these observations, we propose that aerobic oxidation of 3 follows a radical pathway (Scheme 6), resembling the radical-chain mechanism for the oxidation of (bpy)PdMe2 under O2 pressure in the presence of AIBN radical initiator.29 This mechanism is similar to the radical autooxidation of main group organometallics;30 however, intermediates featuring Ni in higher oxidation states are accessible.
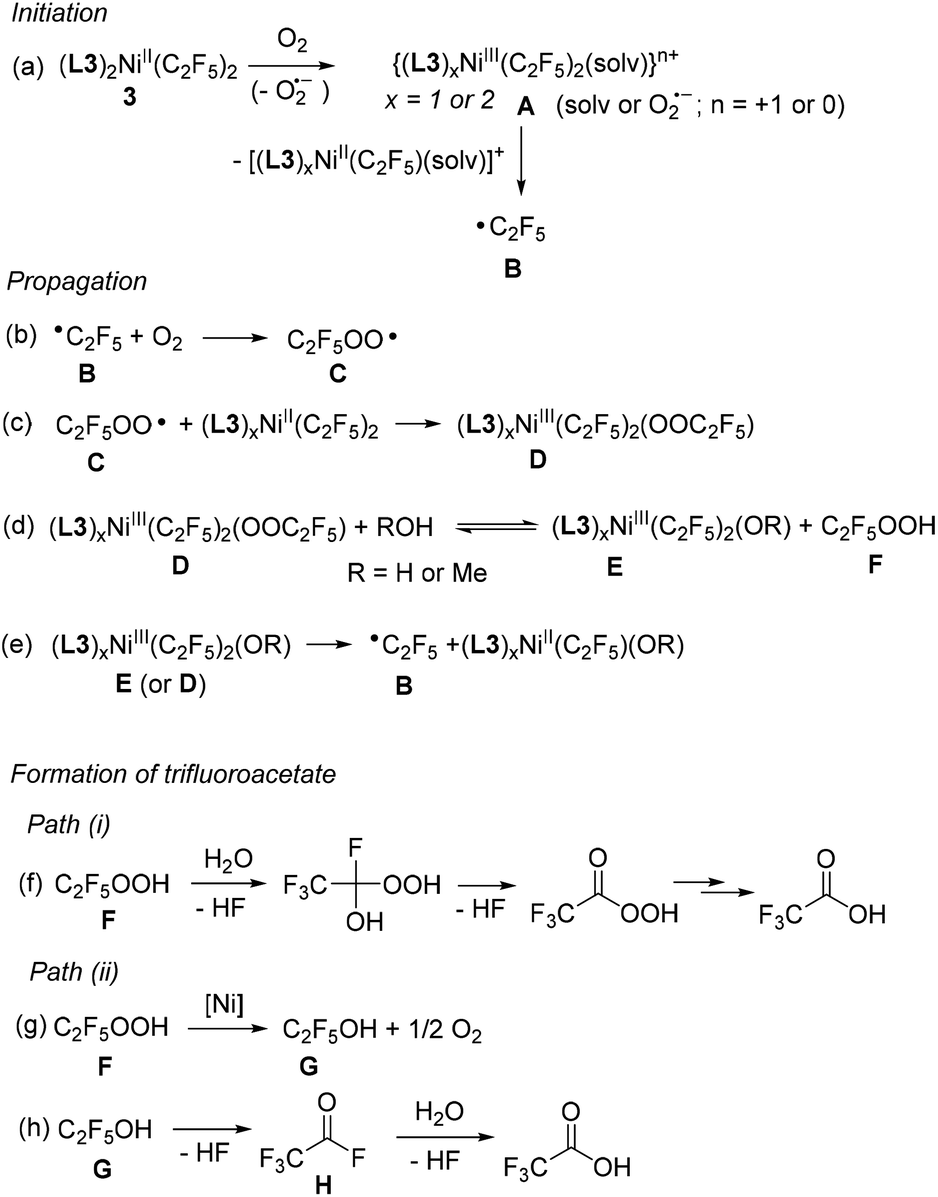 | ||
| Scheme 6 Proposed mechanism for aerobic oxidation of 3. | ||
The initiation step involves the formation of an unstable NiIII species, whose presence was confirmed by an EPR experiment (Fig. 3). Due to steric congestion and the mutual trans-configuration of two strong trans-influencing perfluoroalkyl groups in the starting material, such complexes are expected to be significantly less stable as compared to the previously reported cis-[(L3)2NiIII(CF3)2]+ and likely to undergo facile, spontaneous NiIII–C bond homolysis to produce a perfluoroalkyl radical B (Scheme 6a). In a propagation sequence, perfluoroalkyl radical B reacts with O2, an efficient radical trap for alkyl radicals, to produce the perfluoroalkylperoxyl radical C (Scheme 6b).31,32
Haloalkyl peroxyl radicals have been reported to form in aerated solutions containing parent haloalkyl radicals, and they are known to act as efficient one-electron oxidants for metal complexes and organic substrates; such electron transfer may occur via an inner sphere mechanism and result in the formation of halogenated alkylperoxide anion.31a,33 For example, oxidation of porphyrin metal complexes by CF3OO˙ radical resulting in one-electron oxidation of a metal center and trifluoroperoxide formation has been previously reported in the literature.31a,34 By analogy, such reactivity between C and an easily oxidized NiII bis(perfluoroalkyl) complex would result in the formation of a NiIII intermediate D (Scheme 6c) or, upon displacement of an electron-poor perfluoroalkylperoxo ligand with a protic solvent, intermediate E and free perfluoroalkyl hydroperoxide (Scheme 6d).35 Further propagation involves the extrusion of perfluoroalkyl radical B from a NiIII intermediate D or E (Scheme 6e) via a Ni–C bond homolysis.
