
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Acridone and quinacridone derivatives with carbazole or phenoxazine substituents: synthesis, electrochemistry, photophysics and application as TADF electroluminophores†
Irena
Kulszewicz-Bajer
 *a,
Matas
Guzauskas
b,
Małgorzata
Makowska-Janusik
c,
Małgorzata
Zagórska
a,
Malek
Mahmoudi
b,
Juozas V.
Grazulevicius
b,
Adam
Proń
*a and
Dmytro
Volyniuk
*b
*a,
Matas
Guzauskas
b,
Małgorzata
Makowska-Janusik
c,
Małgorzata
Zagórska
a,
Malek
Mahmoudi
b,
Juozas V.
Grazulevicius
b,
Adam
Proń
*a and
Dmytro
Volyniuk
*b
aFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: ikulsz@ch.pw.edu.pl
bDepartment of Polymer Chemistry and Technology, Kaunas University of Technology, Barsausko 59, LT-51423, Kaunas, Lithuania. E-mail: dmytro.volyniuk@ktu.lt
cFaculty of Science and Technology, Jan Dlugosz University, Al. Armii Krajowej 13/15, 42-200 Częstochowa, Poland
First published on 8th August 2022
Abstract
Six acridone (quinacridone) derivatives containing either carbazole or phenoxazine substituents were designed and synthesized with the aim of elucidating the effect of the donor (D) and acceptor (A) linking pattern (D–A, D–A–D or D–π–A–π–D) on their photophysical and electrochemical properties. These new electroactive compounds combine reversible electrochemical oxidation with excellent luminescent properties. Their electrochemically determined ionization potentials (IPs) are in the range from 5.09 eV to 5.45 eV, higher for derivatives with carbazole donors as compared to phenoxazine ones. The measured electron affinities (EAs) are in the range from −2.53 eV to −2.64 eV with the exception of the quinacridone derivative showing EA of −3.03 eV. Their vacuum-deposited films emit radiation in a wide spectral range from sky-blue to red. Compounds with carbazole moieties (compounds 1, 2 and 6 in the subsequent text) showed prompt fluorescence and aggregation-caused quenching. Photoluminescent quantum yields (PLQYs) of their toluene solutions reached values up to 69%. Compounds containing phenoxazine moieties (compounds marked as 3–5) demonstrated thermally activated delayed fluorescence (TADF) and aggregation-induced emission enhancement (AIEE). Their neat films showed PLQYs of 35%. Quinacridone disubstituted with carbazole (compound 6) showed the highest hole mobility reaching 2.53 × 10−3 cm2 V−1 s−1 at electric field of 3.6 × 105 V cm−1. Carbazolyl disubstituted acridone (compound 2) and phenoxazinyl monosubstituted acridone (compound 3) turned out to be ambipolar compounds showing reasonably balanced electron and hole mobilities. The appropriate combination of redox, transport and luminescent properties makes the studied compounds suitable candidates for optoelectronic applications. Test OLEDs fabricated from 3 exhibited maximum external quantum efficiencies reaching 16.7%. Finally, an excellent agreement between the experimental results and those obtained by DFT calculations should be stressed. The basics for selection according to the user needs of either D–A, D–A–D or D–π–A–π–D types of molecular structures of TADF/AIEE luminophores are provided in this study.
1. Introduction
Acridone and quinacridone have been known and studied for decades. Acridone has been used as a building block in the synthesis of a variety of compounds known for their pharmacological activity.1–4 Quinacridone, in turn, is an industrial pigment.5 However, strong revival in the chemistry of these two compounds has been observed in the past decade. A plethora of low and high molecular mass compounds containing acridone or quinacridone units were synthesized, exhibiting peculiar electronic, optoelectronic or electrochromic properties, frequently unmatched by other groups of electroactive organic compounds. Acridone- or quinacridone-based donor–acceptor copolymers and terpolymers were used as components of photovoltaic cells6–9 and photodetectors.10,11 D–A compounds with acridone or quinacridone acceptors were examined as components of active layers in organic field effect transistors (OFETs) due to their excellent electrical transport properties.12 Several acridone and quinacridone derivatives were found to exhibit interesting electrochemistry13–15 including electrochromism.16 Appropriately functionalized quinacridones were also tested as photocatalysts17,18 and materials for non-linear optical applications.19 However, the capability of generating the thermally activated delayed photoluminescence (TADF), reported for some acridone and quinacridone derivatives, seems to be the most interesting feature of these compounds,20–22 leading to their applications as components of organic light emitting diodes (OLEDs), characterized by high external quantum efficiencies.23,24Phenoxazine units are, in turn, widely used as building blocks in various organic electroactive materials such as components of dye-sensitized solar cells25 or hole transporting materials in perovskite solar cells.26 However, as in the case of acridone and quinacridone derivatives, their most interesting property is the TADF effect, frequently observed in molecules containing phenoxazine donors.27–32 Similarly carbazole is a popular donor in organic molecules and macromolecules for photovoltaics33,34 and electroluminescence.35–37 Thus, phenoxazine or carbazole units in combination with acridone (quinacridone) seem especially interesting as building blocks of various types luminescent and electroactive molecules. For this reason, we undertook a combined theoretical and experimental study devoted to the design, synthesis and characterization of five acridone and one quinacridone donor–acceptor derivatives with two types of donors, namely phenoxazine and carbazole of possible use in optoelectronics. Quantum chemical calculations are especially important in this respect because they play a predictive role, demonstrating that appropriate combination of donors (carbazole, phenoxazine) and acceptors (acridone, quinacridone) may result in the elaboration of new electroactive and luminescent compounds of controllable redox properties, i.e. characterized by appropriate ionization potential (IP) and electron affinity (EA). These parameters together with electrical transport properties are of crucial importance for any electronic or optoelectronic application. Thus, experimental investigations involved detailed electrochemical characterization of the synthesized compounds with the goal to confront theoretical IP and EA values with the experimental ones as well as to elucidate their photophysical properties. The studied compounds showed good charge injecting properties with relatively low ionization potentials and high electron affinities and efficient charge transport (hole-only or bipolar). The latter unequivocally indicated that all studied luminophores exhibited either thermally activated delayed fluorescence (TADF) (so-called E-type delayed fluorescence) or aggregation-induced emission enhancement (AIEE). The emission was very sensitive to molecular structure and media. Emission colour of their vacuum-deposited films could be tuned over a wide spectral range from sky-blue to red. They have the potential for application in non-doped and doped OLEDs. 2-Phenoxazine-N-hexylacridone (indicated as compound 3 in the subsequent text) deserves a special emphasis since test light emitted diodes (OLEDs) with active layers consisting of 3 molecularly dispersed in 1,3-bis(N-carbazolyl)benzene (mCP) or 2,4,6-tris[3-(diphenylphosphinyl)phenyl]-1,3,5-triazines (PO-T2T) exhibited maximum EQEs of 13 and 16.7%, respectively. The study provides basics for the selection of either D–A, D–A–D or D–π–A–π–D types of TADF molecules according to the user needs.
2. Experimental section
Detailed description of the synthesis of new luminophores and techniques used for their characterization can be found in the ESI.† This involves:• Description of all reagents and procedures used in the preparation of the studied compounds together with spectroscopic (NMR, IR) data of intermediate and final products as well as their elemental analyses;
• Description of cyclic voltammetry (CV) experiments;
• Optical measurements, charge carriers mobility, OLEDs fabrication and characterization.
• Methodology of quantum chemical calculations.
3. Results and discussion
3.1. Synthesis
The synthetic pathways leading to the studied compounds, namely 2-(3,6-tert-butylcarbazole)-N-hexylacridone, 1; 2,7-bis(3,6-tert-butylcarbazole)-N-hexylacridone, 2; 2-phenoxazine-N-hexylacridone, 3; 2,7-bis-phenoxazine-N-hexylacridone, 4; 2,7-bis[3-((phenoxazine)phenyl)-N-hexylacridone], 5; 2,9-bis(3,6-tert-butylcarbazole)-N,N′-dioctylquinacridone, 6 is presented in Scheme 1.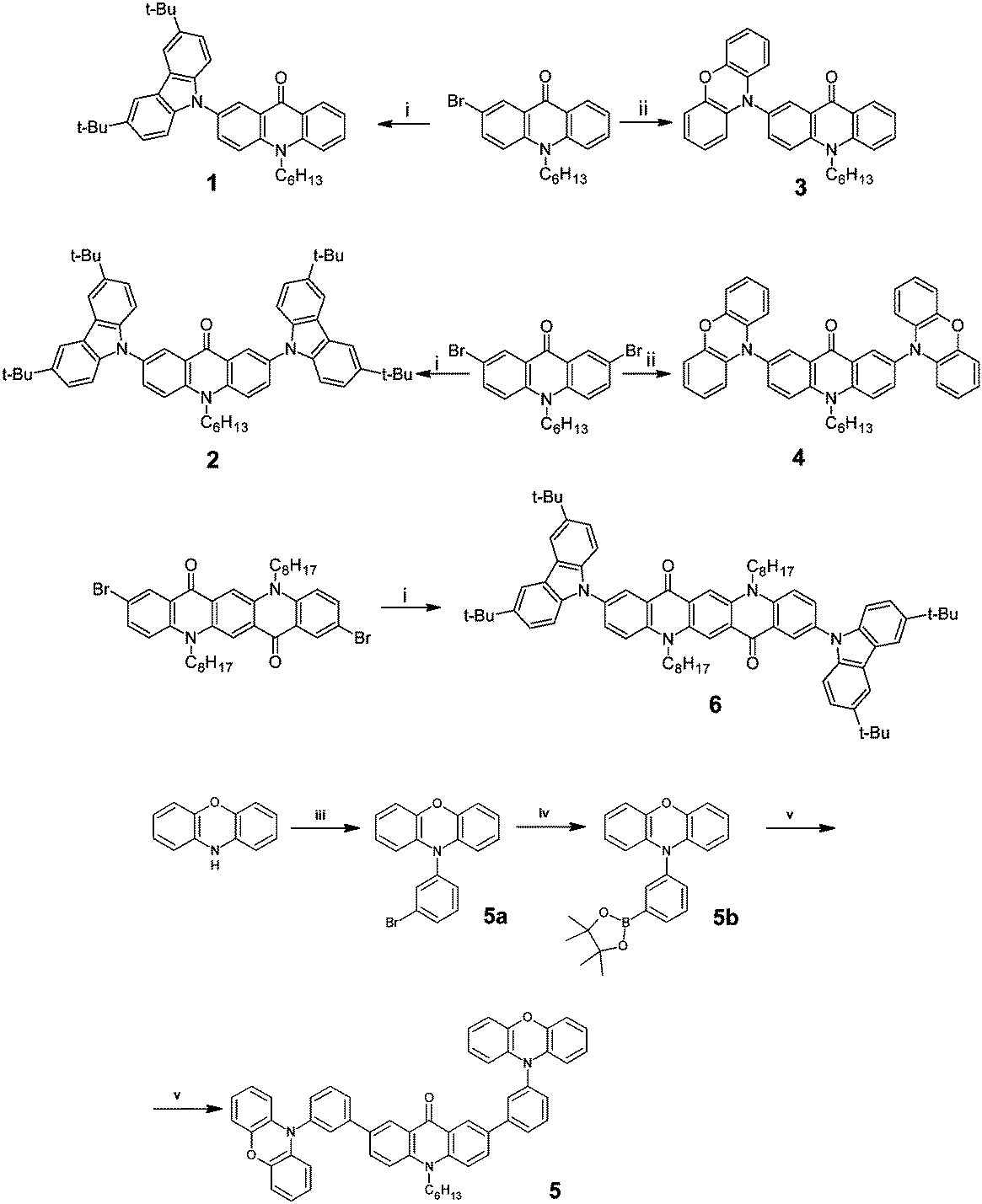 | ||
| Scheme 1 Synthesis of acridone (1–4) and quinacridone (6) derivatives: (i) 3,6-di-tert-butylcarbazole, CuI, trans-1,2-cyclohexanediamine, K3PO4, dioxane, 110 °C; (ii) phenoxazine, Pd(dba)2, t-Bu3P, t-BuONa, toluene, 90 °C. Synthetic pathway to 5: (iii) 1-bromo-3-iodobenzene, Pd2(dba)3, Xantphos, t-BuONa, toluene, 80 °C; (iv) n-BuLi, 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane, THF, −78 °C; (v) 2,7-dibromo-N-hexylacridone, Pd(PPh3)4, 2 M K2CO3, toluene, dioxane. | ||
In the applied procedures N-hexylacridone or N,N-dioctylquinacridone were brominated using bromine in acetic acid/sodium acetate medium as a bromination agent. The obtained bromoderivatives were then reacted with 3,6-di-tert-butylcarbazole in the presence of CuI/trans-1,2-cyclohexanediamine catalyst leading to: (i) compound 1 with (97% yield); (ii) compound 2 (64% yield) and (iii) compound 6 (61% yield). It should be noticed that the use of this catalyst results in a significant decrease of the reaction temperature with concomitant increase of the reaction yield as compared to commonly used procedures.23,38,39 Compounds 3, 4 were synthesized via Buchwald–Hartwig amination coupling of acridone bromoderivatives and phenoxazine using Pddba2/t-Bu3P as a catalyst. The target products were obtained in high yields: 92% in the case of 3 and 82% in the case of 4. Compound 5 was synthesized in a three-step procedure.
