
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The templating effect of diammonium cations on the structural and optical properties of lead bromide perovskites: a guide to design broad light emitters†
Rossella
Chiara
a,
Marta
Morana
 a,
Giulia
Folpini
b,
Andrea
Olivati
bc,
Benedetta
Albini
d,
Pietro
Galinetto
d,
Laura
Chelazzi
e,
Samuele
Ciattini
e,
Elvira
Fantechi
e,
Stefano A.
Serapian
a,
Annamaria
Petrozza
b and
Lorenzo
Malavasi
*a
a,
Giulia
Folpini
b,
Andrea
Olivati
bc,
Benedetta
Albini
d,
Pietro
Galinetto
d,
Laura
Chelazzi
e,
Samuele
Ciattini
e,
Elvira
Fantechi
e,
Stefano A.
Serapian
a,
Annamaria
Petrozza
b and
Lorenzo
Malavasi
*a
aDepartment of Chemistry and INSTM, University of Pavia, Via Taramelli 16, Pavia, 27100, Italy. E-mail: lorenzo.malavasi@unipv.it; Tel: +39 382 987921
bCenter for Nano Science and Technology @ PoliMi, Istituto Italiano di Tecnologia, 20133 Milan, Italy
cPhysics Department, Politecnico di Milano, Piazza L. da Vinci, 32, 20133 Milano, Italy
dDepartment of Physics, University of Pavia, Via Bassi 6, Pavia, 27100, Italy
eCentro di Cristallografia Strutturale, Polo Scientifico di Sesto Fiorentino, Via della Lastruccia No. 3, 50019 Sesto Fiorentino Firenze, Italy
First published on 2nd August 2022
Abstract
The templating role in the formation of 2D metal halide perovskites of diammonium cations, such as 1,3-phenylenediammonium (1,3-PDA), 1,3-xylylenediammonium (1,3-XDA), 1,4-phenylenediammonium (1,4-PDA), and 1,4-xylylenediammonium (1,4-XDA) cations, has been investigated. Single-crystal X-ray diffraction confirms the formation of 2D Dion-Jacobon (DJ) perovskites for all the cations except for the 1,3-XDA cation leading to a so-called 0D perovskitoid. The analysis of the structural data showed a higher distortion degree for the systems containing a shorter cation, namely 1,4-PDA. A detailed spectroscopic investigation, with both static and time-resolved photoluminescence spectroscopy, revealed a broadband emissive component at room temperature with hundreds of nanometers of bandwidth when 1,3-PDA and 1,4-PDA cations are present, while a narrow peak is found for the longer cation, i.e. 1,4-XDA. A broadband emission arises in this last sample as well by decreasing temperature. Such a broad emission, as indicated by the analysis of the power and temperature dependence of the PL, can be attributed to trap-mediated excitonic recombination or STEs, and the trend in emissive properties can be correlated with the different levels of octahedral distortions. The novel systems reported in this work enlarge the family of broadband emitters and add further insights to develop efficient perovskite-based broadband and white light emitters.
Introduction
Two-dimensional (2D) perovskites have been proposed in the last decade as alternatives to their 3D counterparts to improve the luminescence efficiency of this class of semiconductors, thanks to the presence of stable excitons with fast radiative decay.1,2 At the same time, the layered architecture gives them stability and relaxes geometrical constraints, giving access to a wider range of chemical compositions. Despite the huge potential, the emissive properties of these materials have been very disappointing, an issue that can be related to a strong defect activity and a lack of knowledge and control of the structure-property relationship. The latter is strongly defined by the interconnection between the organic and the inorganic moieties driving the stiffness and distortion of the structure and thus its defectiveness. Investigating the templating action of the organic cation is thus critical.The most investigated families of 2D perovskites are the Ruddlesden-Popper (RP) and Dion-Jacobson (DJ) phases of general formula  and A′An−1MnX3n+1, respectively, where A′ and A are organic cations and n represents the number of staggered inorganic layers made of metal M and halide X.3 Most of the investigated organic spacers for 2D perovskites, to date, are monoammonium cations, while diammonium cations have been less explored.3 In the latter case, and in particular for short cations, the DJ phases are generally formed, where the inorganic layers are stacked in an eclipsed fashion on top of each other, differently from the RP phases where they are staggered.3 In addition, 2D perovskites are not always stabilized by diammonium cations, and 1D structures, characterized by linear chains of octahedra, have been also often observed. For example, in the series of linear cations of the general formula NH3(CH2)mNH32+ (m = 4, 7, 8, 10, 12), the cations with even carbon-chain lengths form 2D perovskites, while those with odd carbon chain lengths form 1D structures.4 Recently, the structural diversity of systems incorporating a diammonium cation has been further expanded towards 3D halide perovskitoids using linear cations such as 1,4-butanediamine (1,4BDA), N,N-dimethyl-1,3-propanediamine (NMPA), or N,N-dimethylethylenediamine (DMEA), providing a structural motif comprising dimers with edge-sharing octahedra which are then connected through corner-sharing bonds to form a 3D network.5 In these systems, the optical properties are close to traditional 3D MHPs with the bandgap being mainly influenced by standard octahedra distortion parameters. The correlation between structural distortions and optical properties in 2D perovskites is a key topic to modulate their properties and design novel and optimized materials. Thanks to the vast work performed on perovskites including monoammonium cations, well-defined parameters have been defined and a careful materials design is now possible.3 On the other hand, such kind of correlation is still partially missing on perovskites including diammonium cations. In this context, the most relevant studies have been carried out on some limited compositions including, for example, 2,2′-(ethylenedioxy)bis(ethylammonium) (EDBE), N1-methylethane-1,2-diammonium (N-MEDA), and 3-(2-ammonioethyl)anilinium (AEA) due to the interest in their very broad (white) emission.6–9 In (EDBE)PbBr4, the strong structural distortion induced by the organic spacer closely affects its defectivity leading to the formation of VF centers whose radiative decay ultimately leads to broadened photoluminescence (PL).6 In the other Pb–Br perovskites, where crystal structures were solved by X-ray diffraction (XRD) on single crystals, the broadband emission was as well correlated to self-trapping of photogenerated carriers through excited-state lattice distortions.9 The work of Smith and co-workers puts in prominence, among other structural parameters, the influence, on the relative intensity of the broad emission, of the distortion along the Pb–Br–Pb bond axis.9 These results on diammonium cations further confirm, also for this type of ditopic ligands, the possibility of inducing slight distortions in the inorganic lattice by modifying the nature of the organic cation and therefore finely modulating the optical properties. However, the set of available structural data from single crystal diffraction – and their correlation with optical properties – on systems containing diammonium cations is still limited. In this work we enlarge the family of Pb–Br layered perovskites by a systematic and rationale investigation of the four diammonium cations represented in Fig. 1 (showing the corresponding amines), namely 1,3-phenylenediammonium (1,3-PDA), 1,3-xylylenediammonium (1,3-XDA), 1,4-phenylenediammonium (1,4-PDA), and 1,4-xylylenediammonium (1,4-XDA).
and A′An−1MnX3n+1, respectively, where A′ and A are organic cations and n represents the number of staggered inorganic layers made of metal M and halide X.3 Most of the investigated organic spacers for 2D perovskites, to date, are monoammonium cations, while diammonium cations have been less explored.3 In the latter case, and in particular for short cations, the DJ phases are generally formed, where the inorganic layers are stacked in an eclipsed fashion on top of each other, differently from the RP phases where they are staggered.3 In addition, 2D perovskites are not always stabilized by diammonium cations, and 1D structures, characterized by linear chains of octahedra, have been also often observed. For example, in the series of linear cations of the general formula NH3(CH2)mNH32+ (m = 4, 7, 8, 10, 12), the cations with even carbon-chain lengths form 2D perovskites, while those with odd carbon chain lengths form 1D structures.4 Recently, the structural diversity of systems incorporating a diammonium cation has been further expanded towards 3D halide perovskitoids using linear cations such as 1,4-butanediamine (1,4BDA), N,N-dimethyl-1,3-propanediamine (NMPA), or N,N-dimethylethylenediamine (DMEA), providing a structural motif comprising dimers with edge-sharing octahedra which are then connected through corner-sharing bonds to form a 3D network.5 In these systems, the optical properties are close to traditional 3D MHPs with the bandgap being mainly influenced by standard octahedra distortion parameters. The correlation between structural distortions and optical properties in 2D perovskites is a key topic to modulate their properties and design novel and optimized materials. Thanks to the vast work performed on perovskites including monoammonium cations, well-defined parameters have been defined and a careful materials design is now possible.3 On the other hand, such kind of correlation is still partially missing on perovskites including diammonium cations. In this context, the most relevant studies have been carried out on some limited compositions including, for example, 2,2′-(ethylenedioxy)bis(ethylammonium) (EDBE), N1-methylethane-1,2-diammonium (N-MEDA), and 3-(2-ammonioethyl)anilinium (AEA) due to the interest in their very broad (white) emission.6–9 In (EDBE)PbBr4, the strong structural distortion induced by the organic spacer closely affects its defectivity leading to the formation of VF centers whose radiative decay ultimately leads to broadened photoluminescence (PL).6 In the other Pb–Br perovskites, where crystal structures were solved by X-ray diffraction (XRD) on single crystals, the broadband emission was as well correlated to self-trapping of photogenerated carriers through excited-state lattice distortions.9 The work of Smith and co-workers puts in prominence, among other structural parameters, the influence, on the relative intensity of the broad emission, of the distortion along the Pb–Br–Pb bond axis.9 These results on diammonium cations further confirm, also for this type of ditopic ligands, the possibility of inducing slight distortions in the inorganic lattice by modifying the nature of the organic cation and therefore finely modulating the optical properties. However, the set of available structural data from single crystal diffraction – and their correlation with optical properties – on systems containing diammonium cations is still limited. In this work we enlarge the family of Pb–Br layered perovskites by a systematic and rationale investigation of the four diammonium cations represented in Fig. 1 (showing the corresponding amines), namely 1,3-phenylenediammonium (1,3-PDA), 1,3-xylylenediammonium (1,3-XDA), 1,4-phenylenediammonium (1,4-PDA), and 1,4-xylylenediammonium (1,4-XDA).
 | ||
| Fig. 1 Starting diamines used in the present paper for the synthesis of Pb–Br perovskites. | ||
The first two spacers have been recently investigated in Pb–I and Pb–Br thin films highlighting their improved moisture resistance (in particular, for 1,4-XDA). In absence of single crystal XRD data, it was concluded that the systems based on 1,4-PDA did not form a well-defined layered structure, due to the lack of flexibility of the PDA spacer, as well as its lower level of penetration into the neighboring perovskite layers, which prevents the formation of a layered phase.10 Optical bandgap was reported to increase with the length of the alkyl chain, but a clear correlation with optical properties was hindered by the lack of any structural data.10 Concerning 1,3-PDA and 1,3-XDA, the first cation has been investigated in Pb–I 2D perovskites as a function of the number of inorganic layers, n (providing the crystal structure), showing the formation of 2D DJ perovskites and the expected increase of the bandgap by increasing n.11 These two cations have not yet been investigated in Pb–Br perovskites, and, in particular, 1,3-XDA-based lead halide systems have not been reported in the current literature.
In the present paper, we report the synthesis of lead bromide 2D perovskites (n = 1) including the four diammonium cations mentioned earlier. We grew single crystals, solved the crystal structures, and report a detailed investigation of the optical properties, providing correlations with the bonding details and the interplay between the organic and inorganic components, which is a crucial step for the understanding of structure–property relationships in diammonium-based 2D perovskites.
Results and discussion
A2PbBr4 2D perovskites (A = 1,3-phenylenediammonium (1,3-PDA), 1,3-xylylenediammonium (1,3-XDA), 1,4-phenylenediammonium (1,4-PDA), and 1,4-xylylenediammonium (1,4-XDA), cfr. Fig. 1) have been prepared by a wet-chemistry route as detailed in the Experimental section. Single crystal XRD (SC-XRD) revealed that the compounds crystallize in triclinic and monoclinic space groups, as reported in Table 1.| Sample | Chemical formula | Space group and lattice volume (Å3) | a, b, c (Å) | α, β, γ (°) |
|---|---|---|---|---|
| (1,3-PDA)PbBr4 | (1,3-C6H10N2)PbBr4 | C2/c | 21.784(7) | 90 |
| Monoclinic | 8.821(3) | 107.214(9) | ||
| 1357.8(8) | 7.398(3) | 90 | ||
| (1,3-XDA)2PbBr6 | (1,3-C8H14N2)2PbBr6 | P21/c | 10.7571(18) | 90 |
| Monoclinic | 14.525(3) | 103.540(4) | ||
| 1299.4(4) | 8.5545(10) | 90 | ||
| (1,4-PDA)PbBr4 | (1,4-C6H10N2)PbBr4 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
7.434(3) | 70.570(19) |
| Triclinic | 8.712(3) | 78.527(19) | ||
| 658.9(4) | 11.028(4) | 89.414(19) | ||
| (1,4-XDA)PbBr4 | (1,4-C8H14N2) PbBr4 | P21/c | 12.3274(7) | 90 |
| Monoclinic | 8.1618(4) | 90.817(2) | ||
| 821.68(7) | 8.1675(4) | 90 |
Not all the samples investigated crystallize as a layered perovskite structure, in particular, this does not occur for the composition including the 1,3-XDA cation, giving the chemical formula (1,3-XDA)2PbBr6. Graphical representations of the crystal structures are reported in Fig. 2.