Further decomposition of perfluoroalkyl hydroperoxide F is likely responsible for the carboxylate formation in the presence of a protic solvent or adventitious water from air via Path (i) or Path (ii) (Scheme 6). While there is currently no reported data on the hydrolytic stability of perfluoroalkyl hydroperoxides, 1,1-dichloroethyl peroxide is known to undergo spontaneous reaction with water to give peracetic acid and eventually acetic acid with the release of HCl.36 Similar decomposition of perfluoroalkyl hydroperoxide Fvia Path (i) would then result in the formation of the corresponding carboxylic acid (Scheme 6f).
Alternatively, considering the ability of many transition metal complexes to catalyze alkylhydroperoxide disproportionation to alcohol and O2, Path (ii) could involve Ni-catalyzed disproportionation of perfluorohydroperoxide to form a corresponding perfluorinated alcohol G (Scheme 6g).37 The ability of 3 to catalyze hydroperoxide decomposition was confirmed in the experiment with cumene hydroperoxide (see ESI†) and was reported for other NiII complexes.38 Both perfluoroethanol and perfluoro-n-propyl alcohol are known to be highly unstable and were characterized only in equilibrium mixtures with HF and the corresponding acyl fluoride (H) formed via thermodynamically favorable HF elimination.39 The formation of easily hydrolyzed acyl fluoride H in the presence of protic solvents or adventitious water is expected to result in the formation of trifluoroacetic acid as a final product (Scheme 6h).40 The formation of a singly-labeled trifluoroacetate as a major product in an experiment that involves 18O2/H2O or O2/H218O is also consistent with the proposed mechanism.41
Depending on the nature of the substrate, the O-transfer reactivity with organic substrates in the presence of 3 and O2 could ultimately be due to the formation of perfluoroalkylperoxo species, which could be present either in high valent Ni-ligated form such as intermediate D13a,15b,15d (or oxo/hydroxo species resulting from subsequent O–O bond cleavage) or as free radicals upon their release into solution, both of which are known to be able to transfer oxygen atom to phosphines.30
Stilbene oxidation in the presence of O2 and 3 (Scheme 5e) could also involve free radical reactivity or high valent Ni-ligated species: epoxide and aldehyde formation has been reported in free-radical alkene oxidation via alkylperoxy radical addition42 or dioxetane intermediates, respectively.43 However, considering that high valent Ni oxo or oxyl species have also been commonly proposed as intermediates in alkene epoxidation, these pathways cannot be clearly distinguished.14a–g
