
Toward real-world applications: promoting fast and efficient photoswitching in the solid state
Fanxi
Sun
a and
Dongsheng
Wang
 *ab
*ab
aSchool of Optoelectronic Science and Engineering, University of Electronic Science and Technology of China, China. E-mail: wangds@uestc.edu.cn
bInstitute of Electronic and Information Engineering of UESTC in Guangdong, Dongguan 523808, China
First published on 11th May 2022
Abstract
Photoresponsive materials show the superiorities to be controlled by light in a non-contact, efficient and precise manner, which are therefore attractive in the fields of therapy, pharmacology, semiconductor engineering and nanorobot. However, the isomerization of photoresponsive molecules in the solid state is always hindered, because their planar and conjugated nature results in intermolecular aggregation. This problem becomes the key challenge blocking further development of photoresponsive materials toward real-world applications. In this perspective, we discussed the potential of various photoresponsive molecules (e.g. azobenzene, spiropyran, diarylethene, donor–acceptor Stenhouse adduct) to switch in the solid state by analyzing the molecular structure and isomerization process. Moreover, recent strategies and directions in promoting fast and efficient isomerization of photoresponsive molecules in the solid state are highlighted.
 Fanxi Sun | Fanxi Sun obtained his master's degree under the supervision of Prof. En-Hua Yang and Dr Cise Unluer at Nanyang Technological University. He then joined the group of Prof. Yonghao Zheng and Prof. Dongsheng Wang at the University of Electronic Science and Technology of China as a PhD candidate, to investigate the photoresponisve molecules in the solid state. |
 Dongsheng Wang | Dongsheng Wang is an Associate Professor at the University of Electronic Science and Technology of China (UESTC) from 2017. He received his PhD degree from Max-Planck-Institute for Polymer Research (MPIP) in 2017. He is working as the head of MPIP-UESTC partner group. His main research interests are focused on photoresponisve molecules and materials. |
1. Introduction
Light, as an elegant external stimulus, has been extensively investigated in controlling chemical reactions,1 biological processes2 and materials functions3 in the past century. Compared with stimuli, including temperature, pH, electric field, magnetic field, gas and vapor, light exhibits unique advantages from two aspects: (a) the irradiation time and light spot are precisely controllable and (b) non-contact control could be realized. Many important properties of photoresponsive materials could be reversibly switched by light, such as hydrophilic properties (hydrophilicity and oleophilicity),4 optical properties (color, refractive index and transmittance),5 mechanical properties (modulus, hardness and viscoelasticity),6 chemical properties (solubility and polarity)7 and electrical properties (conductivity and dielectric properties)8 (Fig. 1). Therefore, photoresponsive materials are attractive in the fields of biomedicine,9 phototherapy,10 actuator,11 chip manufacturing,12 information storage,13,14 aerospace,15 security16 and microfluidic technology.17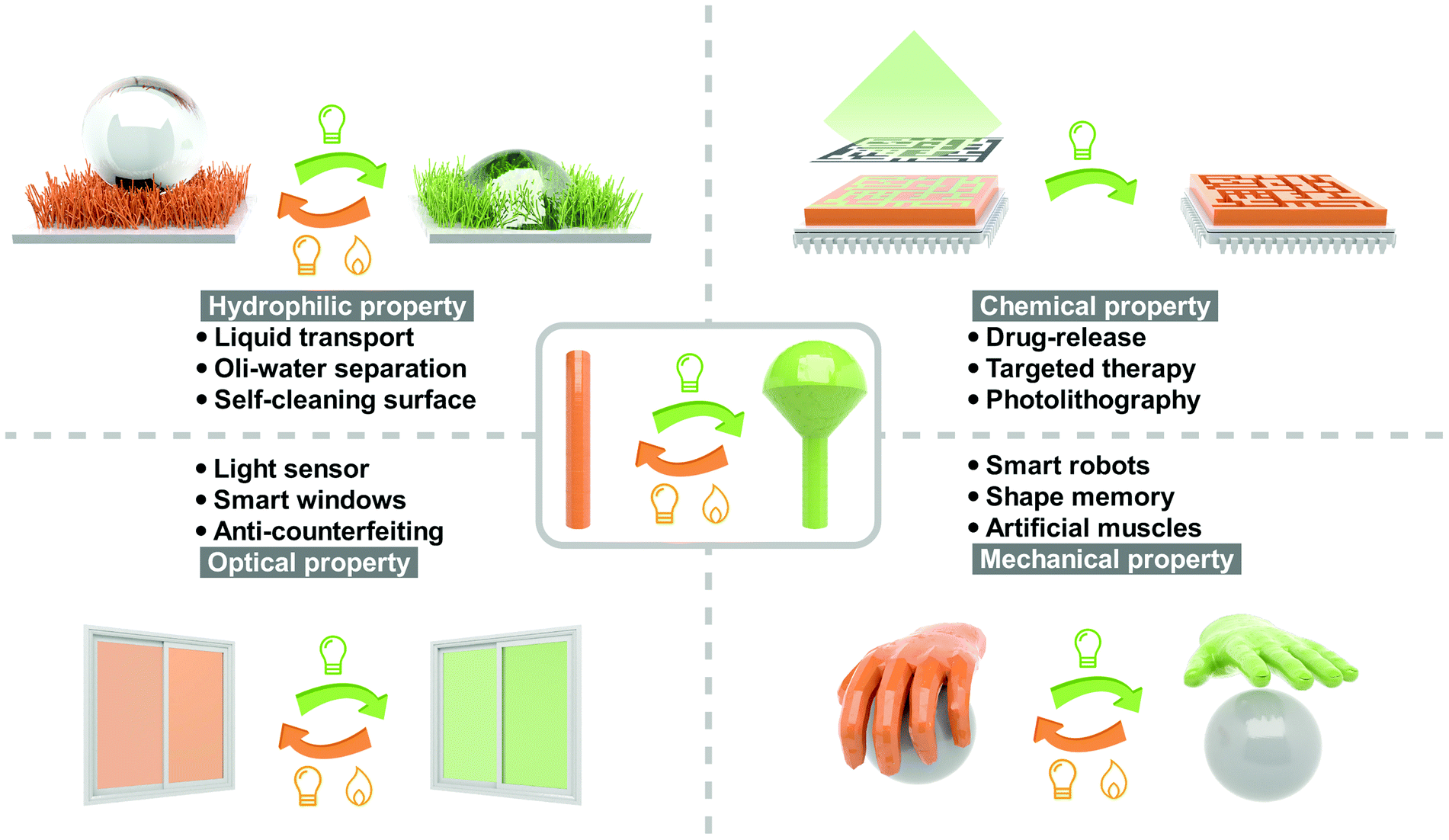 | ||
| Fig. 1 Schematic illustration of the applications of photoresponsive materials. | ||
The light-controllable switching of materials’ properties originate from the isomerization of photoresponsive molecules. Conventional photoresponsive compounds exhibit fast, reversible and efficient isomerization in solution, which is always restricted in the solid state due to π–π stacking of the chromophores and mobility confinement in the dense crystalline packing manner.18 For instance, azobenzene, as the most widely investigated photoswitch, undergoes efficient isomerization between trans and cis in solutions, which is usually hindered in the solid state by the H- (parallel interaction of the chromophores) or J-aggregation (head to tail arrangement of the chromophores).19 This undoubtedly limits the developments and applications of solid-state photoresponsive materials: on the one hand, the slow photoisomerization in the solid state is not conducive to the fast response demand; on the other hand, the long-time irradiation with high intensity may cause the bleaching of photoresponsive molecules. Therefore, seeking to improve the photoisomerization properties in the solid state is important and urgent.
Generally, the isomerization properties of photoswitches in the solid state are closely interrelated to the intrinsic and extrinsic aspects. The intrinsic relationship is contributed by the inherent chemical structure and isomerization mechanism of photoresponsive molecules. The extrinsic reason comes from the surrounded environment, which improves the molecular distribution and avoids intermolecular aggregation. Providing enough free space for isomerization is the philosophy to fabricate solid-state photoresponsive materials with fast and efficient photoresponsiveness. From this perspective, we discuss the potential of various photoresponsive molecules to be switched in the solid state by analyzing the molecular structure and isomerization process. Recent progress in the development of solid-state photoresponsive materials is highlighted.
2. Photoresponsive molecules and isomerization models
Photoresponsive molecules are normally switched between two photostationary states (PSS), by controlling light irradiation. The chemical structure changes through a series of bond rotation and dissociation/formation during the isomerization process, which switches the optical, electronic and geometrical properties of the photoresponsive molecules. The chemical differences between PSS could be identified by characteristic methods such as ultraviolet-visible (UV-Vis) absorption spectroscopy and nuclear magnetic resonance (NMR) spectroscopy. The chemical structure variation is closely interrelated to their isomerization properties in the solid state due to the limited intrinsic mobility. Therefore, photoresponsive molecules with various isomerization models were introduced in this section to discuss their potential in fabricating solid-state photoresponsive materials.2.1 trans–cis isomerization
Sometimes, trans–cis isomerism is also referred to as E–Z (E: Entgegen; Z: Zusammen in German), configurational and geometrical isomerism. The two isomers show the same chemical formula but different configurations. Generally, the substituent groups of cis diastereomer face the same orientation of double bond, which faces the opposite in trans. The trans–cis isomerization results in the variation of planarity, polarity and conjugation property of photoresponsive molecules.19,20![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N transforms into the rotation-free N–N first, followed by the rotation of the C–N–N–C dihedral angle.21 The process is quite different in the inversion theory, where the NN remains, but the C–NN straightens to 180°, generating a linear transition state.22 Under UV light irradiation. The planarity and conjugation of cis Azo are relatively lower than trans because the steric effect of phenyl group results in torsion of diazene. The reversed cis-to-trans isomerization occurs under the treatment by visible light or heat. The isomerization between trans and cis reversibly switches the molecular absorbance, geometrical size and polarity.19
N transforms into the rotation-free N–N first, followed by the rotation of the C–N–N–C dihedral angle.21 The process is quite different in the inversion theory, where the NN remains, but the C–NN straightens to 180°, generating a linear transition state.22 Under UV light irradiation. The planarity and conjugation of cis Azo are relatively lower than trans because the steric effect of phenyl group results in torsion of diazene. The reversed cis-to-trans isomerization occurs under the treatment by visible light or heat. The isomerization between trans and cis reversibly switches the molecular absorbance, geometrical size and polarity.19
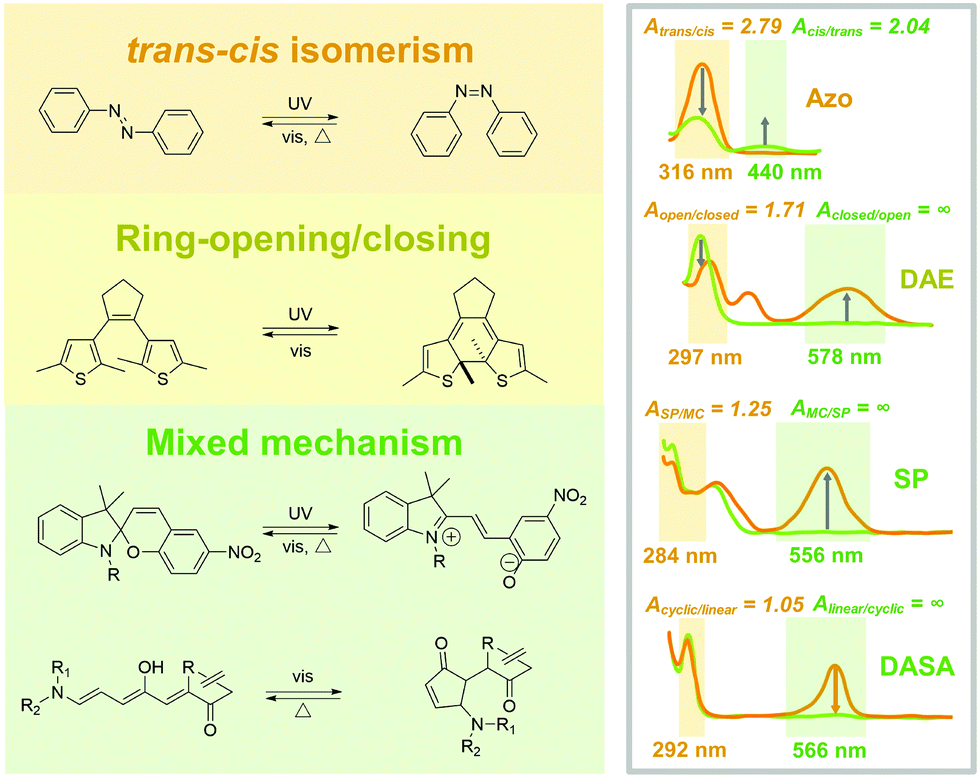 | ||
| Fig. 2 Schematic illustration of chemical structure (left) and absorbance spectra (right) of the PSS of photoresponsive molecules (from top to bottom): azobenzene (Azo), dithienylethene (DAE), spiropyran (SP) and donor–acceptor Stenhouse adduct (DASA). | ||
Typical trans Azo exhibits a π–π* transition band in UV light and n–π* absorption in the visible light region. The trans-to-cis isomerization sharply decreases the π–π* while slightly increasing the n–π* bands. During the photoisomerization process, light irradiation induces electron transition for both isomers and generates an unstable transient state, which further switches to the photostationary state by thermal relaxation. Therefore, the forward and backward isomerizations are always in competition. Studying the difference between the absorbance value at the induced light wavelength of trans and cis Azo (Atrans/cis and Acis/trans) is a quick method to determine the tendency of isomerization. Azo exhibits the Atrans/cis = 2.79 (316 nm) and Acis/trans = 2.04 (440 nm) in good solvents, indicating that trans-to-cis and cis-to-trans isomerization could be effectively induced by UV light and visible light irradiation (Fig. 2).20
However, when applied in a solid, Azo meets some critical problems limiting the photoisomerization: (1) the planarity and conjugated nature of trans Azo contribute to a strong intermolecular π–π interaction, which hinders the necessary molecular mobility for isomerization; (2) isomerization between trans and cis induces large conformational changes, while a free volume of around 120-250 Å is needed,23 therefore, the isomerization is easy to be confined in the packed solids. To address this problem, functional groups could be introduced onto the ortho-position of phenyl, which lowers the planarity of trans Azo and, therefore, relieves the intermolecular aggregation in solids.20
Meanwhile, recent studies have been extended to the derivatives of Azo, such as arylazoisoxazole,24,25 azoheteroarene,26 azopyrimidine,27 and diaryl hydrazones.28,29 For example, in azoheteroarene, one of the benzene rings was replaced with azoarenes, enabling the possible formation of intramolecular hydrogen bonds, which exhibited high stability of cis isomer, solid-state photochromism, and reversible light-induced phase transition.26
2.2 Ring-opening/closing isomerism
Ring-opening/closing isomerism originates from the dissociation and formation of chemical bonds in photoresponsive molecules. The ring-opening and -closing during the isomerization often generates an obvious variation of conjugation for the entire molecules, which sharply switches the color and absorbance.Due to the varied conjugation, a superhigh Aclosed/open = ∞ (578 nm) is calculated for DAE, indicating the high efficiency of backward photoisomerization induced by visible light. Moreover, compared with that of Azo, slight conformation changes exist during the isomerization (1.1 Å on horizon and 0.7 Å on vertical).30,31 Therefore, DAE is attractive to be applied in solid-state photoresponsive materials.
2.3 Mixed mechanisms
Different from Azo and DAE, many photoresponsive molecules exhibit a multi-stage photoswitching process, where the isomerism of trans–cis, ring-opening/closing, and bond-forming/breaking are included. The isomerization procedure based on the mixed mechanism is always complex and involves plenty of intermediates and transient states. In this case, photoresponsive molecules usually require a relatively larger free volume for photoisomerization. Fortunately, due to multiple intermediates, the isomerization property of the photoswitches is closely interrelated to the surrounding physicochemical environment, which has potential to be well-controlled by carefully adjusting the specific transition step.SP shows the conformation in SC under dark with lower ground state energy. The SC-to-MC isomerization is a sequential process of bond dissociation, molecular rearrangement and electron configuration. Briefly, UV light irradiation induces the cleavage of the Cspiro–O bond and follows the rotation of the central C–C bond, which finally isomerizes to the MC state. The isomerization between SC and MC reversibly switches the geometric size and polarity of SP.37
Similar to that of DAE, SP exhibits a high AMC/SC = ∞ (556 nm). Therefore, the MC-to-SC isomerization could be effectively triggered by visible light, resulting in a fast and efficient negative photochromism. However, the obvious conformation changes during the isomerization process might limit the applications of SP in solid-state photoresponsive materials. Especially, the forward SC-to-MC photoisomerization with relatively lower ASC/MC = 1.25 (284 nm) is possible to be limited in solid.38 In this case, increasing irradiation time or intensity might cause photobleaching of SP, which does not benefit the requirement of durability.
Different from SP, linear DASA is a thermally stable isomer, whereas the forward isomerization is triggered by visible light (UV light for SP). 15 intermediates are involved in the isomerization process between linear and cyclic DASA.43 Briefly, visible light triggers the trans–cis isomerization of CC on the triene π-bridge, followed by the formation of the five-membered ring through a 4π-electrocyclic conrotatory rearrangement, which finally isomerizes to the cyclic form via tautomerization. The formation of the five-membered ring is a critical step in the isomerization process. This step is under the assistance of the intramolecular proton transfer from hydroxyl (triene π-bridge) to carbonyl (electron-withdrawing moiety).44–46
Compared with the other photoswitches, DASA exhibits the advantage that the forward isomerization is under control of the visible light. Moreover, the superhigh Alinear/cyclic = ∞ (566 nm) indicates that linear-to-cyclic isomerization could be effectively induced by visible light.47 However, the backward isomerization of DASA only occurs under heat, making it difficult to design photoresponsive materials fully controllable by light. Although the conformation change of DASA during the isomerization process is significant; the photoisomerization properties still have potential for improvement by catalyzing the critical step, such as by the intramolecular proton transfer.
3. Strategy to improve photoisomerization in solids
Isomerization of photoresponsive molecules requires a certain amount of free space, which is limited in solid-state materials (Fig. 3). Therefore, the philosophy to achieve fast and efficient photoswitching in the solid state is to improve the molecular distribution and supply the necessary spatial mobility.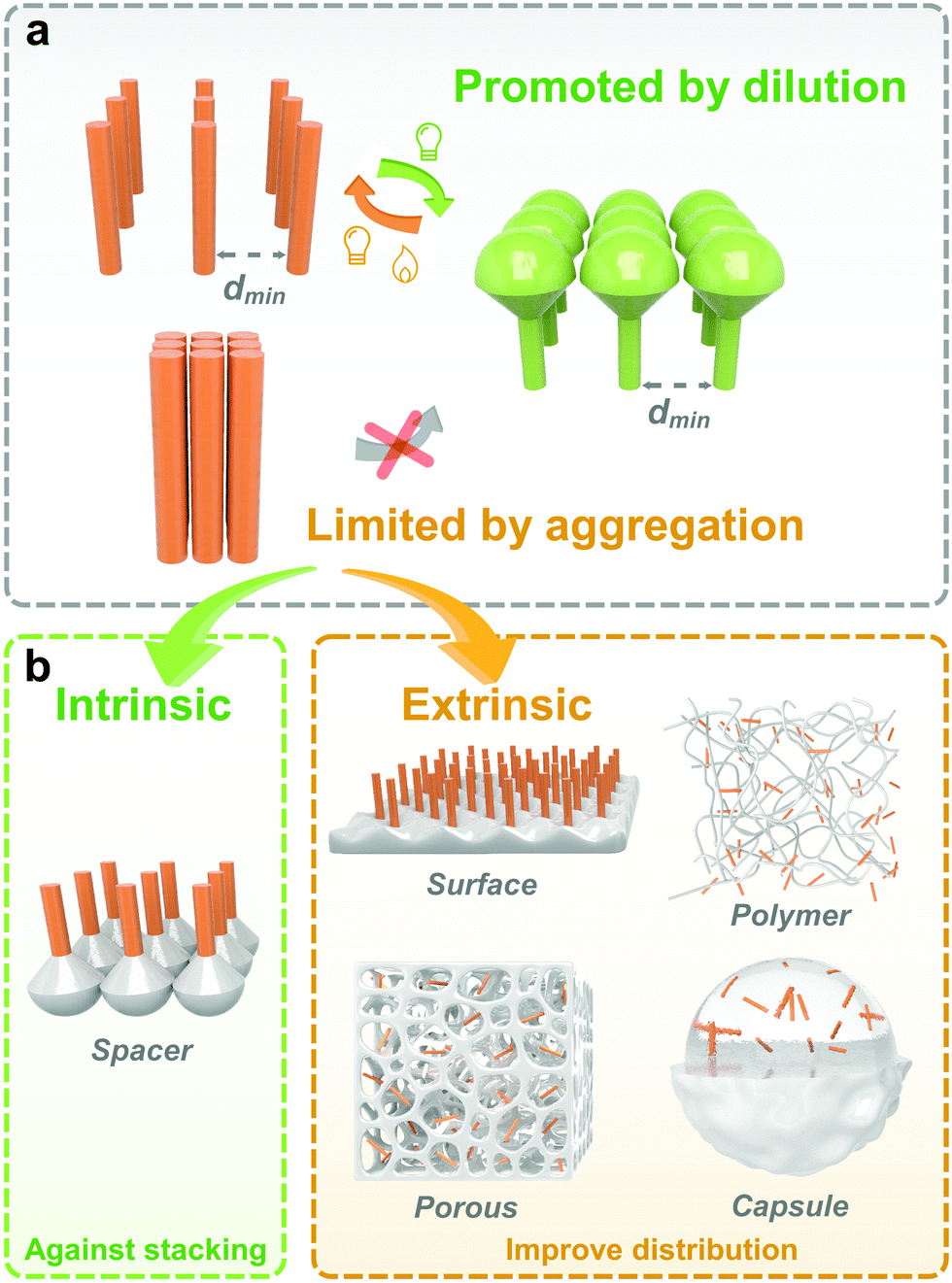 | ||
| Fig. 3 (a) Schematic illustration of the isomerization of photoresponsive molecules in diluted and packed distributing states; (b) intrinsic and extrinsic strategies to improve photoisomerization in the solid state. | ||
The molecular distribution could be improved either through intrinsic or extrinsic strategies. Intrinsic methods include the selection and chemical modification of photoresponsive molecules. Functional groups and spacers are introduced to improve molecular mobility. For the extrinsic methods, a second phase is often introduced to act as a “solvent-like” phase, which prevents intermolecular aggregation and promotes photoisomerization.
3.1 Intrinsic methods: introducing functional groups and spacers
For the photoresponsive molecules with a significant conformation change during the isomerization process, introducing functional groups and spacers helps the distribution of the photoswitches in the solid state and decreases intermolecular aggregation (Fig. 4). The introduction of bulky groups changes the planarity of photoswitches (i.e., Azo), which weakens the π–π stacking in the solid, making the isomerization free to occur.20 Moreover, the grafting of long and soft alkyl chains increases the distance between the photoresponsive molecules in the solid state.48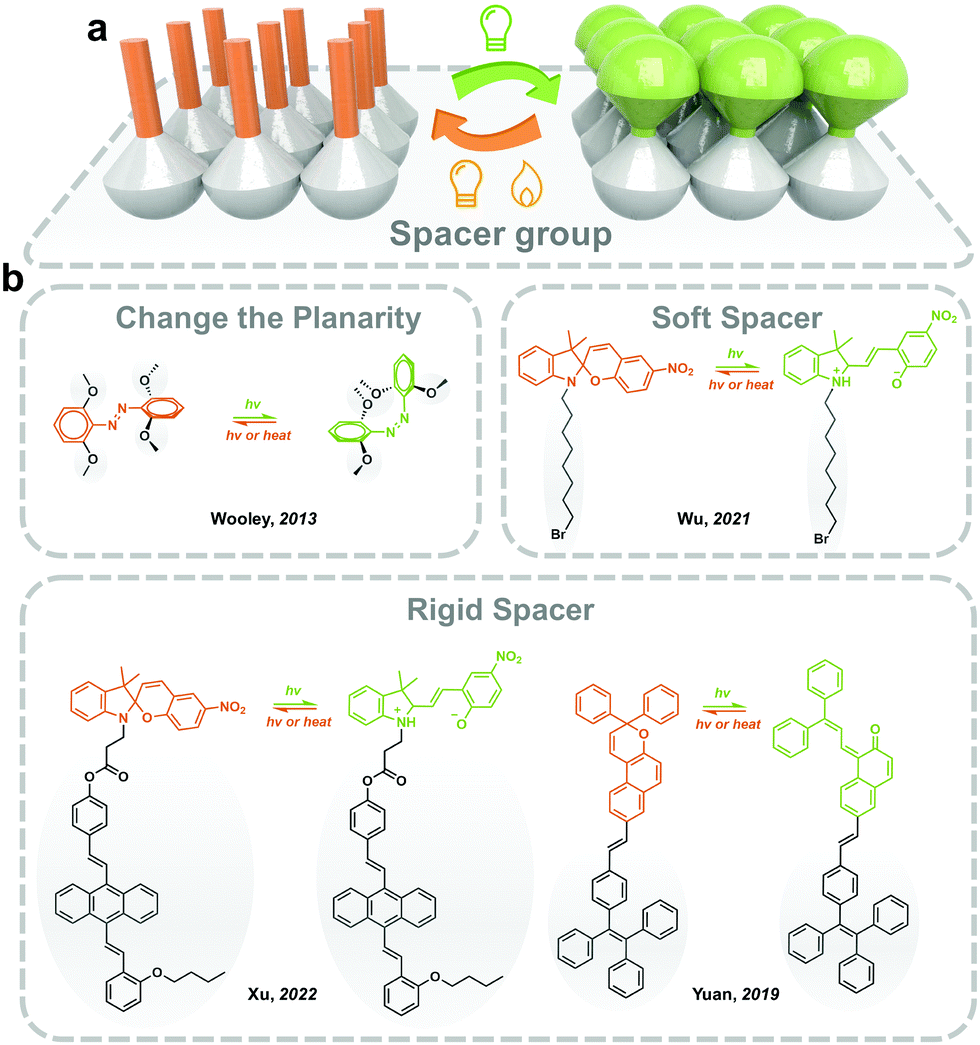 | ||
| Fig. 4 (a) Schematic illustration of the strategy to modify photoswitches with spacer group; (b) schematic illustration of examples of reported spacer groups facilitating photoisomerization in the solid state. | ||
Generally, the planarity and conjugated structure of trans Azo induces intermolecular aggregation in the solid state and lowers the molecular energy of trans isomer, which hinders the trans-to-cis isomerization. Woolley and coworkers reported substituting methoxyl groups at the tetra-ortho-position of Azo (mAzo), which distorts the conjugated structure of trans isomer, generating a certain dihedral angle between the aromatic rings. The mAzo, therefore, exhibits weaker π–π stacking in solid.20 Meanwhile, substitution by halogen (i.e. F and Cl) reported by Hecht and coworkers showed similar results. Moreover, the ortho-substitution of functional groups obviously red-shifts the absorbance spectra and results in a separation of the n–π* transition band between trans and cis isomers. Therefore, the trans-to-cis isomerization could be induced by long-wavelength-light (i.e. visible and near-infrared light), which benefits the durability of solid-state photoresponsive materials.49 Wu and coworkers reported applying mAzo in photoresponsive polymeric materials (which will be discussed in Section 3.4).50 A similar modification strategy also works for azopyrimidine, a derivative of Azo where phenyl is replaced by pyrimidine. The introduced methoxyl lowers the melting point of azopyrimidines, ensuring partial trans-to-cis isomerization in the solid state under the control of UV light irradiation.27
Introducing soft and non-planar groups with large geometrical sizes could effectively eliminate intermolecular compaction in solids. One of the most widely applied spacers is the alkyl chain. Wu and coworkers reported grafting flanking alkyl chains with different lengths (n = 4, 8 and 12) onto the nitrogen of the indole ring to prepare a T-type SP, which exhibits fast and efficient photochromism in solids. The photoisomerization of the T-type SP could be precisely controlled by introducing a halide at the end of the alkyl substitute. The intermolecular electron-donating effect formed between the halide and the cationic nitrogen of the indole ring, stabilizes the MC isomer in solid.48 In addition, tetraphenylethene (TPE) has been introduced into photoresponsive molecules for providing necessary free space as well as achieving aggregation-induced emission (AIE) property. Tian and coworkers functionalized TPE to the indole nitrogen of SP through a flexible covalent ester chain. The twisted TPE offers enough molecular mobility for the isomerization of SP in solids.51 They also reported fabricating a solid-state SP photoswitch with light-controllable fluorescence switching between three colors.52 Similar approaches are also applied for prompting the isomerization of naphthopyran and fulgide in the solid state.53,54
Supramolecular host-guest complex with high flexibility but a relatively stable chemical structure could provide sufficient intrinsic mobility for photoresponsive molecules. Lin and coworkers fabricated a series of photoresponsive rotaxanes using SP as one of the stoppers of the axle. Fast SC-to-MC isomerization of SP occurs in the solid state when the macrocyclic molecule exists on the axle. With the introduced TPE in the macrocyclic molecule, reversible fluorescence switching controlled by UV and visible light irradiation was realized in the solid powder form.55
(1) Further improving the kinetics of photoisomerization in solid. Modified with a bulky spacer or placeholder group enables photoswitches' free volume to isomerize. However, the intermolecular aggregation is not completely prevented in solids, and the photoswitches still need to overcome the interaction to reach another photostationary state. Therefore, the kinetics are not as high as that in the solution. Introducing the well-controlled supramolecular system might be applicable to further improve the flexibility of the surrounding aggregation, while the combined model of photoswitches and supramolecules needs to be carefully studied and simulated.
(2) Morphology collapses during repeated isomerization. In crystalline photoresponsive materials, notable morphology change on the crystal surface could be observed by double-probe atomic force microscopy after repeated photoisomerization.56 This phenomenon happens even for photoresponsive molecules with slight conformation changes (i.e. DAE), which will be more obvious for the photoswitches with significant conformation changes (i.e. azopyrimidine derivative).27 This problem might contribute to uncertainties in practical applications, which is difficult to be solved without the assistance of a fixed framework.
3.2 Extrinsic methods: assembling a monolayer on the surface
The formation of a monolayer on the surface helps the dispersion of photoresponsive molecules on the x–y plane while preventing the stacking at the z-axis, which provides enough free space for photoisomerization (Fig. 5). Moreover, the molecular density on the x–y plane could be precisely controlled by applying spacer molecules. The photoresponsive molecules are introduced on the surface of the substrate either by chemical anchoring or physisorption. Surfaces with various chemical environments, including glass57 and silicon wafers,58 metal59 and metallic oxide,60 polymers61 and two-dimensional materials62 have been widely used as the substrates to reveal the isomerization property of photoswitches. Meanwhile, the hydrophilicity, optical property63 and conductivity64 of the surface could be controlled by light. | ||
| Fig. 5 (a) Schematic illustration of the strategy to assemble monolayer on surface; (b) schematic illustration of examples of reported methods to form monolayer of photoresponsive molecules on surface. | ||
Gold as a chemically inert metal shows a stable surface, which is attractive for the immobilization of photoswitches. Molecules with functional groups, including thiol and carboxyl are used as the linkers between the gold surface and photoresponsive molecules. Zenobi and coworkers introduced a series of hydrazone monolayers onto the gold surface by varying the lengths of the thiolate linkers. The trans–cis photoisomerization of hydrazone improves with the increasing carbon spacer length, which saturates after the spacer length reaches a certain value.67 This indicates that the interaction between the surface and photoresponsive molecules closely affects the isomerization property.
As discussed above, silane is reactive with a surface abundant in hydroxyl groups, and thiol groups work on the gold surface. A general method for introducing a monolayer of photoresponsive molecules involves fabricating an adhesive layer with functional groups on the substrate's surface. Polydopamine (PDA) exhibits strong adhesiveness to most surfaces, including chemically inert materials such as polytetrafluoroethylene and polydimethylsiloxane (PDMS).68,69 The formation of PDA generated amino groups on the surface, which could be used for the immobilization of photoswitches.70 Wang and coworkers reported fabricating a PDA layer on the surface of silica nanoparticles, followed by grafting DASA through the reaction with intermediates. DASA exhibits obviously improved photoisomerization between linear and cyclic molecules compared with those in thin films.71 However, due to its dark brown nature, the optical switching on the PDA-coated surface is not significant.
The aggregation of photoresponsive molecules on the z-axis is solved by forming a monolayer on the surface. However, the molecular distribution on the x–y plane also plays an important role in the photoisomerization on the surface. Without the limitation of grafting density on the x–y plane, the tightly “shoulder to shoulder” molecular arrangement would also hinder the photoisomerization process. The aggregation on the x–y plane could be solved by introducing placeholder groups and fixing the anchor point sites aforehand in a diluted manner. Nakagawa and coworkers involved a macrocyclic amphiphile tethering Azo unit to overcome the x–y plane aggregation. The crown conformer (o-carboxymethylated calix [4] resorcinarene) on the silica plate ensured sufficient free volume for the trans–cis isomerization of the tethered Azo, obtaining 87% of cis Azo under the irradiation of UV light.72 Innovatively, Dai and co-workers modified Au NPs with capped cyclodextrin first, which was used for trapping the Azo terminated poly(N-isopropylacrylamide) by host-guest interaction. The catalyst with tunable catalytic activity was prepared under the irradiation of UV and visible light.73
By coordinating platinum(II) to both ends of an Azo derivative, Han and coworkers reported deposited Azo on the surface of graphene to fabricate a diluted monolayer. The light-triggered mobilization of platinum was observed by annular dark-field scanning transmission electron microscopy, indicating that the photoisomerization of Azo overcomes the intermolecular van der Waals interactions.62 Similar non-covalent approaches are reported for investigating the photoisomerization mechanism on the surface of carbon nanotubes.74 Gopakumar and coworkers immobilized carboxyl-functionalized Azo and arylazopyrazole on highly oriented pyrolytic graphite via hydrogen bonding and investigated the molecular arranging behavior during the trans–cis isomerization. The configuration changing on the free phenyl was clearly observed by scanning tunnel microscopy.75 Nørgaard and co-workers designed a well-defined molecular junction by assembling a dihydroazulene monolayer, which was sandwiched between Au/Ti (bottom) and transparent reduced graphene oxide (top). The solid-state molecular junction could switch the conductance of the device by controlling light and heat.64
(1) It is awkward to control the density of a monolayer on the surface. On the one hand, the diluted monolayer ensures sufficient molecular mobility and generates fast and efficient photoswitching on the surface. However, the photoswitching properties of the monolayer are finite due to the low content of the photoresponsive molecules. On the other hand, increasing the density of photoresponsive molecules causes aggregation on the x–y plane, which limits the photoswitching on the surface. To overcome this shortage, choosing and modifying photoswitches to improve the differential of the properties between photostationary states is helpful. For example, introducing hydrophobic moieties (i.e. –CF3) at the end of Azo switches the wettability on the surface to a wider range. When the Azo is in trans, the hydrophobic group heads up and generates a hydrophobic surface, which hides into the monolayer and increases the hydrophilicity when Azo is in cis.57 Moreover, choosing SP and DASA with zwitterionic photostationary states is also useful.
(2) Durability is an important issue that cannot be ignored. In most cases, the monolayer of photoswitches is formed on the outermost surface of the substrate, which therefore must face the mechanical force, air, and humidity during the practical application. Due to the low content of photoresponsive molecules, the monolayer is easy to be damaged, which induces the loss of photoswitching properties on the surface. One strategy to solve this problem is fabricating a protecting layer on the top of the monolayer. Moreover, damages due to surface scratches and other mechanical forces could be alleviated by some rational engineering designs on the substrate.76
3.3 Extrinsic methods: materials with porous structure
Materials with porous structures exhibit high porosity and inner space, enabling them able to carry and immobilize goods, including gas,77 catalysts,78 drugs79 and photoswitches.80 Because of the large specific surface area, photoresponsive molecules embedded in the pores could be well distributed, forming mono or few layers on the surface of skeletons (Fig. 6). This is similar to the transition from two-dimensional to three-dimensional space, the monolayer formed on the surface of the substrate (discussed in Section 3.2) extends and crimps plenty of times and finally forms the photoresponsive molecules loaded porous materials. Beyond this, compared with the substrate surface, which exposes the outermost layer to external environments, the skeletons of porous materials provide a natural protecting barrier for the inner phase. However, the porous structure does not always improve the photoswitching in solid. The small pore size also hinders photoisomerization because of the confinement environment. Therefore, the physicochemical environment of pores should be well-controlled for fabricating solid-state photoresponsive materials.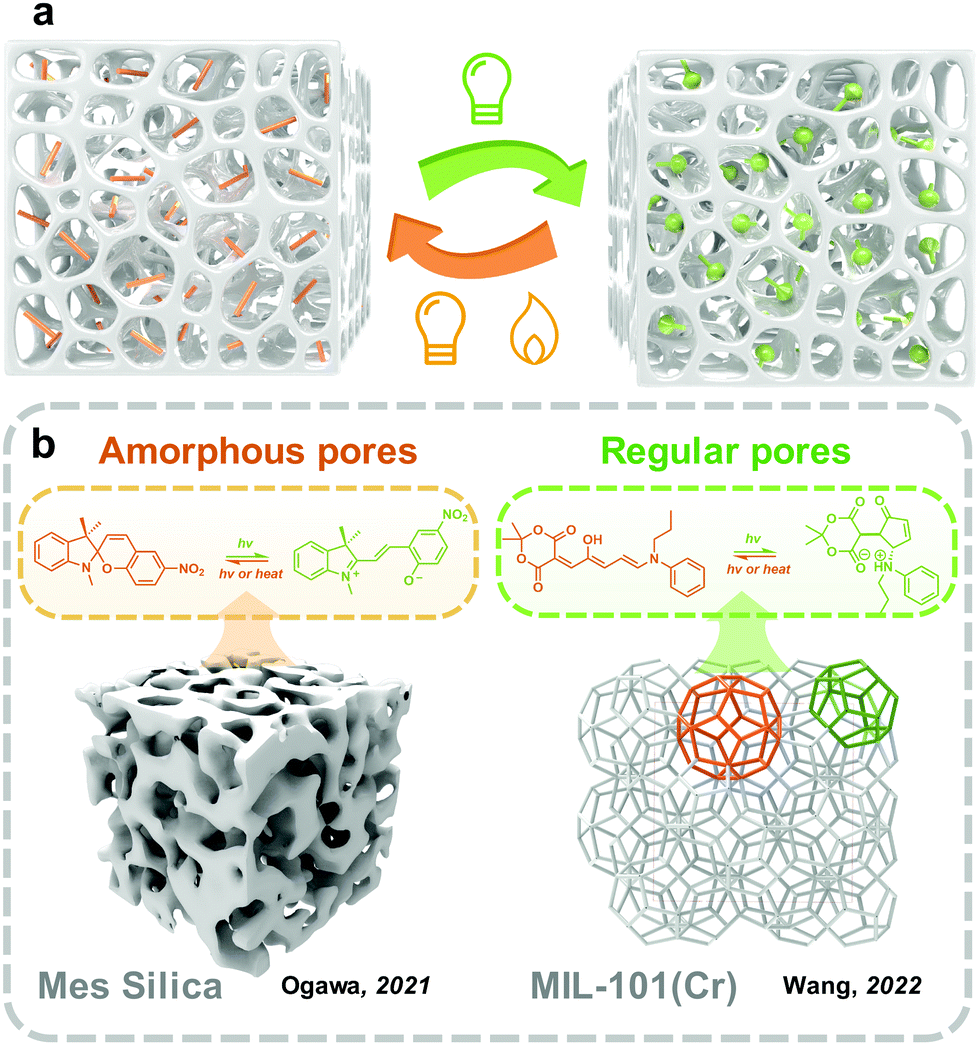 | ||
| Fig. 6 (a) Schematic illustration of the strategy to embed photoswitches into porous structure; (b) schematic illustration of examples of reported photoswitches in amorphous and regular pores. | ||
Mesoporous silica as a hard porous substrate has been extensively applied in many applications including catalysis,85 membrane technology,86 and controlled release.87 The fabrication of mesoporous silica materials through the sol–gel process is convenient and low-cost.88 In photoswitches were loaded into the nanopores of mesoporous silica to fabricate solid-state photoresponsive materials in recent years.89,90 The integration of photoresponsive molecules is similar to the fabrication of the monolayer on the surface discussed in the above section. Ogawa and coworkers reported embedding MC SP in the nanopores of mesoporous silica through a liquid assistance method.91 This hybrid material was further migrated to two organophilic clays, which were also used as a surfactant to control the molecular diffusion in the nanopores and further alleviate the hygroscopic phenomenon during applications. With the assistance of large free volume provided by mesoporous silica, the confined MC-to-SC isomerization efficiency in the solid state was promoted to 92%, which was further increased to 98% with the assistance of organophilic clay, but there was a slight decrease in the rate constant of SC-to-MC isomerization. Andrieu-Brunsen and coworkers grafted SP through the reaction of aminosilane onto the internal surface of mesoporous silica films.92 The SP in porous silica film exhibits fast forward photoisomerization under UV irradiation within 30 s. As a comparison, the authors also prepared a nonporous dense silica film and grafted it with a single monolayer of SP under the same condition. The isomerization of the SP in the nonporous film was non-detectable. Besides, the SP grafted porous silica film further realized the elegant surface water control and gating technology under UV and visible light irradiation.
Polymeric materials with porous structures have been applied for supporting and dispersing photoswitches, such as sponges,93 nanofibers94 and liquid crystal networks.95 These integrated materials have multiple application scenarios, including oil-water separation,93 heavy metal removal96 and wearable UV sensors.97 Lu and coworkers reported immobilizing SP-contained polymers onto the surface of a melamine sponge. The SP-modified sponge showed the SC-to-MC isomerization accomplished in 30 min under the irradiation of UV light (λ = 365 nm), which exhibited a color change from pale pink to deep purple. Although no more optical properties were discussed, the SP-modified sponge achieved an obvious reversible wettability switching between amphiphilic and superhydrophobic.93 Xiang and coworkers coated SP-grafted PDMS to a melamine sponge through dip-coating and thermocuring technology.84 The SC-to-MC isomerization was accomplished in 160 s under UV light irradiation, while the dark relaxation took more than 70 min.
In addition to the above mainstream substrates, photoswitches could be immobilized on the cotton fabrics. Dan and coworkers anchored SP on the cotton fabrics through thiol-ene click chemistry.63 The pale-yellow cotton fabric immediately turns purple within 10 s irradiation of UV light (λ = 365 nm) (SC-to-MC isomerization). Meanwhile, the purple cotton fabric showed a fast color fading rate under dark (5 min), green light irradiation (within 50 s) and heating (90 °C for 5 s).
The first report on loading photoswitches into the nanopores of the molecular sieve was in 1997 by Caro and coworkers.102 Azo loaded in zeolites showed alignment and was parallel to straight nanochannels because of the rod-like conformation. Since then, the strategy of using zeolites as the substrate to fabricate solid-state photoresponsive materials has been widely investigated.103–105 Although the concept of MOFs was proposed in 1995,106 the first example incorporating photoswitches and MOFs was reported in 2010 by Fujita and coworkers.107 The stilbene was soaked into the nanopores of [{(ZnI2)3(tris(4-pyridyl)triazine)2}·x(C6H5NO2)]n. In this work, MOFs behave as a catalyst to promote the photoisomerization of stilbene in solutions, and the photoisomerization in solid-state was not investigated.
Till now, MOFs/COFs have become one of the most promising candidates for integrating photoswitches. Several reviews have summarized the developments and applications of the solid-state photoresponsive materials fabricated through this strategy.108–112 Shustova and coworkers classified the embedding of photoswitches in three methods, guest, linker and backbone (Fig. 7). Photoresponsive molecules as the guests are directly loaded into the nanopores of MOFs by soaking,113 subliming,114 or grinding.115 These convenient methods are applicable to most photoswitches. Photoresponsive molecules with relatively low melting points (e.g. Azo and SP) could be embedded into the MOFs using melting or gas phase methods (sublimation and resublimation), which was first reported by Kim and coworkers.116 This method involved systematic studies by Ruschewitz and coworkers in the past decade while investigating the isomerization properties of Azo, fluorine-substituted Azo, SP and spirooxazine in the nanopores of MOFs.117–120 Ke and coworkers introduced fabricating non-interpenetrated single-crystalline hydrogen-bonded crosslinked organic frameworks by heteromeric carboxylic acid dimers, termed HCOF-101.121 It is the first time that carboxylic acid building blocks are hydrogen-bonded to form a porous crystalline network. Hydrazone was introduced into the nanopores of HCOF-101, which achieved 70% trans-to-cis isomerization under 442 nm light irradiation. To further improve the distribution, photoresponsive molecules could be introduced as a linker or backbone. By partially confining the photoswitches, the intermolecular aggregation could be further excluded. However, these methods are suitable for the photoresponsive molecules with tiny conformation variation during isomerization, such as naphthalenediimide,122 DAE123 and viologen.124 These molecules usually show fast (<30 s) and efficient (>85%) photoisomerization in the nanopores of MOFs/COFs.125,126
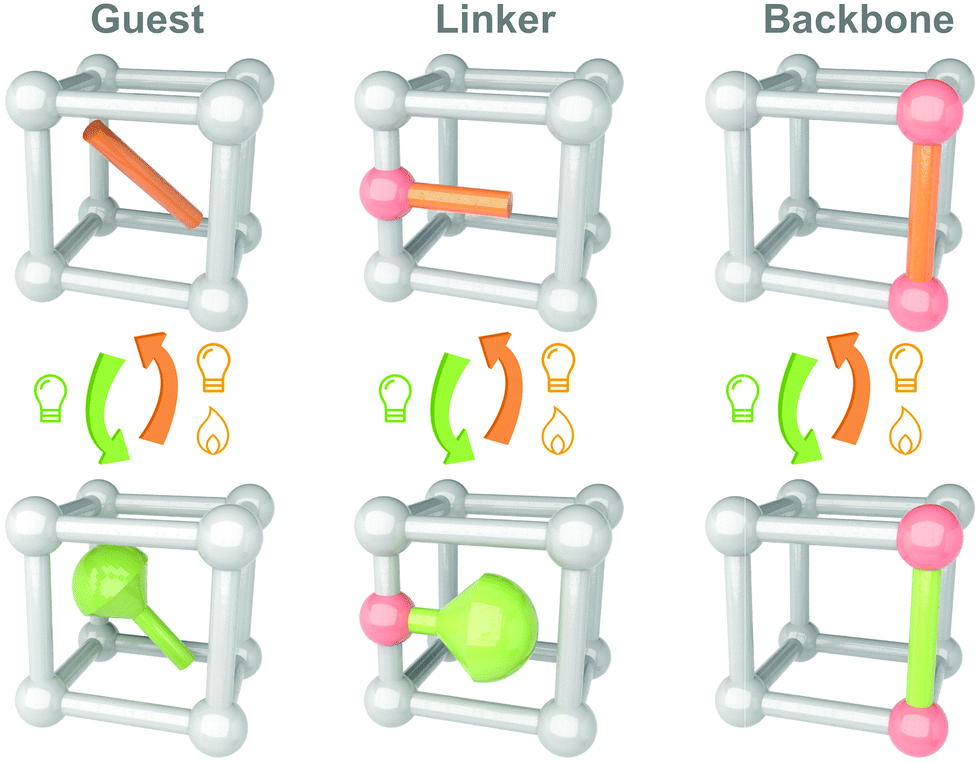 | ||
| Fig. 7 Schematic illustration of the strategy to integrate photoswitches with MOFs/COFs as guest, linker and backbone. | ||
Photoresponsive molecules loaded in the nanopores of MOFs are surrounded by the skeletons, therefore, MOFs are thought to be “solvent-like”.119 In this case, the isomerization properties of photoswitches are closely affected by the physicochemical environment in the nanopores, including the geometrical size, morphology and polarity. Wang and coworkers reported loading DASA with various electron-donating and -withdrawing moieties into the nanopores of two MOFs (MIL-68(Al) and MIL-101(Cr)) with different parameters. DASA exhibits hindered linear-to-cyclic and promoted cyclic-to-linear isomerization in MIL-68(Al), while the isomerization properties in MIL-101(Cr) follow similar rules in solutions. The interaction between photoswitches and skeletons of MOFs was demonstrated to be correlated with the isomerization kinetics and efficiency.113
The embedding of photoresponsive molecules into the nanopores of COFs is similar, Jiang and coworkers prepared photoresponsive COFs by synthesizing aldehyde-functionalized DAE via the Suzuki coupling reaction, which was functionalized as the backbone. Furthermore, a porphyrin photosensitizer was introduced into the COFs. In addition to the as-excepted reversible photochromism and fluorescence-switching functionality of the solid-state materials, the COFs exhibited photocatalytic activities toward singlet oxygen evolution.127
(1) Further improving the durability of the photoswitches in the nanopores. Although protected by the skeletons of porous materials, the mechanical and thermal stability could improve, and the durability of photoresponsive molecules is worthy to be further improved to ensure practical applications. Firstly, the diffusion of photoswitches occurs during the repeated isomerization process and the photoresponsive molecules tend to vibrate to the state with the lowest energy. This leads to the intermolecular aggregation in the nanopores, locked by the rigid “traps” or escape from the porous structure.128,129 Therefore, while providing sufficient free space for isomerization, excessive molecular mobility needs to be limited. This might be solved by introducing surfactants, immobilizing photoswitches on the skeletons and nanopores designing (i.e. window control, post-modification). In addition, due to the hydroxyl-enrichment on the surface, many materials with porous structures are hydrophilic, which have the potential to uptake water molecules from the air.130,131 However, this could be a “disaster” for some moisture-sensitive photoswitches. For example, linear DASA coordinates with water molecules and stabilizes at a cyclic state, which does not switch back in the dark.132 This needs to be considered at an early stage of design.
(2) Tunning the isomerization properties of photoswitches by controlling the physicochemical environment in nanopores. It could be interesting to understand the relationships between the isomerization properties of loaded photoresponsive molecules and the physicochemical environment in nanopores. For the materials with regular nanopores, the physicochemical environment could be well-designed and controlled. For example, the size, morphology and polarity of the nanopores of MOFs could be tuned by varying the chemical structure of organic ligands.133 This is helpful for the systematic fabrication of solid-state photoresponsive materials.
3.4 Extrinsic methods: complexing with polymers
Incorporating photoswitches with polymers is a convenient and efficient strategy to realize the mass production of solid-state photoresponsive materials. Long and soft polymer chains help the dispersion of photoresponsive molecules and improve photoisomerization in the solid state (Fig. 8). However, due to the mobility of polymers, the intermolecular aggregation of photoresponsive molecules could not be completely prevented. The introduction strategies of photoswitches into polymeric materials mainly include chemical modification and physical complexing.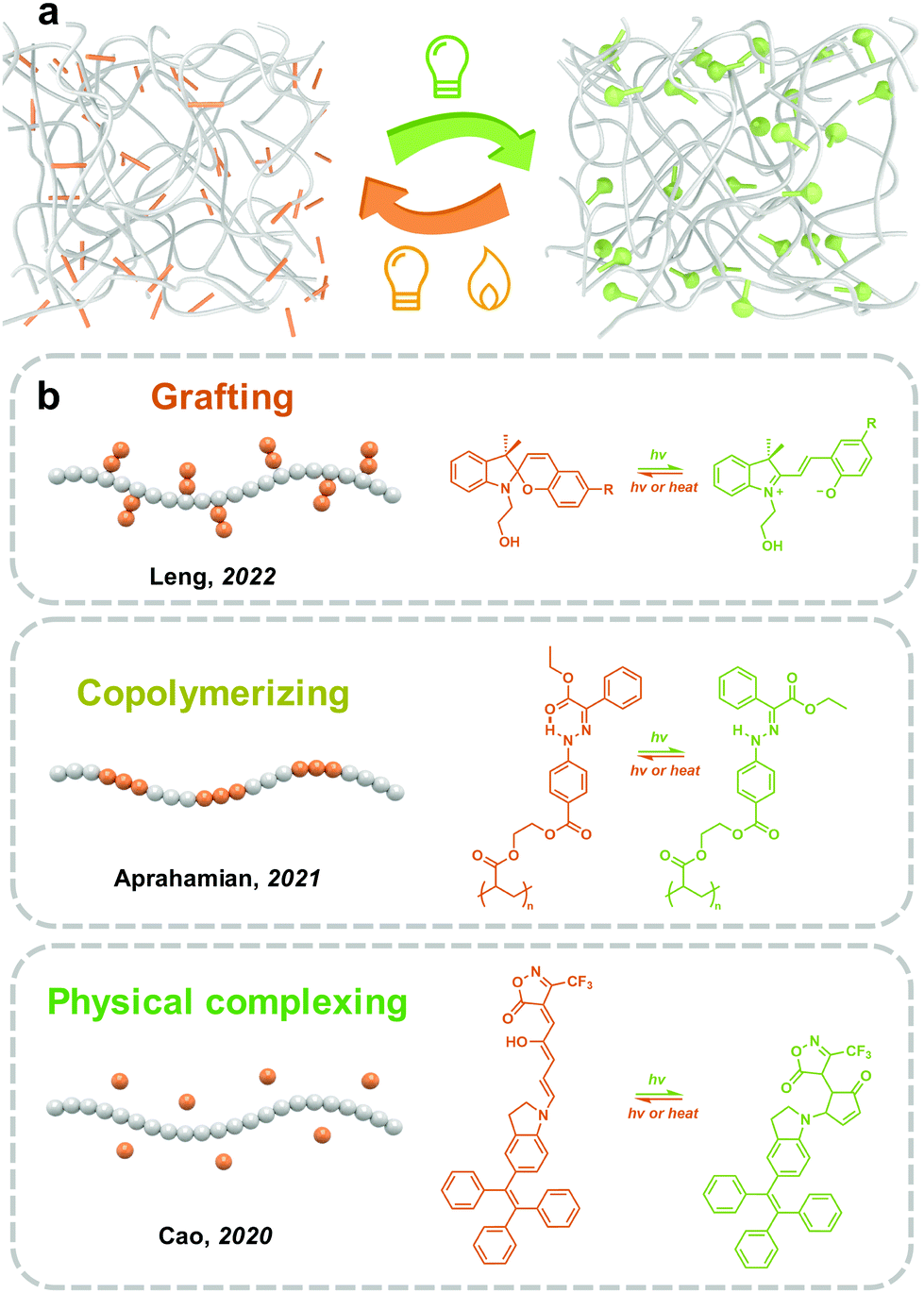 | ||
| Fig. 8 (a) Schematic illustration of the strategy to complex photoswitches in the polymers; (b) schematic illustration of examples of introducing photoswitches into polymer matrix by grafting, copolymerizing and physical complexing. | ||
Studies on the isomerization properties of photoswitches in polymers are mainly focused on solutions because the distribution assisted by polymers does not completely prevent the intermolecular aggregation of photoresponsive molecules. Wu and coworkers reported synthesizing photoresponsive polymers by Azo-modified monomers. Two Azo molecules with planar (Azo) and non-planar (mAzo) trans isomers were investigated.50 While the Azo- and mAzo-polymers exhibited similar trans–cis isomerization properties in solutions, the trans-to-cis photoisomerization of Azo-polymers was significantly hindered in films due to the intermolecular aggregation of Azo. On the other hand, because of the non-planar chemical structure, the forward photoisomerization of mAzo-polymers in films is fast and efficient.50 These indicate that the design of the chemical structure of photoswitches is important for the fabrication of solid-state photoresponsive materials.
However, DASA with photoisomerization mechanisms similar to SP exhibits irreversible isomerization in PMMA. Cao and coworkers synthesized the third generation of DASA using tetra-phenylethylene as the electron-donating moieties, which showed reversible and efficient linear-to-cyclic isomerization in solutions. After doping into PMMA films, the linear-to-cyclic isomerization was accomplished in 30 min under visible light irradiation. However, the backward isomerization was difficult to proceed to the initial state.142 Besides, irreversible isomerization appears to be a common phenomenon of DASA embedded in the polymer matrixes.143
Photoresponsive polymer materials with a light-controlled phase transition have attracted the interest of researchers in the past decade.24–26 Similar to temperature changes that induce melting and freezing of water, light could trigger the phase transition of solid-state polymers by shifting the Tg and melting point (Tm). The solid-to-liquid phenomenon of Azo derivative was first observed by Okui and Han in 2012.144 However, polymers as chain-dominating materials are difficult to crystallize as small molecules.145 In 2017, a series of Azo-containing polymers with obvious solid-to-liquid transition were prepared by Wu and co-workers. Azo with a flexible spacer as a side chain was grafted to polyacrylate, namely azopolymer. The Tg of trans azopolymer is 48 °C, which decreases to −10 °C after UV light irradiation.146 The huge decrease in Tg goes across the room temperature (25 °C), which therefore induces a solid-to-liquid transition and switches the azopolymer from the glassy state to the rubbery state. In 2021, Aprahamian and co-workers prepared a series of hydrazone-containing polymers with multistage reversible Tg, ranging from 21–37 °C to 86–103 °C.147 Till now, phase-transition materials have been widely used in transfer printing, self-healing148 and solar- and thermal energy harvesting.149,150
The phase transition phenomenon of Azo-containing polymers during the isomerization may increase the molecular mobility of photoswitches, which generates a self-accelerating effect the photoisomerization. Venkataramani and co-workers synthesized a series of derivatives of arylazoisoxazole, and demonstrated reversible trans–cis isomerization (4-fluoro-, 3-bromo- and 4-methoxy-substituted derivatives) in the solid state. Besides, partial phase transition upon UV light irradiation could be observed, due to the variation of π–π interactions, N⋯H–C and interactions between azo-N and benzene ring during the process of isomerization.26 Similar analogs of arylazoisoxazole with different alkyl chains (n = 1, 5, 6, 9) were applied as photoresponsive adhesives by Ravoo and co-workers. Owing to the light-induced reversible phase transition, a fast UV response cohesive change reached within 0.5 s.24 Moreover, the azopolymer could also be applied as a light-switchable polymer adhesive, which can be implemented under both dry and wet conditions.151
(1) Toward photoresponsive plastics. The photoresponsive polymeric materials are limited in polymer solutions, films and soft matters (hydrogels and elastomers) because they could support relatively high molecular mobility. However, mechanical strength is one of the basic requirements for polymeric materials in real-world applications. Plastics with high tensile modulus (>1 GPa) meet plenty of applications in human life, such as decoration, packaging and building.152 Therefore, fabricating photoresponsive plastics, in the other words, achieving polymeric materials with macroscopic hardness and microscopic softness is important.
3.5 Extrinsic methods: solvent-in-solid
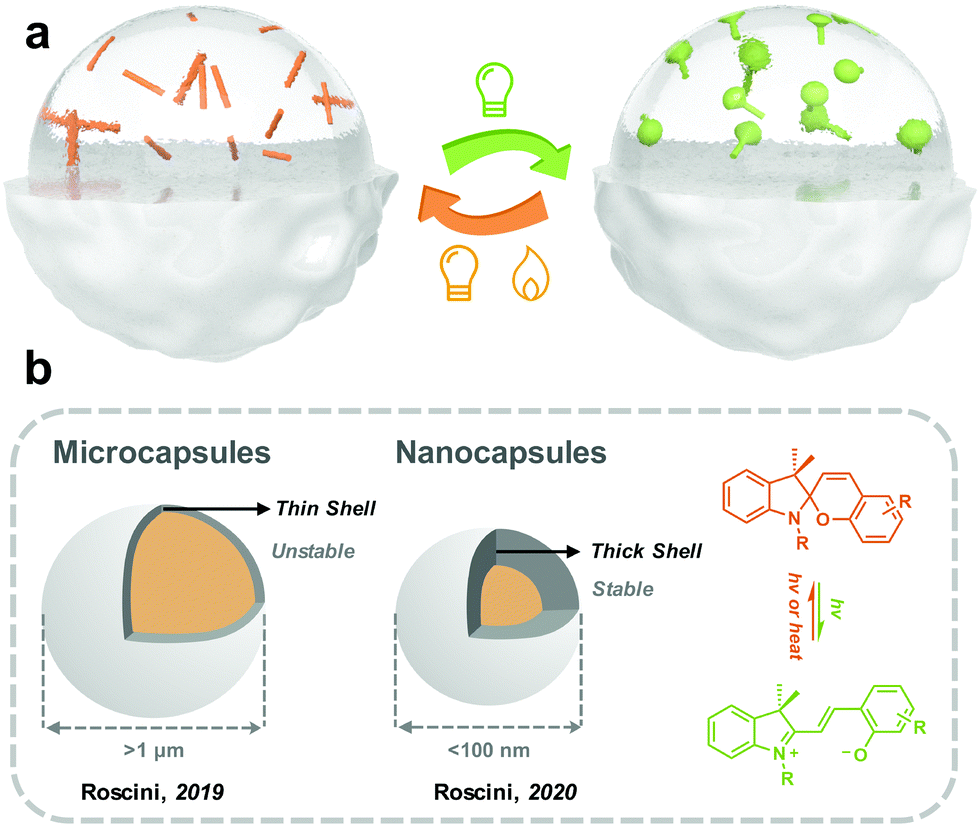 | ||
| Fig. 9 (a) Schematic illustration of the strategy to load photoswitches into the solvent-containing capsules; (b) schematic illustration of examples of reported photoswitches in solvent-containing microcapsules and nanocapsules. | ||
The outmost layer of the microcapsules is usually made of hard polymers with good transparency, mechanical strength and sealing properties. Meanwhile, the polymers should not be dissolved by the inner solvent. Regarding the selection of the solvent, the first is to guarantee fast and efficient photoisomerization of target photoswitches; second, it should be a poor solvent for the shell polymers; finally, solvents with high boiling point and low volatility would be preferred.
Roscini and coworkers started fabricating solid-state photoresponsive materials by solvent-in-solid strategy in 2016. Nonanoic acid was applied as the solvent for SP, which was encapsulated by PMMA through a reaction-free emulsion-solvent evaporation method. At room temperature (below the melting point of nonanoic acid), the SP in the microcapsules was dominated by SC, which efficiently isomerized to MC under the irradiation of UV light. The reversed MC-to-SC isomerization of SP is effectively triggered by visible light irradiation at 60 °C (beyond the melting point of nonanoic acid). This work demonstrated the possibility of preparing solid-state photoresponsive materials through a universal, straightforward and modification-free strategy.153 They extended the similar design philosophy by investigating multiple combinations of cores (SP and solvent) and shells (polymers), and photoresponsive microcapsules with distinct photochromic properties (isomerization kinetics and colors) were obtained.154 Mahdavian and coworkers reported spironaphthoxazine@oleic acid@ PMMA microcapsules, which were used for polymeric coating and non-woven cotton fabric.155
Nanoemulsions can generate droplets with a diameter between 10 to 100 nm, which are therefore more kinetically stable than microcapsules.156 Roscini and coworkers first applied nanoemulsions to encapsulate the T-type spirooxazine to fabricate photochromic films. The photoresponsive nanoemulsions were further enclosed in a hydrophilic polymeric matrix (polyvinyl alcohol), generating a highly transparent film. The photoisomerization behavior of this solid film is superfast and closed to that in the diluted solution. Besides, the photochromic films exhibit excellent fatigue resistance, making them attractive in smart windows.157
(1) Seeking suitable shell materials. First, polymers with strong mechanical stability are preferred, ensuring the intact of the wall against inside pressure and outside forces; second, the thickness of the wall should prevent solvent leakage as well as maintain high transmittance; third, the shell should be insensitive to the temperature changes, melting or extension are prohibited under high temperature, and cooling shrinkage also needs to be avoided.
(2) Further decrease in the diameter of the capsules. So far, the diameter of most photoresponsive capsules is above 1 μm because, the huge surface tension constraints are difficult to overcome by mechanical agitation. A larger particle size brings worse stability and a lower shell thickness/radius ratio, which requires a thicker shell to maintain the internal pressure. Further decreasing the photoresponsive capsule size to between 10 nm and 100 nm could increase the kinetical stability.156
4. Challenges still exist
4.1 Photoresponsive materials with strong mechanical stability
Mechanical stability meets the key requirement of solid-state materials when applied in practical terms. However, seldom works are designed for chasing photoresponsive materials with strong mechanical stability. Because the isomerization of photoresponsive molecules requires certain intrinsic mobility, many solid-state photoresponsive materials are in the form of films and soft matter (Fig. 10).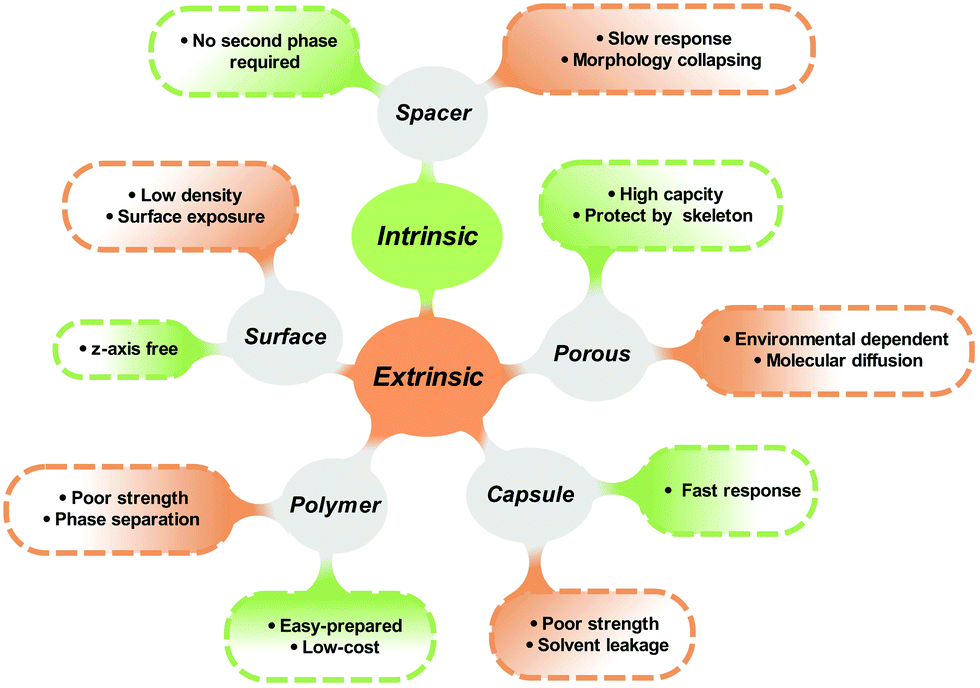 | ||
| Fig. 10 Schematic illustration of the advantages and disadvantages of each strategy. | ||
Photoswitches could be performed as solid powders with the chemical modification and under the assistance of MOFs/COFs, which are made of small crystals and need to be incorporated with another phase to turn into solid blocks with designated shapes. In this case, the introduced phase contributes to the mechanical stability of the photoresponsive materials.
Appropriate incubating methods and molecular structures are able to form crystals with big crystal sizes. However, the formation of the crystals is driven by weak intramolecular forces (i.e. H-bond or π–π interaction), which is not sufficient to endow a strong bonding manner to obtain strong mechanical strength.
Surface modification is a potential way to harvest both, photochromic properties and mechanical strength, by immobilizing the photoresponsive molecules on the surface of the substrate with strong mechanical strength. However, as discussed in the above section, the outmost layer of the materials still needs protection to face the mechanical force, oxygen and water (Fig. 10).
Polymer matrices might be the most promising candidates for preparing photoresponsive solid with good mechanical strength. However, the hard polymer is usually made of rigid chains (high Tg), which might confine the space for efficient isomerization. This is why most cases show fast and efficient isomerization in polymer matrices are limited to “soft” species.
Similarly, for the solvent-in-solid strategy, the shell materials should bear both strong mechanical strength and good sealing properties, maintaining the structural integrity of the capsules, and preventing solvent leakages and oxygen/water penetration.
4.2 Materials responsive to natural light
For the solid-state photoresponsive materials in practical applications, the light irradiating conditions (e.g. intensity, wavelength) are not as good as they are in the laboratory. Therefore, photoresponsive materials under the control of natural light might be attractive. Natural light is a complex mixture of light with different wavelengths, including UV, visible, and NIR components. Therefore, for most photoresponsive molecules with both forward and backward isomerization induced by light, such as Azo, SP, DAE and anthracene, the isomerization would not efficiently occur under natural light because of the competition between forward and backward isomerization. In this case, DASA with the visible-light-induced linear-to-cyclic isomerization and heat-induced backward isomerization is attractive to be controlled by natural light.Besides, red-shifting the induced light is also important for the solid-state photoresponsive materials. Most photoswitching occurs under UV light irradiation. However, due to the inevitable drawbacks such as low penetrability, photobleaching effect and unhealthy for humans, UV light is hard to fulfill the demands of real-world applications. In recent years, plenty of works have been reported on red-shifting the induced light by molecular design and chemical modification, which is applicable to most photoswitches.20,50
4.3 Photoresponsive materials with high thickness
The molecular mobility is sharply decreased with the increase in depth in solid-state materials. Moreover, limited by the penetrability of the induced light, the light intensity could be very weak in the deep solid-state materials because of the absorbance and scattering. Therefore, achieving photoresponsive materials with high thickness remains a challenge.With the assistance of polymers, photoresponsive materials with high thickness have been realized as soft matters, including hydrogels and elastomers. However, preparing the “hard” photoresponsive materials with a certain thickness is still difficult. Rationally, several points need to be fulfilled as mentioned below to fabricate photoresponsive materials with high thickness, (1) the long-wavelength induced is preferred due to the good penetrability; (2) the substrate should be light-penetrable toward a specific wavelength range; (3) molecules with negative photochromism are preferred because the discoloration process initiated from the outmost layer would not or less hinder the light penetrating to inner space, ensuring the fast and efficient photoisomerization.
Author contributions
F. Sun and D. Wang wrote the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We gratefully acknowledge the financial support from the International Cooperation in Science and Technology Innovation of Sichuan (2021YFH0128) and Guangdong Basic and Applied Basic Research Foundation (2019A1515110678). Finally, A tribute to the project of building MPG-UESTC Partner Group.Notes and references
- N. Iqbal, J. Jung, S. Park and E. J. Cho, Angew. Chem., 2014, 126, 549–552 CrossRef.
- L. Schmermund, V. Jurkaš, F. F. Özgen, G. D. Barone, H. C. Büchsenscḧtz, C. K. Winkler, S. Schmidt, R. Kourist and W. Kroutil, ACS Catal., 2019, 9, 4115–4144 CrossRef CAS.
- D. Habault, H. Zhang and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7244–7256 RSC.
- Y. Qing, C. Yang, N. Yu, Y. Shang, Y. Sun, L. Wang and C. Liu, Chem. Eng. J., 2016, 290, 37–44 CrossRef CAS.
- Y. Ke, J. Chen, G. Lin, S. Wang, Y. Zhou, J. Yin, P. S. Lee and Y. Long, Adv. Energy Mater., 2019, 9, 1902066 CrossRef CAS.
- Y. Xia, Y. He, F. Zhang, Y. Liu and J. Leng, Adv. Mater., 2021, 33, 2000713 CrossRef CAS PubMed.
- P. Klán, T. Solomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- Z. Deng, R. Yu and B. Guo, Mater. Chem. Front., 2021, 5, 2092–2123 RSC.
- A. Y. Rwei, W. Wang and D. S. Kohane, Nano Today, 2015, 10, 451–467 CrossRef CAS PubMed.
- X. Deng, Z. Shao and Y. Zhao, Adv. Sci., 2021, 8, 2002504 CrossRef CAS PubMed.
- J. Chen, F. K.-C. Leung, M. C. Stuart, T. Kajitani, T. Fukushima, E. van der Giessen and B. L. Feringa, Nat. Chem., 2018, 10, 132–138 CrossRef CAS PubMed.
- J. A. Lv, Y. Liu, J. Wei, E. Chen, L. Qin and Y. Yu, Nature, 2016, 537, 179–184 CrossRef CAS PubMed.
- Y. Zhuang, X. Ren, X. Che, S. Liu, W. Huang and Q. Zhao, Adv. Photonics, 2020, 3, 014001 Search PubMed.
- L. Mao, Z. Wang, Y. Duan, C. Xiong, C. He, X. Deng, Y. Zheng and D. Wang, ACS Nano, 2021, 15, 10384–10392 CrossRef PubMed.
- K. M. Lee, H. Koerner, R. A. Vaia, T. J. Bunning and T. J. White, Soft Matter, 2011, 7, 4318–4324 RSC.
- Z. Li, X. Liu, G. Wang, B. Li, H. Chen, H. Li and Y. Zhao, Nat. Commun., 2021, 12, 1363 CrossRef CAS PubMed.
- H. K. Bisoyi and Q. Li, Acc. Chem. Res., 2014, 47, 3184–3195 CrossRef CAS PubMed.
- S. Varghese and S. Das, J. Phys. Chem. Lett., 2011, 2, 863–873 CrossRef CAS PubMed.
- H. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- S. Samanta, A. A. Beharry, O. Sadovski, T. M. McCormick, A. Babalhavaeji, V. Tropepe and G. A. Woolley, J. Am. Chem. Soc., 2013, 135, 9777–9784 CrossRef CAS PubMed.
- J. L. Magee, W. Shand Jr and H. Eyring, J. Am. Chem. Soc., 1941, 63, 677–688 CrossRef CAS.
- D. Y. Curtin, E. J. Grubbs and C. G. McCarty, J. Am. Chem. Soc., 1966, 88, 2775–2786 CrossRef CAS.
- T. Naito, K. Horie and I. Mita, Macromolecules, 1991, 24, 2907–2911 CrossRef CAS.
- L. Kortekaas, J. Simke, D. W. Kurka and B. J. Ravoo, ACS Appl. Mater. Interfaces, 2020, 12, 32054–32060 CrossRef CAS PubMed.
- D. T. Nguyen, M. Freitag, C. Gutheil, K. Sotthewes, B. J. Tyler, M. Böckmann, M. Das, F. Schlüter, N. L. Doltsinis, H. F. Arlinghaus, B. J. Ravoo and F. Glorius, Angew. Chem., Int. Ed., 2020, 59, 13651–13656 CrossRef CAS PubMed.
- P. Kumar, A. Srivastava, C. Sah, S. Devi and S. Venkataramani, Chem. – Eur. J., 2019, 25, 11924–11932 CrossRef CAS PubMed.
- E. Procházková, J. Filo, L. M. Čechová, M. Dračínský, I. Císařová, Z. Janeba, I. Kawamura, A. Naito, I. Kuběna and P. Nádaždy, Dyes Pigm., 2022, 199, 110066 CrossRef.
- G. Accorsi, A. L. Capodilupo, R. M. Claramunt, G. J. Clarkson, A. Farrán, F. G. Gatti, S. León and S. Venturi, New J. Chem., 2021, 45, 12471–12478 RSC.
- S. Crespi, N. A. Simeth and B. König, Nat. Rev. Chem., 2019, 3, 133–146 CrossRef CAS.
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed.
- S. Kobatake, T. Yamada, K. Uchida, N. Kato and M. Irie, J. Am. Chem. Soc., 1999, 121, 2380–2386 CrossRef CAS.
- R. J. Li, J. Tessarolo, H. Lee and G. H. Clever, J. Am. Chem. Soc., 2021, 143, 3865–3873 CrossRef CAS PubMed.
- L. Sicard, F. Lafolet, M. Boggio-Pasqua, C. Bucher, G. Royal and S. Cobo, ChemPhysChem, 2022, 23, e202200004 CrossRef CAS PubMed.
- J. Qi, C. Chen, X. Zhang, X. Hu, S. Ji, R. T. Kwok, J. W. Lam, D. Ding and B. Z. Tang, Nat. Commun., 2018, 9, 1848 CrossRef PubMed.
- H. B. Cheng, S. Zhang, E. Bai, X. Cao, J. Wang, J. Qi, J. Liu, J. Zhao, L. Zhang and J. Yoon, Adv. Mater., 2021, 34, 2108289 CrossRef PubMed.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- Y. Duan, H. Zhao, C. Xiong, L. Mao, D. Wang and Y. Zheng, Chin. J. Chem., 2021, 39, 985–998 CrossRef CAS.
- G. Cottone, R. Noto, G. La Manna and S. L. Fornili, Chem. Phys. Lett., 2000, 319, 51–59 CrossRef CAS.
- S. Helmy, F. A. Leibfarth, S. Oh, J. E. Poelma, C. J. Hawker and J. Read de Alaniz, J. Am. Chem. Soc., 2014, 136, 8169–8172 CrossRef CAS PubMed.
- S. Helmy, S. Oh, F. A. Leibfarth, C. J. Hawker and J. Read de Alaniz, J. