DOI:
10.1039/D2SC07037E
(Edge Article)
Chem. Sci., 2023,
14, 7036-7043
Substituent effects on paratropicity and diatropicity in π-extended hexapyrrolohexaazacoronene†
Received
23rd December 2022
, Accepted 4th June 2023
First published on 6th June 2023
Abstract
Research into the application of antiaromatic compounds as molecular materials is an attractive strategy in the development of electronic materials. Antiaromatic compounds have traditionally been considered to be unstable, and thus, the creation of stable antiaromatic compounds has been sought in the field of organic chemistry. Recently, some studies have been reported on the synthesis, isolation, and elucidation of the physical properties of compounds with stability and definitive antiaromatic properties. In general, antiaromatic compounds are considered to be more susceptible to substituents due to their inherently narrow HOMO–LUMO gap compared to aromatic compounds. However, there have been no studies examining substituent effects in antiaromatic compounds. In this study, we have developed a synthetic method to introduce various substituents into π-extended hexapyrrolohexaazacoronene (homoHPHAC+), one of the stable and clearly antiaromatic compounds, and investigated the substituent effects on the optical, redox, and geometrical properties and paratropicity of a series of compounds. In addition, the properties of the two-electron oxidized form, homoHPHAC3+, were investigated. Control of electronic properties by introducing substituents into antiaromatic compounds provides a new design guideline for molecular materials.
Introduction
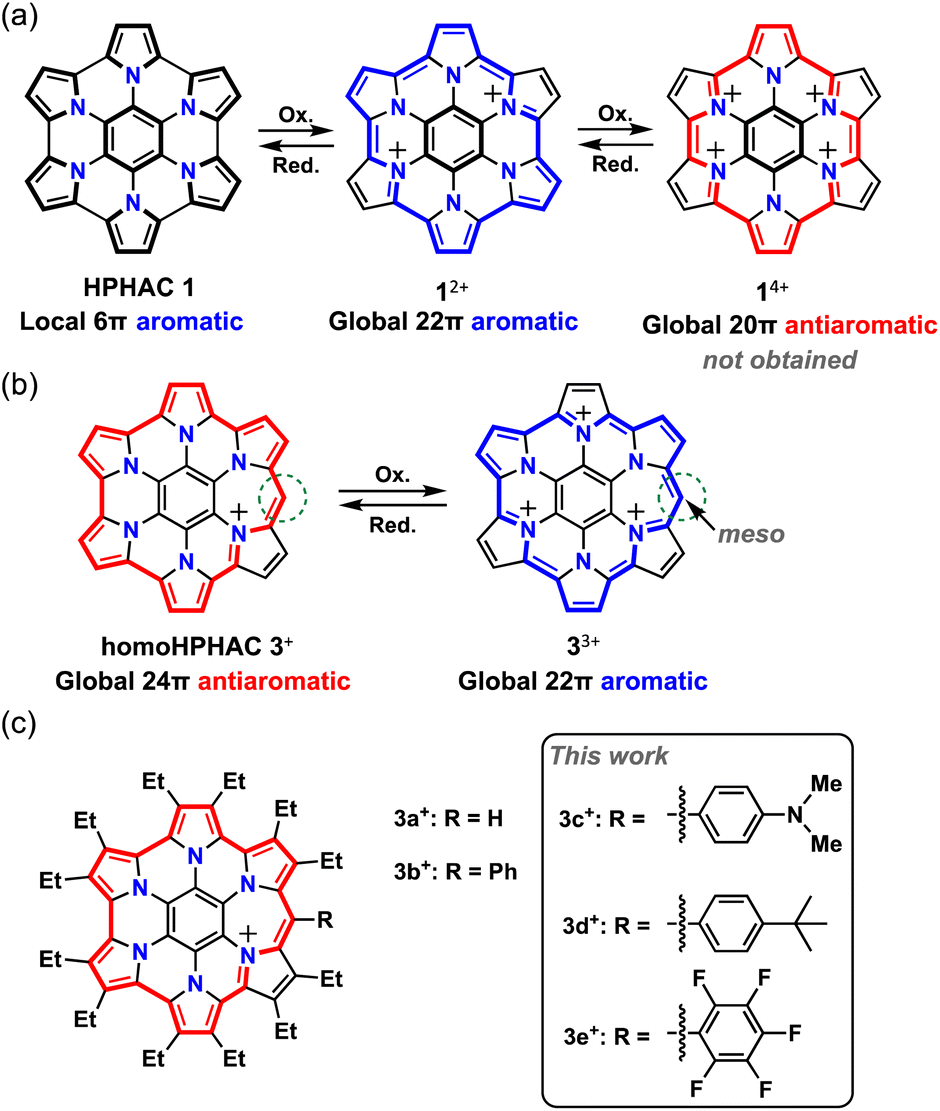
Aromaticity is an important concept in chemistry. When a molecule has cyclic π-conjugation with (4n + 2) π-electrons, it becomes aromatic and is more stable compared to acyclic π-conjugated molecules.1–3 Therefore, such compounds are widely used in our surroundings, including in medicine, plastics, dyes, and organic electronics. A molecule with 4n π-electrons, on the other hand, becomes antiaromatic; such molecules are known to decompose or undergo structural change to give compounds that have lost their antiaromatic properties. Recent studies have revealed that antiaromatic compounds exhibit optical absorption in the near-infrared region,4 high charge transport properties,5 and amphoteric redox properties,6 but there are still limited examples of such compounds.
Several research groups, including ours, have studied the synthesis and characterisation of hexapyrrolohexaazacoronene (HPHAC), a nitrogen-containing polycyclic aromatic compound, using pyrroles.7–10 Composed of electron-rich pyrroles, HPHACs are easily oxidised, and their dications exhibit aromaticity based on macrocyclic conjugation, although their antiaromatic tetracations have not yet been successfully isolated and characterized (Fig. 1a). In general, their properties can be switched between ‘aromatic’ and ‘antiaromatic’ via redox, as their aromaticity depends on the number of π-electrons. With this in mind, homoHPHAC, a π-extended analogue of HPHAC with an embedded methine carbon, was synthesized, and its antiaromatic properties as a cation and aromatic properties as a trication were revealed (Fig. 1b).9 Because homoHPHACs are originally monocations and the delocalization of cations/π-electrons over the π-system is a major factor in the antiaromaticity and stability, we hypothesized that the introduction of substituents might be effective for controlling the optical and redox properties as well as paratropicity of homoHPHAC+.3 Furthermore, because antiaromatic compounds have an intrinsically narrow HOMO–LUMO gap, the investigation of substituent effects, which are usually not great for aromatic compounds,3c is important for applications such as organic electronic materials (OEMs). To the best of our knowledge, no studies have examined the substituent effects in antiaromatic compounds.11
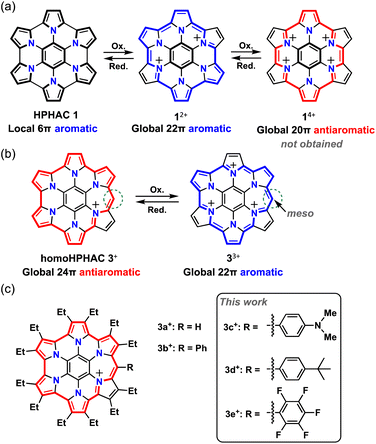 |
| | Fig. 1 (a) Chemical structures of HPHAC 1 and its cationic species. (b) Resonance structures of antiaromatic homoHPHAC 3+ and aromatic 33+. (c) HomoHPHAC 3a+–3e+ with the various substituents studied in this work. | |
In this study, we synthesized homoHPHAC monocations and trications with various substituents and investigated their effects on the optical, redox, and geometrical properties and para(dia)tropicity of a series of homoHPHAC mono(tri)cations. Friedel–Crafts-type reactions were used to enable the introduction of various substituents.
Results and discussion
Synthesis of monocations 3c+, 3d+, and 3e+
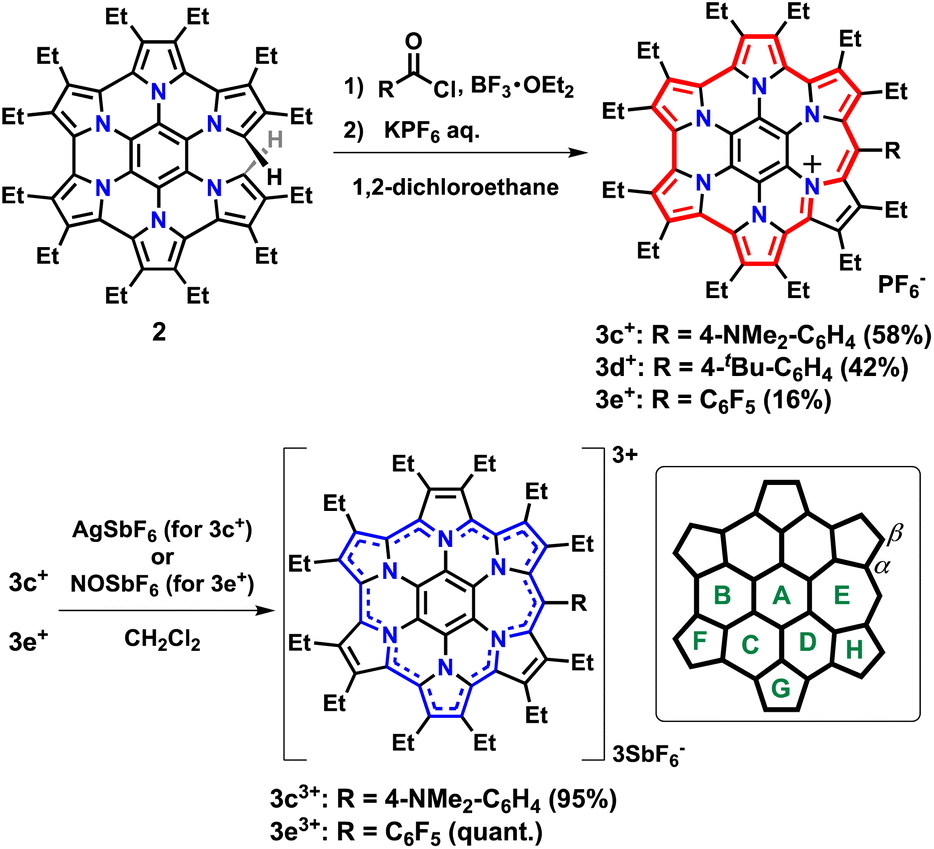
Three substituents were chosen to emphasize the electronic contribution to homoHPHAC 3c–3e; the electron-donating 4-dimethylaminophenyl group, the electron-withdrawing pentafluorophenyl group, and the 4-tert-butylphenyl group, which exhibits intermediate properties (Fig. 1c). In the case of 3c+, it is also interesting to see how the dimethylamino group affects the resonance structure of the oxidised species. The previous research has shown that σ-bond formation may occur at the meso-position of homoHPHAC+ and at the para-position of the aryl substituent in their neutral radical states.9a Therefore, aryl substituents with a functional group at the para-position were introduced at the meso-position for kinetic protection.
To increase the utility of the previous synthetic method,9a which facilitates the introduction of various functional groups, a Friedel–Crafts-type reaction was investigated using commercially available benzoyl chlorides and partially unfused HPHAC (secoHPHAC 2) (Scheme 1). The reaction was carried out in 1,2-dichloroethane under reflux conditions in the presence of BF3·OEt2 as the Lewis acid; the solution of the reaction mixture changed from yellow to brown. After treatment with an aqueous solution of KPF6, homoHPHAC 3c+[PF6−], 3d+[PF6−], and 3e+[PF6−] were obtained in 58%, 42%, and 16% yield, respectively. The yields are lower compared to the previous Vilsmeier-type method (88% for 3b+[PF6−]).9a The target compounds were identified using 1H NMR, 13C NMR, and high-resolution MS (Fig. S3a–g and S5a–c in the ESI†). In addition, the crystal structures of all compounds were successfully revealed.
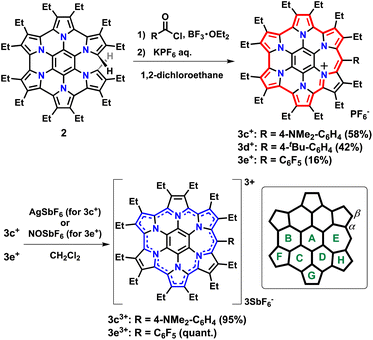 |
| | Scheme 1 Synthesis of homoHPHAC 3c+–3e+ and 3c3+–3e3+ from secoHPHAC 2 and ring labeling for the NICS calculations. | |
Crystal structures of 3c+, 3d+, and 3e+
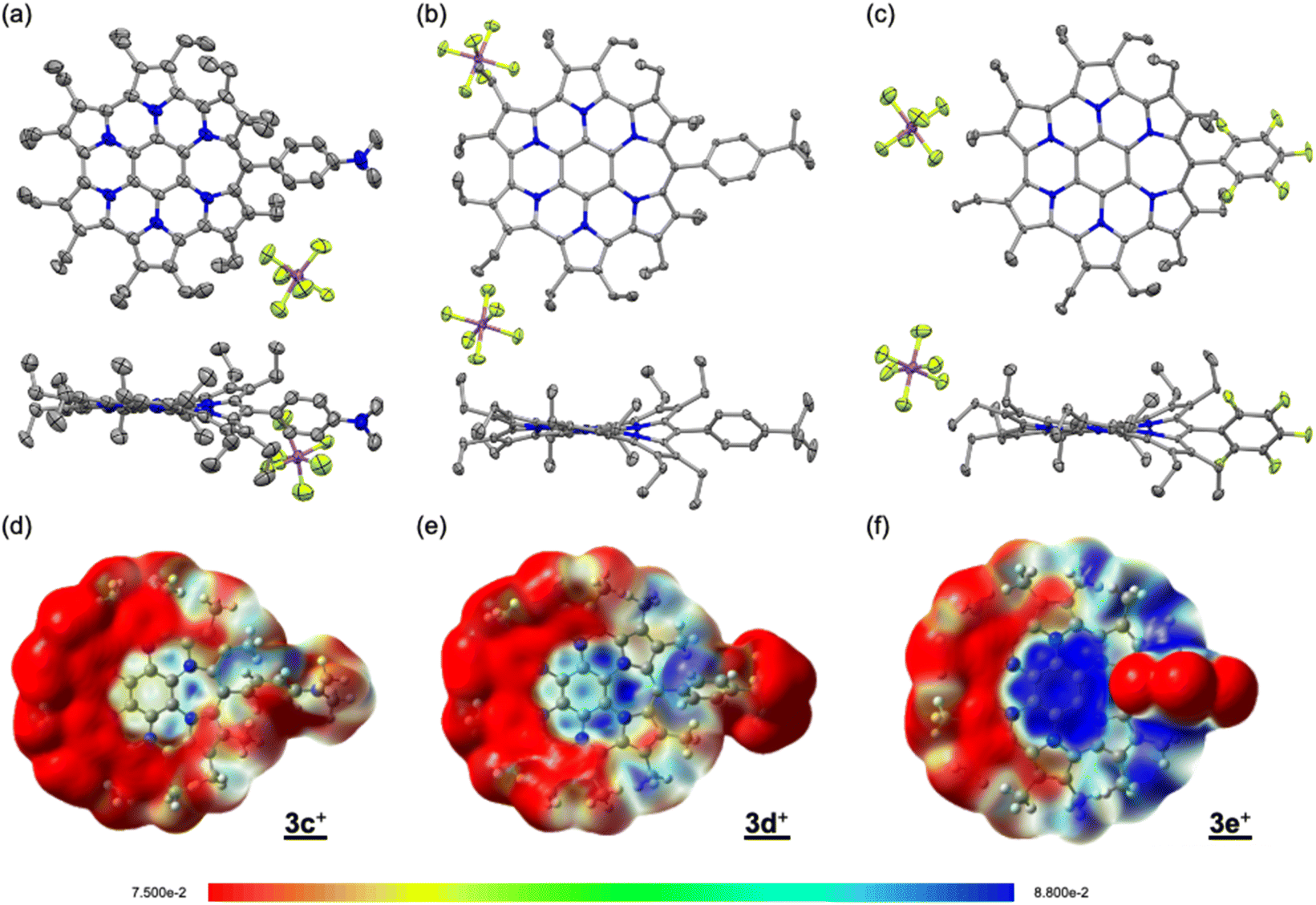
Single crystals were obtained by vapour diffusion of hexane into mixed solutions of anisole and acetone (3c+), ethylacetate (3d+), or chloroform (3e+) as SbF6− salts after treatment with an aqueous solution of NaSbF6. X-ray crystallography analysis of 3c+[SbF6−], 3d+[SbF6−], and 3e+[SbF6−] revealed the target structures containing a seven-membered ring with a methyne linkage (Fig. 2a–c and S6a–c in the ESI†). As observed for previously reported 3b+[SbF6−],9a the structures are all slightly twisted due to steric repulsion between the aryl and ethyl substituents. The quinoidal resonance structure in the H pyrrole ring is observed as the double-bondedness of Cβ–Cβ and the single-bondedness of Cα–Cβ, which differ from the bond lengths in the F and G rings with a typical pyrrole framework (Fig. S6d in the ESI†). This result suggests a macrocyclic conjugation of 3+, as depicted in Scheme 1. Indeed, the averaged values of harmonic oscillator model of aromaticity (HOMA)12 in the six pyrrole moieties are calculated to be 0.76 (3c+), 0.78 (3d+), and 0.69 (3e+), which are sufficiently smaller than the reported typical pyrrole with values of 0.85–0.93 (Fig. S6e in the ESI†).12b
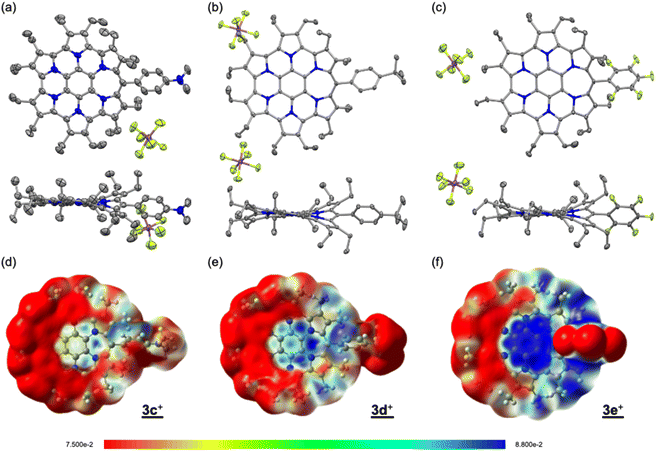 |
| | Fig. 2 Crystal structures (top and side views) of (a) 3c+[SbF6−], (b) 3d+[SbF6−], and (c) 3e+[SbF6−]. Thermal ellipsoids are scaled to 50% probability. Solvent molecules and disordered atoms with lower occupancy are omitted for clarity. ESP plots of (d) 3c+, (e) 3d+, and (f) 3e+ calculated at the B3LYP/6-31G(d) level of theory, in which the Et groups have been replaced with Me groups to simplify the calculations. | |
To discuss the electronic contribution of the aryl substituent, the torsion angles between the mean planes of the aryl substituent and the homoHPHAC skeleton are important.13 The values are 46.80° (3c+), 39.17° (3d+), and 45.00° (3e+), which are roughly similar to that of 3b+ (52.95°) in the crystal state. These results indicate that all the aryl substituents in 3c+–3e+ likely influence the π-electron system of homoHPHAC+ by an inductive effect, not a resonance effect.
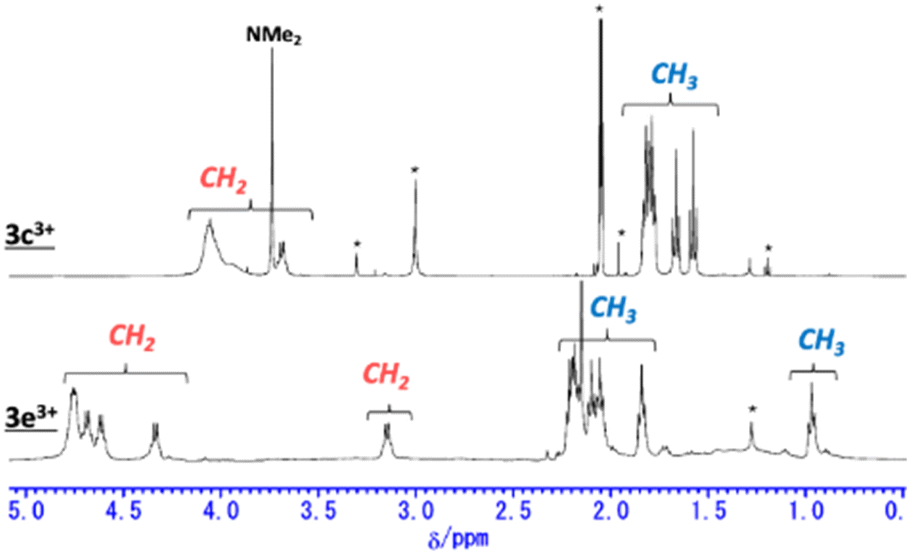
NMR spectroscopy
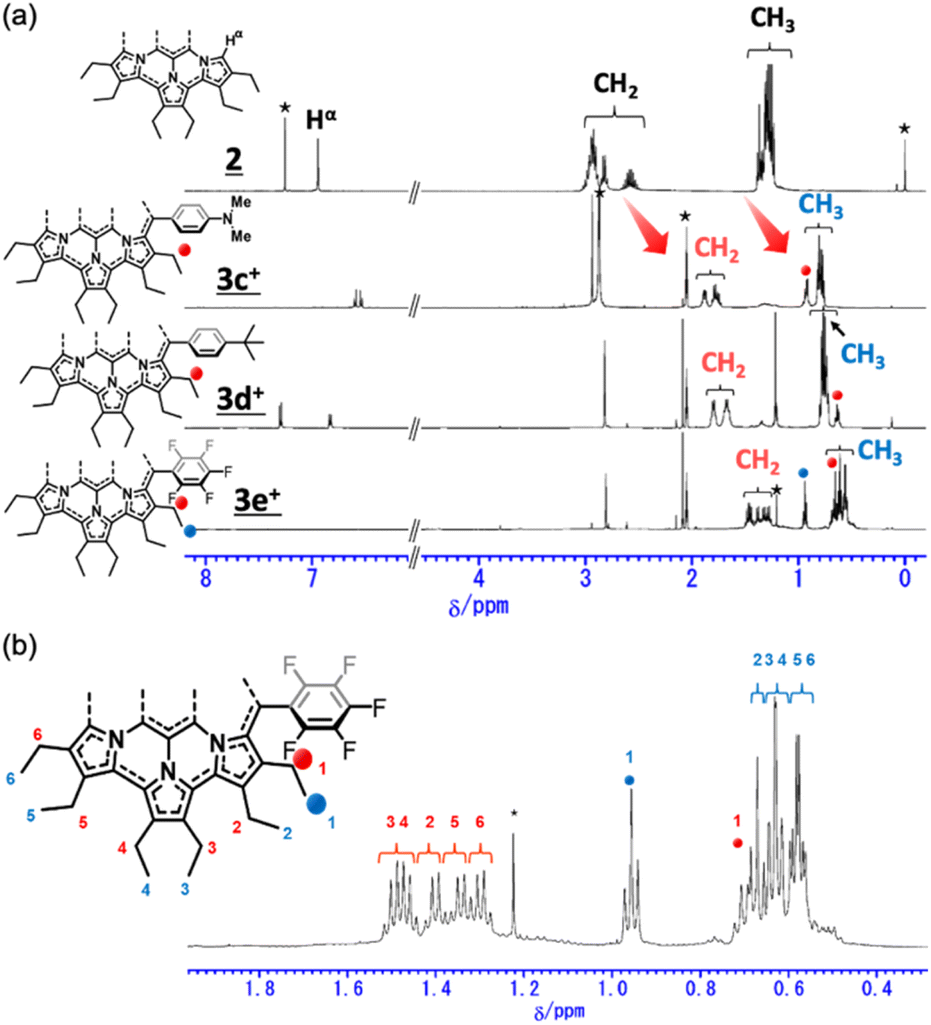
The 1H NMR spectra of secoHPHAC 2 and homoHPHAC 3c+–3e+are shown in Fig. 3a. Disappearance of the signals at the α-position of the pyrroles, Hα, was observed upon cyclisation. Despite being a monocation, the signals for the ethyl groups of 3c+–3e+ were clearly observed at higher field relative to those of 2, among which the signals of 3e+ were observed at the highest field. This is strong evidence of a paratropic ring current, and the paratropicity of 3e+ appears to be the strongest of the three compounds. For a more detailed analysis, 1H–1H COSY and NOESY measurements were performed to assign the well-resolved signals of 3e+ (Fig. S4c and d in the ESI†). The methylene signals next to the meso-position, which are marked with a red dot (labeled as 1 in red), shifted significantly to higher field than the other methylene signals, due to the ring current effect of the aryl substituents in close proximity. On the other hand, the methyl signals of 3e+ next to the meso-position, which are marked with a blue dot (labeled as 1 in blue), were observed at relatively low field, which possibly indicates the existence of C–H⋯F interactions.14 The detailed assignments of 3e+ are shown in an enlarged view of the spectrum (Fig. 3b). Except for the signals mentioned above that reflect their local environment, the signals of the ethyl groups close to the aryl group were observed on the lower-field side, while those far from the aryl group appeared on the higher-field side. Although the signals are mainly observed on the higher-field side due to the strong paratropicity of 3e+, this result reflects the inductive effect, i.e., electron-withdrawing effect, of the pentafluorophenyl group. The electrostatic surface potential (ESP) plots support this tendency; the ethyl groups close to the meso-position are covered in blue (less surface potential) and the far sides in red (Fig. 2d–f). Notably, there appears to be a correlation between the strength of the electron-withdrawing ability of the substituents and the shifts to higher field, i.e., paratropicity.