At the same time, the oxidation of alcohols likely involves the interaction of a Ni center directly with a substrate as no reaction was observed under free radical conditions in the absence of Ni.
Detection of mixed-valent NiIV/NiII species
In an attempt to intercept high-valent intermediates, complex 3 was exposed to dry O2 at −78 °C in CH2Cl2–MeCN solution. After 2 minutes, a purple paramagnetic solution formed, which showed an EPR signal (Fig. S106†) analogous to that in Fig. 3. Further slow crystallization from this solution for 2 weeks at −80 °C produced blue crystals of complex 5-MeCN (Scheme 7 and Fig. 4). SC-XRD analysis of 5-MeCN revealed the formation of a binuclear complex with two inequivalent Ni atoms bridged by three fluorine atoms; where one Ni atom is bound to three pentafluoroethyl groups, while another Ni atom coordinates to a κ2-bound L3 and one MeCN molecule. The analogous reaction in CH2Cl2-THF led to the crystallization of a similar THF-solvated 5-THF (Fig. 4). The formation of bridging fluoride also confirms the presence of an extracted individual fluorine atom during aerobic oxidation of 3.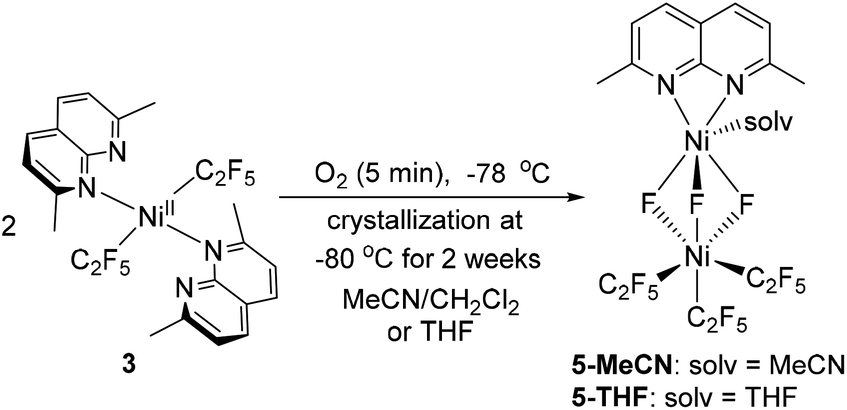 | ||
| Scheme 7 Formation of 5-MeCN and 5-THF. | ||
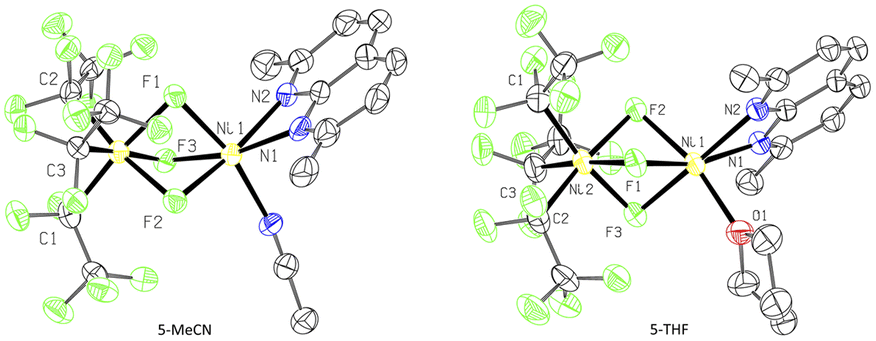 | ||
| Fig. 4 ORTEP of 5-MeCN (left) and 5-THF (right) at the 50% probability level.51 | ||
Complexes 5-THF and 5-MeCN show distinctly different Ni1–F bonds around two Ni atoms,44 with shorter Ni2–F bonds (1.910(4)–1.914(4) Å for 5-MeCN, 1.9066(15)–1.9136(17) Å for 5-THF) surrounding the Ni(C2F5)3 center and longer Ni1–F bonds (1.968(6)–1.971(7) Å for 5-MeCN and 1.998(17)–2.043(16) for 5-THF) at the (L3)Ni(solv) center (Table 1). The Ni1–N bonds at the (L3)Ni(solv) center (2.097(2)–2.106(6) Å) are similar to those present in paramagnetic NiII complexes 2a–4a (2.100(1)-2.182(2) Å) and are also similar to NiII–N bonds in high spin NiII complexes with N-donor ligands (2.045–2.095 Å),45 while somewhat shorter Ni–N bonds may be expected from high-valent Ni complexes.46 All three Ni2–C distances are similar (1.968(6) Å, 1.969(7) Å, and 1.971(7) Å in 5-MeCN), suggesting pseudooctahedral coordination expected from NiIV rather than a Jahn–Teller distorted NiIII,46,47 and similar to NiIV–CF3 distances reported in the known NiIV complexes (1.960–2.016 Å).46 Based on the highly inequivalent coordination properties of each Ni center, complexes 5-MeCN and 5-THF can be assigned as NiIV/NiII mixed valent species, further supported by comparing the DFT-calculated relative stability of NiIV/NiIIvs. the antiferromagnetically coupled NiIII/NiIII assignment for 5-MeCN.
