
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsomerization-induced fluorescence enhancement of two new viologen derivatives: mechanism insight and DFT calculations†
Xiuping
Yin
a,
Xinxing
Li
ab,
Xuyi
Li
a,
Malgorzata
Biczysko
 *ab,
Shourong
Zhu
a,
Jiaqiang
Xu
a and
Yue-Ling
Bai
*a
*ab,
Shourong
Zhu
a,
Jiaqiang
Xu
a and
Yue-Ling
Bai
*a
aCollege of Science, Shanghai University, 99 Shangda Road, Shanghai 200444, China. E-mail: yuelingbai@shu.edu.cn; biczysko@shu.edu.cn
bInternational Center for Quantum and Molecular Structures, Department of Physics, College of Science, Shanghai University, 99 Shangda Road, Shanghai 200444, China
First published on 12th June 2023
Abstract
The dark-colored viologen radical cations are unstable in air and easily fade, thus greatly limiting their applications. If a suitable substituent is introduced into the structure, it will have the dual function of chromism and luminescence, which will broaden its application field. Here, Vio1·2Cl and Vio2·2Br were synthesized by introducing aromatic acetophenone and naphthophenone substituents into the viologen structure. The keto group (–CH2CO–) on the substituents is prone to isomerize into the enol structure (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) COH–) in organic solvents, especially in DMSO, resulting in a larger conjugated system to stabilize the molecular structure and enhance fluorescence. The time-dependent fluorescence spectrum shows obvious keto-to-enol isomerization-induced fluorescence enhancement. The quantum yield also increased significantly (T = 1 day, ΦVio1 = 25.81%, ΦVio2 = 41.44%; T = 7 days, ΦVio1 = 31.48%, and ΦVio2 = 54.40%) in DMSO. The NMR and ESI-MS data at different times further confirmed that the fluorescence enhancement was caused by isomerization, and no other fluorescent impurities were produced in solution. DFT calculations show that the enol form is almost coplanar throughout the molecular structure, which is conducive to stabilizing the structure and enhancing fluorescence. The fluorescence emission peaks of the keto and enol structures of Vio12+ and Vio22+ were at 416–417 nm and 563–582 nm, respectively. The fluorescence relative oscillator strength of Vio12+ and Vio22+ enol structures is significantly higher than that of keto structures (f value changes from 1.53 to 2.63 for Vio12+ and from 1.62 to 2.81 for Vio22+), indicating stronger fluorescence emission of the enol structure. The calculated results are in good agreement with the experimental results. Vio1·2Cl and Vio2·2Br are the first examples of isomerization-induced fluorescence enhancement of viologen derivatives, which shows strong solvatofluorochromism under UV light, making up for the disadvantage that it is easy for a viologen radical to fade in air, and providing a new strategy for designing and synthesizing viologen materials with strong fluorescence.
COH–) in organic solvents, especially in DMSO, resulting in a larger conjugated system to stabilize the molecular structure and enhance fluorescence. The time-dependent fluorescence spectrum shows obvious keto-to-enol isomerization-induced fluorescence enhancement. The quantum yield also increased significantly (T = 1 day, ΦVio1 = 25.81%, ΦVio2 = 41.44%; T = 7 days, ΦVio1 = 31.48%, and ΦVio2 = 54.40%) in DMSO. The NMR and ESI-MS data at different times further confirmed that the fluorescence enhancement was caused by isomerization, and no other fluorescent impurities were produced in solution. DFT calculations show that the enol form is almost coplanar throughout the molecular structure, which is conducive to stabilizing the structure and enhancing fluorescence. The fluorescence emission peaks of the keto and enol structures of Vio12+ and Vio22+ were at 416–417 nm and 563–582 nm, respectively. The fluorescence relative oscillator strength of Vio12+ and Vio22+ enol structures is significantly higher than that of keto structures (f value changes from 1.53 to 2.63 for Vio12+ and from 1.62 to 2.81 for Vio22+), indicating stronger fluorescence emission of the enol structure. The calculated results are in good agreement with the experimental results. Vio1·2Cl and Vio2·2Br are the first examples of isomerization-induced fluorescence enhancement of viologen derivatives, which shows strong solvatofluorochromism under UV light, making up for the disadvantage that it is easy for a viologen radical to fade in air, and providing a new strategy for designing and synthesizing viologen materials with strong fluorescence.
1. Introduction
Viologens (1,1′-disubstituted 4,4′-bipyridinium, V2+), acting as strong electron acceptors, can undergo two reversible reduction steps to generate an intensely colored radical cation (V˙+) or a neutral form (V0) under appropriate external stimuli,1–3 such as light, heat, electricity or guest molecules, have been getting much attention. Because of the visible color change during the charge transfer (CT)/electron transfer (ET) process, viologens have been widely studied and applied in the fields of photochromic and4 electrochromic materials,5,6 molecular electronics,7 redox sensors,8,9 host–guest complexes,10,11 as well as energy storage.12–14However, because the deep-colored viologen radical cations are not very stable and easily fade in air,15 anaerobic and non-H2O environments are usually used to avoid the strong radical-quenching effect of H2O and O2, which will greatly limit their applications.16 If chromism and fluorescence are concentrated on the viologen molecules at the same time, fluorescence recognition can be used to compensate for the disadvantage of viologen radical instability, which will broaden the potential applications of viologen materials. However, the current reports about viologen fluorescent materials mostly focus on polymers17 and inorganic hybrid materials,18,19 while examples of a viologen itself with strong fluorescence are rarely reported. In addition, the fluorescence behavior of viologens in solution has always been a controversial topic in the past and it is frequently reported that a viologen itself has no fluorescence in fluid solution unless fluorescent pyridinone impurities are produced due to oxidation.20–22 So far, there has been no in-depth study on the luminescence mechanism and the molecular structure of viologen derivatives and their fluorescence properties. Here, we expect to introduce aromatic fluorophores of acetophenone and naphthophenone substituents into the viologen structure, which is conducive to expanding the conjugated system to stabilize the molecular structure and enhance fluorescence. Moreover, molecules containing a larger conjugated structure often exhibit interesting emission enhancement, and emission switching due to isomerization or aggregation.23 This unique isomerization induced fluorescence enhancement phenomenon is worthy of further investigation, and can also guide the synthesis of fluorescent viologens.
Herein, we designed and synthesized two new viologen derivatives, named Vio1·2Cl and Vio2·2Br, which were obtained by the reaction of trans-1,2-bis(4-pyridyl)ethylene (dpe) with chloroacetophenone and bromoacetonaphthone, respectively (Scheme 1). The carbonyl group on the acetophenone and naphthophenone substituent in Vio12+ and Vio22+ is prone to isomerization to an enol structure to form large conjugated systems in organic solvents, which will enhance the fluorescence emission. Time-dependent fluorescence results showed that Vio12+ and Vio22+ in organic solvents, especially DMSO, showed obvious fluorescence enhancement with the increase of time, and the fluorescence lifetime and quantum yield (QY) also increased with the increase of enol components (T = 1 day, ΦVio1 = 25.81%, ΦVio2 = 41.44%; T = 7 days, ΦVio1 = 31.48%, and ΦVio2 = 54.40%). Time-dependent 1H NMR (nuclear magnetic resonance) showed that the enol component of Vio12+ and Vio22+ increased with time in d-DMSO, and no new species with an unknown structure were produced. Time-dependent electrospray ionization mass spectrometry (ESI-MS) data also showed that no fluorescent impurities were produced over time. The density functional theory (DFT) calculations show that the enol form of Vio12+ and Vio22+ enhances the migration of π electrons in the system; the enol structure has a more stable structure, and stronger fluorescence emission. As far as we know, Vio1·2Cl and Vio2·2Br are the first examples of isomerization-induced fluorescence enhancement of viologen derivatives, and the unique fluorescence behavior and the relationship between the luminescence mechanism and structure are explored in detail here.