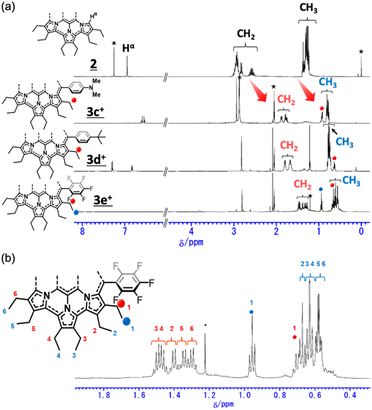 |
| | Fig. 3 Comparison of the 1H NMR spectra of (a) secoHPHAC 2 (in CDCl3) and homoHPHAC 3c+–3e+ (in acetone-d6), and (b) an enlarged view of the spectrum of 3e+. | |
DFT evaluation of aromaticity
Aromaticity and its conceptual counterpart, antiaromaticity are not exactly defined, and thus, should be evaluated from multiple perspectives. The magnitude of (anti)aromaticity is typically estimated in terms of criteria such as energetic,15 geometric,12 magnetic,16–18 and electronic19 aspects, which are usually correlated with each other. Since the aryl substituents affect the paratropic ring current of homoHPHACs+, nucleus-independent chemical shift (NICS)16,17 and anisotropy of the induced current density (ACID)18 calculations were performed for the optimized structures. For comparison, NICS calculation of secoHPHAC 2 was also performed. Table 1 summarizes the averaged NICS(±1)zz values calculated at the GIAO/HF/6-311+G(d,p)//B3LYP/6-31G(d) level of theory. Except for 3c+, the values at the B, C, D, and E rings are drastically increased upon cyclisation, while those of the A, F, G, and H rings become less negative, strongly indicating global paratropic ring current. It should be pointed out that the positive changes compared to the values of secoHPHAC 2 are the most evident for 3e+ among the four compounds including pristine homoHPHAC 3a+. Again, there appears to be a correlation between the strength of the electron-withdrawing ability of the substituents and the global paratropicity, as anticipated from the 1H NMR spectra. The ACID plots also support the presence of a global counterclockwise paratropic ring current in 3c+–3e+, in which 3e+ is most pronounced. The central benzene moieties possess a clockwise ring current as observed in the previous system (Fig. S7 in the ESI†).9,20
Table 1 The averaged NICS(±1)zz values of secoHPHAC 2, homoHPHAC monocation 3a+,9a3c+–3e+ and trications 3a3+,9a3c3+ and 3e3+, in which the Et groups were replaced with H atoms to simplify the calculations (GIAO/HF/6-311+G(d,p)//B3LYP/6-31G(d) level)
|
|
A
|
B
|
C
|
D
|
E
|
F
|
G
|
H
|
|
2
|
−4.8 |
−1.9 |
−3.7 |
−4.0 |
— |
−1.5 |
−2.4 |
−4.6 |
|
3a+
|
−5.1 |
−1.8 |
3.8 |
3.8 |
−2.3 |
−1.9 |
−2.3 |
−1.7 |
|
3c+
|
−29.7 |
3.0 |
−3.6 |
−3.0 |
3.2 |
−22.5 |
−27.5 |
−17.0 |
|
3d+
|
−10.3 |
31.4 |
26.2 |
21.7 |
32.5 |
−17.4 |
−19.0 |
−6.0 |
|
3e+
|
7.2 |
57.6 |
47.1 |
37.0 |
50.3 |
−15.7 |
−15.1 |
3.1 |
|
3a3+
|
−1.5 |
−2.6 |
−2.6 |
−4.1 |
−5.4 |
−5.4 |
−3.7 |
−2.8 |
|
3c3+
|
−30.0 |
3.0 |
−3.0 |
−3.6 |
−3.2 |
−17.0 |
−27.5 |
−22.5 |
|
3e3+
|
−59.6 |
−42.5 |
−45.3 |
−36.4 |
−29.8 |
−27.1 |
−38.2 |
−31.6 |
Optical properties
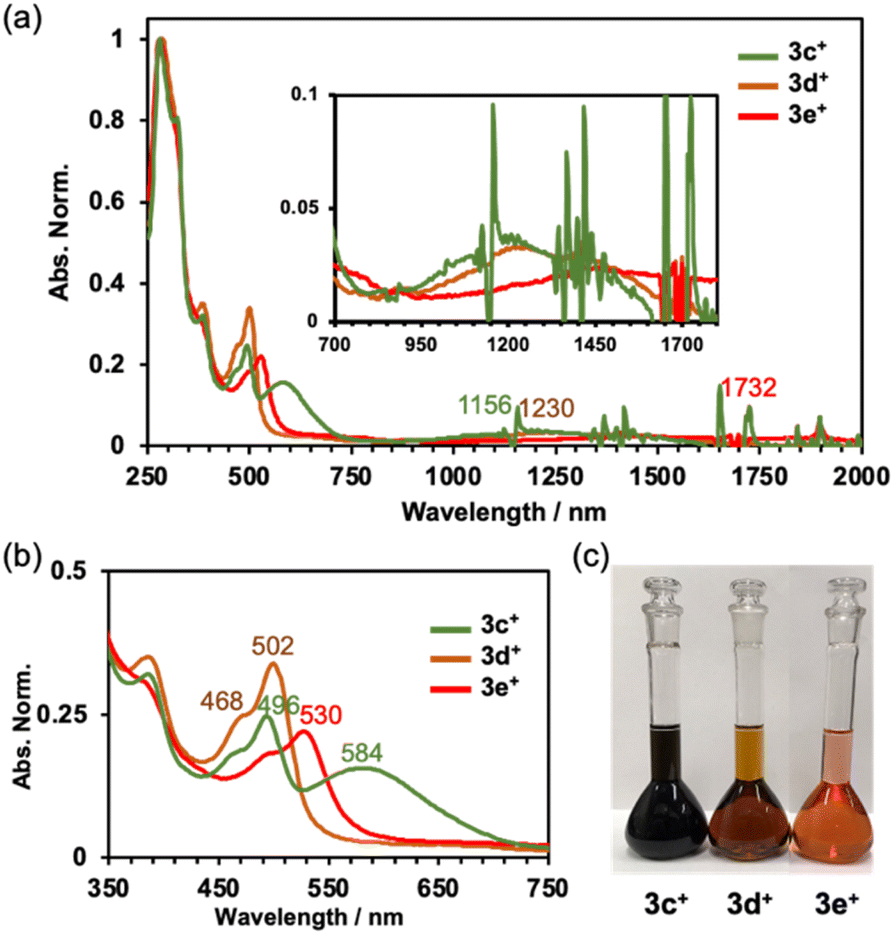
The absorption spectra of 3c+–3e+ are shown in Fig. 4a. As observed previously,9a the characteristic absorptions for the antiaromatic compounds were confirmed to be broad, weak absorptions in the NIR region around 750 to 1800 nm. Time-dependent DFT (TD-DFT) calculations at the B3LYP/6-31G+(d) level of theory indicate small oscillator strengths (f) for the HOMO → LUMO transition (3c+: 1337 nm, f = 0.0291, 3d+: 1388 nm, f = 0.0288, and 3e+: 1756 nm, f = 0.0251) (Fig. S9a–c in the ESI†). The differences in the colour of the solutions are well illustrated by the absorption spectra in the visible light range (Fig. 4b and c). As the electron-withdrawing ability of the substituent increases, the spectrum shifts towards longer wavelength (496 nm (3c+), 502 nm (3d+), and 530 nm (3e+)). Dimethylamino-substituted 3c+ possesses two distinct absorption maxima in the visible region. The TD-DFT calculations suggest that the absorption around 550–750 nm consists of HOMO − 3–HOMO − 1 → LUMO transitions, but only HOMO − 2 and HOMO − 1 of 3c+ have large orbitals on the aryl group. Other orbitals related to the absorption around 550–750 nm extended to the homoHPHAC skeletons (Fig. S9a–c and S10b–d and Table S9 in the ESI†), indicating a charge transfer (CT) transition from the dimethylaminophenyl group to the homoHPHAC skeleton in 3c+. In fact, positive solvatochromism was observed mainly for the second peak at around 584 nm in the cyclohexane, benzene, chloroform, methanol, and dimethyl sulfoxide solutions, which was also supported by a relatively large dipole moment of 5.95 D calculated at the B3LYP/6-31G(d) level of theory (Fig. S8a and b in the ESI†).