Complexes 5-THF and 5-MeCN likely result from a bimolecular alkyl group transfer in a follow-up reactivity after initial aerobic oxidation to NiIII. Similarly, Me group transfer was reported during the oxidation of PdMe2 complexes with N-donor ligands by ferrocenium or O2, leading to the formation of [(L′)PdIVMe3]+ and [(L′)PdIIMe(solv)]+ (L′ = 4,4′-bis-tert-butyl-2,2′-bipyridine or N,N′,N′′-trimethyl-1,4,7-triazacyclononane).48 Such alkyl group transfer has not been reported in the aerobic oxidation of Ni complexes; however, the formation of mixed trialkyl NiIV complexes by the direct radical attack at a NiIII occurred when radicals were generated thermally using conventional free-radical initiators.47b Similarly, the alkyl radical attack at the metal center leading to its one-electron oxidation a new M-alkyl bond formation has been studied for other transition metal complexes.35,49 Therefore, the formation of 5 represents a rare example of alkyl group transfer between Ni complexes induced by aerobic oxidation. Although complexes 5-solv may not be directly involved in O-transfer reactivity, they demonstrate that mixed-valent complex formation may be viewed as a way to trap the C2F5 radical (as well as the HF byproduct) in the absence of a substrate. Interestingly, warming up the initially obtained purple solution obtained at low-temperature reaction of 3 with O2 still results in the formation of 3a as confirmed by SC-XRD, and trifluoroacetate formation is detected by NMR in the mixtures with protic solvents.50
| Interatomic distance (Å) | 5-MeCN | 5-THF |
|---|---|---|
| a Atom numbering is according to Fig. 4. b N. A. – not applicable. | ||
| Ni1–Ni2 | 2.6705(14) | 2.6554(6) |
| Ni1–F1 | 2.045(4) | 1.9982(17) |
| Ni1–F2 | 2.026(4) | 2.0439(16) |
| Ni1–F3 | 2.006(4) | 2.0118(16) |
| Ni1–N1 | 2.102(5) | 2.097(2) |
| Ni1–N2 | 2.106(6) | 2.108(2) |
| Ni1–O1 | N. A.b | 2.040(2) |
| Ni1–N3 | 2.020(5) | N. A.b |
| Ni2–F1 | 1.910(4) | 1.9136(17) |
| Ni2–F2 | 1.913(4) | 1.9066(15) |
| Ni2–F3 | 1.912(4) | 1.9135(18) |
| Ni2–C1 | 1.972(7) | 1.967(3) |
| Ni2–C2 | 1.968(6) | 1.948(3) |
| Ni2–C3 | 1.969(7) | 1.966(3) |
Conclusions
Aerobic oxidation of perfluoroalkyl complexes containing perfluoroethyl or perfluoropropyl groups shows distinctly different reactivity from the analogous trifluoromethyl complexes, which can be attributed to greater steric congestion imposed by longer-chain perfluoroalkyl groups. By contrast to trifluoromethyl complexes, stable NiIII could not be obtained, although transient NiIII intermediates were observed by EPR spectroscopy. In the absence of an external substrate, perfluorocarboxylate complexes were formed by the transfer of O-atoms from O2 to the perfluoroalkyl group. Isotopic labeling studies suggest that in the absence of added water, O2 serves as a major source of both O-atoms in the carboxylate product via dioxygenase-like reactivity, while in the presence of excess water, only one of the O-atoms originates from O2.Oxygenation of phosphine, sulfide, and stilbenes by O2 as a terminal oxidant was also achieved that was promoted by longer-chain perfluoroalkyl Ni complexes. Aerobic oxidation of alcohols was also observed under stoichiometric conditions.
Overall, these results demonstrate that longer-chain perfluoroalkyl metal complexes may show drastically different reactivity from their commonly studied trifluoromethyl analogs. In the case of Ni complexes with naphthyridine ligands, such reactivity led to the observation of dioxygenase-like O-atom transfer from dioxygen under mild conditions.
Data availability
Data supporting this article have been uploaded as ESI.†Author contributions
SD and JK designed the study, interpreted the data, and wrote the manuscript; RG and JB performed ligand synthesis; SV, EK and RF collected and refined crystallographic data; MR helped to carry out the mass spectrometry measurements; JK performed EPR experiments and computational analysis. All authors contributed in manuscript writing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Instrumental Analysis section and Engineering section for technical support, the HPC facility for access to computational resources, and OIST for funding. R. R. F. performed crystal structure determination within the assignment for the Kazan Scientific Center of RAS. The authors thank Dr Ayumu Karimata for GPC analysis.Notes and references
- X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef CAS PubMed.
- (a) G. B. Shul'pin, J. Chem. Res., 2002, 2002, 351–353 CrossRef; (b) A. S. Hay, J. Org. Chem., 1962, 27, 3320–3321 CrossRef CAS; (c) S. S. Stahl, Angew. Chem., Int. Ed., 2004, 43, 3400–3420 CrossRef CAS PubMed; (d) L. Que and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef CAS PubMed.
- (a) J. S. Valentine, Chem. Rev., 1973, 73, 235–245 CrossRef CAS; (b) D. D. Wick and K. I. Goldberg, J. Am. Chem. Soc., 1999, 121, 11900–11901 CrossRef CAS; (c) V. V. Rostovtsev, L. M. Henling, J. A. Labinger and J. E. Bercaw, Inorg. Chem., 2002, 41, 3608–3619 CrossRef CAS PubMed; (d) J. R. Khusnutdinova, P. Y. Zavalij and A. N. Vedernikov, Organometallics, 2007, 26, 2402–2413 CrossRef CAS; (e) R. A. Taylor, D. J. Law, G. J. Sunley, A. J. P. White and G. J. P. Britovsek, Angew. Chem., Int. Ed., 2009, 48, 5900–5903 CrossRef CAS PubMed; (f) A. R. Petersen, R. A. Taylor, I. Vicente-Hernández, P. R. Mallender, H. Olley, A. J. P. White and G. J. P. Britovsek, J. Am. Chem. Soc., 2014, 136, 14089–14099 CrossRef CAS PubMed; (g) L. Xu, D. P. Solowey and D. A. Vicic, Organometallics, 2015, 34, 3474–3479 CrossRef CAS; (h) H. E. Zeitler, W. A. Kaminsky and K. I. Goldberg, Organometallics, 2018, 37, 3644–3648 CrossRef CAS.
- (a) M. M. Konnick, I. A. Guzei and S. S. Stahl, J. Am. Chem. Soc., 2004, 126, 10212–10213 CrossRef CAS PubMed; (b) J. M. Keith, R. P. Muller, R. A. Kemp, K. I. Goldberg, W. A. Goddard and J. Oxgaard, Inorg. Chem., 2006, 45, 9631–9633 CrossRef CAS PubMed; (c) D. Wang, A. B. Weinstein, P. B. White and S. S. Stahl, Chem. Rev., 2018, 118, 2636–2679 CrossRef CAS PubMed.
- R. Neumann and M. Dahan, Nature, 1997, 388, 353–355 CrossRef CAS.
- (a) C. M. Frech, L. J. W. Shimon and D. Milstein, Helv. Chim. Acta, 2006, 89, 1730–1739 CrossRef CAS; (b) J. M. Praetorius, D. P. Allen, R. Wang, J. D. Webb, F. Grein, P. Kennepohl and C. M. Crudden, J. Am. Chem. Soc., 2008, 130, 3724–3725 CrossRef CAS PubMed; (c) E. C. Keske, O. V. Zenkina, A. Asadi, H. Sun, J. M. Praetorius, D. P. Allen, D. Covelli, B. O. Patrick, R. Wang, P. Kennepohl, B. R. James and C. M. Crudden, Dalton Trans., 2013, 42, 7414–7423 RSC.
- (a) Z. M. Heiden and T. B. Rauchfuss, J. Am. Chem. Soc., 2007, 129, 14303–14310 CrossRef CAS PubMed; (b) D. Bridget Williams, W. Kaminsky, J. M. Mayer and K. I. Goldberg, Chem. Commun., 2008, 4195–4197 RSC; (c) S. Chowdhury, F. Himo, N. Russo and E. Sicilia, J. Am. Chem. Soc., 2010, 132, 4178–4190 CrossRef CAS; (d) K. E. Allen, D. M. Heinekey, A. S. Goldman and K. I. Goldberg, Organometallics, 2014, 33, 1337–1340 CrossRef CAS; (e) M. Feller, E. Ben-Ari, Y. Diskin-Posner, R. Carmieli, L. Weiner and D. Milstein, J. Am. Chem. Soc., 2015, 137, 4634–4637 CrossRef CAS PubMed; (f) P. Abril, M. P. del Río, J. A. López, A. Lledós, M. A. Ciriano and C. Tejel, Chem.–Eur. J., 2019, 25, 14546–14554 CrossRef CAS PubMed.
- (a) N. Kitajima and Y. Moro-oka, Chem. Rev., 1994, 94, 737–757 CrossRef CAS; (b) E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed; (c) N. Gagnon and W. B. Tolman, Acc. Chem. Res., 2015, 48, 2126–2131 CrossRef CAS PubMed; (d) C. E. Elwell, N. L. Gagnon, B. D. Neisen, D. Dhar, A. D. Spaeth, G. M. Yee and W. B. Tolman, Chem. Rev., 2017, 117, 2059–2107 CrossRef CAS PubMed.
- (a) M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Chem. Rev., 2004, 104, 939–986 CrossRef CAS PubMed; (b) T. L. Poulos, Chem. Rev., 2014, 114, 3919–3962 CrossRef CAS PubMed; (c) S. Sahu and D. P. Goldberg, J. Am. Chem. Soc., 2016, 138, 11410–11428 CrossRef CAS PubMed; (d) V. A. Larson, B. Battistella, K. Ray, N. Lehnert and W. Nam, Nat. Rev. Chem., 2020, 4, 404–419 CrossRef CAS PubMed; (e) B. Battistella and K. Ray, Coord. Chem. Rev., 2020, 408, 213176 CrossRef CAS.
- (a) J. A. Kovacs, Acc. Chem. Res., 2015, 48, 2744–2753 CrossRef CAS PubMed; (b) A. T. Fiedler and A. A. Fischer, JBIC, J. Biol. Inorg. Chem., 2017, 22, 407–424 CrossRef CAS PubMed.
- (a) D. Kim, J. Cho, Y.-M. Lee, R. Sarangi and W. Nam, Chem.–Eur. J., 2013, 19, 14112–14118 CrossRef CAS PubMed; (b) T. Corona, S. K. Padamati, F. Acuña-Parés, C. Duboc, W. R. Browne and A. Company, Chem. Commun., 2017, 53, 11782–11785 RSC; (c) P. Liebing, F. Oehler, M. Wagner, P. F. Tripet and A. Togni, Organometallics, 2018, 37, 570–583 CrossRef CAS; (d) P. Kumar, S. V. Lindeman and A. T. Fiedler, J. Am. Chem. Soc., 2019, 141, 10984–10987 CrossRef CAS PubMed.