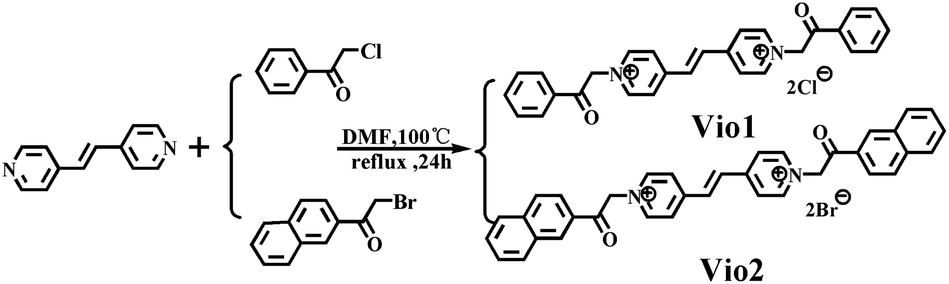 | ||
| Scheme 1 The synthesis and structure of Vio1·2Cl and Vio2·2Br. | ||
2. Experimental
2.1. Chemicals
2-Bromo-2′-acetonaphthone (Aladdin, 98%), 2-chloroacetophenone (Xiya Reagent, 99%), and trans-1,2-bis(4-pyridyl) ethylene (Aladdin, 99%). All other chemicals were used as received without further purification. Solutions were prepared with sub-boiled water distilled in an all-quartz apparatus. All other organic solvents were supplied by Sinopharm Chemical Reagent Co., Ltd.2.2. Synthesis of Vio1·2Cl
trans-1,2-Bis(4-pyridyl)ethylene (0.364 g, 2 mmol) and 2-chloroacetophenone (0.773 g, 5 mmol) were dissolved in anhydrous DMF (N,N-dimethylformamide, 6 mL) (Scheme 1). And then the mixture solution was refluxed for 24 h at 100 °C. A pale yellow precipitate gradually formed during the reaction process, and after cooling to room temperature, the mixture was centrifuged, the precipitate was washed 3–5 times with anhydrous DMF and acetone, and dried in a vacuum at 80 °C for 8 h. The light-yellow powder (0.752 g) was collected, and the yield is 76.5%. 1H NMR, 13C NMR (dimethyl sulfoxide (d-DMSO), 25 °C, Fig. S1 and S2†) and IR (infrared spectroscopy, Fig. S3†) spectra are provided in the ESI.†2.3. Synthesis of Vio2·2Br
The synthesis method of Vio2·2Br is similar to that of Vio1·2Cl except 2-chloroacetophenone was used instead of 2-bromo-2′-acetonaphthone (1.245 g, 5 mmol). The bright yellow powder (0.955 g) was collected, the yield is 70.2%. 1H NMR, 13C NMR (d-DMSO, 25 °C, Fig. S4 and S5†) and IR (Fig. S6†) spectra are provided in the ESI.†2.4. Synthesis of the Vio1·2Cl crystal (Cry1)
The crystal of Vio1·2Cl (yellow block) was obtained by the solvent evaporation method. Vio1·2Cl powder (20 mg, 0.0407 mmol) was dissolved in 10 mL methanol (MeOH), and filtered, and then the filtrate was slowly evaporated in a 25 mL beaker at room temperature. The yellow block crystal suitable for X-ray diffraction was obtained in 3 days. Yield: 85%. Elemental analysis calcd (%), C28H24N2O2Cl2: C, 68.436; H, 4.923; N, 5.701. Found: C, 68.593; H, 4.945; N, 5.686.2.5. Synthesis of the Vio2·2Br crystal (Cry2)
The crystal of Vio2·2Br (orange block) was also obtained by the solvent evaporation method at room temperature. Vio2·2Br powder (10 mg, 0.0147 mmol) was dissolved in a mixed solvent of 12 mL (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) MeOH and acetonitrile. The orange block crystal suitable for X-ray diffraction was obtained in a week. Yield: 81%. Elemental analysis calcd (%), C36H28N2O2Br2: C, 63.546; H, 4.148; N, 4.178. Found: C, 63.440; H, 4.143; N, 4.186.
:1) MeOH and acetonitrile. The orange block crystal suitable for X-ray diffraction was obtained in a week. Yield: 81%. Elemental analysis calcd (%), C36H28N2O2Br2: C, 63.546; H, 4.148; N, 4.178. Found: C, 63.440; H, 4.143; N, 4.186.
2.6. Instrumentation
1H NMR and 13C NMR spectra were obtained on a Bruker AV 500 MHz spectrometer. The elemental analyses (C, H, and N) were obtained on a Vario EL III analyzer. Infrared spectra were obtained on an AVATAR 370 FTIR. Ion chromatography was measured on a Metrohm Eco IC. UV-vis absorption spectra were obtained using a Puxi TU-1900 spectrometer equipped with a 1.0 cm quartz cell. Pure solvents were used as references in the test. If not mentioned, all spectral data were collected immediately after mixing. Electron paramagnetic resonance (EPR) spectra were recorded by using a JES-FA 200 spectrometer fitted with a DICE ENDOR accessory, an EN801 resonator, and an ENI A-500 rf power amplifier. The fluorescence spectra were collected at room temperature on a PerkinElmer LS-55. The photoluminescence (PL) QY (Φ) was collected on a transient steady-state fluorescence spectrometer (Edinburgh Instruments, FLS 1000).2.7. X-ray crystallography
X-ray diffraction data were collected using a Bruker Apex II CCD diffractometer with a graphite-monochromated Mo Kα radiation source (λ = 0.71073 Å) at 296 K. The structures were solved by direct methods and refined by full-matrix least squares calculations (F2) by using the SHELXTL software. All non-H atoms were refined in an anisotropic approximation against F2 for all reflections. All H atoms were placed at their calculated positions and refined in the isotropic approximation. The crystallographic data and structural correction parameters of Vio1·2Cl and Vio2·2Br are summarized in Table S1.†2.8. Computational section
The quantum mechanical (QM) calculations on ground and excited state properties of fluorophores Vio12+ and Vio22+ have been performed at the density functional theory (DFT)24 and its time-dependent extension (TD-DFT) levels of theory,25 respectively. Due to the possibility of long-distance charge transfer, the CAM-B3LYP26,27 density functional has been chosen together with the SNSD basis,28 which was developed for spectroscopic studies of medium-to-large molecular systems. Moreover, due to the conformational flexibility of Viol2+ and Vio22+, the DFT and TD-DFT computations have been performed including Grimme’s dispersion corrections,29 in its D3 formulation in conjunction with the Becke–Johnson (BJ) damping.30 Vertical excitation energies (VE) and excited state equilibrium geometries have been calculated with TD-DFT, followed by TD-DFT harmonic frequency computations, in order to assure the nature of stationary points as local minima. In order to match experimental conditions, the polarizable continuum model (PCM) in its conductor-like screening formulation, C-PCM,31 has been applied to simulate a solvent environment. These computational models have been successfully applied for spectroscopic studies of other fluorescent conjugated chromophores.32–34 All computations have been performed with the Gaussian 16 suite.353. Results and discussion
Vio1·2Cl and Vio2·2Br were synthesized and characterised by single crystal X-ray diffraction analysis, 1H NMR, 13C NMR, IR and element analysis (Fig. 1, S1–S6 and Table S1†). 1H NMR indicates that free Vio12+ and Vio22+ exist in the keto form in the initial state (Fig. S1 and S3†).36 The keto structure of the acetyl group (–CH2CO–) on the substituent is prone to isomerize into the enol structure (–CHCOH–)16,36 with a larger conjugated system in alkaline solution and organic solvents, and with the increase of time, the enol form component increases, resulting in enhanced fluorescence.
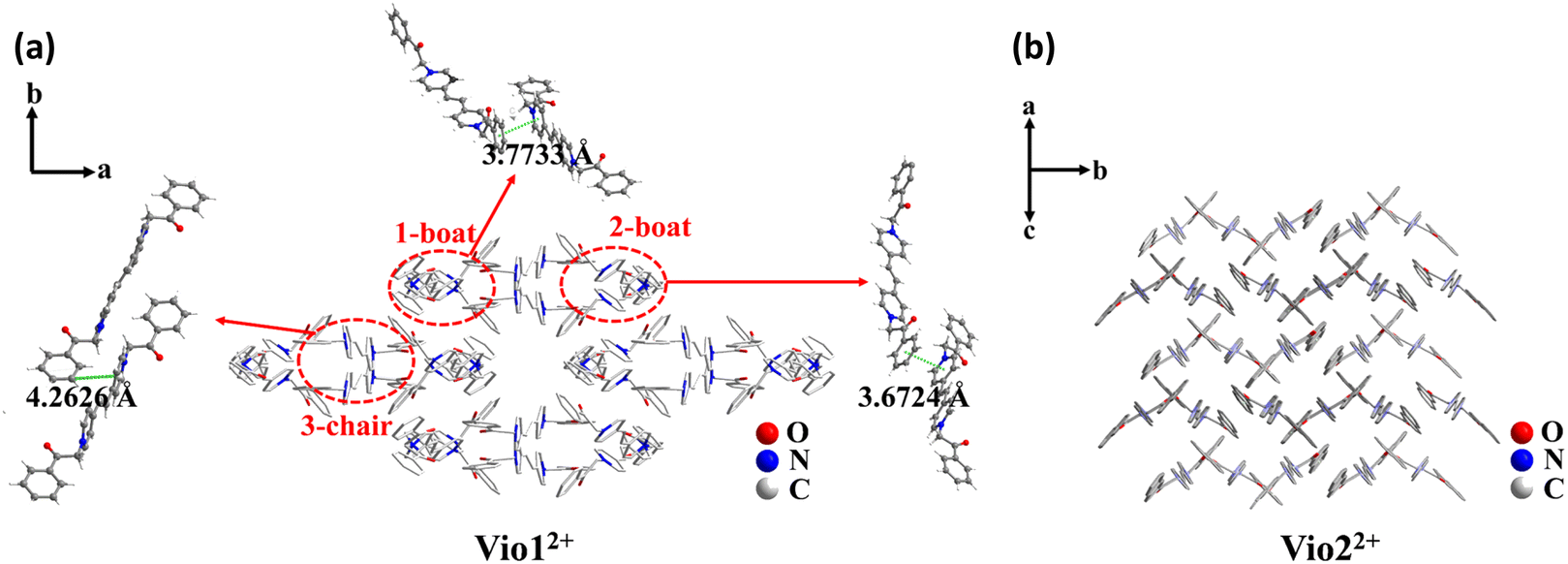 | ||
| Fig. 1 The stacking structure of Vio12+ (a) and Vio22+ (b) and the intermolecular interaction of Vio12+. | ||
3.1. Crystal structure
The single crystal X-ray diffraction measurements evidence that Vio1·2Cl and Vio2·2Br belong to the monoclinic system and P21/c space group, respectively (Table S1†). For Vio1·2Cl, there are three Vio12+ molecules and six Cl− counterions in the asymmetric unit. The Vio12+ molecules exist in two different configurations of a chair and boat, and the molecules of the same configuration interact in pairs (Fig. 1 and S7†). The π–π distance between the pyridine ring and the benzene ring is 3.6724 and 3.7733 Å for boat configuration molecules, while the distance is 4.2626 Å between chair configuration molecules (Fig. 1a). The interaction between chair molecules is weaker than that between boat molecules, but the Vio12+ molecules with both configurations are prone to exist as dimers through the intermolecular interactions.For Vio2·2Br, the asymmetric unit of Vio2·2Br contains one and a half Vio22+ molecules and three Br− counterions. The Vio22+ molecules exhibit only one chair configuration in the structure, and the molecules are perpendicular and parallel to each other to form a grid-like structure and no dimer formation (Fig. 1b). It should be mentioned that dimers are easily formed in Vio12+, while not in Vio22+, probably because the steric hindrance of the naphthophenone substituent of Vio22+ is much larger than that of the acetophenone of Vio12+.37
3.2. Solvatochromism and EPR
Sensitive chromic behavior is the most basic characteristic of a viologen, and the solvochromic effect is usually one of its important properties. Vio12+ and Vio22+ are also solvent sensitive, being colorless in H2O, light purple and blue in MeOH, EtOH (ethanol) and DMSO (Fig. 2 and S8†), respectively. The UV-vis (ultraviolet-visible) spectra showed that the maximum absorption wavelengths (λmax) of Vio12+/Vio22+ in MeOH, EtOH, and DMSO are 554/565 nm, 573/582 nm, 595/603 nm (Fig. S9, S10 and Table S2†). Therefore, in the UV-vis spectrum of the same solvent, the maximum absorption wavelength of Vio22+ has a significant red shift compared with that of Vio12+, which is due to the fact that Vio22+ has a larger conjugate system. It can be seen that the maximum absorption wavelength in different solvents is related to the polarity of the solvent, and shows a blue-shift in higher solvent polarity. The relationship between the maximum absorbance wavelength (λ) and polarity (ENT) was obtained by using the empirical formula: λ = aENT + b, where a, b are −80.2 ± 15.9, 619.5 ± 9.0 for Vio12+; −101.1 ± 9.3, 644.6 ± 5.3 for Vio22+. Vio22+ shows a more sensitive solvatochromic effect because it has a larger negative value a. Previous work has demonstrated that acetophenone-substituted viologen derivatives are sensitive to bases and solvents. In alkaline solutions or weakly alkaline organic solvents, the keto structure will be partially isomerized to an enol structure, and be reduced to free radicals to show different colors.22 Therefore, there is a conversion equilibrium between divalent cations, free radicals and/or dimers in solution. When the solution is diluted, the equilibrium moves to the direction of keto-to-enol conversion and free radical formation. The absorption peak could change and gradually increase, and the dilution effect is weaker than the conversion effect. When the solution is further diluted, the color of the solution becomes lighter and the intensity of the absorption peak gradually decreases. The conversion and equilibrium rates depend on solvent polarity and its electron donating capacity. For MeOH and EtOH with stronger polarity and weaker electron donating ability, viologen reaches the conversion equilibrium faster, while for DMSO with relatively weaker polarity and stronger electron donating ability,16,36 the conversion equilibrium rate will be slower, which was proved by the UV-vis spectra of Vio12+ and Vio22+ at different concentrations (Fig. S9 and S10†).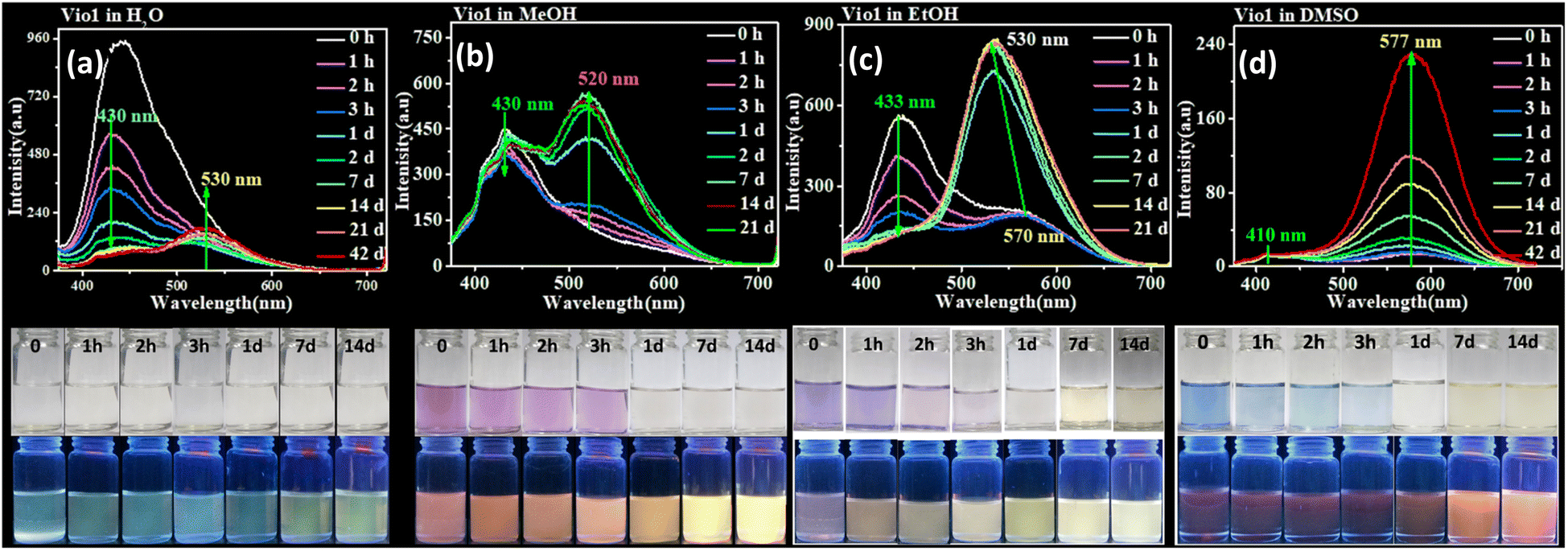 | ||
| Fig. 2 Fluorescence emission spectra of Vio1·2Cl (0.2 mmol L−1) in H2O (a), MeOH (b), EtOH (c), and DMSO (d) at different times (from the initial state to 42 days). λex = 365 nm (top). The photograph of Vio1·2Cl (0.2 mmol L−1) in H2O, MeOH, EtOH, and DMSO solvents under natural light and 365 nm UV light at different times (from the initial state to 14 d) (bottom). | ||
It has been known for a long time that MV2+ is reduced to MV˙+ in alkaline aqueous and alcoholic solutions to show color.7 To prove that the color in organic solvents arises from the viologen radicals of Vio1˙+ and Vio2˙+, EPR tests were performed. Taking Vio12+ as an example, the colorless Vio12+ aqueous solution is EPR silent, while the colored DMSO solution has a weaker EPR signal of s free radical at g = 2.0020, indicating the generation of trace amounts of free radicals Vio1˙+ in DMSO. MeOH and EtOH solutions are EPR silent, perhaps due to too few radicals produced in solution, indicating that the electron donating ability of DMSO is stronger than that of MeOH and EtOH. When NaOH was added to the H2O and DMSO solvents, the solutions become darker and produce a strong free radical signal (Fig. S11†), which illustrates that free radicals are chromogenic species in solutions.
3.3. Time-dependent fluorescence
Normally, all viologen solutions will fade due to the oxidation of free radicals by oxygen in air, and lose the function of solvent recognition by the naked eye, which greatly limits their further applications. Vio12+ and Vio22+ also have the same disadvantages. As shown in Fig. 2 and S8,†Vio12+ and Vio22+ in MeOH, EtOH, and DMSO showed different colors under natural light; one day later, the solution has completely faded and cannot be recognized. But interestingly, the discolored solution can continue to be distinguished by solvatofluorochromism under 365 nm UV light because the fluorophore was introduced into the structure of Vio12+ and Vio22+, and the introduced fluorophore is easily isomerized in organic solvents to enhance fluorescence.To explore the luminescence mechanism, the time-dependent fluorescence behavior of Vio12+ and Vio22+ in different solvents in air were investigated in detail. As shown in Fig. 2a and b, the initial spectra of Vio12+ show an emission peak at ∼430 nm, accompanied by a weak shoulder peak which appears at 520–530 nm in H2O and MeOH (Fig. 2a and b). The emission intensities of the ∼430 nm peak decrease linearly with increasing time in aqueous solution, and they slightly decreased in MeOH solution. The ∼530 nm peak increases slightly and is still very weak in H2O within 42 days, while in MeOH solution, the emission peak at ∼520 nm increases rapidly one day after the solution faded completely, and then slowly increased with increasing time and reached a dynamic equilibrium after 7 days. For EtOH solution, the ∼430 nm emission peak also decreases linearly with increasing time, and the difference is that the weak shoulder peak appears at ∼570 nm in the initial state. 1 day later, the solution faded and the emission peak at ∼570 nm was blue-shifted to ∼530 nm and rapidly intensified (Fig. 2c). After 2 days, the solution reached a kinetic equilibrium and the fluorescence intensity remained basically unchanged. It is well known that MV2+ mainly exists in the form of a dimeric aggregate, (MV2+)2 in strong protic solvents, involving a dynamic equilibrium between MV2+ and (MV2+)2.22 Here, H2O is a neutral protic solvent, which cannot promote the conversion of the keto structure to the enol structure of Vio12+, but tends to form dimers. Therefore, the peaks at ∼430 and ∼530 nm may be attributed to the emission of divalent cations Vio12+ and dimers (Vio12+)2, respectively. In the protic solvent MeOH, the two emission peaks are similar to those in water, also corresponding to divalent cations and dimers. The crystal structure obtained in MeOH solution also shows that Vio12+ mainly exists in the form of a dimer. Different from that in MeOH, there is a peak at ∼570 nm in the initial state in EtOH, which can be assigned to the enol structure emission of Vio12+; the solution fades after 1 day, and the peak blue shifts to ∼530 nm which can also be assigned to the dimer emission. Compared with EtOH, MeOH is a stronger protic solvent, and the aggregation rate of Vio12+ in MeOH may be higher than the conversion rate, so the ∼570 nm emission peak of the enol form was not found. 1 day later, the rapid enhancement of fluorescence was attributed to the fading of the solution, the disappearance of Vio1˙+ radicals and the disappearance of their quenching effect on fluorescence, which was caused by the oxidation of oxygen in air.
As shown in Fig. 2d, different from Vio12+ in protic solvents, Vio12+ has only one obvious emission peak at ∼577 nm in DMSO, which gradually increases with increasing time, and does not reach a dynamic equilibrium within 42 days. The emission peak at ∼410 nm is very weak and basically unchanged, and there is no emission peak at ∼530 nm. Since DMSO has a certain weak basicity and is an aprotic solvent, it is easier to promote the isomerization of the keto to enol form rather than dimerization,38 so the peak at ∼577 nm can be attributed to the enol emission of Vio12+. The subsequent time-dependent 1H NMR and DFT calculations also support this speculation.
Vio22+ has almost no fluorescence in H2O, so it has not been studied. The crystal structure analysis of Vio22+ shows that the formation of dimers is not easy because of the larger steric hindrance effect of naphthophenone substituents.39 Therefore, there is no dimer emission peak (∼530 nm) in MeOH and EtOH, and only one emission peak at ∼430 nm in MeOH, coming from the keto structure emission of Vio22+. In EtOH, there are two emissions, at ∼430 and ∼571 nm, which can be assigned to the Vio22+ keto structure and enol structure emission, respectively. Similar to Vio12+, there is a strong emission peak of the enol structure at ∼577 nm in DMSO. The fluorescence intensity gradually increases with time in MeOH, EtOH and DMSO solutions. On one hand, the colored radicals were gradually oxidized by oxygen, the solution faded, and the fluorescence quenching effect of radicals was gradually weakened. On the other hand, the continuous structure conversion of the keto-to-enol form, led to a gradual increase of the enol form composition and enhanced fluorescence. However, in EtOH and DMSO solutions, the conversion equilibrium was still not reached within 14 days, and the conversion rate was very slow (Fig. S12†). From the above results, it is obvious that the enol form of Vio12+ and Vio22+ has stronger fluorescence.
From the UV-vis and PL spectra, it is found that Vio12+ and Vio22+ have a larger stack shift (Δλ = 62–135 nm and ΔSt > 2572–5600 cm−1) in DMSO (Table S3†), so the low background interference and high detection sensitivity are beneficial to the application of fluorescence sensing. It should be mentioned that the fluorescence lifetime and QY were also tested in several solvents. The results show that the DMSO solution has the highest fluorescence QY and the longest lifetime, and the QY increases with time, T = 1 day, ΦVio1 = 25.81%, ΦVio2 = 41.44%; T = 7 days, ΦVio1 = 31.48%, τ = 3.80 ns, ΦVio2 = 54.40%, and τ = 3.68 ns. Indeed, the DMSO solutions of Vio12+ and Vio22+ can maintain strong fluorescence for extended periods (>12 month) in air. This demonstrates that Vio12+ and Vio22+ have stable excited states under ambient conditions in DMSO, and low rates of intersystem crossing to the reactive triplet state.40 The fluorescence lifetime of Vio12+ and Vio22+ far exceeds that of reported viologen derivatives,41 and it is also the first viologen derivative found to have fluorescence enhancement due to isomerization.
In order to verify the effect of light sources (natural light and 365 nm UV light) on the fluorescence or isomerization of viologen solution, we also carried out UV illumination experiments on the viologen solution. As we envisioned, UV irradiation can accelerate the conversion of the ketone to the enol form and fluorescence enhancement in the viologen solution. As shown in Fig. S13 and S14,† the fluorescence gradually increases with the increase of UV lamp irradiation time and the maximum emission peak at ∼577 nm remains unchanged in DMSO solution for Vio12+ and Vio22+, indicating that no other new fluorescent impurities were produced in the solution under UV light excitation. The difference between the two compounds is that Vio12+ is more sensitive to UV light, while the fluorescence intensity of Vio22+ gradually increases with time even if placed in the dark, its conversion rate will be faster under UV lamp irradiation. The changes in fluorescence intensity suggest that UV illumination plays an important role in the structural transformation of the keto to he enol form and the fluorescence enhancement in DMSO solution.
3.4. Time-dependent 1H NMR, ESI-MS and FTIR
According to the UV-vis spectra and time-dependent fluorescence changes, we can speculate that Vio12+ mainly undergoes the conversion of the keto-to-enol configuration and intramolecular or intermolecular electron transfer to generate free radicals in organic solutions (Scheme 2). For Vio22+, the dimer is not easily formed due to the large steric hindrance of the naphthophenone substituents, and no emission peak at ∼530 nm appears. Therefore, the fluorescence enhancement of Vio12+ and Vio22+ in solution mainly originates from isomerization rather than the generation of fluorescent impurities. The reason is that keto structure is prone to isomerization into enol structure due to the existence of active hydrogen on –CH2CO– groups,36 which can be easily converted into enol groups (–CHCOH–) by the pseudobase solvent effect, which makes its structure has larger conjugated system and more stable. To further confirm our speculation, time-dependent 1H NMR and ESI-MS were characterized in solution.
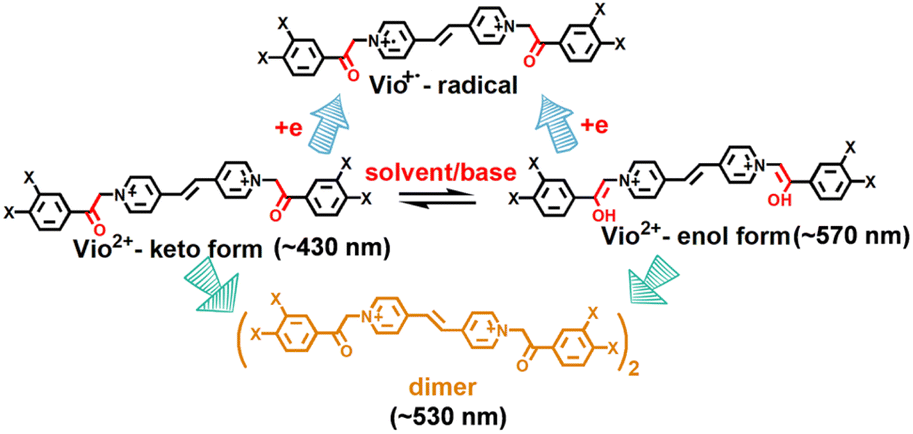 | ||
| Scheme 2 The reaction mechanism of Vio12+ and Vio22+ in solvents. | ||
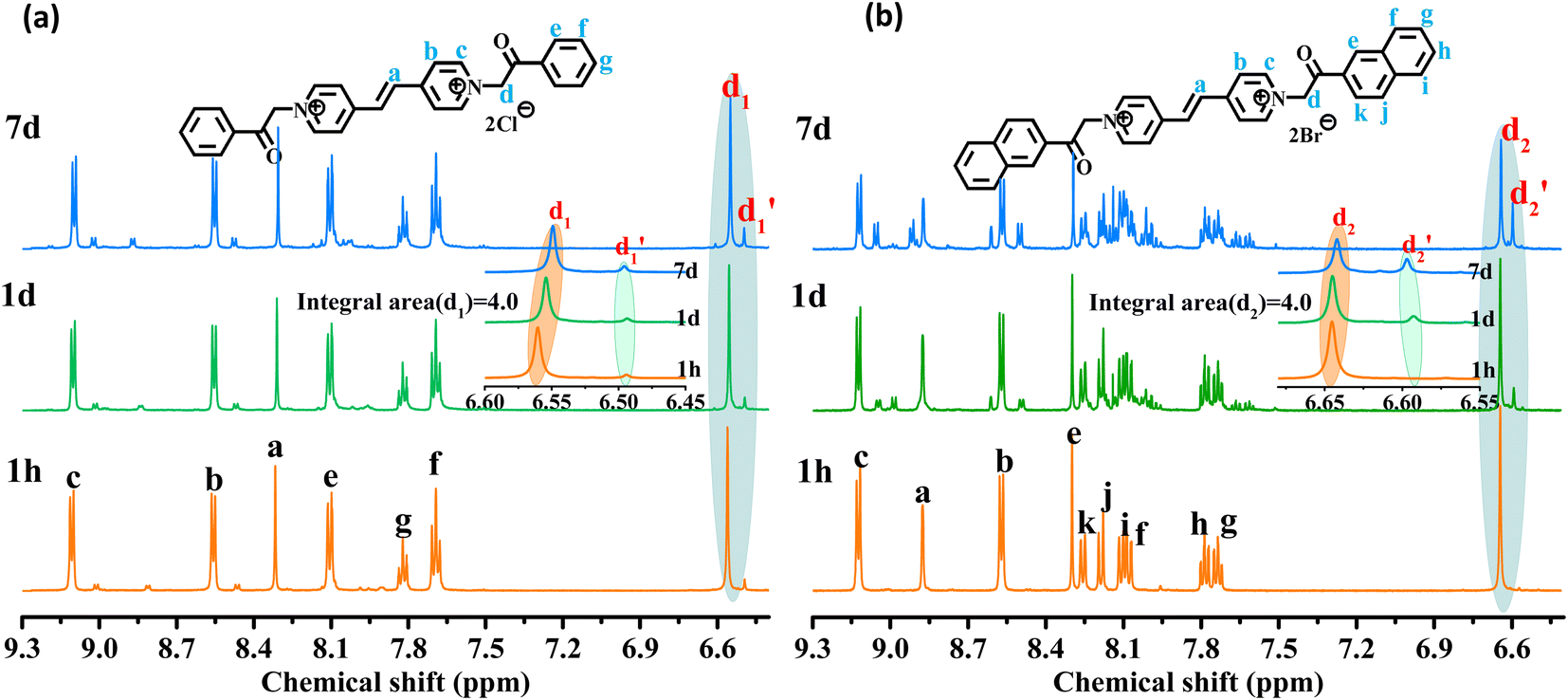
As shown in Fig. 3, the 1H NMR spectrum of d-DMSO solution within 1 h showed the chemical shifts of the methylene hydrogens of Vio12+ and Vio22+ were split into two 6.50 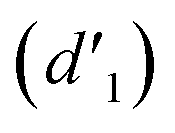 and 6.60 ppm
and 6.60 ppm 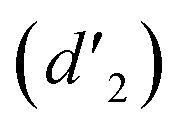 and were assigned to the H peak of the enol form,42,43 which are very weak for Vio12+ and almost invisible for Vio22+, indicating that only trace amounts of the enol configuration were generated within one hour. However, the intensity of
and were assigned to the H peak of the enol form,42,43 which are very weak for Vio12+ and almost invisible for Vio22+, indicating that only trace amounts of the enol configuration were generated within one hour. However, the intensity of 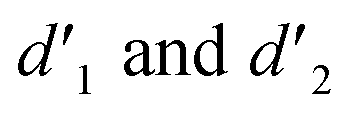 gradually increased after 1 day and 7 days, indicating that the enol form gradually increased with time. The growth rate of
gradually increased after 1 day and 7 days, indicating that the enol form gradually increased with time. The growth rate of 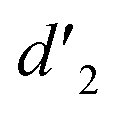 is obviously stronger than that of
is obviously stronger than that of 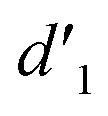 , indicating that the conversion rate of Vio22+ is higher than that of Vio12+ at the same time, perhaps because the greater steric hindrance of the substituent of Vio22+ makes the intermolecular interaction weaker, which is conducive to the conversion.
, indicating that the conversion rate of Vio22+ is higher than that of Vio12+ at the same time, perhaps because the greater steric hindrance of the substituent of Vio22+ makes the intermolecular interaction weaker, which is conducive to the conversion.
 | ||
| Fig. 3 1H NMR spectra of Vio12+ (a) and Vio22+ (b) at different times (1 h, 1 d, and 7 d) in d-DMSO solution. | ||
It should be mentioned that there are 4 methylene hydrogens in the keto structure, and only 2 alkene hydrogens in the enol structure. Therefore, the change of the content of keto (d/4) and enol (d′/2) in the solution can be measured by the integrated area of d and d′, and the content of the enol and the fluorescence intensity at different times are summarized in Table 1. It can be seen that the enol content of Vio12+ increases from 14.17% to 23.95% from 1 h to 7 days, the fluorescence intensity increased 3.8 times, and the corresponding QY increased to 31.84%, while for Vio22+, the enol content increased from 6.11% to 42.86% from 1 h to 7 days, the fluorescence intensity increased 6.9 times, and the QY increased to 54.40%. The time-dependent 1H NMR results showed that there was no obvious new species produced, so the fluorescence enhancement of DMSO solution with time should be mainly due to the generation of the isomerized enol configuration, and the peak at ∼577 nm should be assigned to the enol form emission.
| 1 h | 1 d | 7 d | ||
|---|---|---|---|---|
| Vio12+ | d 1 (keto) integrated area | 4.00 | 4.00 | 4.00 |
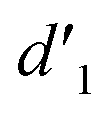 (enol) integrated area (enol) integrated area |
0.33 | 0.45 | 0.63 | |
Keto/enol 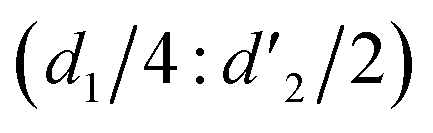 |
4:0.66 |
4:0.90 |
4:1.26 |
|
| Enol (%) | 14.17 | 18.37 | 23.95 | |
| Fluorescence intensity | 14.61 | 22.73 | 55.34 | |
| Quantum yield (%) | — | 25.81 | 31.48 | |
| Vio22+ | d 2 (keto) integrated area | 4.00 | 4.00 | 4.00 |
 (enol) integrated area (enol) integrated area |
0.13 | 0.66 | 1.50 | |
Keto/enol 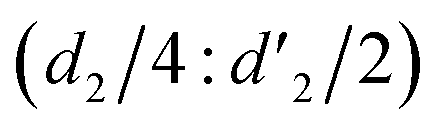 |
4:0.26 |
4:1.32 |
4:3.00 |
|
| Enol (%) | 6.11 | 24.81 | 42.86 | |
| Fluorescence intensity | 8.86 | 20.41 | 61.51 | |
| Quantum yield (%) | — | 41.44 | 54.40 | |
In order to further verify the luminescence mechanism and determine whether new species are generated, the time-dependent ESI-MS of Vio12+ and Vio22+ was also studied. As shown in Fig. 4 and S15,† the m/z of 419 and 210 correspond to the viologen radical (M˙+), and double charge ion peak (M2+/2) of Vio12+ in H2O and MeOH within 1 h, respectively, and a few fragment peaks were found. With increasing time, the peaks of the free radical and fragments gradually weakened on the 7th day. After the 14th day, all fragment and the free radical peaks completely disappeared, with only one double charge ion peak of a divalent cation, indicating that only Vio12+ existed in solution. The difference is that there are DMSO solvent peaks and more fragmented peaks in DMSO solution within the first hour.
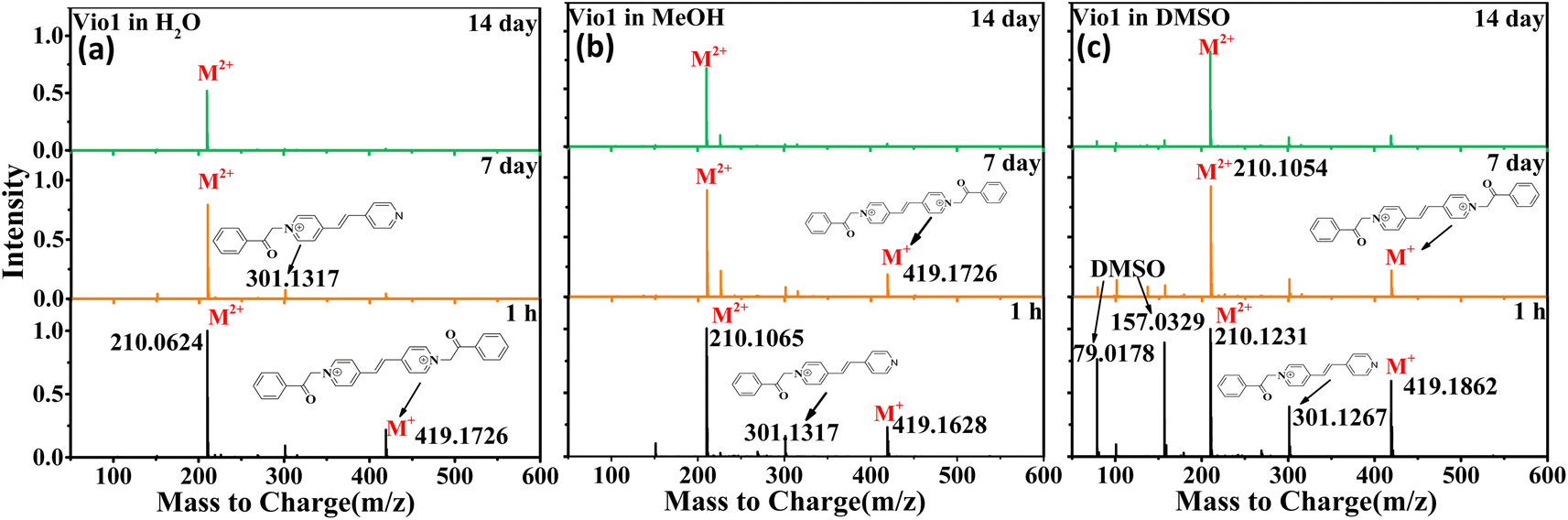 | ||
| Fig. 4 ESI-MS of Vio12+ in different solvents (H2O, MeOH and DMSO) at different times (1 h, 7 d, and 14 d). | ||
For Vio22+, the ESI-MS results were similar to Vio12+ except that few fragment peaks did not completely disappear in DMSO after 14 days (Fig. S15†). It should be noted here that the double charge ion peak of divalent cations should come from the keto and/or enol form. The ESI-MS spectrum cannot determine the specific molecular configuration, but it can be shown that no new luminescent species are formed in the solution over time. Therefore, this further illustrates that the enhanced fluorescence in solution with time should mainly come from the conformational transition.
The double charge ion peak abundances of divalent cations in different solvents shows different changes with time. For Vio12+, the ionic abundance of M2+/2 decreased significantly with time in H2O and MeOH, from 1 to 0.791 and 0.928 on the 7th day, and to 0.523 and 0.675 on the 14th day, respectively, while in DMSO, the decrease was not obvious, only from 1 to 0.824 until the 14th day (Fig. 4 and Table S4†). The obvious decrease of ion abundance in H2O and MeOH should be mainly due to the dimerization of divalent cations, while in DMSO, dimerization is not easy, and the slightly decreased ion abundance may be due to the fact that there are still small amounts of free radicals, benzene and fragments. The double charge ion peak abundances of Vio22+ did not change significantly after 14 days in MeOH, EtOH and DMSO, respectively, indicating that no dimerization occurred because of the large steric effect of the naphthophenone substituents, and the reduced ion abundance was attributed to the presence of fragmentation peaks. This result is consistent with the crystal structure and time-dependent fluorescence spectroscopic data.
In situ FTIR is an effective tool to characterize isomerization of the keto to the enol form. Due to the better volatility of ethanol, FTIR of Vio12+ and Vio22+ was performed on the ethanol solutions. In order to promote the conversion and improve the conversion rate, an appropriate amount of NaOH solution was added to the ethanol solution and it was kept for one day. As shown in Fig. S16,† the FTIR spectra of Vio12+ and Vio22+ have obvious keto carbonyl groups at ∼1680 nm−1 and ∼1630 nm−1 in the initial state, while the characteristic peaks of the carbonyl group are very weak and almost disappear after 1 day, and then the strong and wide absorption peak of the enol form appears at 3200–3400 nm−1 and 1500–1300 nm−1, which further proves that Vio12+ and Vio22+ are indeed prone to the conversion of the keto to the enol form under alkaline conditions.
3.5. DFT and TD-DFT calculations
The starting ground state molecular structures of keto viologens have been constructed according to the X-ray crystallographic results, and also used as starting points to form enolic structures with two configurations: boat and chair. For the latter the optimal orientation of the migrating hydrogen atoms has been extensively tested by a relaxed scan. Such ground state keto and enol forms of Vio12+ and Vio22+ have been further optimized without any constraints (Fig. S17†). The structural diagrams of the ground and excited states of different configurations of Vio12+ and Vio22+ (Fig. S18 and S19†) and their corresponding energies (Tables S5 and S6†) were optimized using the dispersion empirical formula B3LYP.As shown in Fig. S18,† the Vio12+ and Vio22+ keto forms are nonplanar, in both ground and excited electronic states, while enolic molecules form structures with high coplanar properties. In the case of Vio12+, the non-planar original keto boat and chair forms also show very similar optical properties. When the Vio12+ keto form is converted to the enol form, its energy is changed by ∼90 kJ mol−1, and the dihedral angle is reduced by about 60° (form 84.19° to 24.98° for the boat configuration; from 87.41° to 23.95° for the chair configuration). A similar phenomenon is also observed in Vio22+, whose energy has a larger change of ∼560 kJ mol−1 and the dihedral angle is reduced by about 80° (from 83.54° to 4.65°) (Tables S5 and S6†).
This suggests that the conversion of the enol form promotes coplanarity throughout the molecular structure, thereby expanding the range of electron transfer in the molecule. It also indicates that the enol form structure is more stable. Furthermore, these changes can be intuitively and clearly explained by analyzing the molecular orbitals and electron densities. Fig. 5 shows the plots of the difference in electron density (ELD) between the ground state and the first excited electronic state for Vio12+ and Vio22+. It is found that electronic excitation mainly occurs on the pyridine ring for the keto structure, while for enolic structures it is extended over the whole molecule. This leads to a more conjugated structure with an increased number of π-bonds. Therefore, increasing the degree of conjugation of the viologen molecule is beneficial to its stability and enhanced fluorescence.
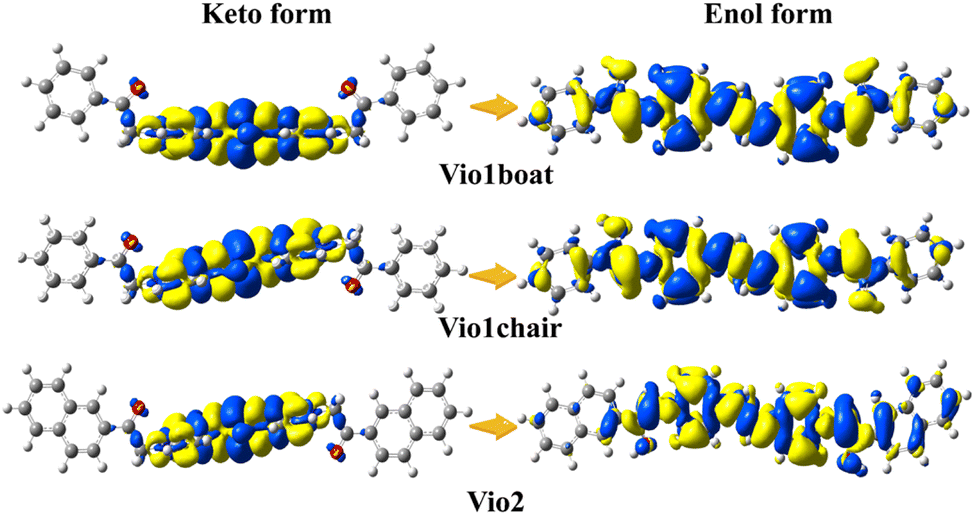 | ||
| Fig. 5 Plots of the difference in electron density (ELD) between the ground state and the first excited electronic state computed at the equilibrium geometry of the ground state. The regions that have lost electron density as a result of the transition are shown in yellow, whereas the blue regions have gained electron density. ELD densities were plotted with an isovalue threshold of 0.001. | ||
Similarly, like in the detailed experimental analysis, we have focused on the Vio12+ molecules in DMSO solution due to their continuously enhanced fluorescence with time. The solvent model applied in this study is expected to correctly account for the bulk solvent effects due to environment polarizability. The absorption electronic spectra have been obtained from vertical excitation energies and transition moments computed at the TD-DFT level, also allowing the indication of the lowest bright electronic states, for which TD-DFT optimizations have been performed. Fluorescence spectra have been obtained as a vertical emission from TD-DFT optimized structures.