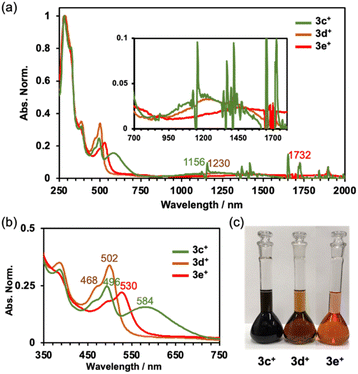 |
| | Fig. 4 (a) Absorption spectra of 3c+–3e+ in CH2Cl2, (b) enlarged view of the spectra in the visible light region, and (c) photographs of the solutions ([3c+–3e+] = 2.1 × 10−5 M in CH2Cl2). | |
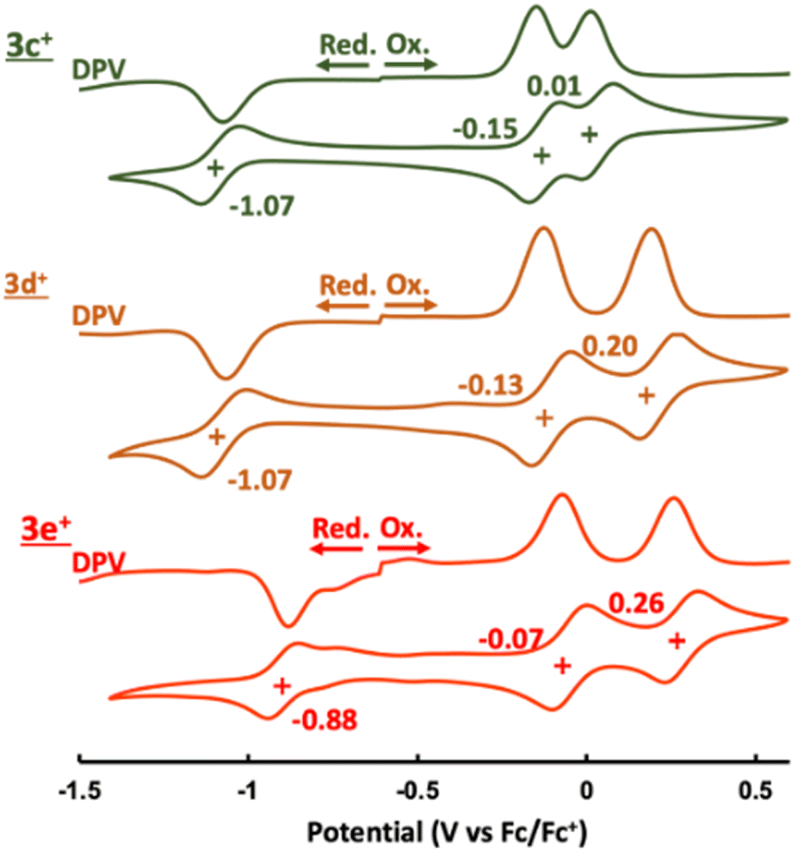
Electrochemistry
The cyclic and differential pulse voltammograms (CV and DPV) of 3c+–3e+ were measured in CH2Cl2 (Fig. 5). Amphoteric redox behavior was clearly observed with the small HOMO–LUMO gaps of 0.92 eV (3c+), 0.94 eV (3d+), and 0.81 eV (3e+), showing two reversible oxidation steps and one reversible reduction step. The reduction waves of 3a+ and 3b+ were irreversible,9a which proved that kinetic protection of both the meso-position of homoHPHAC+ and the para-position of the aryl substituent were important to prevent side reactions such as σ-dimer formation between two radical species. Attempts to isolate a one-electron reductant, i.e., neutral radical, have not been successful to date due to the extremely low oxidation potentials. Reflecting the strong electron-withdrawing ability of the pentafluorophenyl group, 3e+ has the lowest LUMO level, which also contributes to the higher oxidation potentials compared with those of 3c+ and 3d+. Notably, the difference between the first and second oxidation potentials of 3c+ (0.16 eV) is much smaller than those of 3a+ (0.32 eV),9a3b+ (0.32 eV),9a3d+ (0.33 eV), and 3e+ (0.33 eV). Therefore, chemical preparation of trication 3c3+ and 3e3+ was then conducted to investigate their electronic state and diatropicity.
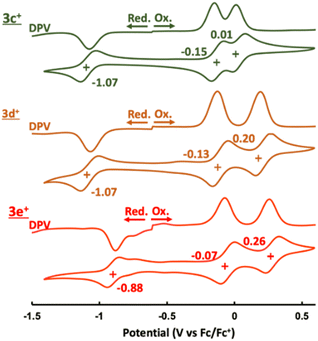 |
| | Fig. 5 CV and DPV of 3c+–3e+ containing 0.1 M nBu4NPF6 in CH2Cl2 at a scan rate of 100 mV s−1 ([3c+–3e+] = 1.0 mM). | |
Preparation and diatropicity of trications 3c3+ and 3e3+
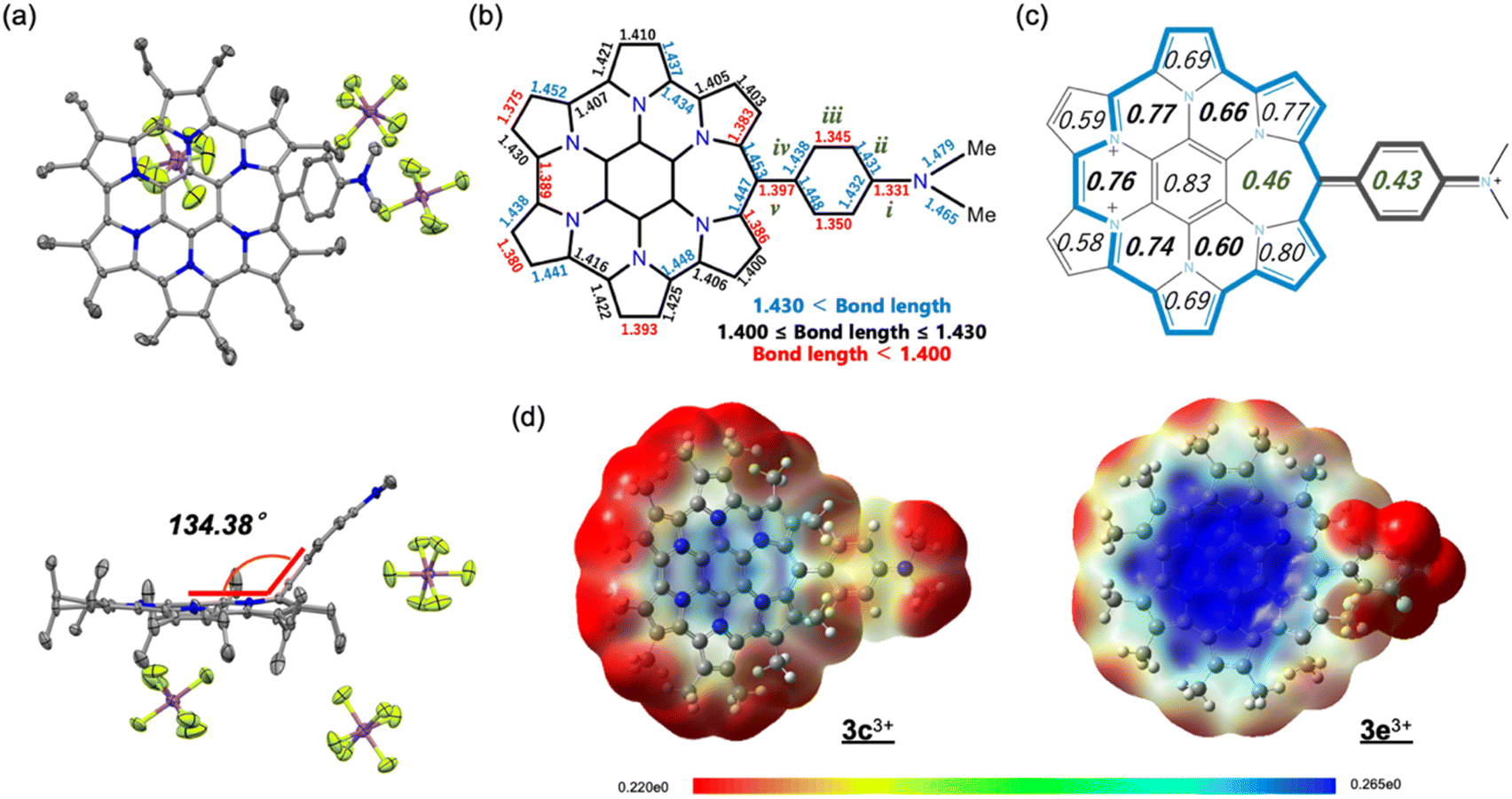
Trications 3c3+ and 3e3+ were prepared almost quantitatively in CH2Cl2 using AgSbF6 and NOSbF6 as the oxidant, respectively (Scheme 1). The change of the absorption spectra was drastic in the oxidation states, as was observed for previous systems (Fig. S9d and e in the ESI†). The isolated compounds were definitively identified using 1H NMR and 13C NMR. As shown in Fig. 6, the 1H NMR signals of all the ethyl protons of 3c3+ and 3e3+ appeared at lower field compared with those of 3c+ and 3e+. This result is thought to be not only due to the change in their oxidation state from monocationic to tricationic, but also switching of their aromaticity from antiaromatic to aromatic.21 The low-field shifts for 3e3+ are qualitatively more pronounced than those of 3c3+. Furthermore, it should be noted that even the signal of the dimethylamino group of 3c+, which is located far from the homoHPHAC π-system, is significantly shifted from 2.49 ppm to 3.74 ppm upon oxidation, suggesting the localisation of the cationic charge on the dimethylamino group. The coupling constant of the aryl substituent supports this prediction; the J values of the two doublets in 3c+ are 8.7 Hz, which is a typical coupling constant value for para-disubstituted benzene, whereas those in 3c3+ are 10.8 Hz, which is in the range for a cis-olefinic one (Fig. S3h in the ESI†). Therefore, 3c3+ is considered to dominantly adopt a quinomethane resonance form with two cations over the homoHPHAC π-system and a monocation on the dimethylamino group. The ESP plots clearly show the difference between 3c3+ and 3e3+ (Fig. 7d).
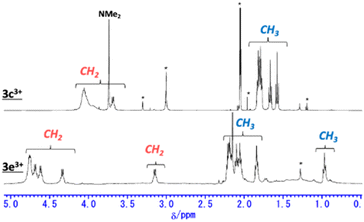 |
| | Fig. 6
1H NMR spectra of 3c3+ in acetone-d6 and 3e3+ in CD2Cl2. | |
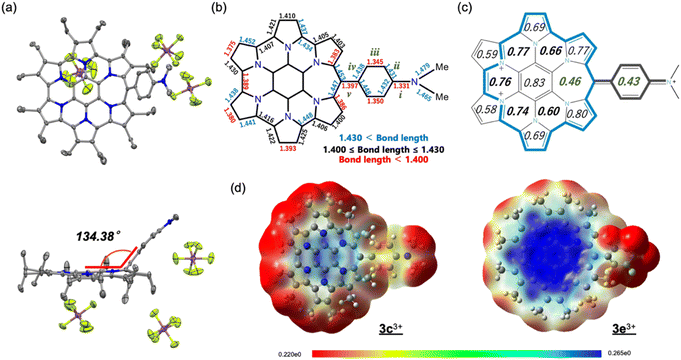 |