- (a) F. Al-Mjeni, T. Ju, T. C. Pochapsky and M. J. Maroney, Biochem, 2002, 41, 6761–6769 CrossRef CAS PubMed; (b) A. T. Fiedler, P. A. Bryngelson, M. J. Maroney and T. C. Brunold, J. Am. Chem. Soc., 2005, 127, 5449–5462 CrossRef CAS PubMed; (c) J. L. Boer, S. B. Mulrooney and R. P. Hausinger, Arch. Biochem. Biophys., 2014, 544, 142–152 CrossRef CAS PubMed; (d) Y.-J. Sun, Q.-Q. Huang and J.-J. Zhang, Dalton Trans., 2014, 43, 6480–6489 RSC; (e) J.-H. Jeoung, D. Nianios, S. Fetzner and H. Dobbek, Angew. Chem., Int. Ed., 2016, 55, 3281–3284 CrossRef CAS PubMed.
- (a) A. Company, S. Yao, K. Ray and M. Driess, Chem.–Eur. J., 2010, 16, 9669–9675 CrossRef CAS; (b) A. T. Higgs, P. J. Zinn and M. S. Sanford, Organometallics, 2010, 29, 5446–5449 CrossRef CAS.
- (a) J. D. Koola and J. K. Kochi, Inorg. Chem., 1987, 26, 908–916 CrossRef CAS; (b) J. F. Kinneary, J. S. Albert and C. J. Burrows, J. Am. Chem. Soc., 1988, 110, 6124–6129 CrossRef CAS PubMed; (c) J. F. Kinneary, T. R. Wagler and C. J. Burrows, Tetrahedron Lett., 1988, 29, 877–880 CrossRef CAS; (d) H. Yoon and C. J. Burrows, J. Am. Chem. Soc., 1988, 110, 4087–4089 CrossRef CAS; (e) H. Yoon, T. R. Wagler, K. J. O'Connor and C. J. Burrows, J. Am. Chem. Soc., 1990, 112, 4568–4570 CrossRef CAS; (f) R. I. Kureshy, N. H. Khan, S. H. R. Abdi, P. Iyer and A. K. Bhatt, J. Mol. Catal. A: Chem., 1998, 130, 41–50 CrossRef CAS; (g) R. I. Kureshy, N. H. Khan, S. H. R. Abdi, S. T. Patel, P. Iyer, E. Suresh and P. Dastidar, J. Mol. Catal. A: Chem., 2000, 160, 217–227 CrossRef CAS; (h) T. Nagataki, Y. Tachi and S. Itoh, Chem. Commun., 2006, 4016–4018 RSC; (i) Y. Morimoto, S. Bunno, N. Fujieda, H. Sugimoto and S. Itoh, J. Am. Chem. Soc., 2015, 137, 5867–5870 CrossRef CAS PubMed; (j) Y. Qiu and J. F. Hartwig, J. Am. Chem. Soc., 2020, 142, 19239–19248 CrossRef CAS PubMed.
- (a) M. T. Kieber-Emmons, R. Schenker, G. P. A. Yap, T. C. Brunold and C. G. Riordan, Angew. Chem., Int. Ed., 2004, 43, 6716–6718 CrossRef CAS PubMed; (b) M. T. Kieber-Emmons, J. Annaraj, M. S. Seo, K. M. Van Heuvelen, T. Tosha, T. Kitagawa, T. C. Brunold, W. Nam and C. G. Riordan, J. Am. Chem. Soc., 2006, 128, 14230–14231 CrossRef CAS PubMed; (c) C. A. Rettenmeier, H. Wadepohl and L. H. Gade, Chem. Sci., 2016, 7, 3533–3542 RSC; (d) A. J. McNeece, K. A. Jesse, J. Xie, A. S. Filatov and J. S. Anderson, J. Am. Chem. Soc., 2020, 142, 10824–10832 CrossRef CAS PubMed.
- (a) F. Tang, N. P. Rath and L. M. Mirica, Chem. Commun., 2015, 51, 3113–3116 RSC; (b) S. M. Smith, O. Planas, L. Gómez, N. P. Rath, X. Ribas and L. M. Mirica, Chem. Sci., 2019, 10, 10366–10372 RSC.
- N. Zhao, A. S. Filatov, J. Xie, E. A. Hill, A. Y. Rogachev and J. S. Anderson, J. Am. Chem. Soc., 2020, 142, 21634–21639 CrossRef CAS PubMed.
- V. Dimitrov and A. Linden, Angew. Chem., Int. Ed., 2003, 42, 2631–2633 CrossRef CAS PubMed.
- S. Deolka, R. Govindarajan, E. Khaskin, R. R. Fayzullin, M. C. Roy and J. R. Khusnutdinova, Angew. Chem., Int. Ed., 2021, 60, 24620–24629 CrossRef CAS PubMed.
- S. Deolka, R. Govindarajan, S. Vasylevskyi, M. C. Roy, J. R. Khusnutdinova and E. Khaskin, Chem. Sci., 2022, 13, 12971–12979 RSC.