In the first step 5a was prepared from phenoxazine and 1-bromo-3-iodobenzene (95% yield), then, in the second step it was transformed into a Suzuki coupling reagent, namely 3-phenoxazine-1-phenylboronic acid pinacol ester 5b (78% yield). The target product 5 was obtained by Suzuki coupling between 5b and 2,7-dibromo-N-hexylacridone (67% yield).
3 and 4 were reported in ref. 40 whereas 1, 2, 5 and 6 are new. Detailed descriptions of all synthetic procedures together with full spectroscopic characterization of the products as well as their elemental analyses can be found in ESI.†
3.2. Redox properties, ionization potentials (IPs) and electron affinities (EAs)
Redox properties of electroactive molecules are of crucial importance because on their basis electron affinities (EAs) and ionization potentials (IPs) can be determined which are predictive parameters for essentially all electronic and optoelectronic applications.In Fig. 1a, a representative cyclic voltammogram (CV) of acridone monosubstituted with carbazole, 1, is presented. The corresponding differential pulse voltammogram (DPV) are given in ESI† (Fig. S1). In general, carbazole moieties undergo reversible oxidation to a radical cation. This can be followed by consecutive irreversible oxidation processes possibly involving oligomerization or polymerization if appropriate coupling positions in the carbazole aromatic rings are available.41 Cyclic voltammogram of 1 reveals however two oxidation processes of reversible and quasi-reversible nature at 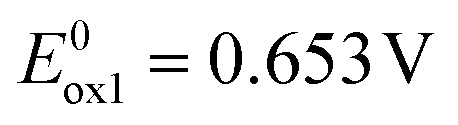 and
and  which essentially excludes dimerization or oligomerization. This is not unexpected since the most probable coupling sites in 1 are blocked by bulky tert-butyl groups. Thus, the first peak can be attributed to the oxidation of the carbazole substituent to a radical cation,38 whereas the second one to the oxidation of the acridone unit. Unsubstituted acridone undergoes quasi-reversible oxidation at 1.020 V.40 Its shift in 1 to a potential which is higher by 0.112 V is a consequence of the fact that the removal of an additional electron from 1 in its radical cation form is more difficult than the abstraction of the first electron from neutral acridone. The reduction of 1 is essentially irreversible and occurs at relatively low potentials, with a maximum of the cathodic peak at −2.399 V vs. Fc/Fc+.
which essentially excludes dimerization or oligomerization. This is not unexpected since the most probable coupling sites in 1 are blocked by bulky tert-butyl groups. Thus, the first peak can be attributed to the oxidation of the carbazole substituent to a radical cation,38 whereas the second one to the oxidation of the acridone unit. Unsubstituted acridone undergoes quasi-reversible oxidation at 1.020 V.40 Its shift in 1 to a potential which is higher by 0.112 V is a consequence of the fact that the removal of an additional electron from 1 in its radical cation form is more difficult than the abstraction of the first electron from neutral acridone. The reduction of 1 is essentially irreversible and occurs at relatively low potentials, with a maximum of the cathodic peak at −2.399 V vs. Fc/Fc+.
 | ||
| Fig. 1 Cyclic voltammograms registered for 1 (a), 2 (b), 5 (c) and 6 (d). Electrolyte: 0.1 M Bu4NBF4/CH2Cl2, scan rate: 50 mV s−1. Photoelectron emission spectra of vacuum-deposited thin layers of 1–6 (e). | ||
2 contains two carbazole substituents symmetrically connected to the acridone central unit, thus it can be considered as a D–A–D molecule. Consecutive oxidations of two carbazole groups occur at potentials 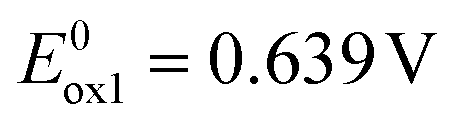 and
and 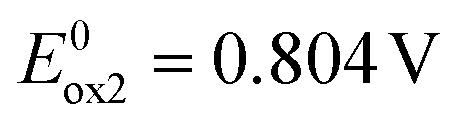 (Fig. 1b). Note that the first oxidation potential is very similar to that registered for the oxidation of 1. An increase of the oxidation potential of the second carbazole unit may indicate some delocalization of the radical cation charge through the whole molecule making the abstraction of the second electron more difficult. Further oxidation of this dication requires polarization to relatively high potentials. As a result the central acridone unit is being irreversibly oxidized as evidenced by an anodic peak with a maximum at E = 1.297 V i.e. at a potential significantly higher than those of the oxidation of neutral acridone and the oxidation of the radical cation form of 1. Irreversible reduction of 2 starts at a low potential of −2.22 V, yielding a cathodic peak without a clear maximum. 2, i.e. 2,7-bis(3,6-tert-butylcarbazole)-N-hexylacridone is slightly easier to oxidize than its unsubstituted analogue (2,7-di(carbazole-9-yl)-N-hexylacridone), whose redox properties are reported in ref. 42. It can be rationalized by weak electrodonating properties of tert-butyl substituents in this compound. 2 is however more resistant to oxidation than diphenylamino- and dinaphthylamino-substituted alkylacridones, whose oxidation starts at potentials lower by 0.42 V.42 Higher resistance against oxidation of acridones with carbazole substituents makes these compounds more suitable for applications in electronic devices operating in ambient conditions.
(Fig. 1b). Note that the first oxidation potential is very similar to that registered for the oxidation of 1. An increase of the oxidation potential of the second carbazole unit may indicate some delocalization of the radical cation charge through the whole molecule making the abstraction of the second electron more difficult. Further oxidation of this dication requires polarization to relatively high potentials. As a result the central acridone unit is being irreversibly oxidized as evidenced by an anodic peak with a maximum at E = 1.297 V i.e. at a potential significantly higher than those of the oxidation of neutral acridone and the oxidation of the radical cation form of 1. Irreversible reduction of 2 starts at a low potential of −2.22 V, yielding a cathodic peak without a clear maximum. 2, i.e. 2,7-bis(3,6-tert-butylcarbazole)-N-hexylacridone is slightly easier to oxidize than its unsubstituted analogue (2,7-di(carbazole-9-yl)-N-hexylacridone), whose redox properties are reported in ref. 42. It can be rationalized by weak electrodonating properties of tert-butyl substituents in this compound. 2 is however more resistant to oxidation than diphenylamino- and dinaphthylamino-substituted alkylacridones, whose oxidation starts at potentials lower by 0.42 V.42 Higher resistance against oxidation of acridones with carbazole substituents makes these compounds more suitable for applications in electronic devices operating in ambient conditions.
6 differs from 2 by its central acceptor unit (quinacridone vs. acridone). Following the same reasoning as in the case of 2, we can attribute the first two reversible oxidation processes at potentials  and
and  to consecutive oxidations of carbazole substituents (see Fig. 1d). It should be noted that the oxidation of carbazole units in 6 occurs at lower potentials and in narrower potential range as compared to 2. This reflects the effect of central acceptor unit which in the case of 6 is more extended and better delocalizes the charge of the radical cation formed in the first oxidation step. The third anodic peak in the cyclic voltammogram of 6 corresponds to reversible oxidation of the quinacridone central unit at
to consecutive oxidations of carbazole substituents (see Fig. 1d). It should be noted that the oxidation of carbazole units in 6 occurs at lower potentials and in narrower potential range as compared to 2. This reflects the effect of central acceptor unit which in the case of 6 is more extended and better delocalizes the charge of the radical cation formed in the first oxidation step. The third anodic peak in the cyclic voltammogram of 6 corresponds to reversible oxidation of the quinacridone central unit at 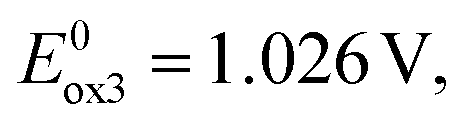 a value which is by ca. 0.4 V higher than the potential of the oxidation of neutral quinacridone.43 Note that the reduction potential of 6 (−1.938 V) is higher than those of 1 and 2, consistent with easier reducibility of quinacridone43 as compared to acridone.40
a value which is by ca. 0.4 V higher than the potential of the oxidation of neutral quinacridone.43 Note that the reduction potential of 6 (−1.938 V) is higher than those of 1 and 2, consistent with easier reducibility of quinacridone43 as compared to acridone.40
Electrochemical data of 3 and 4 are reported in our previous paper.40 In general, phenoxazine derivatives of acridone are easier to oxidize than the corresponding carbazole derivatives but their reduction potential is similar. Both phenoxazine substituents in 5 undergo reversible oxidation at the same potential of E0 = 0.293 V, followed by irreversible oxidation of the acridone central unit at E = 1.097 V (Fig. 1c). Reduction of the acridone unit in 5 starts at −2.23 V, yielding a cathodic peak without a clear maximum, as in the case of 2 (compare Fig. 1b and c).