 | ||
| Fig. 2 Crystal structures of (a) (1,3-PDA)PbBr4, (b) (1,3-XDA)2PbBr6, (c) (1,4-PDA)PbBr4, and (d) (1,4-XDA)PbBr4. | ||
As mentioned, (1,3-XDA)2PbBr6 does not possess the perovskite structure, being constituted by a slightly distorted isolated octahedra, corresponding to a 0D perovskitoid. The crystal cohesion is then given by the aromatic cations whose NH3+ groups interact with both equatorial and axial Br atoms. To the best of our knowledge, this is one of the first 0D bromide perovskite derivatives including a diammonium cation, while chloride-containing 0D systems have been reported in the past.12 (1,3-PDA)PbBr4, (1,4-PDA)PbBr4, and (1,4-XDA)PbBr4 occur as DJ phases with n = 1, according to the general formula A′An−1MnX3n+1. They all comprise layers of PbBr6 octahedra separated by layers of organic cations. The aromatic cations interact with the inorganic framework in the terminal mode. The interaction between the NH3+ terminal group and the inorganic framework is known to affect the structure and properties of hybrid halide perovskites.13,14 In the case of diammonium cations, it was already reported that some cations cannot give origin to layered perovskite structures. For example, even-membered alkyl chains with ammonium groups at both ends form layered perovskite-type hybrid structures, both in bromide and iodide compounds whereas odd-membered chains do not.15 The layered perovskite structure is adopted only when the chains are kinked so that the hydrogens on both the ends of the organic molecules can hydrogen bond with the halides of the inorganic layers.15 Moving away from linear chains, it has been shown that cations with fused aromatic rings can give the layered perovskite structure only when the cation is able to tilt and hydrogen bond to the halides.15 More generally, aromatic cations where the ammonium groups may have no degree of freedom do not necessarily give the layered perovskite motif, and 1D motifs are observed for 4,4′-MDAPbI415 [MDA = methylenedianilinium (H3NC6H4CH2C6H4NH3)], (1,4PDA)PbI4,16 whereas the aforementioned (1,3-PDA)PbBr4, (1,4-PDA)PbBr4, and (1,4-XDA)PbBr4, together with (1,3-PDA)PbI4,17 (1,3-PDA)PbCl412 and (AEA)PbBr4 form a layered perovskite structure.18 The XRD powder patterns obtained on the crushed crystals are reported in Fig. 3 superimposed to the calculated patterns from the single crystal XRD data.
 | ||
| Fig. 3 XRD powder patterns of (a) (1,3-PDA)PbBr4, (b) (1,3-XDA)2PbBr6, (c) (1,4-PDA)PbBr4, (d) (1,4-XDA)PbBr4 superimposed to the calculated patterns from SC-XRD (vertical red bars). | ||
We can further explore the interaction between the inorganic framework and the organic cation using the penetration depth, defined as the distance between the N atom of the amino group and the plane of the terminal halides, according to ref. 14. The NH3+ penetration affects both the distortion between the octahedra, in terms of deviation from 180° of the Pb–X–Pb angle, and within the octahedra, in terms of the octahedral elongation length (〈λoct〉), and their bond angle variance (σoct2), as defined by Robison et al. (1971).19 By comparing the present data for the 2D perovskites, and the above mentioned reported systems including diammonium cations, it is possible to note that the Pb–X–Pb angle increases with increasing penetration with a linear trend from chloride to iodide (Fig. S1, ESI†).14 It is worth noting that (1,4-PDA)PbBr4 falls outside this trend, probably because the PDA cation is short and rigid, and because of the increasing strength of the hydrogen bond from I to Br to Cl.20,21 For the present 2D DJ perovskites, the values of the average Pb–Br–Pb angles are 142.95° for (1,3-PDA)PbBr4, 142.12° for (1,4-PDA)PbBr4, and 150.21° for (1,4-XDA)PbBr4, indicating therefore a greater distortion for the two short diammonium cations with respect to 1,4-XDA. The plots correlating the octahedral elongation and bond angle variance of present 2D perovskites, and other DJ systems based on diammonium cations where crystal structure data are available, are reported in Fig. 4a and b.
 | ||
| Fig. 4 (a) Octahedral elongation and (b) bond angle variance as a function of NH3 penetration. PDA= phenylenediammonium; AEA = 3-(2-ammonioethyl)anilinium; XDA= xylylenediammonium; AMC = 1,4-bis(ammoniomethyl)cyclohexane. Values for (AEA)PbBr4 and (1-4,AMC)PbI4 are calculated from ref. 17 and 18, respectively, and their molecular structures are reported in Fig. S1 (ESI†). | ||
It is clear that the octahedral elongation and bond angle variance of this group of compounds decrease as the NH3+ penetration increases. This effect may be a consequence of the flexibility of the organic cation, in particular of the degrees of freedom of the substituents on the aromatic ring. In fact, the 1,3-PDA cation, where all the atoms except the hydrogens are forced to lie in the same plane, induces a larger distortion among all the investigated halides.12,22 Notably, the 1,4-PDA cation induces a distortion within the octahedra similar to the 1,3-PDA cation, whilst the distortion decreases for the 1,4-XDA that is more flexible since the nitrogen atoms are not bound to stay in the same plane as the aromatic ring. The smallest distortion is shown by 1,4-AMC cation, which is the aliphatic analogue of 1,4-XDA reported in ref. 17, suggesting that a conjugate ring provides more rigid structural constraints.
To address the possibility that additional flexibility could be leading to the formation of stronger H⋯Br hydrogen bonds via differences in the electrostatic potential, we carried out a series of density functional theory (DFT) calculations on isolated diammonium cations 1,3-PDA2+, 1,4-PDA2+, and 1,4-XDA2+ in the gas phase; on isolated [PbBr6]4− and [Pb2Br11]7− in the gas phase (retaining distortions present in each parent perovskite); and of the three systems (1,3-PDA)PbBr4, (1,4-PDA)PbBr4, and (1,4-XDA)PbBr4 (see the ESI† for details; all calculations are available electronically).