- C.-P. Zhang, H. Wang, A. Klein, C. Biewer, K. Stirnat, Y. Yamaguchi, L. Xu, V. Gomez-Benitez and D. A. Vicic, J. Am. Chem. Soc., 2013, 135, 8141–8144 CrossRef CAS PubMed.
- (a) L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC; (b) A. Okuniewski, D. Rosiak, J. Chojnacki and B. Becker, Polyhedron, 2015, 90, 47–57 CrossRef CAS; (c) D. Rosiak, A. Okuniewski and J. Chojnacki, Polyhedron, 2018, 146, 35–41 CrossRef CAS.
- Y. Yamaguchi, H. Ichioka, A. Klein, W. W. Brennessel and D. A. Vicic, Organometallics, 2012, 31, 1477–1483 CrossRef CAS.
- (a) V. V. Grushin and W. J. Marshall, J. Am. Chem. Soc., 2006, 128, 4632–4641 CrossRef CAS PubMed; (b) R. E. Douthwaite, M. L. H. Green, P. J. Silcock and P. T. Gomes, Organometallics, 2001, 20, 2611–2615 CrossRef CAS; (c) I. Kieltsch, G. G. Dubinina, C. Hamacher, A. Kaiser, J. Torres-Nieto, J. M. Hutchison, A. Klein, Y. Budnikova and D. A. Vicic, Organometallics, 2010, 29, 1451–1456 CrossRef CAS.
- (a) X. Meng, H. Hou, G. Li, B. Ye, T. Ge, Y. Fan, Y. Zhu and H. Sakiyama, J. Organomet. Chem., 2004, 689, 1218–1229 CrossRef CAS; (b) G. Hu, N. Xiao, L. Wang, L. Shen, X. Li, H. Xu, L. Han and X. Xiao, J. Solid State Chem., 2019, 270, 247–251 CrossRef CAS; (c) J. Z. Travis, C. J. LaRose and R. L. LaDuca, Inorg. Chim. Acta, 2017, 456, 158–170 CrossRef CAS.
- A. Das, S. Chakraborty and S. K. Mandal, Chem.–Asian J., 2021, 16, 2257–2260 CrossRef CAS PubMed.
- (a) K. Reszka and C. F. Chignell, Free Radical Res. Commun., 1991, 14, 97–106 CrossRef CAS PubMed; (b) G. R. Buettner, Free Radical Biol. Med., 1987, 3, 259–303 CrossRef CAS PubMed; (c) I. Yamazaki and L. H. Piette, J. Biol. Chem., 1990, 265, 13589–13594 CrossRef CAS.
- The generation of free radicals during aerobic oxidation of 3 is also consistent with its ability to initiate polymerization of methylacrylate. Thus, reacting 1.5 μmol 3 and 1.5 μmol of O2 with 1.5 mmol of methylacrylate for 16 h at 60 °C resulted in the formation of polymethylacrylate in 74% yield, while no polymerization was observed in the presence of O2 without 3 or in the presence of 3 under N2 with exclusion of O2.
- L. Boisvert, M. C. Denney, S. K. Hanson and K. I. Goldberg, J. Am. Chem. Soc., 2009, 131, 15802–15814 CrossRef CAS PubMed.
- A. G. Davies, J. Chem. Res., 2008, 2008, 361–375 CrossRef.
- (a) P. Neta, R. E. Huie and A. B. Ross, J. Phys. Chem. Ref. Data, 1990, 19, 413–513 CrossRef CAS; (b) P. W. N. M. Van Leeuwen, H. Van Der Heijden, C. F. Roobeek and J. H. G. Frijns, J. Organomet. Chem., 1981, 209, 169–182 CrossRef CAS; (c) J. K. Thomas, J. Phys. Chem., 1967, 71, 1919–1925 CrossRef CAS; (d) L. C. T. Shoute, Z. B. Alfassi, P. Neta and R. E. Huie, J. Phys. Chem., 1994, 98, 5701–5704 CrossRef CAS.
- The bimolecular rate constant of an O2 reaction with alkyl radicals is typically ∼ 109 M−1 s−1. See ref. 31a.
- (a) J. E. Packer, T. F. Slater and R. L. Willson, Life Sci., 1978, 23, 2617–2620 CrossRef CAS PubMed; (b) J. E. Packer, R. L. Willson, D. Bahnemann and K.-D. Asmus, J. Chem. Soc., Perkin Trans. 2, 1980, 296–299 RSC; (c) D. Brault and P. Neta, J. Phys. Chem., 1982, 86, 3405–3410 CrossRef CAS; (d) S. Mosseri, P. Neta and P. Hambright, J. Phys. Chem., 1989, 93, 2358–2362 CrossRef CAS; (e) D. Brault and P. Neta, J. Phys. Chem., 1984, 88, 2857–2862 CrossRef CAS; (f) D. Brault and P. Neta, J. Phys. Chem., 1983, 87, 3320–3327 CrossRef CAS.
- D. Brault and P. Neta, J. Phys. Chem., 1987, 91, 4156–4160 CrossRef CAS.