For the calculations of IP and EA of 1–6 from the electrochemical data we have taken the potentials of the onsets of the first oxidation and first reduction peaks. They are listed in Table 1. As seen from these data, 1–5 exhibit similar and low reduction potential and, by consequence, low electron affinities (EA) in the range 2.50–2.65 eV. The only exception is 6 containing significantly more extended quinacridone core. Its reduction potential is by ca. 400–500 mV higher as compared to 1–5 leading to |EA| exceeding 3 eV.
| Compound | E red,onset [V] | E ox,onset [V] | EAelb [eV] | IPelc [eV] | IPUPSd [eV] |
|---|---|---|---|---|---|
| a Potential vs. Fc/Fc+. b Calculated according to equation: EA = e(Ered,onset + 4.8) [eV]. c Calculated according to equation: IP = −e(Eox,onset + 4.8) [eV]. d Measured by photoelectron spectroscopy in air (Fig. 1e). | |||||
| 1 | −2.27 | 0.651 | −2.53 | 5.45 | 5.51 |
| 2 | −2.22 | 0.637 | −2.58 | 5.44 | 5.48 |
| 3 | −2.23 | 0.317 | −2.57 | 5.12 | 5.33 |
| 4 | −2.16 | 0.332 | −2.64 | 5.13 | 5.3 |
| 5 | −2.23 | 0.294 | −2.57 | 5.09 | 5.46 |
| 6 | −1.77 | 0.600 | −3.03 | 5.40 | 5.55 |
The studied derivatives differ, however, in the onsets of their oxidation potentials. Phenoxazine-functionalized acridones (3–5) exhibit Eox1onset values lower by 300–320 mV as compared to carbazole-substituted ones (1, 2 and 6). Consequently, their IPs are lower, only barely exceeding 5.0 eV (see Table 1).
Finally, it is instructive to verify whether a correlation exists between IP values derived from the electrochemical data (IPel) and those obtained by photoelectron spectroscopy (IPUPS). The photoelectron emission spectra registered for 1–6 are shown in Fig. 1e whereas IPUPS values, calculated on their basis, are collected in Table 1.
It should be stressed that in both cases different physical quantities are measured. IPel, determined for compounds dissolved in an nonaqueous solvent, corresponds to the ionization energy which is modified by electrostatic interactions between the ionized molecule and its polarizable environment (Psol). In the case of IPUPS, the measurements are carried out for surfacial molecules in a thin solid film. In this case the ionized molecule interacts with the solid polarizable environment (Pc), and the polarization energy again modifies the ionization energy. IPUPS values are higher than the IPel ones but the observed values for phenoxazine derivatives are systematically lower than those measured for carbazole ones (Table 1). It should also be noted that the difference between IPel and IPUPS is slightly more pronounced in the case of phenoxazine derivatives. This is not unexpected since Psol and Pc energies usually are not equal, which inevitably leads to differences in the IP values determined by CV and UPS. However, in a series of papers Sworakowski et al.44–46 demonstrated that a clear correlation can be found between the measured redox potentials (and by consequence IPel) and IPUPS for a large number of organic semiconductors of low molecular and high molecular nature.
3.3. Photophysical properties
Absorption spectra of 1–6 are characterized by bands attributable to their D and A segments i.e. acridone (Acr) and tert-butylcarbazole (tCz) for 1 and 2, acridone (Acr) and phenoxazine (PhNZ) for 3–5 and quinacridone (QAcr) and tCz in the case of 6. This can be clearly deduced from Fig. 2a and b where the spectra of toluene solutions of 1–6 are presented together with those registered for Acr, QAcr, PhNZ and tCz dissolved in the same solvent. Bands characteristic of the D and A moieties are marked by thin vertical lines: at 389 nm for Acr, at 290 nm (π → π*) and 307–335 nm (n → π*) for tCz; at 313 nm for PhNZ and at 290 and 504 nm for QAcr. Slight red-shifts and broadening of these bands are observed in the spectra of all studied compounds (1–6), especially in the low-energy regions. This observation can apparently be explained by the formation of intramolecular charge transfer (ICT) states due to interactions between the electron-donating and electron-accepting moieties. These issues are discussed in greater detail in the next subsection (Quantum chemical calculations).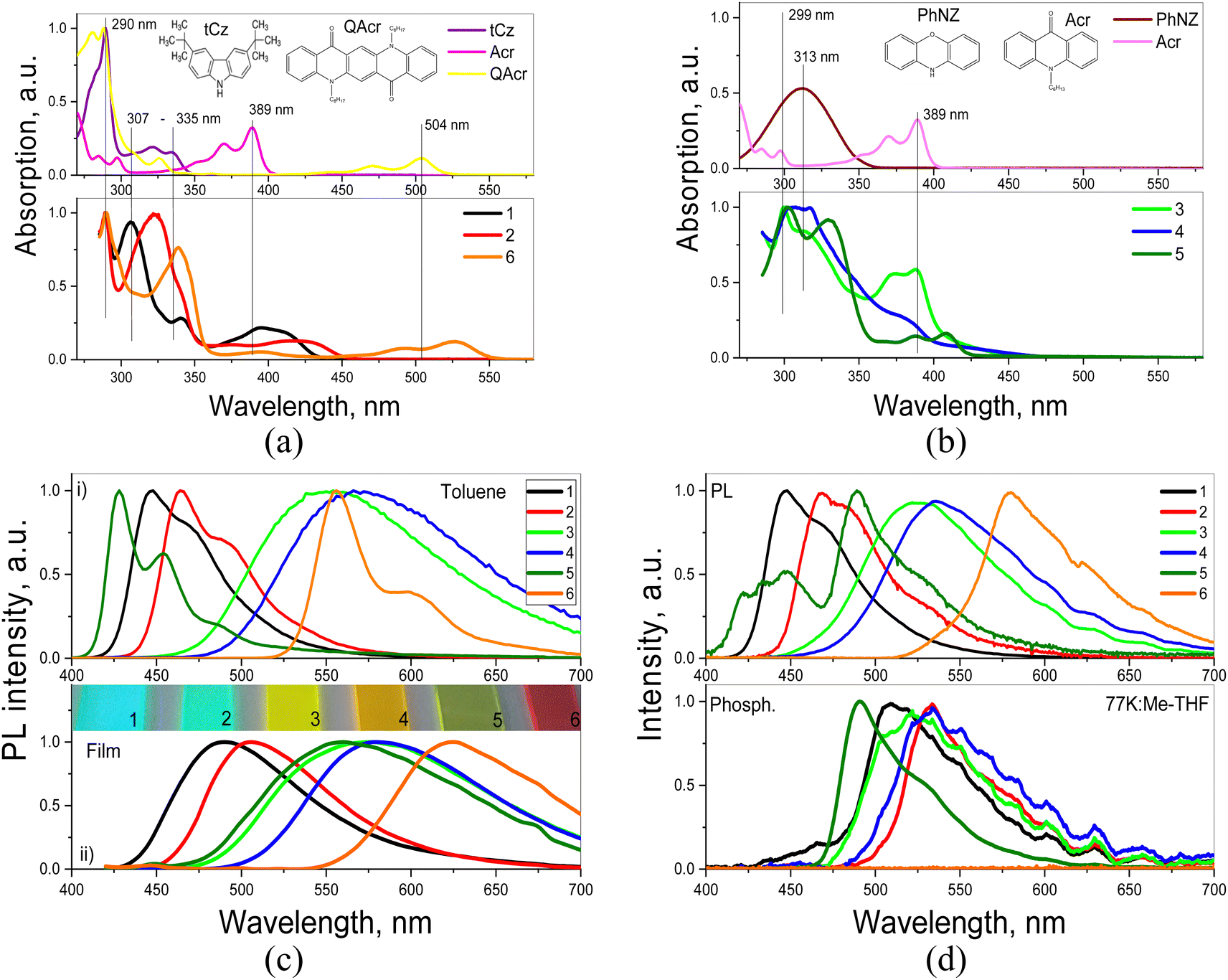 | ||
| Fig. 2 Absorption spectra (a and b) of toluene solutions of 1–6, together with the spectra of tCz, QAcr, PhNZ, Acr shown for comparison; photoluminescence (PL) spectra (c) of 1–6 dissolved in toluene and their vacuum-deposited films; and PL and phosphorescence (Phosph.) spectra (d) of 1–6 dissolved in Me-THF, recorded at 77 K. The delay time of 1 ms after excitation was used recording phosphorescence spectra. The excitation wavelength of 330 nm was used. Inset of (c) shows photos of vacuum-deposited films under UV excitation. | ||
Absorption spectra of solid thin films of 1–6 are similar to those of the corresponding toluene solutions (Fig. S2a, ESI†). However, they are characterized by even larger broadening. Two origins of these effects can be proposed either aggregation or stronger ICT. The latter seems more plausible, taking into account the ICT-induced shapes (non-structured) of the corresponding PL spectra of all studied compounds (Fig. 2c). In contrast, specific features of PL spectra of toluene solutions of 1, 2 and 5, 6 originate from recombination of the locally excited (LE) states formed mainly by electron-accepting moieties (acridone or quinacridone). 3 and 4 demonstrate ICT-shaped, PL spectra both in toluene solutions and in the solid-state, which is apparently caused by strong D–A interactions. These problems are also additionally discussed in the subsection Quantum chemical calculation. PL decays (typically few nanoseconds) registered for toluene solutions of 1–6 are presented in Fig. S2b (ESI†).
In the PL spectrum of 10% molecular dispersion of 5 in 1,3-bis(N-carbazolyl)benzene (mCP) two bands are observed at 452 and 525 nm, resulting from recombination of LE and ICT states respectively (see Fig. 3 and Table 2). In the case of the dispersions of the remaining compounds (1–4, 6) only one band can be distinguished. In all cases the PL spectra of molecular dispersions in the host of low polarity (dielectric constant of mCP = 2.84)47 undergo a hypsochromic shift as compared to the corresponding spectra of thin films of neat compounds (compare Fig. 2c and 3a). This trend additionally proves the predominance of ICT character of the excited states in solid films of 1–6. Time-resolved spectroscopic studies show long-lived emission of molecular dispersions of compounds 3–5 in mCP under vacuum conditions (see Fig. 3b). The attribution of this long-lived emission to fluorescence is supported by the absence of visible changes in the shape of PL spectra before and after evacuation (Fig. 3c). Evacuation-induced increase of the intensity of PL spectra of solutions and molecular dispersions of 3–5 strongly indicates the triplet origin of that delayed fluorescence (Fig. 3c and d). As previously demonstrated in ref. 48, the long-lived fluorescence of 3 and 4 is TADF in nature. This supposition is in a very good agreement with the observed small singlet–triplet splitting, in the studied compounds, as derived from the high energy onsets of their fluorescence and phosphorescence spectra (Fig. 2d and Table 2).
 | ||
| Fig. 3 Normalized PL spectra of molecular dispersions (10 wt% in mCP) of compounds 1–6 (a); PL decays of these dispersions (b); normalized PL spectra of the dispersion of compound 3 (10 wt% in mCP) in air and under vacuum (c); PL spectra of toluene solutions of compound 3: as prepared (in air), after deoxygenation and reoxygenation (d). | ||
| Compound | λ tol.MAX, nm | λ solidMAX, nm | λ solidMAX, nm | PLQY, % | E S1, eV | E T1, eV | ΔEST |
|---|---|---|---|---|---|---|---|
| Media | Tol. | Film of neat comp. | Mol. disp. in mCP | Tol./mol. disp. in mCP | Me-THF at 77 K | ||
| a Shoulder. First singlet (ES1) and triplet (ET1) energies were taken at the onset of the high-energy edges of photoluminescence (fluorescence) and phosphorescence spectra, respectively, registered for Me-THF solutions of 1–6 at the temperature of liquid nitrogen (77 K). PLQYs of 1–6 measured in oxygen-free conditions. | |||||||
| 1 | 448, 469 | 490 | 462 | 69/66 | 2.92 | 2.58 | 0.34 |
| 2 | 464, 490a | 506 | 479 | 61/68 | 2.79 | 2.46 | 0.33 |
| 3 | 551 | 565 | 525 | 1.5/35 | 2.68 | 2.59 | 0.09 |
| 4 | 571 | 581 | 541 | 2/31 | 2.59 | 2.53 | 0.06 |
| 5 | 429, 454, 488a | 558 | 452, 525 | 8.6/15.4 | 3.02 | 2.64 | 0.38 |
| 6 | 555, 600 | 625 | 594 | 92/35 | 2.31 | — | — |
In addition, to support the conclusion on TADF nature of the delayed fluorescence the PL decay measurements were performed for the films of compound 3 in air and vacuum conditions at the different temperatures (Fig. 4a–d). The PL decay curves of TADF compounds are characterized by the component prompt fluorescence in nanoseconds range and by the component microseconds range. The delayed fluorescence intensity increased with the increase of the temperature (Fig. 4d). This observation proves that thermally activated process was responsible for the delayed fluorescence. TADF properties of compound 3 are in good agreement with its small singlet-triplet splitting of 0.06 eV and with low activation energy (Ea) of 12 meV of reverse intersystem crossing (RICS) (Fig. 4e and f). Ea value was obtained by fitting the temperature dependence of RICS rates of compound 3 shown in Fig. 5f using the Arrhenius dependence k = A × exp(−Ea/kBT), where Ea is activation energy, kB is Boltzmann constant and A is the frequency factor involving the spin–orbit coupling constant.49
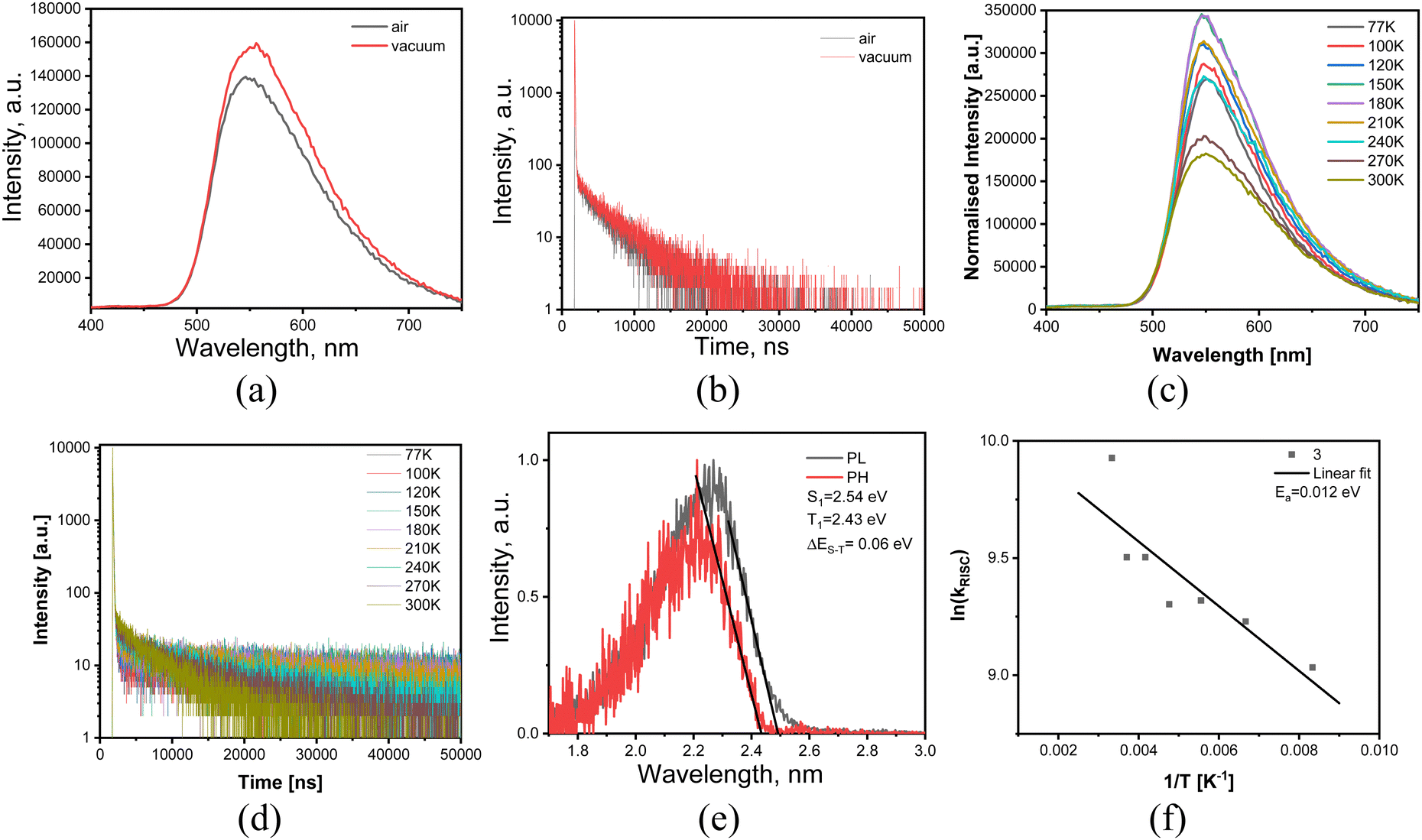 | ||
| Fig. 4 PL spectra (a) and PL decay curves (b) recorded in vacuum and air at room temperature; PL and phosphorescence spectra (c) recorded at 77 K; PL spectra (d) and PL decay curves recorded at the different temperatures in an inert atmosphere; RISC rate as the function of temperature (e). | ||
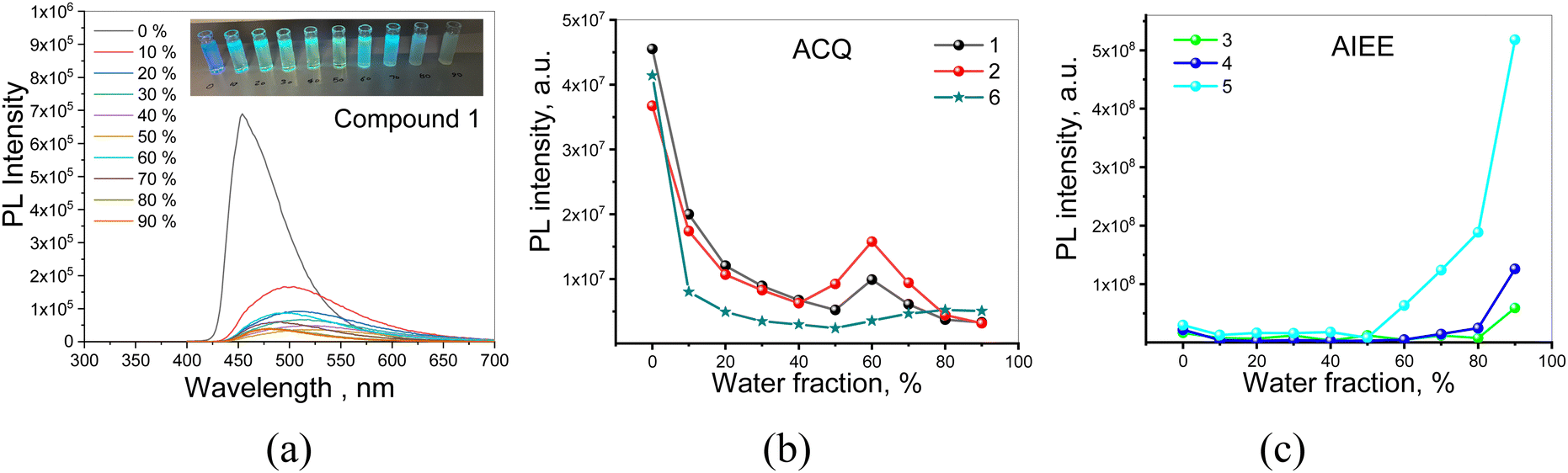 | ||
| Fig. 5 PL spectra of the dispersions of compound 1 in THF/water mixtures with various water fractions (a). Inset shows photographs of the dispersions under UV excitation. Integrated areas of PL spectra versus water volume fractions for the dispersions of the compounds in the mixtures of THF and water (b and c). | ||
PLQY values, determined for 1–6 dissolved in toluene or dispersed (10 wt%) in mCP, are collected in Table 2. Solutions of 1, 2 and 6, in toluene, which exhibit fluorescence generated by recombination of LE states, are characterized by relatively high PLQY values up to 92% (the PLQY values were measured using integrated sphere and recalculated using IN2/Iair coefficients which expresses the PL intensity ratio determined for deoxygenated and air-equilibrated solutions). Compounds 3–5 are characterized by rather low PLQY values in solutions and significantly increased PLQY values of their dispersion in solid matrices. This behaviour should be attributed to AIEE.50 More detailed information on PLQY of 1–6 is provided by theoretical calculation presented in the subsequent subsection. To study the photophysical properties of compounds 1–6 in solid-state in more detail PL measurements for their dispersions in the THF and water mixtures were provided (Fig. 5 and Fig. S3, ESI†).
Emission intensities (integrated area of PL spectra) of the dispersions of 1–6 in the THF–water mixtures with water fractions from 0 to 90% are shown in Fig. 5a and Fig. S3 (ESI†). Decrease in the emission intensity of compound 1 with increasing water fractions in the solution mixture was observed (Fig. 5a and b). Such observation is typical for compounds exhibiting aggregation-caused quenching (ACQ).51 ACQ was also detected for compounds 2 and 6 (Fig. 5b and Fig. S3, ESI†). In the case of the dispersions of compounds 3–5 in the THF–water mixtures, the emission intensity constantly increased with the increase of water fraction, due to the increasing amount of the solid aggregates (Fig. 5c and Fig. S3, ESI†). This observation indicates that compounds 3–5 exhibit AIEE.
3.4. Quantum chemical calculations
Quantum chemical calculations were carried out with the goal to support experimental results. In particular, they were aimed at the determination of the geometries of all molecules studied (1–6), elucidation of the frontier orbitals spatial distribution and calculation of their energies. Finally, theoretical calculations of IPs and EAs values were performed, considered as a theoretical support of the electrochemical and UPS data.As already stated, each molecule consists of either one donor (carbazole or phenoxazine) and one acridone-type acceptor (molecules 1 and 3) or two donors (carbazole or phenoxazine) and one acceptor (acridone or quinacridone, molecules 2, 4 and 6, respectively). Molecule 5 is different in the sense that its acceptor part (acridone) is connected to two donor parts (phenoxazine) via 1,3-phenylene linkers. As derived from the geometry optimization procedure, donor and acceptor parts of each molecule are flat but they are mutually twisted. Geometries of all investigated molecules relaxed in vacuum and in dichloromethane are presented in ESI† (Fig. S9). In the case of 1–4 and 6 dihedral angles between the planes of the donor and the acceptor moieties were calculated for molecules relaxed in vacuum and in dichloromethane, i.e. the same solvent which was used in electrochemical experiments. They are listed in Table S1 presented in ESI.† In the case of 5 (also relaxed in vacuum and in dichloromethane) the listed angles correspond to the torsion of planar acceptor and donor moieties with respect to the plane of 1,3-phenylene linker. From these data, it is clear that the twist angles in carbazole containing molecules are smaller by ca. 25° as compared to the corresponding angles in molecules with phenoxazine substituents (compare molecules 1, 3 and molecules 2, 4). This relationship is seen for molecules relaxed in vacuum as well as in dichloromethane. Additionally, in all cases, the solvent effect enhances the twisting of the acceptor part of the molecule with respect to the donor part but, again, it is more pronounced for the molecules with carbazole donors than for those with phenoxazine ones. Atom coordinates of all investigated molecules are listed in ESI.†
Electronic parameters calculated for 1–6 are collected in Table 3. Note that HOMO levels of acridones substituted with carbazole (molecules 1, 2) are lower lying than the corresponding levels of acridones functionalized with phenoxazine (molecules 3, 4, 5). 6, which is the only quinacridone derivative studied, exhibits the low-lying level of HOMO, characteristic of the carbazole-substituted molecules, as well as the lowest-lying LUMO level of all examined molecules. As a result, significantly smaller band gap is observed for this compound as compared to the other carbazole derivatives (1 and 2), consistent with experimental electrochemical data. The solvent effect leads to an increase of the band gap for all investigated molecules, except 6. It is due to more pronounced decrease of the HOMO level and only a slight decrease of the LUMO level. Solvent molecule interactions increase the electric dipole moment of 1–5 molecules and decrease the polarity of 6, which is still very small as compared to the value calculated for molecules 1–5. Carbazole derivatives (1, 2) are less polar than phenoxazine ones (3–5) whose dipole moment is directed along the short axis of the planar acridone moiety. The dipole moment of 6 is perpendicular to the plane of the quinacridone moiety and the charges are symmetrically distributed around the quinacridone segment (see Fig. S9 in ESI† for the electron distribution density in this molecule). Additionally, the aliphatic chains are perpendicular to the quinacridone plane. As a result, the polarity of 6 is negligible.
| Molecule | HOMO [eV] | LUMO [eV] | ΔEHOMO–LUMO [eV] | μ [D] | IPvertical [eV] | IPadiabatic [eV] | EAvertical [eV] | EAadiabatic [eV] | |
|---|---|---|---|---|---|---|---|---|---|
| a Dichloromethane, DCM. | |||||||||
| 1 | Vacuum | −5.34 | −2.07 | 3.27 | 6.22 | 6.51 | 6.43 | 0.71 | 0.83 |
| DCMa | −5.49 | −2.17 | 3.32 | 8.53 | 5.51 | 5.43 | 2.13 | 2.28 | |
| 2 | Vacuum | −5.32 | −2.20 | 3.12 | 6.26 | 6.23 | 6.17 | 0.95 | 1.09 |
| DCM | −5.43 | −2.25 | 3.18 | 8.81 | 5.42 | 5.36 | 2.22 | 2.37 | |
| 3 | Vacuum | −5.04 | −2.16 | 2.88 | 7.41 | 6.49 | 6.65 | 0.78 | 0.54 |
| DCM | −5.23 | −2.21 | 3.02 | 9.99 | 5.30 | 5.12 | 2.17 | 2.31 | |
| 4 | Vacuum | −5.12 | −2.37 | 2.75 | 8.00 | 6.14 | 6.02 | 1.09 | 1.22 |
| DCM | −5.25 | −2.33 | 2.92 | 10.97 | 5.27 | 5.13 | 2.29 | 2.44 | |
| 5 | Vacuum | −5.00 | −2.18 | 2.82 | 7.23 | 5.98 | 6.37 | 0.93 | 0.53 |
| DCM | −5.19 | −2.19 | 3.00 | 9.98 | 5.27 | 5.10 | 2.17 | 2.31 | |
| 6 | Vacuum | −5.31 | −2.70 | 2.61 | 0.21 | 9.19 | 9.69 | 1.58 | 1.16 |
| DCM | −5.38 | −2.77 | 2.61 | 0.18 | 5.37 | 5.32 | 2.75 | 2.89 | |
Distributions of frontier orbitals within different parts of the studied molecule, together with their energies, determine spectroscopic and redox properties of electroactive compounds. These distributions are also strongly related to the molecular geometry. As already stated, in the case of 3 and 4 their donor and acceptor parts are almost perpendicular. The same applies to 5, where the phenoxazine unit is almost perpendicular to the 1,3-phenylene linker. For these reasons HOMO and LUMO of 3, 4 and 5 are located on different parts of the molecules, HOMO being essentially limited to the phenoxazine unit whereas HOMO−1, LUMO and LUMO+1 to the acridone one (see Fig. 6 for the shapes and distribution of frontier orbitals).
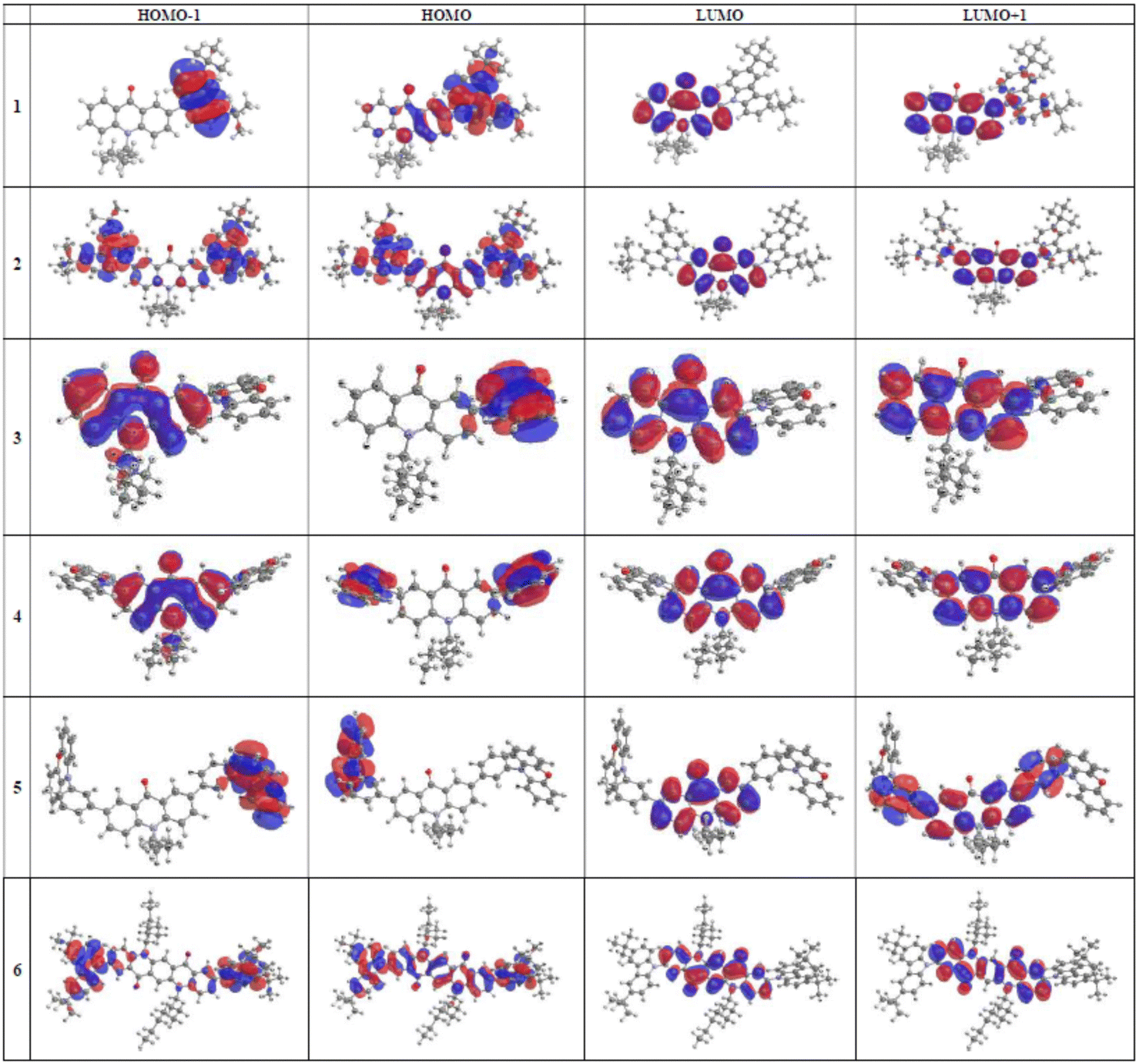 | ||
| Fig. 6 Frontier orbitals of the 1–6 molecules in vacuum calculated by DFT/B3LYP-GD3BJ/6-311++G(d,p) method. | ||
The spatial separation of HOMO and LUMO orbitals in 3, 4 and 5 is manifested in the calculated transitions. It inhibits transitions of dominant HOMO → LUMO configurations i.e. S0 → S1 transitions which in the case of these compounds exhibit either zero or negligible oscillator strength (see ESI,† Table S2). The lowest energy transition of non-negligible oscillator strength is HOMO−1 → LUMO in the case of 3 whereas for 4 and 5 it corresponds to the HOMO−2 → LUMO transition. Smaller dihedral angle in carbazole derivatives of acridone and quinacridone significantly changes the distribution of frontier orbitals. Although LUMO in the case of 1, 2 and 6 is limited to the acridone (quinacridone) unit, HOMO extends from the carbazole substituent to the acridone (quinacridone) moiety. This means that no separation in space of frontier orbitals exists in these molecules and the lowest energetic transition (S0 → S1) of a HOMO → LUMO configuration is not inhibited. As demonstrated by the calculations (see ESI,† Table S2) these transitions exhibit reasonably high oscillator strengths, although the transitions of the highest oscillator strength correspond to HOMO−2 → LUMO, HOMO → LUMO+1 and HOMO → LUMO+2 for 1, 2 and 6, respectively.
It is instructive to confront these calculations with experimental findings. First, it should be expected that the photoluminescence quantum yield (PLQY) of phenoxazine derivatives (compounds 3, 4 and 5) should be significantly smaller than that of carbazole ones (1, 2 and 6) since, as evoked by the Kasha rule, small oscillator strength of the S0 → S1 transition hinders the fluorescence. This is indeed the case, since PLQY of 1, 2 and 6 measured for solutions in toluene is over one order of magnitude higher than that determined for 3, 4 and 5 (see Table 2). Moreover, experimental UV-vis spectra of 3, 4 and 5 (Fig. 2) clearly show that the absorption bands of the lowest energy significantly exceed the expected energy of the HOMO → LUMO, (S0 → S1), indicating that they originate from S0 → Sn transition, n >1.
To the contrary, in the case of UV-vis spectra of 1, 2 and 6 the lowest energy bands clearly correspond to the energy of the HOMO → LUMO transition. Finally, the calculations are fully consistent with the observed voltamperometric behavior of the studied compounds. Strict spatial separation of HOMO and LUMO, with HOMO being located on the phenoxazine moiety, results in one-step oxidation of both substituents in 4 and 5 which are being oxidized independently at the same potential. Extension of HOMO orbitals over the whole molecule in carbazole derivatives results, in turn, in a two-step oxidation of carbazole moieties in 2 and 6, since the charge formed in the oxidation of one carbazole unit is then transmitted to the second substituent via delocalization, making its consecutive oxidation more difficult.
In Table 3 vertical and adiabatic ionization potentials (IPs) and electron affinities (EAs) calculated for 1–6 molecules are presented. They were evaluated from the differences of the total energies of the neutral molecules and their respective radical-ions. By comparing IPvertical with IPadiabatic it can be concluded that for all molecules relaxing in dichloromethane the values of IPadiabatic are lower. An excellent agreement between IPadiabatic and the experimental values derived from electrochemical investigations (IPel) should be pointed out (compare the data collected in Tables 1 and 3). It involves not only the same trend but also close proximity of the experimentally determined and calculated values. The values of EAadiabatic are consistently lower than experimental EAel but the trend within the series of compounds 1–6 is the same.
3.5. Charge transport properties
Electrical transport in emissive layers of OLEDs is of crucial importance for proper operation of the device. In particular, electroluminophores in one-component layers or hosts in guest/host OLEDs should exhibit ambipolar transport properties. In view of applications of 1–6 as electroluminophores in OLEDs, their charge transport properties were examined by the time of flight (TOF) technique. The experiments were carried out using diode-type indium tin oxide (ITO)/thick organic layer/Al samples. Dispersive TOF current transients but with observed transit times (ttr) in log–log scales were recorded either under applied positive (for holes) or negative (for electrons) voltages to the side of optically transparent electrode (ITO) (see Fig. 7a and Fig. S4 in ESI†). In these conditions compounds 1, 4–6 turned out unipolar exhibiting measurable mobility for holes only. 2 and 3 were bipolar exhibiting both p-type (hole) and n-type (electron) transport (Fig. 7b). | ||
| Fig. 7 TOF signals for holes (a) in the vacuum-deposited layer of compound 6 and charge mobilities as the function of electric field (b) measured for vacuum-deposited layers of 1–6. | ||
Taking ttr values from the TOF current transients, hole and electrons mobilities were estimated at the different electric fields displaying their hole-only or bipolar charge transport (see Fig. 7b). The highest value of hole mobility μh = 2.53 × 10−3 cm2 V−1 s−1 at E = 3.6 × 105 V cm−1 was found for the quinacridone derivative (compound 6, see Table 4). This was not unexpected since high hole mobilities, measured either in the field effect transistor52,53 or diode configurations,54 were reported for various quinacridone core-containing compounds. Hole mobilities of the remaining compounds were from over one order (4) to two orders (1) of magnitude lower (Fig. 7b). The measured hole mobility values are typical of donor–acceptor compounds exhibiting TADF in which the molecular segments are highly twisted.55,56 Among acridone derivatives (1–5), those containing phenoxazine donors (3–5) showed slightly higher hole mobilities than the compounds with carbazole donors (1, 2).
| Compound | Holes | Electrons | ||||
|---|---|---|---|---|---|---|
| μ h (cm2 V−1 s−1) | μ 0 (cm2 V−1 s−1) | β (cm V−1)1/2 | μ e (cm2 V−1 s−1) | μ 0 (cm2 V−1 s−1) | β (cm V−1)1/2 | |
| a Taken from the TOF measurements (Fig. 7), hole (μh) and electron (μe) mobilities at electric field (E) of 3.6 × 105 V cm−1. b Mobilities at absent electric field (μ0). c Field dependence parameter (β) of a Poole–Frenkel type mobility: μ = μ0 × exp(β × E1/2). | ||||||
| 1 | 1.25 × 10−5 | 1.89 × 10−7 | 7.75 × 10−3 | — | — | — |
| 2 | 1.59 × 10−5 | 1.91 × 10−8 | 11.1 × 10−3 | 1.8 × 10−5 | 1.75 × 10−8 | 11.4 × 10−3 |
| 3 | 4.62 × 10−5 | 1.77 × 10−7 | 5.49 × 10−3 | 1.3 × 10−5 | 6.6 × 10−8 | 8.55 × 10−3 |
| 4 | 8.5 × 10−5 | 2.17 × 10−7 | 10 × 10−3 | — | — | — |
| 5 | 2.39 × 10−5 | 6.8 × 10−7 | 6.13 × 10−3 | — | — | — |
| 6 | 2.53 × 10−3 | 4.48 × 10−5 | 6.8 × 10−3 | — | — | — |
Results of fitting of the Poole–Frenkel electric field dependences of experimental charge carrier mobilities, led to relatively high field dependence parameters. β values differed within the range from 5.49 × 10−3 for 3 to 11.1 × 10−3 for 2 as determined at room temperature for all investigated compounds (1–6) (Table 4). They can be attributed to the relatively strongly dispersive transport. The weakest dispersity was observed for compound 3, characterized by the lowest β value (Fig. S4, ESI†).572 and 3 turned out to be bipolar, exhibiting at the same electric field similar electron mobilities of 1.8 × 10−5 and 1.3 × 10−5 cm2 V−1 s−1, respectively. Moreover, μe values of 2 and 3 were comparable to their μh values indicating well-balanced hole and electron transport. This property is of crucial importance for the use of these compounds in electroluminescent devices such as OLEDs.
3.6. Electroluminescence
The results of spectroscopic, electrochemical and electrical transport investigations supported by quantum chemical calculations seem to indicate that the synthesized acridone and quinacridone derivatives are very well suited for a variety of applications such as electrochromic materials, components of active layers in ambipolar or p-channel field effect transistors and electroluminophores in light-emitting diodes. In the research described here we focused on this last application. Thus, 1–6 were used as emitters in test OLEDs of the following structure: ITO/MoO3 (0.3 nm)/NPB (40 nm)/mCP (4 nm)/EML (24 nm)/TSPO1 (4 nm)/TPBi (40 nm)/LiF (0.3 nm)/Al (120 nm), where EML stands for “emissive layer”. Chemical formulae of particular components of the devices are shown in Fig. 8. Two types of emissive layers were fabricated by vacuum deposition: (i) non-doped, i.e. consisting of neat compounds 1–6 and in the subsequent text termed as A1–A6 series, the number being related to the number of the synthesized compound; (ii) doped, i.e. 10 wt% molecular dispersions of a given compound in a mCP matrix, subsequently abbreviated as B1–B6 series, the number corresponding to the number of the synthesized compound. To assure good injection of holes from the anode (ITO) and electrons from the cathode (Al), additional layers were deposited (MoO3/LiF) as well as hole/electron transporting layers (NPB/TPBi). They were selected on the basis of their energy levels (see Fig. 8). Hole/electron (mCP/TSPO1) blocking layers were also carefully chosen for ensuring recombination of hole–electron pairs within the EML. They additionally acted as exciton blocking layers due to their high triplet energy levels that can prevent from exciton leakages to the neighbouring layers. Therefore, it can be claimed that the observed electroluminescence is fully attributed to the emission of EMLs (Fig. 9a).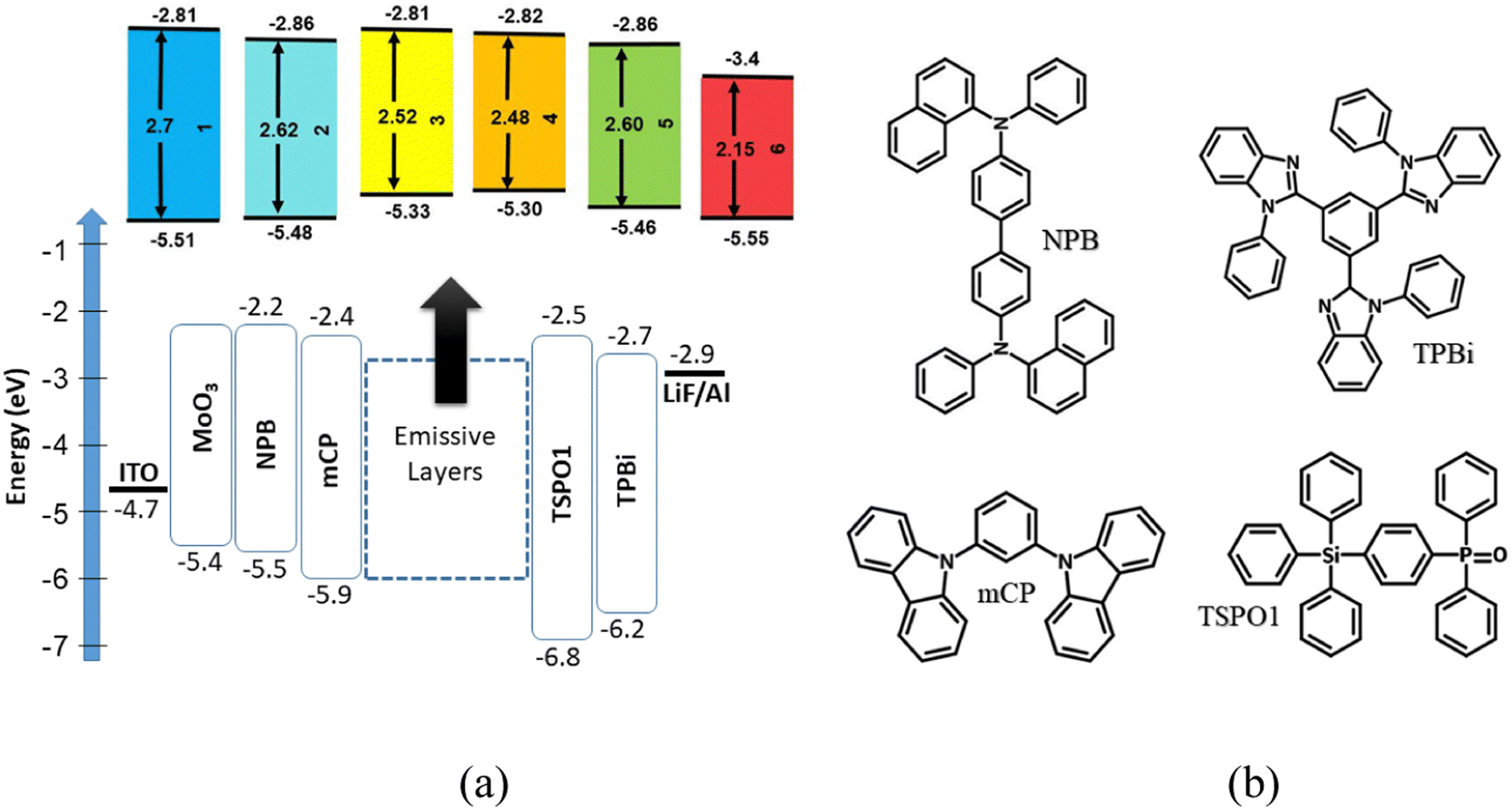 | ||
| Fig. 8 Equilibrium energy diagram (a) and chemical structures of selected components of test OLEDs (b). | ||
 | ||
| Fig. 9 EL spectra recorded at 10 V (a) and CIE colour diagram (b) with the corresponding CIE coordinates. Current density and brightness as the function of applied voltages (c), EQE versus current density plots (d) and TREL curves of device B3 (e). | ||
Indeed, the positions and shapes of the obtained EL spectra closely remind the corresponding PL spectra of neat (non-doped) and molecularly dispersed in mCP (doped) films of 1–6 (compare Fig. 9a and 4a). EL spectra were stable under different voltages for the majority of devices except B2, B5 and A6 (Fig. S5 in ESI†). Possible explanation of these differences has to involve several factors. Emissions from these luminophores are of LE and ICT character (see Fig. 3c and d). Thus, their LE/ICT emission ratio can differ, depending on the type of the excitation source (optical vs. electrical), leading to meaningful differences between the PL and EL spectra. B5 device can be considered here as an instructive example. Its EL emission is mainly of LE character (high energy spectrum with structured bands) whereas LE and ICT emissions equally contribute to the PL spectrum of 5 molecularly dispersed in mCP (Fig. 4a).
Functionalization of acridone or quinacridone with only two types of donors, namely tCz and PhNZ, results in a series electroluminophores whose emission spectra cover large part of the visible spectrum from blue to orange. In particular, non-doped and doped EMLs of tCz substituted acridone (compounds 1 and 2 in devices A1, A2, B1 and B2) emit blue electroluminescence of similar CIE 1931 colour coordinates (see Fig. 9b, Fig. S6 and Table 5, Table S1, ESI†). A3 and A4, i.e. devices with non-doped EMLs consisting of PhNZ substituted acridone emit green (CIE1931 of (0.336; 0.567)) and yellow (CIE1931 of (0.439; 0.541)) light, respectively. The observed bathochromic shift of the spectra of A3 and A4 as compared to those recorded for A1 and A2 is attributed to stronger donor–acceptor interactions in acridone-phenoxazine derivatives. However, dispersions of 3 and 4 in mCP (devices B3 and B4) yield EL spectra which are hypsochromically shifted with respect to the spectra measured for A3 and A4 devices with CIE1931 colour coordinates characteristic of blue region (see Table 5). This colour change can be attributed to the luminophore interactions with the mCP host, a phenomenon somehow similar to solvatochromism. Interestingly, yellow versus blue electroluminescence was obtained for EMLs in A5 and B5 devices, respectively, i.e. layers exhibiting different electroluminescence mechanisms. This difference cannot be attributed to solvatochromism but rather to a different combination of ICT and LE emissions, as previously reported for indolocarbazole-type emitters.58,59 Thus, weakening of donor–acceptor interactions in compound 5 dispersed in mCP as compared to the film of neat luminophore (undoped EML) results in switching from ICT to LE emission, which is more intensive under electrical excitations than under optical ones.
| Device name | EML | λ EL,c nm | V ON,c V | L MAX,c cd m−2 | CEMAX,c cd A−1 | PEMAX,c lm W−1 | EQEMAX,c % | CIE 1931,cx; y |
|---|---|---|---|---|---|---|---|---|
| a Device structure is ITO/MoO3/NPB/mCP/EML/TSPO1/TPBi/LiF/Al. b Device structure is ITO/MoO3/TCTA/EML/TSPO1/TPBi/LiF/Al. c Collected parameters are: EL maximum (λEL); turn-on voltage (VON); maximum brightness (LMAX); maximum current (CEMAX); power (PEMAX) and external quantum (EQEMAX) efficiencies; colour coordinates (CIE 1931). | ||||||||
| B1a | 1(10%):mCP | 458 | 5.0 | 4270 | 6.7 | 3.8 | 4.0 | 0.146; 0.149 |
| B2a | 2(10%):mCP | 459 | 6.4 | 2150 | 1.7 | 0.7 | 1.2 | 0.170; 0.208 |
| B3a | 3(10%):mCP | 505 | 4.7 | 7950 | 37.1 | 19.8 | 13.0 | 0.212; 0.475 |
| B4a | 4(10%):mCP | 548 | 3.5 | 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 520 520 |
23.5 | 20.5 | 7.2 | 0.399; 0.567 |
| B5a | 5(10%):mCP | 426, 455 | 5.4 | 1820 | 0.8 | 0.4 | 0.5 | 0.196; 0.216 |
| B6a | 6(10%):mCP | 587 | 6.0 | 4010 | 1.4 | 0.5 | 0.6 | 0.497; 0.436 |
| C3b | 3(50%):POT2T | 520 | 4.6 | 6820 | 52.5 | 28.5 | 16.7 | 0.26; 0.54 |
Undoped and doped EMLs of the only quinacridone derivative studied in this research (6) yields orange, close to red, electroluminescence (see Fig. 9 for the spectra and Table 5 for CIE 1931 coordinates). Unexpectedly, a high energy band peaked at ca. 490 nm can be observed in the EL spectrum of device A6. This band was not present in the photoluminescence spectrum of the film of neat 6 and its nature was not yet clarified. Tentatively, this band could be attributed to the electroplex emission of 6, taking into account that it appeared only under electrical excitation. Its presence could be exploited in the development of white OLEDs, as previously proposed.58
Concluding this part of the paper, non-doped EMLs, consisting of neat 1–6 compounds (devices A1–A6) demonstrated CIEx values in a rather wide range from 0.15 to 0.439. Expectably, the CIEx range of EMLs fabricated as dispersions of 1–6 in mCP (devices B1–B6) was narrower (0.17 to 0.399), which could be ascribed to lowering of their polarity in the matrix. CIEy values varied in the ranges from 0.161 to 0.57 and from 0.149 to 0.567 for A1–A6 and B1–B6 devices, respectively. These results clearly show that using only two donor and two acceptor building blocks in the synthesis of D–A, D–A–D or D–π–A–π–D compounds, it is possible to fabricate electroluminophores covering large part of the visible spectrum.
The lowest value of the turn on voltage (3.5 V) was measured for device B4 (Fig. 9c and Table 5), consistent with the lowest ionization potential of 4 (IP = 5.3 eV) as determined by UPS and its second highest hole mobility (see Tables 1 and 4). Despite high hole mobility and relatively good charge injecting properties of 6, devices fabricated with the use of this compound (A6 and B6) demonstrated rather high turn on voltages. This could be tentatively ascribed to electroplex formation, as judged from their EL spectra. Large differences in the maximum brightness values of the fabricated devices are related to different emitter efficiencies and to varying sensitivity of the human eye to different emission colours (Table 5).
Outstanding TADF/AIEE emissive and bipolar charge-transporting properties of compound 3 should be pointed out (see Tables 2 and 4). As a result, A3 and B3 demonstrated the highest maximum EQEs of 2.3% and 13%, respectively (Fig. 9d, Fig. S7 and Table 5, Table S1, ESI†). The transient electroluminescence (TREL) signal reaching milliseconds helps to prove the significant participation of triplet excitons in electroluminescence of the device B3 based on the TADF emitter 3 (Fig. 9e). OLEDs based on compound 3 after the additional optimization can apparently show better output parameters in comparison to those described in the present manuscript. To support this claim, we fabricated OLED using an electron-transporting host and slightly modified device structure (Table 5). Improved maximum extremal quantum efficiency of 16.7% was obtained for device C3 with the emitting layer of compound 3 dispersed in the host PO-T2T in comparison to that of device B3 (Fig. S8, ESI†). Such improvement is mainly related to electron-transporting properties of PO-T2T.60 The lower electron mobility in comparison to hole mobility of compound 3 can be compensated by good electron-transporting properties of PO-T2T in the light-emitting layer of device C3 leading to improved hole–electron balance within light-emitting layer of device C3 (Table 5).
Emissive properties of compound 4 were comparable to those of compound 3, however, devices A4 and B4 showed ca. twice lower maximum EQE efficiencies of 1.32 and 7.2%, respectively. This result could be tentatively explained by unbalanced hole–electron transport within the EML, since, in contrast to bipolar 3, compound 4 was found to be a hole-only conductor (Fig. 7 and Table 4). Compound 1 is also an interesting case. Although it did not show TADF due to relatively high singlet-triplet splitting of 0.34 eV (Table 2), devices A1 and B1 demonstrated, high maximum EQEs of 1.6 and 4%, respectively, i.e. values which could be considered very promising for simple fluorescent OLEDs (Table 5). These relatively high efficiencies were mainly related to high PLQY value of 1 in the solid state (66%, Table 2). Indeed, despite high PLQY values of the films of the doped compounds 1 (66%) and 2 (68%) (Table 2), the EQEs of OLEDs based on these compounds were relatively low. This result is not surprising since the compounds were not characterized by TADF (Fig. 3b). Thus, they do not allow harvesting of triplets in OLEDs. As a result, their maximum theoretical EQE values should not be higher than 5% according to the formula:61
| ηext = γ × ΦPL × χ × ηout | (1) |
The maximum EQE of 4% of device B1 based on the compound 1 is very close to the theoretical maximum of ηext = 4.65% taking γ = 1, χ = 0.25 (as for prompt fluorescence compounds) and ηout = 0.3 (Table 5). OLEDs based on compounds 3 and 4 exhibiting TADF showed considerably higher EQE values due to their efficiency of exciton production χ = 1. Thus, our result demonstrates the effect of the mode of linking of the donor and acceptor moieties (D–A, D–A–D or D–π–A–π–D) on the electroluminescent properties of the compounds.
4. Conclusion
To summarize, we have demonstrated that using only one acceptor (acridone) and two donors (tert-butyl carbazole or phenoxazine) it was possible to design and synthesize a series of D–A, D–A–D and D–π–A–π–D compounds of interesting and reversible electrochemistry and tunable emissive properties. Photoluminescence of their vacuum-deposited films covered a large part of visible spectrum from sky-blue to red. Replacement of the acridone moiety by quinacridone (compound 6) allowed the extension of the emission spectral range to red of the vacuum-deposited film. Compounds 3 (phenoxazinyl substituted acridone), 4 (phenoxazinyl disubstituted acridone) and 5 (phenoxazinyl disubstituted acridone through the phenyl bridges) exhibited excellent emissive properties due to the AIEE and TADF effects. This property, combined with the ambipolar character of 3 which facilitated balanced electron/hole transport, made this compound an excellent candidate for its application in active layers of OLEDs as an electroluminophore. The fabricated test diode exhibited maximum external quantum efficiency of 16.7%. Finally, an excellent agreement between the experimental and theoretically predicted results was obtained pointing out the adequacy of the applied quantum chemical calculations.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
I. K. B., M. Z. and A. P. acknowledge financial support from National Science Centre, Poland (NCN, Grant No. 2019/33/B/ST5/00582). M. G. and D. V. acknowledge the Research Council of Lithuania (Project “ELOS” No. S-MIP-21-30). This research was carried out with the support of the Interdisciplinary Centre for Mathematical and Computational Modelling at the University of Warsaw (ICM UW) under computational allocation no G85-922.References
- H. T. Nguyen, M. C. Lallemand, S. Boutefnouchet, S. Michel and F. Tillequin, J. Nat. Prod., 2009, 72, 527–539 CrossRef CAS PubMed.
- P. Singh, J. Kaur, P. Kaur and S. Kaur, Bioorg. Med. Chem., 2009, 17, 2423–2427 CrossRef CAS PubMed.
- V. Nadaraj, S. Thamarai Selvi and S. Mohan, Eur. J. Med. Chem., 2009, 44, 976–980 CrossRef CAS PubMed.
- R. Hegde, P. Thimmaiah, M. C. Yerigeri, G. Krishnegowda, K. N. Thimmaiah and P. J. Houghton, Eur. J. Med. Chem., 2004, 39, 161–177 CrossRef CAS PubMed.
- A. Shakhnovich and J. Belmont, Chem. Inkjet Inks, 2009, pp. 101–122 Search PubMed.
- J. Su, X. Wen, W. Chen, Y. Miao, F. Li and Y. Wang, New J. Chem., 2018, 42, 5005 RSC.