| | Fig. 7 (a) Crystal structures (top and side views) of 3c3+[SbF6−]3. Thermal ellipsoids are scaled to 50% probability. Solvent molecules and disordered atoms are omitted for clarity. (b) Selected bond lengths (Å) and (c) HOMA values of the crystal structure of 3c3+[SbF6−]3. (d) ESP plots of 3c3+ and 3e3+ calculated at the B3LYP/6-31G(d) level of theory, in which the Et groups are replaced with Me groups to simplify the calculations. | |
For comparison of the diatropicity of the trications, NICS(±1)zz values were calculated for 3c3+ and 3e3+ (Table 1). HomoHPHAC 3e3+ shows large negative values at all points, as observed for 3a3+ and 3b3+.9a On the other hand, much less-negative and nearly-zero values are calculated in the case of 3c3+, especially for the B, C, D, and E rings. For aromatic compounds, the NMR signals of the inner core provide the most direct evidence due to the shifts toward higher field based on the diatropic ring current. In the present case, the 13C NMR signals are important to evaluate the aromaticity in terms of magnetic properties. Three signals of the central benzene moieties were observed in the high-field region at 99.3, 98.1, and 92.7 ppm for 3e3+, indicating global diatropicity based on the 22π-cyclic conjugation as depicted in Scheme 1, whereas those for 3c3+ were observed at relatively lower field compared with 3e3+ (107.2, 103.8, and 92.6 ppm). In the ACID plots, there is a compensation between the differently oriented currents on the outer rim versus those on the individual inner rings (Fig. S7b in the ESI†). In 3e3+, the intensity on the outer rim of the homoHPHAC skeleton appears stronger. On the other hand, that on the outer rim is close to those on the inner rings in 3c3+, leading to the much less global diatropicity.20
Crystal structure of 3c3+
Fortunately, single crystals of 3e3+ were obtained by vapour diffusion of hexane into its chloroform and anisole mixed solution. The molecular structure of 3e3+[SbF6−]3 was determined by X-ray diffraction analysis (Fig. 7a and S6f in the ESI†). The most significant difference from the structure of monocation 3e+[SbF6−] is that the aryl substituent is largely bent away from coplanarity with the homoHPHAC skeleton. The angle between the two mean planes is 134.38°. The bond lengths of the dimethylaminophenyl moiety confirm the quinomethane resonance form, as suggested by the coupling constant in the 1H NMR spectrum. The bonds labelled as “i”, “iii”, and “v” (i: 1.331 Å, iii: 1.345 and 1.350 Å, v: 1.397 Å) are predominantly shorter than the bonds “ii” and “iv” (ii: 1.431 and 1.432 Å, iv: 1.438 and 1.448 Å) in the crystal structure (Fig. 7b).22 Because of the double bondedness of the “v” bond (although it is rather long for a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond due to the steric hindrance of the adjacent ethyl substituents), sufficient homo(cyclic)conjugation at the meso-position is not able to form on homoHPHAC3+, leading to the lower global paratropicity of 3e3+. The HOMA value of 0.43 at the aryl substituent also shows decreased aromaticity (Fig. 7c). In agreement with the NICS calculations shown in Table 1, the aromaticity of the F ring is the smallest among the F, G, and H rings.
C double bond due to the steric hindrance of the adjacent ethyl substituents), sufficient homo(cyclic)conjugation at the meso-position is not able to form on homoHPHAC3+, leading to the lower global paratropicity of 3e3+. The HOMA value of 0.43 at the aryl substituent also shows decreased aromaticity (Fig. 7c). In agreement with the NICS calculations shown in Table 1, the aromaticity of the F ring is the smallest among the F, G, and H rings.
Conclusions
We developed versatility in the synthesis of homoHPHAC via Friedel–Crafts-type reaction of secoHPHAC and commercially available benzoyl chlorides, which enabled the investigation of substituent effects on antiaromatic compounds. The NMR and DFT calculations show that the global paratropicity of 3e+ with an electron-withdrawing pentafluorophenyl group substituent is the strongest among the compounds reported here, while the global diatropicity of 3e3+ is the strongest. These results indicate that delocalisation of cations/π-electrons enhanced by the substituent promotes cyclic conjugation, leading to strong paratropicity and diatropicity for the monocation and trication, respectively. The effect of a substituent on the electronic structure is essentially the same, regardless of whether it is aromatic or antiaromatic. However, because antiaromatic compounds have a high-lying HOMO and low-lying LUMO and thus have a small HOMO–LUMO gap, the effects appear to be relatively larger in antiaromatic compounds. The present study will provide new design guide for organic electronic materials, which should merit further research on antiaromatic compounds.
Data availability
Experimental procedures, NMR and MS spectra, and computational details and results are available in the ESI.† Details of the crystal structures have been deposited in the Cambridge Crystallographic Data Centre with CCDC 2232450–2232453 for 3c+, 3d+, 3e+, and 3c3+, respectively.
Author contributions
M. T. and H. U. designed the project. T. T. performed most of the experimental works except for X-ray measurement and analysis. S. M., K. O., and H. U. performed X-ray measurement and analysis. M. T. and T. T. prepared the overall manuscript. All authors, including K. O., S. M., and H. U., contributed by commenting on the manuscript. M. T. and H. U. supervised the overall research.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JSPS KAKENHI (JP20H02725, JP19K05422, and JP23H03964). M. T. acknowledges the Takahashi Industrial and Economic Research Foundation and the Tokyo Ohka Foundation for the Promotion of Science and Technology for financial support.
Notes and references
- P. v. R. Schleyer, Introduction: Aromaticity, Chem. Rev., 2001, 101, 1115 CrossRef CAS PubMed.
-
(a)
M. Solà, A. I. Boldyrev, M. K. Cyrański, T. M. Krygowski and G. Merino, Aromaticity and Antiaromaticity, Wiley-VCH, 2023 Search PubMed;
(b)
Aromaticity, ed. I. Fernández, Elsevier, 2021 Search PubMed;
(c)
R. Gleiter and G. Haberhauer, Aromaticity and Other Conjugation Effects, Wiley-VCH, 2012 Search PubMed.
-
(a) H. Szatylowicz, A. Jezuita and T. M. Krygowski, On the Relations Between Aromaticity and Substituent Effect, Struct. Chem., 2019, 30, 1529–1548 CrossRef CAS;
(b) T. M. Krygowski and B. T. Stępień, Sigma- and pi-Electron Delocalization: Focus on Substituent Effects, Chem. Rev., 2005, 105, 3482–3512 CrossRef CAS PubMed;
(c) T. M. Krygowski, K. Ejsmont, B. T. Stępień, M. K. Cyrański, J. Poater and M. Solà, Relation between the Substituent Effect and Aromaticity, J. Org. Chem., 2004, 69, 6634–6640 CrossRef CAS PubMed.
-
(a) T. Ito, Y. Hayashi, S. Shimizu, J.-Y. Shin, N. Kobayashi and H. Shinokubo, Gram-Scale Synthesis of Nickel(II) Norcorrole: The Smallest Antiaromatic Porphyrinoid, Angew. Chem., Int. Ed., 2012, 51, 8542–8545 CrossRef CAS PubMed;
(b) S. Cho, Z. S. Yoon, K. S. Kim, M.-C. Yoon, D.-G. Cho, J. L. Sessler and D. Kim, Defining Spectroscopic Features of Heteroannulenic Antiaromatic Porphyrinoids, J. Phys. Chem. Lett., 2010, 1, 895–900 CrossRef CAS;
(c) S. Mori and A. Osuka, Aromatic and Antiaromatic Gold(III) Hexaphyrins with Multiple Gold-Carbon Bonds, J. Am. Chem. Soc., 2005, 127, 8030–8031 CrossRef CAS PubMed.
-
(a) J. L. Marshall, K. Uchida, C. K. Frederickson, C. Schütt, A. M. Zeidell, K. P. Goetz, T. W. Finn, K. Jarolimek, L. N. Zakharov, C. Risko, R. Herges, O. D. Jurchescu and M. M. Haley Indacenodibenzothiophenes, Synthesis, Optoelectronic Properties and Materials Applications of Molecules with Strong Antiaromatic Character, Chem. Sci., 2016, 127, 5547–5558 RSC;
(b) T. Nishinaga, T. Ohmae, K. Aita, M. Takase, M. Iyoda, T. Arai and Y. Kunugi, Antiaromatic Planar Cyclooctatetraene: A Strategy for Developing Ambipolar Semiconductors for Field Effect Transistors, Chem. Commun., 2013, 49, 5354–5356 RSC.
-
(a) D. T. Chase, A. G. Fix, S. J. Kang, B. D. Rose, C. D. Weber, Y. Zhong, L. N. Zakharov, M. C. Lonergan, C. Nuckolls and M. M. Haley, 6,12-Diarylindeno[1,2-b]fluorenes: Syntheses, Photophysics, and Ambipolar OFETs, J. Am. Chem. Soc., 2012, 134, 10349–10352 CrossRef CAS PubMed;
(b) J.-Y. Shin, T. Yamada, H. Yoshikawa, K. Awaga and H. Shinokubo, An Antiaromatic Electrode-Active Material Enabling High Capacity and Stable Performance of Rechargeable Batteries, Angew. Chem., Int. Ed., 2014, 53, 3096–3101 CrossRef CAS PubMed.
- M. Lazerges, M. Jouini, P. Hapiot, P. Guiriec and P.-C. Lacaze, Mechanism of Pyrrolyl Oxidation in Star-Shaped Compounds, J. Phys. Chem. A, 2003, 107, 5042–5048 CrossRef CAS.
-
(a) M. Takase, V. Enkelmann, D. Sebastiani, M. Baumgarten and K. Müllen, Annularly Fused Hexapyrrolohexaazacoronenes: An Extended π System with Multiple Interior Nitrogen Atoms Displays Stable Oxidation States, Angew. Chem., Int. Ed., 2007, 46, 5524–5527 CrossRef CAS;
(b) Y. Sasaki, M. Takase, S. Mori and H. Uno, Synthesis and Properties of NitroHPHAC: The First Example of Substitution Reaction on HPHAC, Molecules, 2020, 25, 2486 CrossRef CAS PubMed;
(c) K. Oki, M. Takase, N. Kobayashi and H. Uno, Synthesis and Characterization of Peralkylated Pyrrole-Fused Azacoronene, J. Org. Chem., 2021, 86, 5102–5109 CrossRef CAS;
(d) Y. Sasaki, M. Takase, N. Kobayashi, S. Mori, K. Ohara, T. Okujima and H. Uno, Radially π-Extended Pyrrole-Fused Azacoronene: A Series of Crystal Structures of HPHAC with Various Oxidation States, J. Org. Chem., 2021, 86, 4290–4295 CrossRef CAS PubMed.
-
(a) K. Oki, M. Takase, S. Mori and H. Uno, Synthesis and Isolation of Antiaromatic Expanded Azacoronene via Intramolecular Vilsmeier-Type Reaction, J. Am. Chem. Soc., 2019, 141, 16255–16259 CrossRef CAS PubMed;
(b) M. Takase, A. Ueno, K. Oki, H. Matsumoto, S. Mori, T. Okujima and H. Uno, Tropo(thio)ne-Embedded HomoHPHACs: Does the Tropylium Cation Induce Global Antiaromaticity in Expanded Hexapyrrolohexaazabenzocoronene?, Chem. Commun., 2022, 58, 3366–3369 RSC.
-
(a) M. Żyła-Karwowska, H. Zhylitskaya, J. Cybińska, T. Lis, P. J. Chmielewski and M. Stępień, An Electron-Deficient Azacoronene Obtained by Radial π-Extension, Angew. Chem., Int. Ed., 2016, 55, 14658–14662 CrossRef;
(b) M. Navakouski, H. Zhylitskaya, P. J. Chmielewski, T. Lis, J. Cybińska and M. Stępień, Stereocontrolled Synthesis of Chiral Heteroaromatic Propellers with Small Optical Bandgaps, Angew. Chem., Int. Ed., 2019, 58, 4929–4933 CrossRef CAS PubMed;
(c) M. Navakouski, H. Zhylitskaya, P. J. Chmielewski, M. Żyła-Karwowska and M. Stępień, Electrophilic Aromatic Coupling of Hexapyrrolylbenzenes. A Mechanistic Analysis, J. Org. Chem., 2020, 85, 187–194 CrossRef CAS PubMed.
- Very recently antiaromatic s-indacene with various substituents at specific positions was reported: S.-J. Jhang, J. Pandidurai, C.-P. Chu, H. Miyoshi, Y. Takahara, M. Miki, H. Sotome, H. Miyasaka, S. Chatterjee, R. Ozawa, Y. Ie, I. Hisaki, C.-L. Tsai, Y.-J. Cheng and Y. Tobe,
s-Indacene Revisited: Modular Synthesis and Modulation of Structures and Orbitals of Hexaaryl Derivatives, J. Am. Chem. Soc., 2023, 145, 4716–4729 CrossRef CAS PubMed.
-
(a) T. M. Krygowski and M. K. Cyrański, Structural Aspects of Aromaticity, Chem. Rev., 2001, 101, 1385–1420 CrossRef CAS PubMed;
(b) T. M. Krygowski, H. Szatylowicz, O. A. Stasyuk, J. Dominikowska and M. Palusiak, Aromaticity from the viewpoint of Molecular Geometry: Application to Planar Systems, Chem. Rev., 2014, 114, 6383–6422 CrossRef CAS.
-
(a) A. Almenningen, O. Bastiansen, L. Fernholt, B. N. Cyvin, S. J. Cyvin and S. Samdal, Structure and Barrier of Internal Rotation of Biphenyl Derivatives in the Gaseous State: Part 1. The Molecular Structure and Normal Coordinate Analysis of Normal Biphenyl and Perdeuterated Biphenyl, J. Mol. Struct., 1985, 128, 59–76 CrossRef CAS;
(b) O. Bastiansen and S. Samdal, Structure and Barrier of Internal Rotation of Biphenyl Derivatives in the Gaseous State: Part 4. Barrier of Internal Rotation in Biphenyl, Perdeuterated Biphenyl and Seven Non-ortho-substituted Halogen Derivatives, J. Mol. Struct., 1985, 128, 115–125 CrossRef CAS.
-
(a) M. Morisue, M. Kawanishi, I. Ueno, T. Nakamura, T. Nabeshima, K. Imamura and K. Nozaki, Evidence of C–F⋯H–C Attractive Interaction: Enforced Coplanarity of a Tetrafluorophenylene-Ethynylene-Linked Porphyrin Dimer, J. Phys. Chem. B, 2021, 125, 9286–9295 CrossRef CAS;
(b) J. D. Dunitz and W. B. Schweizer, Molecular Pair Analysis: C–H···F Interactions in the crystal Structure of Fluorobenzene? And Related Matters, Chem.–Eur. J., 2006, 12, 1381–6845 CrossRef PubMed;
(c) V. R. Thalladi, H.-C. Weiss, D. Bläser, R. Boese, A. Nangia and G. R. Desiraju, C–H···F Interactions in the Crystal Structures of Some Fluorobenzenes, J. Am. Chem. Soc., 1998, 120, 8702–8710 CrossRef CAS.
- R. M. K. Cyrański, Energetic Aspects of Cyclic Pi-Electron Delocalization: Evaluation of the Methods of Estimating Aromatic Stabilization Energies, Chem. Rev., 2005, 105, 3773–3811 CrossRef.
-
(a) P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. v. E. Hommes, Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS;
(b) Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- R. Gershoni-Poranne and A. Stanger, Magnetic Criteria of Aromaticity, Chem. Soc. Rev., 2015, 44, 6597–6615 RSC.
-
(a) R. Herges and D. Geuenich, Delocalizaton of Electrons in Molecules, J. Phys. Chem. A, 2001, 105, 3214–3220 CrossRef CAS;
(b) D. Geuenich, K. Hess, F. Köhler and R. Herges, Anisotropy of the Induced Current Density (ACID), a General Method To Quantify and Visualize Electronic Delocalization, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed.
- F. Feixas, E. Matito, J. Poater and M. Solà, Quantifying Aromaticity with Electron Delocalisation Measures, Chem. Soc. Rev., 2015, 44, 6434–6451 RSC.
-
(a) M. Baumgarten, L. Gherghel, M. Wagner, A. Weitz, M. Rabinovitz, P.-C. Cheng and L. T. Scott, Corannulene Reduction: Spectroscopic Detection of All Anionic Oxidation States, J. Am. Chem. Soc., 1995, 117, 6254–6257 CrossRef CAS;
(b) C. Liu, M. E. Sandoval-Salinas, Y. Hong, T. Y. Gopalakrishna, H. Phan, N. Aratani, T. S. Herng, J. Ding, H. Yamada, D. Kim, D. Casanova and J. Wu, Macrocyclic Polyradicaloids with Unusual Super-ring Structure and Global Aromaticity, Chem, 2018, 4, 1586–1595 CrossRef CAS;
(c) S. Furukawa, M. Fujita, Y. Kanatomi, M. Minoura, M. Hatanaka, K. Morokuma, K. Ishimura and M. Saito, Double Aromaticity Arising from σ- and π-Rings, Commun. Chem., 2018, 1, 60 CrossRef;
(d) S. Escayola, N. P. Vedin, A. Poater, H. Ottosson and M. Solà, In the Quest of Hückel–Hückel and Hückel-Baird Double Aromatic Tropylium (Tri)cation and Anion Derivatives, J. Phys. Org. Chem., 2022, e4447 Search PubMed.
-
(a) A. Muranaka, S. Ohira, D. Hashizume, H. Koshino, F. Kyotani, M. Hirayama and M. Uchiyama, [18]/[20]π Hemiporphyrazine: A Redox-Switchable Near-Infrared Dye, J. Am. Chem. Soc., 2012, 134, 190–193 CrossRef CAS PubMed;
(b) T. Y. Gopalakrishna, J. S. Reddy and V. G. Anand, An Amphoteric Switch to Aromatic and Antiaromatic States of a Neutral Air-Stable 25π Radical, Angew. Chem., Int. Ed., 2014, 53, 10984–10987 CrossRef CAS PubMed;
(c) M. D. Peeks, T. D. W. Claridge and H. L. Anderson, Aromatic and Antiaromatic Ring Currents in a Molecular Nanoring, Nature, 2017, 541, 200–203 CrossRef CAS PubMed;
(d) S. Dong, T. Y. Gopalakrishna, Y. Han and C. Chi, Cyclobis(7,8-(para-quinodimethane)-4,4'-triphenylamine) and Its Cationic Species Showing Annulene-Like Global (Anti)Aromaticity, Angew. Chem., Int. Ed., 2019, 58, 11742–11746 CrossRef CAS PubMed;
(e) H. Ochiai, K. Furukawa, H. Nakano and Y. Matano, Doubly Strapped Redox-Switchable 5,10,15,20-Tetraaryl-5,15-diazaporphyrinoids: Promising Platforms for the Evaluation of Paratropic and Diatropic Ring-Current Effects, J. Org. Chem., 2021, 86, 2283–2296 CrossRef CAS PubMed;
(f) H. Gao, F. Wu, Y. Zhao, X. Zhi, Y. Sun and Z. Shen, Highly Stable Neutral Corrole Radical: Amphoteric Aromatic–Antiaromatic Switching and Efficient Photothermal Conversion, J. Am. Chem. Soc., 2022, 144, 3458–3467 CrossRef CAS PubMed.
- Due to the disorder of 4-dimethylaminophenyl group in the crystal structure of 3c+[SbF6−], direct comparison of their bond lengths is avoided.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2232450–2232453. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc07037e |
| ‡ Current address: Department of Molecular and Macromolecular Chemistry, Graduate School of Engineering, Nagoya University, Nagoya 464-8603, Japan. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Toranosuke
Takata
a,
Kosuke
Oki‡
a,
Shigeki
Mori
*ab,
Toranosuke
Takata
a,
Kosuke
Oki‡
a,
Shigeki
Mori