- A. Sauer, H. Cohen and D. Meyerstein, Inorg. Chem., 1988, 27, 4578–4581 CrossRef CAS.
- S. Gaeb and W. V. Turner, J. Org. Chem., 1984, 49, 2711–2714 CrossRef CAS.
- R. R. Hiatt, K. C. Irwin and C. W. Gould, J. Org. Chem., 1968, 33, 1430–1435 CrossRef CAS.
- (a) J. A. Howard and J. H. B. Chenier, Can. J. Chem., 1976, 54, 390–401 CrossRef CAS; (b) H. Sigel, K. Wyss, P. Waldmeier and R. Griesser, J. Coord. Chem., 1974, 3, 235–247 CrossRef CAS.
- A. F. Baxter, J. Schaab, J. Hegge, T. Saal, M. Vasiliu, D. A. Dixon, R. Haiges and K. O. Christe, Chem.–Eur. J., 2018, 24, 16737–16742 CrossRef CAS PubMed.
- C. J. Young and S. A. Mabury, in Reviews of Environmental Contamination and Toxicology Volume 208: Perfluorinated alkylated substances, ed. P. De Voogt, Springer New York, New York, NY, 2010, pp. 1–109 Search PubMed.
- The possible mechanism of the formation of doubly labeled trifluoroacetate is discussed in detail in the ESI.†.
- Y. Wu, X. Tang, J. Zhao, C. Ma, L. Yun, Z. Yu, B. Song and Q. Meng, ACS Sustainable Chem. Eng., 2020, 8, 1178–1184 CrossRef CAS.
- (a) G.-Z. Wang, X.-L. Li, J.-J. Dai and H.-J. Xu, J. Org. Chem., 2014, 79, 7220–7225 CrossRef CAS PubMed; (b) A. Rajagopalan, M. Lara and W. Kroutil, Adv. Synth. Catal., 2013, 355, 3321–3335 CrossRef CAS.
- To examine the possiblity of alternative assignments for fluorine atoms (e. g. hydroxide), when the type of bridging atoms was changed from fluorine to oxygen, the R-factors increased significantly. In this case, the volume of the thermal ellipsoids unnaturally decreased and positive residual peaks appeared very close to the nucleus. This usually indicates that an element with more electrons should be assigned (in this case, fluorine provided the best results). Moreover, when replacing fluorine with oxygen, no peaks of residual density are observed that could be attributed to the accompanying hydrogen atoms, although the quality of the data and their resolution are sufficient to expect them to appear.
- (a) M. Garai, D. Dey, H. R. Yadav, A. R. Choudhury, N. Kole and B. Biswas, Polyhedron, 2017, 129, 114–122 CrossRef CAS; (b) I. Ucar, O. Z. Yesilel, A. Bulut, H. Olmez and O. Buyukgungor, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, m947–m949 CrossRef CAS.
- F. D'Accriscio, P. Borja, N. Saffon-Merceron, M. Fustier-Boutignon, N. Mézailles and N. Nebra, Angew. Chem., Int. Ed., 2017, 56, 12898–12902 CrossRef PubMed.
- (a) E. A. Meucci, S. N. Nguyen, N. M. Camasso, E. Chong, A. Ariafard, A. J. Canty and M. S. Sanford, J. Am. Chem. Soc., 2019, 141, 12872–12879 CrossRef CAS PubMed; (b) J. R. Bour, D. M. Ferguson, E. J. McClain, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2019, 141, 8914–8920 CrossRef CAS PubMed.
- (a) M. P. Lanci, M. S. Remy, W. Kaminsky, J. M. Mayer and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 15618–15620 CrossRef CAS PubMed; (b) J. R. Khusnutdinova, F. Qu, Y. Zhang, N. P. Rath and L. M. Mirica, Organometallics, 2012, 31, 4627–4630 CrossRef CAS.
- (a) J. H. Espenson, Acc. Chem. Res., 1992, 25, 222–227 CrossRef CAS; (b) J. K. Kochi, Acc. Chem. Res., 1974, 7, 351–360 CrossRef CAS; (c) N. Shaham, A. Masarwa, Y. Matana, H. Cohen and D. Meyerstein, Eur. J. Inorg. Chem., 2002, 2002, 87–92 CrossRef.
- Allowing the purple reaction mixture obtained by low temperature oxidation of 3 with O2 to warm up to RT only for 3–5 minutes led to the formation of yellow solution, from which a mixed F-bridged/TFA-bridged complex 6 (see ESI†), [Ni(C2F5)3(μ-F)(μ-OOCCF3)2Ni(κ2-L3)(THF)] was obtained by crystallization at −80 °C for 3 days. This complex is likely formed via F-displacement by trifluoroacetate accumulated in solution during room temperature reaction.
- Deposition Numbers 2241816–2241824 and 2204584 contain the supplementary crystallographic data for this paper..
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2241816–2241824 and 2204584. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01861j |
| This journal is © The Royal Society of Chemistry 2023 |