- C. Li, N. Zheng, H. Chen, J. Huang, Z. Mao, L. Zheng, C. Weng, S. Tan and G. Yu, Polym. Chem., 2015, 6, 5393 RSC.
- C. Wang, W. Chen, S. Chen, S. Zhao, J. Zhang, D. Qiu and Y. Wang, New J. Chem., 2012, 36, 1788–1797 RSC.
- I. Javed, Z. Zhang, T. Peng, T. Zhou, H. Zhang, M. Issa Khan, Y. Liu and Y. Wang, Sol. Energy Mater. Sol. Cells, 2011, 95, 2670–2676 CrossRef CAS.
- K. H. Lee, D. S. Leem, J. S. Castrucci, K. B. Park, X. Bulliard, K. S. Kim, Y. W. Jin, S. Lee, T. P. Bender and S. Y. Park, ACS Appl. Mater. Interfaces, 2013, 5, 13089–13095 CrossRef CAS PubMed.
- K. H. Lee, G. H. Lee, D. S. Leem, J. Lee, J. W. Chung, X. Bulliard, H. Choi, K. B. Park, K. S. Kim, Y. W. Jin, S. Lee and S. Y. Park, J. Phys. Chem. C, 2014, 118, 13424–13431 CrossRef CAS.
- H. Li, C. Gu, L. Jiang, L. Wei, W. Hu and H. Fu, J. Mater. Chem. C, 2013, 1, 2021–2027 RSC.
- K. D. Thé, C. Radford, M. Parvez and T. C. Sutherland, Phys. Chem. Chem. Phys., 2015, 17, 20903 RSC.
- B. K. Sharma, A. M. Shaikh, N. Agarwal and R. M. Kamble, RSC Adv., 2016, 6, 17129–17137 RSC.
- H. T. Le, R. Saleah, N. Kungwan, M. P. Nghiem, F. Goubard and T. T. Bui, ChemistrySelect, 2020, 5, 15180–15189 CrossRef CAS.
- T. Takeda, H. Sugihara, Y. Suzuki, J. Kawamata and T. Akutagawa, J. Org. Chem., 2014, 79, 9669–9677 CrossRef CAS PubMed.
- M. Xu, K. Kong, H. Ding, Y. Chu, S. Zhang, F. Yu, H. Ye, Y. Hu and J. Hua, J. Mater. Chem. C, 2020, 8, 930 RSC.
- P. Yang, S. Guo, X. Yu, F. Zhang, B. Yu, H. Zhang, Y. Zhao and Z. Liu, Ind. Eng. Chem. Res., 2019, 58, 9636–9643 CrossRef CAS.
- J. Jia, Y. Li, W. Wang, C. Luo, L. Han, Y. Li and J. Gao, Dyes Pigm., 2017, 146, 251–262 CrossRef CAS.
- Y. Mei, D. Liu, J. Li, H. Li and W. Wei, J. Mater. Chem. C, 2021, 9, 5885–5892 RSC.
- A. A. Awasthi, N. Gupta, T. Siddiqui, P. Parab, D. K. Palit, S. Bose and N. Agarwal, J. Chem. Sci., 2019, 131, 94 CrossRef.
- M. Mamada, K. Inada, T. Komino, W. J. Potscavage, H. Nakanotani and C. Adachi, ACS Cent. Sci., 2017, 3, 769–777 CrossRef CAS PubMed.
- Q. T. Siddiqui, A. A. Awasthi, P. Bhui, P. Parab, M. Muneer, S. Bose and N. Agarwal, RSC Adv., 2019, 9, 40248–40254 RSC.
- J. Xu, X. Wu, J. Guo, Z. Zhao and B. Z. Tang, J. Mater. Chem. C, 2021, 9, 15505–15510 RSC.
- L. Li, X. Dai, X. Liao, X. Zang, J. Huang, H. Zhang, X. Yin and Y. Hong, Sol. Energy, 2021, 225, 173–183 CrossRef CAS.
- N. Cai, F. Li, Y. Atong Chen, R. Luo, T. Onghui Hu, F. Lin, S.-M. Yiu, D. Liu, D. Lei, Z. Zhu and A. K.-Y. Jen, Angew. Chem., Int. Ed., 2021, 60, 20437–20442 CrossRef CAS PubMed.
- A. Kumar Gupta, Z. Zhang, E. Spuling, M. Kaczmarek, Y. Wang, Z. Hassan, I. D. W. Samuel, S. Bräse and E. Zysman-Colman, Mater. Adv., 2021, 2, 6684 RSC.
- Y. P. Zhang, X. Liang, X. F. Luo, S. Q. Song, S. Li, Y. Wang, Z. P. Mao, W. Y. Xu, Y. X. Zheng, J. L. Zuo and Y. Pan, Angew. Chem., Int. Ed., 2021, 60, 8435–8440 CrossRef CAS PubMed.
- J.-X. Chen, Y.-F. Xiao, K. Wang, D. Sun, X.-C. Fan, X. Zhang, M. Zhang, Y. i-Zhong Shi, J. Yu, F. eng-Xia Geng, C.-S. Lee and X.-H. Zhang, Angew. Chem., Int. Ed., 2021, 60, 2478–2484 CrossRef CAS PubMed.
- W. Yang, Y. Yang, X. Cao, Y. Liu, Z. Chen, Z. Huang, S. Gong and C. Yang, Chem. Eng. J., 2021, 415, 128909 CrossRef CAS.
- Y. Y. Jing, X. D. Tao, M. X. Yang, X. L. Chen and C. Z. Lu, Chem. Eng. J., 2021, 413, 127418 CrossRef CAS.
- G. Li, A. Klimash, E. Zysman-Colman, I. D. W. Samuel idws, P. Address, G. Copley, S. Krotkus, T. Matulaitis, S. Diesing, E. Archer, C. Keum, D. B. Cordes, A. M. Z. Slawin, M. C. Gather and I. D. W. Samuel, Front. Chem., 2021, 1, 572862 Search PubMed.
- S. Do Sung, M. Soo Kang, I. Taek Choi, H. Mo Kim, H. Kim, M. Hong, H. Kyu Kim and W. In Lee, Chem. Commun., 2014, 50, 14161–14163 RSC.
- Y. Zhao, X. Wang, T. Yu, Z. C. Wei, X. Liu and J. Chem, New J. Chem., 2017, 41, 4702 RSC.
- X. Yang, X. Xu and G. Zhou, J. Mater. Chem. C, 2015, 3, 913–944 RSC.
- P. Heimel, A. Mondal, F. May, W. Kowalsky, C. Lennartz, D. Andrienko and R. Lovrincic, Nat. Commun., 2018, 9, 4990 CrossRef PubMed.
- A. Zampetti, A. Minotto and F. Cacialli, Adv. Funct. Mater., 2019, 29, 1807623 CrossRef.
- P. Pander, R. Motyka, P. Zassowski, M. Lapkowski, A. Swist and P. Data, J. Phys. Chem. C, 2017, 121, 11027–11036 CrossRef CAS.
- C. Wang, S. Chen, K. Wang, S. Zhao, J. Zhang and Y. Wang, J. Phys. Chem. C, 2012, 116, 17796–17806 CrossRef CAS.
- I. Kulszewicz-Bajer, M. Zagorska, M. Banasiewicz, P. A. Gunka, G. Gunka, P. Toman, B. Kozankiewicz, G. Wiosna-Salyga and A. Pron, Phys. Chem. Chem. Phys., 2020, 22, 8522 RSC.
- K. Karon and M. Lapkowski, J. Solid State Electrochem., 2015, 19, 2601–2610 CrossRef CAS.
- M. Vasylieva, P. Pander, B. K. Sharma, A. M. Shaikh, R. M. Kamble, F. B. Dias, M. Czichy and P. Data, Electrochim. Acta, 2021, 384, 138347 CrossRef CAS.
- P. Kurzep, Ł. Skórka, S. Skórka, M. Zagorska, P. A. GúNka, M. Banasiewicz, B. Kozankiewicz and I. Kulszewicz-Bajer, RSC Adv., 2017, 7, 8627–8632 RSC.
- J. Sworakowski, J. Lipiński and K. Janus, Org. Electron., 2016, 33, 300–310 CrossRef CAS.
- J. Sworakowski and K. Janus, Org. Electron., 2017, 48, 46–52 CrossRef CAS.
- J. Sworakowski, Synth. Met., 2018, 235, 125–130 CrossRef CAS.
- E. Skuodis, O. Bezvikonnyi, A. Tomkeviciene, D. Volyniuk, V. Mimaite, A. Lazauskas, A. Bucinskas, R. Keruckiene, G. Sini and J. V. Grazulevicius, Org. Electron., 2018, 63, 29–40 CrossRef CAS.
- I. Kulszewicz-Bajer, M. Zagorska, M. Banasiewicz, P. A. Guńka, P. Toman, B. Kozankiewicz, G. Wiosna-Salyga and A. Pron, Phys. Chem. Chem. Phys., 2020, 22, 8522–8534 RSC.
- A. E. Nikolaenko, M. Cass, F. Bourcet, D. Mohamad and M. Roberts, Adv. Mater., 2015, 27, 7236–7240 CrossRef CAS PubMed.
- Z. Zhao, H. Aoke Zhang, W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888–9907 CrossRef CAS PubMed.
- Y. Zuo, X. Wang and D. Wu, J. Mater. Chem. C, 2019, 7, 14555 RSC.
- T. Marszalek, I. Krygier, A. Pron, Z. Wrobel, P. M. W. Blom, I. Kulszewicz-Bajer and W. Pisula, Org. Electron., 2019, 65, 127–134 CrossRef CAS.
- C. Wang, D. Chen, W. Chen, S. Chen, K. Ye, H. Zhang, J. Zhang and Y. Wang, J. Mater. Chem. C, 2013, 1, 5548–5556 RSC.
- H. D. Pham, S. M. Jain, M. Li, S. Manzhos, K. Feron, S. Pitchaimuthu, Z. Liu, N. Motta, H. Wang, J. R. Durrant Bd and P. Sonar, J. Mater. Chem. A, 2019, 7, 5315–5323 RSC.
- H. Lee, K. J. Kim, Y. J. Moon, Y. K. Kim and T. Kim, Org. Electron., 2020, 84, 105816 CrossRef CAS.
- D. Gudeika, J. H. Lee, P. H. Lee, C. H. Chen, T. L. Chiu, G. V. Baryshnikov, B. F. Minaev, H. Ågren, D. Volyniuk, O. Bezvikonnyi and J. V. Grazulevicius, Org. Electron., 2020, 83, 105733 CrossRef CAS.
- S. Heun and P. M. Borsenberger, Chem. Phys., 1995, 200, 245–255 CrossRef CAS.
- D. Luo, X. L. Li, Y. Zhao, Y. Gao and B. Liu, ACS Photonics, 2017, 4, 1566–1575 CrossRef CAS.
- E. Jatautiene, J. Simokaitiene, G. Sych, D. Volyniuk, K. Ivaniuk, P. Stakhira, V. Fitio, H. Petrovska, V. Savaryn, Y. Nastishin and J. V. Grazulevicius, Appl. Mater. Today, 2021, 24, 101121 CrossRef.
- Y. Zuo, X. Wang and D. Wu, J. Mater. Chem. C, 2019, 7, 14555 RSC.
- T. Tsutsui, MRS Bull., 1997, 22(6), 39–45 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc02270b |
| This journal is © The Royal Society of Chemistry 2022 |