Fig. 6a summarizes the photophysical processes. The fluorescence emission peaks of the keto and enol structures of Vio12+ and Vio22+ were at 416–417 nm and 563–582 nm, respectively. As the Vio12+-boat/chair keto form changes to the enol form, a stimulated emission band peaking at ∼416 nm starts to develop, along with a red shift of the signal (from 416 nm to 563 nm and 417 nm to 564 nm for the chair and boat forms). For Vio22+, the simulated emission peak also shows an obvious red shift from 417 nm to 582 nm from the keto to the enol structure. The corresponding theoretical emission λmax is listed in Table S7,† the spectra obtained with broadening by Gaussian distribution functions is shown in Fig. 6b. In consequence, the excitation energy is lowered, resulting in a significant red shift in the fluorescence emission wavelength. Moreover, the fluorescence relative oscillator strength of Vio12+ and Vio22+ enol structures is significantly higher than that of keto structures (f value changes from 1.53 to 2.63 for Vio12+ and from 1.62 to 2.81 for Vio22+), which can be directly correlated with stronger fluorescence emission for the enol structure (Table S7†). The good match between the calculated spectra of keto forms and observed fluorescence wavelengths at 410 nm, as well as between the computed spectra of the enol form and the experimental band at 577 nm, which increases the intensity with time, strongly supports the hypothesis of the spectral changes due to the keto-to-enol transitions. It is worth noting that the keto and enol structures of the boat and chair configurations of Vio12+ in the simulated fluorescence spectra have very similar fluorescence curves, so the two configurations of Vio12+ have no effect on their fluorescence.
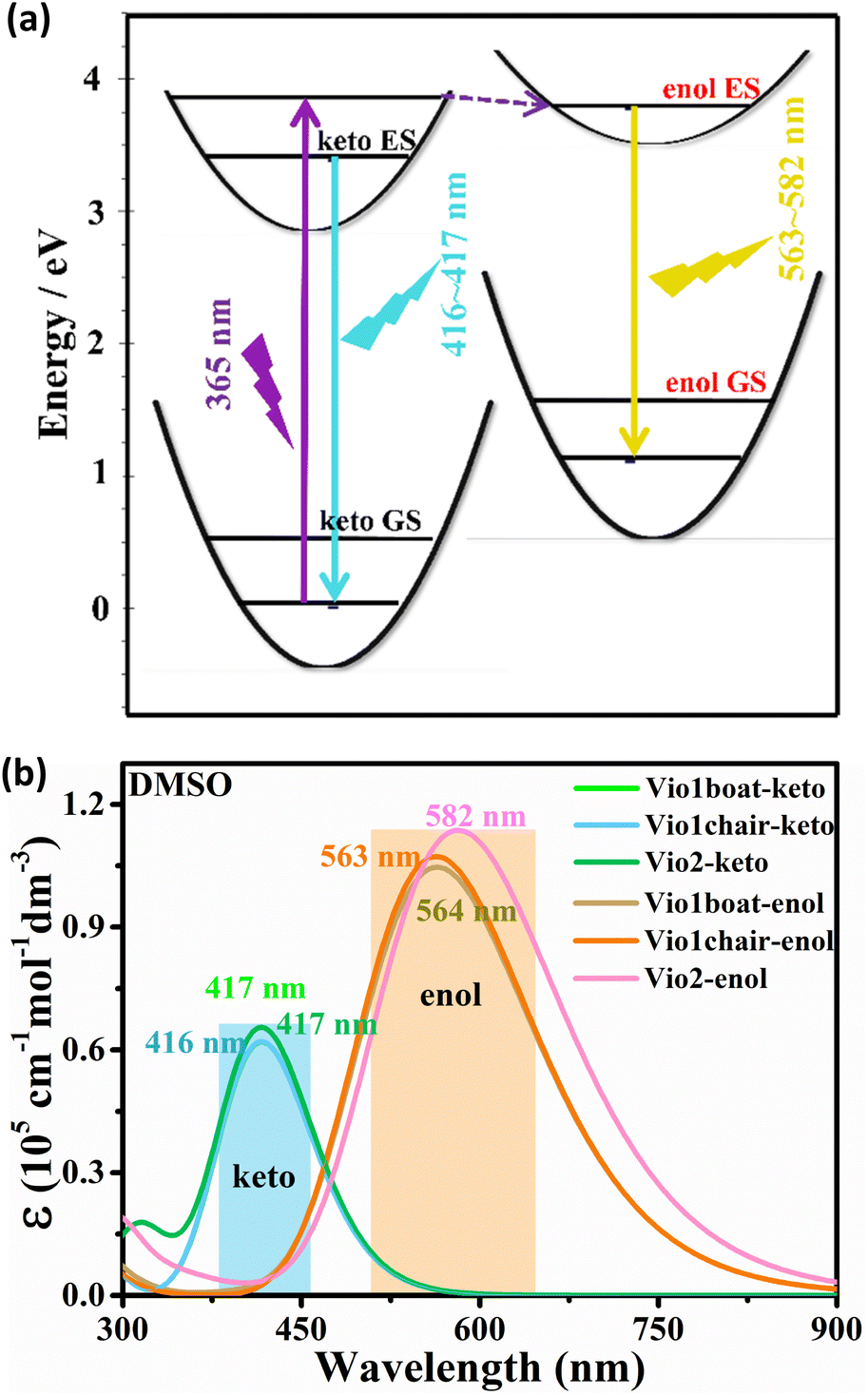 | ||
| Fig. 6 (a) Diagram of the fluorescence luminescence mechanism of a viologen. (b) Simulation of fluorescence spectra of Vio12+ and Vio22+ in DMSO solvent based on TD-DFT calculation results. | ||
3.6. Sensing applications
The sensitive solvatochromism and strong fluorescence properties inspire us to explore the applications of Vio1·2Cl and Vio2·2Br as multipurpose sensing materials. Since Vio1·2Cl has better solubility and more obvious color difference due to solvatofluorochromism, Vio1·2Cl is taken as an example to study its portable sensing applications. A colorless piece of filter paper with Vio1·2Cl dispersed can be easily obtained by dropping or soaking it in a Vio1·2Cl aqueous solution. The dry test filter paper can undergo a high-contrast color change immediately upon contact with different alkaline organic solvents because of the generation of free radicals (Fig. S20 and S21†), which can easily and effectively sense and recognize different alkaline reagents. In addition, this test strip is also sensitive to ammonia and temperature (Fig. S21a†). When encountering NH3 gas, it will turn blue immediately, and HCl gas can make it fade back to the initial state (Fig. S21b†). When the temperature reaches 60 °C, the filter paper strip can change color, and with the increase in temperature, the color deepens, and it fades immediately at room temperature (Fig. S21c†). The test strip has excellent chromism reversibility and circularity for NH3/HCl and temperature. However, after the test strip fades, it is no longer possible to distinguish the solvent type of the test strips. Here, solvatofluorochromism can make up for this shortcoming (Fig. 7).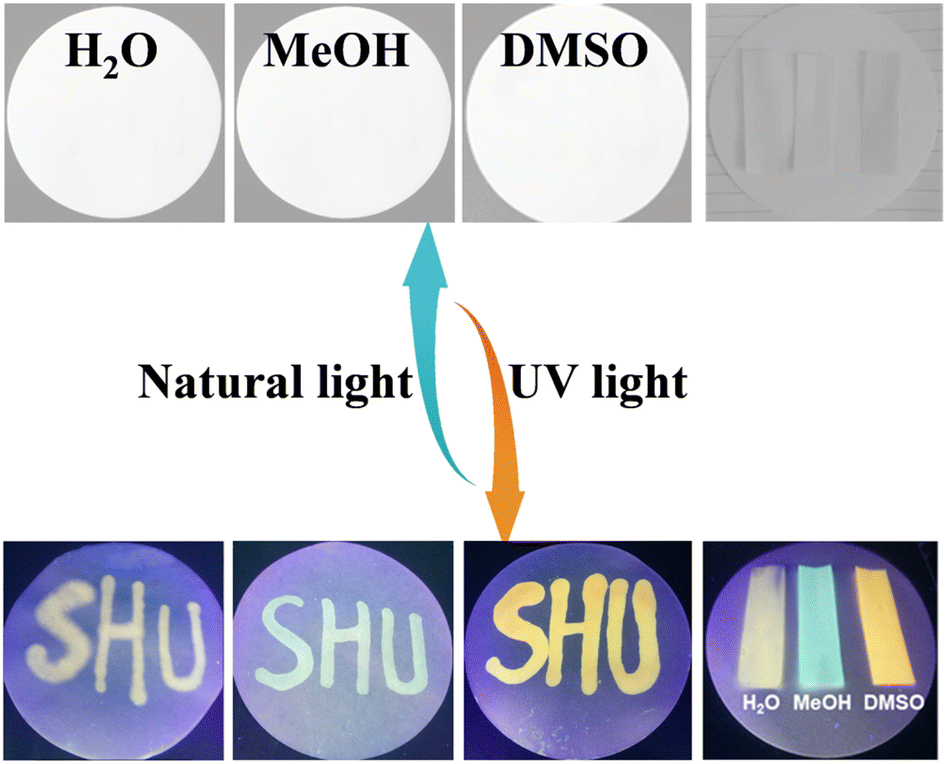 | ||
| Fig. 7 The photos of the Vio1·2Cl dispersed test papers after treatment with different organic solvents under natural light and UV light. | ||
4. Conclusions
In conclusion, we have designed and synthesized two new viologen derivatives of Vio1·2Cl and Vio2·2Br by introducing the acetophenone and acetonaphthone substituents, which shows sensitive chromism and fluorescence enhancement in solvents because these two substituents are prone to keto-to-enol isomerization in organic solvents, especially in DMSO. After isomerization to enol configuration, the conjugated system of viologen structure will increase, which will make these two compounds more stable and enhance fluorescence emission. And their strong solvatofluorochromism under UV light can also make up for the disadvantage of viologen radical fading in air. Experimental characterization and DFT calculations proved that fluorescence enhancement of Vio1·2Cl and Vio2·2Br in solvent is mainly related to the structural isomerization of the keto (∼430 nm) to enol form (∼570 nm), rather than the appearance of fluorescent impurities. The unique fluorescence enhancement phenomenon and the sensitive (color/luminescence) response properties to solvents, temperature, and NH3/HCl make them possible to have a potential application. This work provides a new strategy for designing multifunctional viologen derivatives.Data availability
CCDC numbers 2218985 (Vio1·2Cl) and 2218984 (Vio2·2Br) contain the supplementary crystallographic data for this paper.† Additional experimental details and data are provided in the ESI,† including NMR, IR, ESI-MS, and crystallographic data.Author contributions
Xiuping Yin conceived and designed this study. Xiuping Yin and Xuyi Li synthesized the compounds. Xiuping Yin performed viscosity and spectroscopic measurements. Malgorzata Biczysko and Xinxing Li performed the quantum chemical calculations. Xiuping Yin and Yue-Ling Bai analyzed the data. Xiuping Yin produced the manuscript. Shourong Zhu, Jiaqiang Xu and Yue-Ling Bai checked and modified the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the National Natural Science Foundation of China (21571126) and by the Shanghai Key Laboratory of High Temperature Superconductors. We thank the Instrumental Analysis and Research Center of Shanghai University for measurements.Notes and references
- M. Kathiresan, B. Ambrose, N. Angulakshmi, D. E. Mathew, D. Sujatha and A. M. Stephan, J. Mater. Chem. A, 2021, 9, 27215–27233 RSC.
- J. A. McCune, M. F. Kuehnel, E. Reisner and O. A. Scherman, Chem, 2020, 6, 1819–1830 CAS.
- J. Ding, C. Zheng, L. Wang, C. Lu, B. Zhang, Y. Chen, M. Li, G. Zhai and X. Zhuang, J. Mater. Chem. A, 2019, 7, 23337–23360 RSC.
- R.-G. Lin, G. Xu, M.-S. Wang, G. Lu, P.-X. Li and G.-C. Guo, Inorg. Chem., 2013, 52, 1199–1205 CrossRef CAS PubMed.
- G. Li, L. Xu, W. Zhang, K. Zhou, Y. Ding, F. Liu, X. He and G. He, Angew. Chem., Int. Ed., 2018, 57, 4897–4901 CrossRef CAS PubMed.
- Y. Zhuang, W. Zhao, L. Wang, F. Li, W. Wang, S. Liu, W. Huang and Q. Zhao, Sci. China: Chem., 2020, 63, 1632–1644 CrossRef CAS.
- X.-Y. Chen, H. Mao, Y. Feng, K. Cai, D. Shen, H. Wu, L. Zhang, X. Zhao, H. Chen, B. Song, Y. Jiao, Y. Wu, C. L. Stern, M. R. Wasielewski and J. F. Stoddart, Angew. Chem., Int. Ed., 2021, 60, 25454–25462 CrossRef CAS PubMed.
- A. Ahadi, H. Alamgholiloo, S. Rostamnia, X. Liu, M. Shokouhimehr, D. A. Alonso and R. Luque, ChemCatChem, 2019, 11, 4803–4809 CrossRef CAS.
- H. Aramoto, M. Osaki, S. Konishi, C. Ueda, Y. Kobayashi, Y. Takashima, A. Harada and H. Yamaguchi, Chem. Sci., 2020, 11, 4322–4331 RSC.
- M. Freitag, L. Gundlach, P. Piotrowiak and E. Galoppini, J. Am. Chem. Soc., 2012, 134, 3358–3366 CrossRef CAS PubMed.
- M. Olesińska, G. Wu, S. Gómez-Coca, D. Antón-García, I. Szabó, E. Rosta and O. A. Scherman, Chem. Sci., 2019, 10, 8806–8811 RSC.
- C. R. Bridges, A. M. Borys, V. A. Béland, J. R. Gaffen and T. Baumgartner, Chem. Sci., 2020, 11, 10483–10487 RSC.
- H. Li, H. Fan, B. Hu, L. Hu, G. Chang and J. Song, Angew. Chem., Int. Ed., 2021, 60, 26971–26977 CrossRef CAS PubMed.
- B. Hu and T. L. Liu, Science, 2021, 372, 788–789 CrossRef CAS PubMed.
- L. Liu, Q. Liu, R. Li, M.-S. Wang and G.-C. Guo, J. Am. Chem. Soc., 2021, 143, 2232–2238 CrossRef CAS PubMed.
- W. Shi, F. Xing, Y.-L. Bai, M. Hu, Y. Zhao, M.-X. Li and S. Zhu, ACS Appl. Mater. Interfaces, 2015, 7, 14493–14500 CrossRef CAS PubMed.
- Z. Wang and N. V. Tsarevsky, Poly. Chem., 2016, 7, 4402–4410 RSC.
- X. Wang, J. Kuang, P. Wu, Z. Zong, Z. Li, H. Wang, J. Li, P. Dai, K. Y. Zhang, S. Liu, W. Huang and Q. Zhao, Adv. Mater., 2022, 34, 2107013 CrossRef CAS PubMed.
- T. Fu, Y.-L. Wei, C. Zhang, L.-K. Li, X.-F. Liu, H.-Y. Li and S.-Q. Zang, Chem. Commun., 2020, 56, 13093–13096 RSC.
- M. Alvaro, H. García, S. García, F. Marquez and J. Scaiano, J. Phys. Chem. B, 1997, 101, 3043–3051 CrossRef CAS.
- T. W. Ebbesen, L. E. Manring and K. S. Peters, J. Am. Chem. Soc., 1984, 106, 7400–7404 CrossRef CAS.
- V. Novakovic and M. Z. Hoffman, J. Am. Chem. Soc., 1987, 109, 2341–2346 CrossRef CAS.
- A. N. Woodward, J. M. Kolesar, S. R. Hall, N.-A. Saleh, D. S. Jones and M. G. Walter, J. Am. Chem. Soc., 2017, 139, 8467–8473 CrossRef CAS PubMed.
- K. Burke, J. Chem. Phys., 2012, 136, 150901 CrossRef PubMed.
- M. E. Casida, J. Mol. Struct.: THEOCHEM, 2009, 914, 3–18 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- R. Kobayashi and R. D. Amos, Chem. Phys. Lett., 2006, 420, 106–109 CrossRef CAS.
- V. Barone, M. Biczysko and J. Bloino, Phys. Chem. Chem. Phys., 2014, 16, 1759–1787 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- A. D. Laurent, C. Adamo and D. Jacquemin, Phys. Chem. Chem. Phys., 2014, 16, 14334–14356 RSC.
- V. Barone, F. Bellina, M. Biczysko, J. Bloino, T. Fornaro, C. Latouche, M. Lessi, G. Marianetti, P. Minei and A. Panattoni, Phys. Chem. Chem. Phys., 2015, 17, 26710–26723 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- X.-R. Xiao, C.-B. Wang and H. T. Tien, J. Mol. Catal., 1984, 23, 9–16 CrossRef CAS.
- M. Hu, F. Xing, Y. Zhao, Y.-L. Bai, M.-X. Li and S. Zhu, ACS Omega, 2017, 2, 1128–1133 CrossRef CAS PubMed.
- M. D. Curtis, J. Cao and J. W. Kampf, J. Am. Chem. Soc., 2004, 126, 4318–4328 CrossRef CAS PubMed.
- V. G. Machado, R. I. Stock and C. Reichardt, Chem. Rev., 2014, 114, 10429–10475 CrossRef CAS PubMed.
- J. Kim, I. A. Levitsky, D. T. McQuade and T. M. Swager, J. Am. Chem. Soc., 2002, 124, 7710–7718 CrossRef CAS PubMed.
- W. G. Santos, D. S. Budkina, V. M. Deflon, A. N. Tarnovsky, D. R. Cardoso and M. D. Forbes, J. Am. Chem. Soc., 2017, 139, 7681–7684 CrossRef CAS PubMed.
- M. Freitag, L. Gundlach, P. Piotrowiak and E. Galoppini, J. Am. Chem. Soc., 2012, 134, 3358–3366 CrossRef CAS PubMed.
- R. Postolachi, R. Danac, N. J. Buurma, A. Pui, M. Balan, S. Shova and C. Deleanu, RSC Adv., 2013, 3, 17260–17270 RSC.
- R. Danac and I. I. Mangalagiu, Eur. J. Med. Chem., 2014, 74, 664–670 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2218984 and 2218985. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02051g |
| This journal is © The Royal Society of Chemistry 2023 |