Focusing on charges calculated for aminic protons and bromide (Fig. S2, ESI†), our single-point energy calculations show that while a systematic charge transfer from Br to –(N)H3+ is expectedly recognized upon perovskite formation by both charge assignment methods, charge distribution remains consistently uniform across atoms of all three isolated diammonium cations, atoms of the [PbBr6]4−/[Pb2Br11]7− octahedra, and perovskites (1,3-PDA)PbBr4, (1,4-PDA)PbBr4, and (1,4-XDA)PbBr4 alike (more details in the ESI†).23,24 Geometry optimization (carried out for all species except [PbBr6]4−/[Pb2Br11]7− octahedra) does not alter the situation: changes observed with respect to single-point energy calculations always remain consistent across all atoms, and charges retain comparably uniform distributions across all three perovskite systems after optimization. Together with the relatively small variation in hydrogen bond lengths observed both experimentally and computationally across (1,3-PDA)PbBr4, (1,4-PDA)PbBr4, and (1,4-XDA)PbBr4, these results lead us to conclude that there should be no systematic differences in electronic distribution between the three perovskites that could drive the formation of stronger or weaker hydrogen⋯halide bonds.
This reasoning is further confirmed by the room temperature Raman spectra for (1,3-PDA)PbBr4, (1,4-PDA)PbBr4, and (1,4-XDA)PbBr4 samples reported in Fig. 5 in the range 70–250 cm−1.
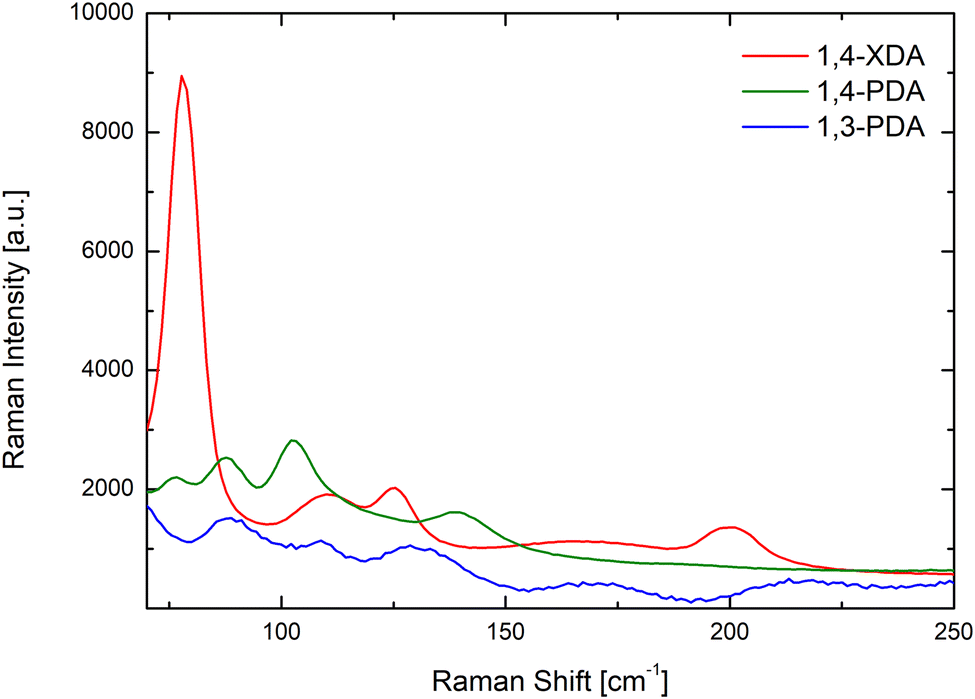 | ||
| Fig. 5 Room temperature Raman spectra of (1,3-PDA)PbBr4 (blue line), (1,4-PDA)PbBr4 (green line) and (1,4-XDA)PbBr4 (red line) excited at 632.8 nm with microscopic resolution. All the spectra are the average over 10 spectra collected along a line of 30 μm. | ||
According to ref. 23, in this energy region, two main vibrating reservoirs should be active: the stretching and bending modes of the Pb–Br cages in the lower energy part and the liberation of the organic cations partially bound to the Pb–Br framework.25 At first, we observe well-defined Raman fingerprints for all the investigated samples thus confirming a high crystal quality. In addition, the data point out that the Raman yields from (1,3-PDA)PbBr4 and (1,4-PDA)PbBr4 are markedly lower with respect to that from (1,4-XDA)PbBr4. This is particularly clear for the mode observed for (1,4-XDA)PbBr4 at 78.5 cm−1 probably associated with the stretching motion of the Pb–Br unit. For (1,3-PDA)PbBr4 and (1,4-PDA)PbBr4, weaker bands are observed at 88 cm−1. This behavior is consistent with the greater distortion affecting the two short diammonium cations with respect to 1,4-XDA. In the region 130–250 cm−1 we observe, in comparison with the octahedral Raman yield, a small Raman activity decreases for 1,4-PDA and 1,4-XDA with respect to 1,3-PDA. Even if a detailed mode attribution is required, this could be due to the smaller NH3+ penetration characterizing (1,3-PDA) PbBr4 sample.
The importance of the flexibility of the organic cation is confirmed by a closer analysis of the octahedral distortion reported in Fig. 6. In this plot, related to the octahedra structural parameters, we could also include those perovskite derivatives not crystallizing as 2D systems. For these systems, the distortion is mostly due to the arrangement of the inorganic framework. In particular, in (1,4-PDA)PbI4, the asymmetric unit contains a Pb atom on a site of 2/m symmetry and two I atoms on mirror planes and thus, by symmetry, the equatorial plane is defined by four equivalent positions for atom I2, whereas the two equivalent positions for atom I1 are necessarily axial.16 The symmetry constraints together with the edge-sharing motif that occurs through the equatorial I atoms give rise to regular octahedra. (1,3-XDA)2PbBr6 comprises isolated and slightly distorted octahedra, characterized by a relatively high angle variance, suggesting a deviation of the Br–Pb–Br angles from 90°. The crystal cohesion is given by a framework of contacts between the amino groups and the bromide atoms. The organic molecule present in the asymmetric unit is tilted by 20° with respect to the (001) plane. Coming to the layered perovskites, the octahedra show large distortion when cations where the nitrogen atoms are bound to sit in the same plane as the aromatic cation, such as the 1,3-PDA and 1,4-PDA, are present. On the other hand, the more flexible 1,4-XDA cation induces only a slight distortion in the PbBr6 octahedra. Analogously with the AEA cation, where one nitrogen atom is bound to the aromatic plane and the other one is not, the octahedra adopts a distortion between those reported for 1,4-XDA and 1,4-PDA.18 Again, based on the present results, the 2D DJ perovskites reported in this work show an octahedral distortion increasing passing from (1,4-XDA)PbBr4 to (1,4-PDA)PbBr4 and to (1,3-PDA)PbBr4, with the last two showing, in particular, a significant increase in the octahedral angle variance with respect to (1,4-XDA)PbBr4.
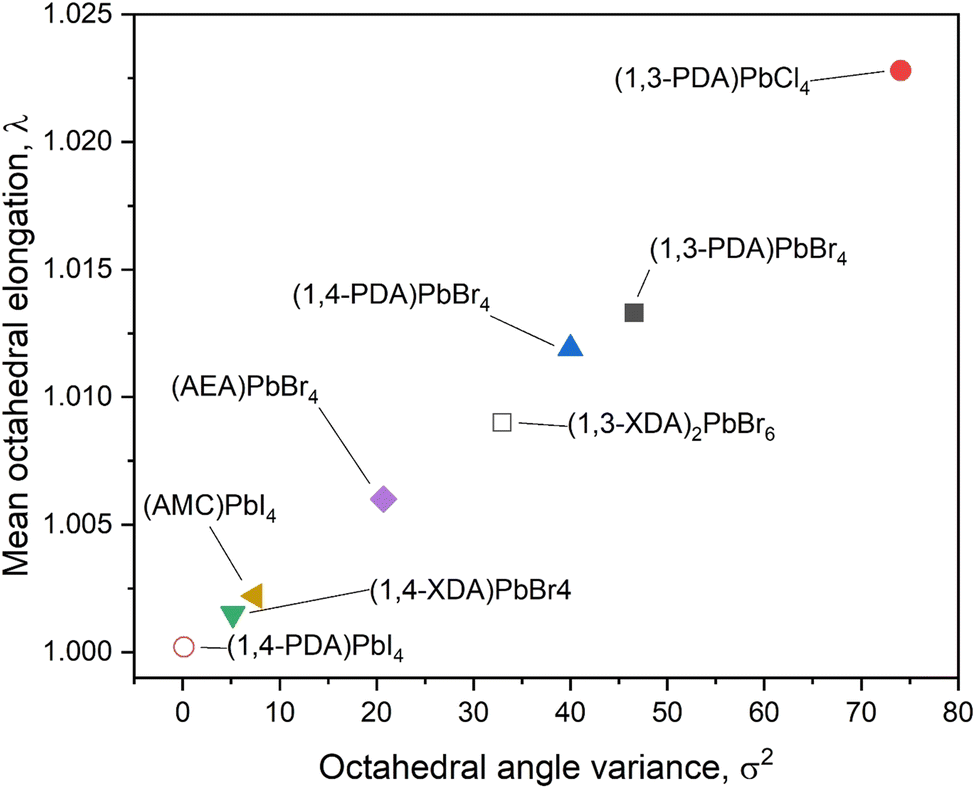 | ||
| Fig. 6 Distortion parameters for the newly synthesized compounds and already reported materials with similar characteristics. Open symbols mark compositions that do not crystallize in a layered perovskite structure. | ||
To further investigate the relationship between the structure of the perovskites, in particular the degree of octahedral distortion, and their optical properties, we performed an in-depth spectroscopic study on the samples which present the 2D DJ structure: (1,3-PDA)PbBr4, (1,4-PDA)PbBr4, and (1,4-XDA)PbBr4.
The materials’ static absorbance and photoluminescence (PL) at room temperature are presented in Fig. 7a. For all samples, the absorbance (solid lines), calculated from the reflectivity of powdered single crystals, shows an absorption edge well close to the UV range (between 380 nm and 420 nm) as expected for two-dimensional Pb bromides.26–28 Furthermore, even at room temperature, a relatively narrow absorption peak can be observed on top of the band edge, consistent with a stable excitonic population, typical of the quantum confinement effect.27
 | ||
| Fig. 7 (a) Absorption (solid lines) and PL (dashed lines) spectra of (1,3-PDA)PbBr4 (blue line), (1,4-PDA)PbBr4 (green line) and (1,4-XDA)PbBr4 (red line) at 297K. (b)–(d) Temperature-dependent photoluminescence spectra of: (b) (1,3-PDA)PbBr4, (c) (1.4-PDA)PbBr4, (d) (1,4-XDA)PbBr4. All spectra are acquired using an excitation wavelength, λex, at 355 nm between 77 and 297 K. | ||
The static PL (dashed lines) measured under pulsed excitation at 355 nm shows a more diverse picture: only (1,4-XDA)PbBr4 shows a narrow peak (≈26 nm bandwidth) centered at 421 nm with a Stokes shift of only a few nm, as expected from excitonic photoluminescence. On the other hand, (1,3-PDA)PbBr4 and (1,4-PDA)PbBr4 show very broad emission peaks centered, respectively, at 650 nm and at 565 nm, with FWHM of hundreds of nm. While the absorption edge of these materials remains in the blue region, their PL shows a very large Stokes shift uncharacteristic of excitonic emission, resulting in bright white or yellow light emission. Such bright and broadband emission is typical of highly distorted 2D materials, where excitonic recombination is mediated by defects or self-trapping resulting in a significant red shift and broadening of the PL peak.7,26
To better understand the nature of the photoluminescence, we measured temperature-dependent PL spectra between 77 and 297 K (Fig. 7b–d). The two samples with a higher (and similar) degree of distortion, (1,3-PDA)PbBr4 and (1,4-PDA)PbBr4, show a similar trend, with a bright and broad feature dominating the emission at all considered temperatures; intriguingly a small narrow feature appears at 420 nm for (1,3-PDA)PbBr4 while cooling down. While hard to identify in static PL measurements due to its comparatively low intensity, this feature is particularly prominent in the first few picoseconds of emission: its spectrum is highlighted in Fig. S3 (ESI†) (inset). At lower temperatures, luminescence efficiency of (1,4-PDA)PbBr4 is boosted. In (1,4-XDA)PbBr4, the PL is still dominated by the narrowband emission at 420 nm, with broadband emission centered at around 575 nm arising as the temperature decreases.
Significant insight into the relationship between light emission and structural parameters can be gleaned from a more careful analysis of the temperature dependence of the PL. First of all, we analyze the temperature dependence of the integrated PL intensity (Fig. 8a), integrating the broadband emission, i.e. the broad emission of (1,3-PDA)PbBr4 (450–1000 nm), (1,4-PDA)PbBr4 (400–900 nm), and (1,4-XDA) PbBr4 (500–1000 nm). For all three materials, the PL intensity decreases from 77 K to room temperature, suggesting the presence of thermally activated non-radiative recombination channels which compete with radiative excitonic recombination. For all three materials, we fit the data with an Arrhenius curve to estimate the activation energy for non-radiative recombination pathways. The obtained activation energies are ΔE1,4-PDA = 95 meV, ΔE1,3-PDA = 39 meV and ΔE1,4-XDA = 71 meV. We note that in the case of (1,4-XDA)PbBr4, the recombination channel might be back transferred to the excitonic state, which indeed is the brightest emissive feature for this material. A higher value of ΔE is desirable for photoluminescence application, indicating a PL more stable at room temperature. Indeed, (1,4-PDA)PbBr4 shows a large ΔE and consistently shows a very promising intense luminescence.29
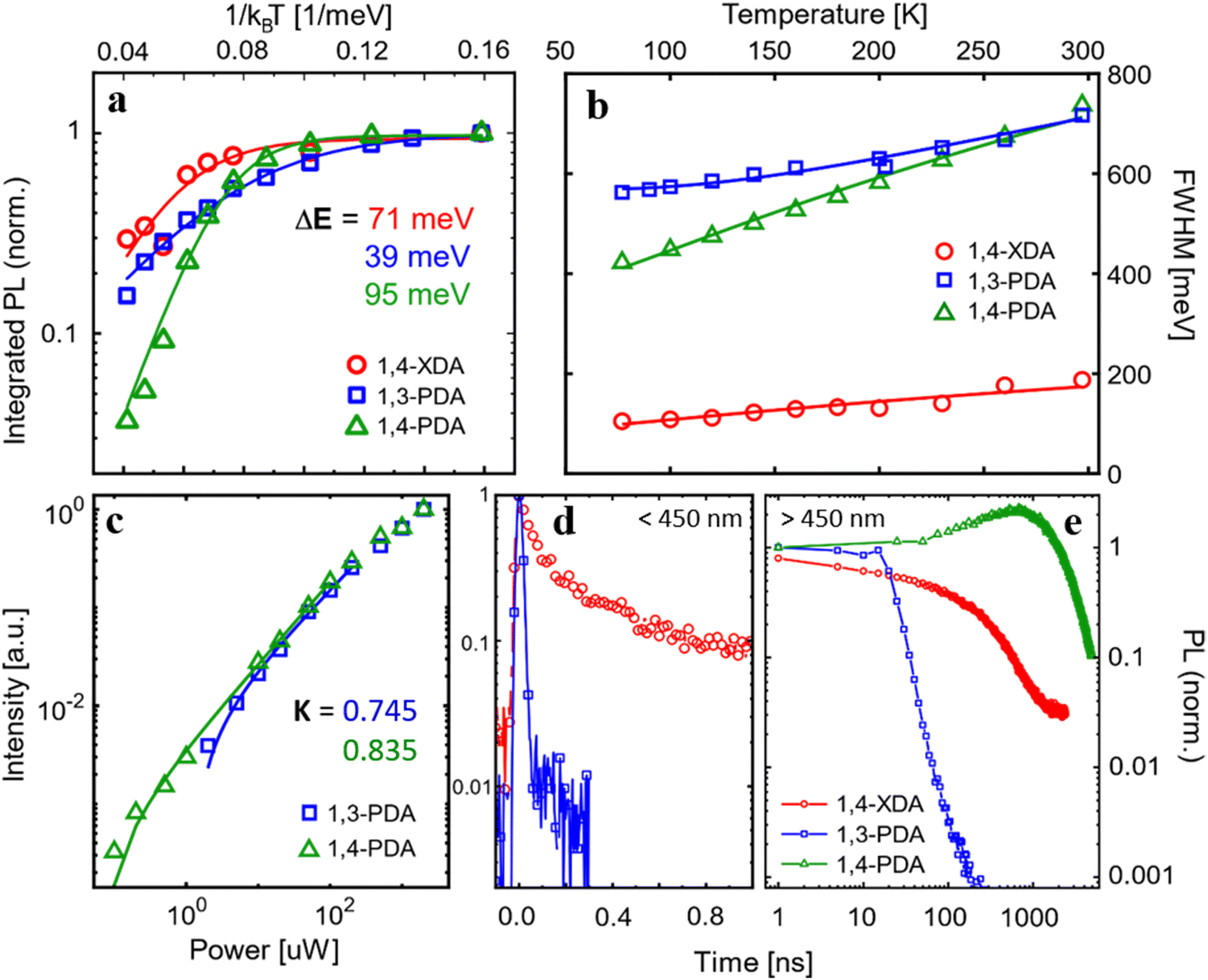 | ||
| Fig. 8 (a) Integrated PL intensity and (b) FWHM as a function of temperature, measured under pulsed excitation (1 kHz) at λex =355 nm. (c) Fluence dependence of the integrated PL intensity at 77 K fitted with a power law (d and e) Time-resolved photoluminescence measured for the (d) narrow band components (<450 nm) and (e) broad emission (>500 nm). The fast decays of excitonic features are measured with ps resolution up to 1 ns, while the long-lived broad emission is acquired up to a window of 10 μs. | ||
We then consider the variation of the luminescence bandwidth, estimated by fitting the main emission component (at 650 nm and 550 nm) for (1,3-PDA)PbBr4 and (1,4-PDA)PbBr4, respectively, and at 420 nm for (1,4-XDA)PbBr4 with a Gaussian and taking its FWHM (Fig. 8b). The broad luminescence of (1,4-XDA)PbBr4 is not considered in this analysis as its superimposition to the narrow component makes it hard to isolate correctly, especially at higher temperatures.
The PL FWHM can be used to evaluate the coupling of the carriers – excitons in this case – with phonons by fitting the FWHM trend vs. temperature with eqn (1), thus obtaining the Huang-Rhys factor and the energy of the involved phonons.30–32
 | (1) |
Further indication of the nature of the broad emission comes from the pump fluence dependence of PL (Fig. S4, ESI†). Fitting the intensity of emission in the function of the incident power with a power law I = PK, we estimate exponential K (Fig. 8c): the values K1,3-PDA = 0.745 and K1,4-PDA = 0.835, lower than 1, indicate a recombination pathway mediated by defects leading to the formation of self-trapped excitons.8,26,35 A purely excitonic recombination on the other hand is expected to yield a power dependence with K = 1. However, it is not possible to compare this power law with the narrow emission of (1,4-XDA)PbBr4: its small Stokes shift results in significant reabsorption in powdered single crystal samples, distorting the measured spectrum. Furthermore, while (1,3-PDA)PbBr4 and (1,4-PDA)PbBr4 have remarkably photostable emissions, (1,4-XDA)PbBr4 is unstable under prolonged intense illumination, even on a scale of a few seconds (Fig. S5, ESI†). The excitonic component decreases faster than the broad emission, making a fluence dependence measurement unreliable.
To validate the idea of different origins for the emission of the samples, we performed time-resolved photoluminescence (trPL) measurement at 77 K. To better show the disparate dynamics of the narrow- and broadband PL features, we measured trPL in different time ranges: high resolution decays in a time range <1 ns (Fig. 8d) where the excitonic decay is fully resolved are coupled with measurements up to 10 μs (Fig. 8e) to fully record the decay of the broadband components. The detailed setup of both high resolution and long temporal window measurements are found in the ESI,† together with the complete trPL maps in both regimes (Fig. S6, ESI†). In sample (1,4-XDA)PbBr4, the difference between the excitonic and broad emission is stark: while the excitonic component at 410 nm decays in τ = 76 ± 2 ps, in line with the expected excitonic lifetimes at 77 K, the decay rate of the broad emission is 3 orders of magnitude slower (τ = 180 ns) which is compatible with a trap- or phonon-mediated process, with a consequent redshift of the PL emission.
Sample (1,3-PDA)PbBr4 also shows a long-lived lifetime for its broad emission at 650 nm (τ = 8.5 ns). Interestingly, in the high-resolution time-resolved measurement, we also observe a narrowband component centered at 420 nm (FWHM = 20 nm), which has a very low intensity in the static PL measurements. This is related to its extremely short lifetime, which is shorter than the response function of instruments indicating τ < 3.5 ns: such rapid PL decay, coupled with the very low total intensity of this narrow band emission, points to emission from free exciton recombination that is swiftly quenched by transfer to states with red-shifted emission. The lifetime of both narrow and broad emission of (1,3-PDA)PbBr4 is almost an order of magnitude faster than the one of the samples (1,4-XDA)PbBr4: this is an indication, supported also by the brighter emission and higher activation energy for non-radiative pathways of the latter, that fast non-radiative recombination is the dominant mechanism in (1,3-PDA)PbBr4.
Lastly, the sample (1,4-PDA)PbBr4, as in the static measures, shows only a broadband emission which presents a decay characterized by a late rise in the intensity (about 1000 ns) followed by a long decay over tens of μs. This could be an indication of a complete and efficient energy transfer from the higher energy-free exciton to a red-shifted self-trapped exciton with an efficient radiative recombination mechanism, which prevents any radiative excitonic recombination to be observed even on fast timescales. Consistently, this sample has the highest ΔE (ΔE1,4-PDA = 95 meV) between the analyzed samples leading to photoluminescence dominated by radiative recombination of carriers both at room temperature and cryogenic temperature. The late rise in PL intensity suggests a back transfer mechanism from the non-radiative recombination sites to the radiative one, resulting in delayed luminescence; this feature is not present in (1,3-PDA)PbBr4 decay which emission is, in fact, dominated by non-radiative pathways, from which a back transfer to the radiative state is hindered.
As we have shown with both static and time-resolved photoluminescence spectroscopy at 77 K, the three materials which form 2D Dion–Jacobson phases all exhibit a broadband emissive component with hundreds of nanometers of bandwidth and a significant shift (100 to 150 nm) from the absorption edge. Such broad emission, as indicated by the analysis of the power and temperature dependence of the PL, can be attributed to trap-mediated excitonic recombination or STEs. In the case of (1,4-XDA)PbBr4, the strongest emission is not the broadband component, but rather a narrowband, short-lived component characterized by a small Stokes shift, which is consistent with the radiative recombination of free excitons; at room temperature, this narrow band feature at 420 nm is the only significant emission.
The different luminescence of the three samples can be rationalized by considering their structural characteristics. The two materials, (1,3-PDA)PbBr4 and (1,4-PDA)PbBr4, with larger values of σ2 and λoct (Fig. 6), show a dominant role in the broadband emission, as expected in materials with significant deviations from ideal octahedral geometry.7 On the other hand, (1,4-XDA)PbBr4, which has relatively undistorted octahedra, but shows significant tilting of the Pb–Br–Pb angle (<150°), exhibits a mixture of narrow and broad band emission, with only the free exciton component persisting at room temperature.8 However, it is interesting to note that the respective degrees of inter- and intra-octahedral distortion are not sufficient to fully explain the luminescence properties we have observed. In particular, (1,3-PDA)PbBr4 has a highly distorted geometry, very similar to (1,4-PDA)PbBr4 in terms of the most commonly used short-hands for octahedral distortion: their values of λoct, σ2 and Dtilt are comparable. However, (1,3-PDA)PbBr4 also shows a small narrowband component at 420 nm, which is quickly transferred (<3 ps) to the broadband feature, but not before radiative-free exciton recombination can occur. No such feature is observed for (1,4-PDA)PbBr4: this behavior may indicate a much more effective transfer to the broadband emissive states which completely overtakes radiative recombination.
It should also be noted that (1,3-PDA)PbBr4 is significantly less emissive than its 1,4 counterpart, pointing to a larger contribution from traps acting as non-radiative recombination centers.
Conclusions
In this study we extended the family of DJ halide perovskite containing diammonium cations, by preparing, solving the structure, and characterizing novel lead bromide materials containing 1,3-phenylenediammonium (1,3-PDA), 1,3-xylylenediammonium (1,3-XDA), 1,4-phenylenediammonium (1,4-PDA), and 1,4-xylylenediammonium (1,4-XDA) cations. We showed that the choice of the organic cation has a great influence on the topology of the structure, determining whether the layered perovskite structure can occur or not. Consistently, the degree of freedom of the organic cation deeply affects the interactions with the inorganic framework, influencing octahedral distortions and thus the optical properties of the compounds. Thanks to detailed structural and optical characterizations, we could link the differences in the optical properties, such as the nature of luminescence, to the structural parameters. In fact, samples with a high degree of distortion (containing the 1,3-PDA and 1,4-PDA cations), i.e. with larger values of σ2 and λoct, show a dominant role in the broadband emission, while the presence of relatively undistorted octahedra when 1,4-XDA cation is the spacer leads to a mixture of narrow and broadband emission. However, the difference in optical properties, and in particular in the intensity of the emission, cannot be completely explained based on common and traditionally used parameters of the distortion in the octahedral layers. This suggests that further investigation into the relationship between structural properties and broadband luminescence in low-dimensional perovskites containing diammonium cations is called for to identify precise design rules allowing development of efficient perovskite-based broadband and white light emitters.Conflicts of interest
The authors declare no competing interests.Acknowledgements
Both M. M. and L. M. thank the CRIST “Centro di Cristallografia Strutturale” (University of Florence) for support in the data collection. S. A. S. wishes to thank Dr Franziska Simone Hegner (Technical University of Munich) for her kind instructions on running DFT calculations for this work.References
- A. Krishna, S. Gottis, M. K. Nazeeruddin and F. Sauvage, Adv. Funct. Mater., 2019, 29, 1806482 CrossRef.
- S. Ghimire and C. Klinke, Nanoscale, 2021, 13, 12394–12422 RSC.
- X. Li, J. M. Hoffman and M. G. Kanatzidis, Chem. Rev., 2021, 121, 2230–2291 CrossRef CAS PubMed.
- A. Lemmerer and D. G. Billing, Dalton Trans., 2012, 41, 1146–1157 RSC.
- X. Li, M. Kepenekian, L. Li, H. Dong, C. C. Stoumpos, R. Seshadri, C. Katan, P. Guo, J. Even and M. G. Kanatzidis, J. Am. Chem. Soc., 2022 DOI:10.1021/jacs.1c11803.
- D. Cortecchia, S. Neutzner, A. R. Srimath Kandada, E. Mosconi, D. Meggiolaro, F. De Angelis, C. Soci and A. Petrozza, J. Am. Chem. Soc., 2017, 139, 39–42 CrossRef CAS PubMed.
- D. Cortecchia, J. Yin, A. Petrozza and C. Soci, J. Mater. Chem. C, 2019, 7, 4956–4969 RSC.
- M. D. Smith and H. I. Karunadasa, Acc. Chem. Res., 2018, 51, 619–627 CrossRef CAS PubMed.
- M. D. Smith, A. Jaffe, E. R. Dohner, A. M. Lindenberg and H. I. Karunadasa, Chem. Sci., 2017, 8, 4497–4504 RSC.
- A. Dučinskas, G. C. Fish, M. A. Hope, L. Merten, D. Moia, A. Hinderhofer, L. C. Carbone, J.-E. Moser, F. Schreiber, J. Maier, J. V. Milić and M. Grätzel, J. Phys. Chem. Lett., 2021, 12, 10325–10332 CrossRef PubMed.
- L. Gao, X. Li, B. Traoré, Y. Zhang, J. Fang, Y. Han, J. Even, C. Katan, K. Zhao, S. Liu and M. G. Kanatzidis, J. Am. Chem. Soc., 2021, 143, 12063–12073 CrossRef CAS PubMed.
- L. Dobrzycki and K. Woźniak, CrystEngComm, 2008, 10, 577–589 RSC.
- X. Li, J. M. Hoffman and M. G. Kanatzidis, Chem. Rev., 2021, 121, 2230–2291 CrossRef CAS PubMed.
- K. Du, Q. Tu, X. Zhang, Q. Han, J. Liu, S. Zauscher and D. B. Mitzi, Inorg. Chem., 2017, 56, 9291–9302 CrossRef CAS PubMed.
- A. Lemmerer and D. G. Billing, CrystEngComm, 2012, 14, 1954–1966 RSC.
- A. Lemmerer and D. G. Billing, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, m597–m601 CrossRef CAS PubMed.
- M. K. Rayner and D. G. Billing, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, m660 CrossRef CAS PubMed.
- M. D. Smith, A. Jaffe, E. R. Dohner, A. M. Lindenberg and H. I. Karunadasa, Chem. Sci., 2017, 8, 4497–4504 RSC.
- K. Robinson, G. V. Gibbs and P. H. Ribbe, Science, 1971, 172, 567–570 CrossRef CAS PubMed.
- T. Steiner, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1998, 54, 456–463 CrossRef.
- L. Brammer, E. A. Bruton and P. Sherwood, Cryst. Growth Des., 2001, 1, 277–290 CrossRef CAS.
- L. Gao, X. Li, B. Traoré, Y. Zhang, J. Fang, Y. Han, J. Even, C. Katan, K. Zhao, S. Liu and M. G. Kanatzidis, J. Am. Chem. Soc., 2021, 143, 12063–12073 CrossRef CAS PubMed.
- A. V. Marenich, S. V. Jerome, C. J. Cramer and D. G. Truhlar, J. Chem. Theory Comput., 2012, 8, 527–541 CrossRef CAS PubMed.
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 1833–1840 CrossRef CAS.
- S. Krishnamurthy, R. Naphade, W. J. Mir, S. Gosavi, S. Chakraborty, R. Vaidhyanathan and S. Ogale, Adv. Opt. Mater., 2018, 6, 1800751 CrossRef.
- B. Febriansyah, T. Borzda, D. Cortecchia, S. Neutzner, G. Folpini, T. M. Koh, Y. Li, N. Mathews, A. Petrozza and J. England, Angew. Chem., Int. Ed., 2020, 59, 10791–10796 CrossRef CAS PubMed.
- X. Li, X. Lian, J. Pang, B. Luo, Y. Xiao, M.-D. Li, X.-C. Huang and J. Z. Zhang, J. Phys. Chem. Lett., 2020, 11, 8157–8163 CrossRef CAS PubMed.
- B. Febriansyah, D. Giovanni, S. Ramesh, T. M. Koh, Y. Li, T. C. Sum, N. Mathews and J. England, J. Mater. Chem. C, 2020, 8, 889–893 RSC.
- H. Peng, Y. Tian, X. Wang, T. Huang, Z. Yu, Y. Zhao, T. Dong, J. Wang and B. Zou, ACS Appl. Mater. Interfaces, 2022, 14, 12395–12403 CrossRef CAS PubMed.
- W. Stadler, D. M. Hofmann, H. C. Alt, T. Muschik, B. K. Meyer, E. Weigel, G. Müller-Vogt, M. Salk, E. Rupp and K. W. Benz, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 10619–10630 CrossRef CAS PubMed.
- R. Zeng, L. Zhang, Y. Xue, B. Ke, Z. Zhao, D. Huang, Q. Wei, W. Zhou and B. Zou, J. Phys. Chem. Lett., 2020, 11, 2053–2061 CrossRef CAS PubMed.
- K. M. McCall, C. C. Stoumpos, S. S. Kostina, M. G. Kanatzidis and B. W. Wessels, Chem. Mater., 2017, 29, 4129–4145 CrossRef CAS.
- J. Luo, X. Wang, S. Li, J. Liu, Y. Guo, G. Niu, L. Yao, Y. Fu, L. Gao, Q. Dong, C. Zhao, M. Leng, F. Ma, W. Liang, L. Wang, S. Jin, J. Han, L. Zhang, J. Etheridge, J. Wang, Y. Yan, E. H. Sargent and J. Tang, Nature, 2018, 563, 541–545 CrossRef CAS PubMed.
- H. Peng, S. Yao, Y. Guo, R. Zhi, X. Wang, F. Ge, Y. Tian, J. Wang and B. Zou, J. Phys. Chem. Lett., 2020, 11, 4703–4710 CrossRef CAS PubMed.
- T. Schmidt, K. Lischka and W. Zulehner, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 8989–8994 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and Methods (including details on DFT calculations), additional figures of photoluminescence measurements, distortion parameters and atomic charge assignments (PDF). Input and output of DFT calculations with Gaussian16 (DFT). CCDC 2174149–2174152. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2tc02113g |
| This journal is © The Royal Society of Chemistry 2022 |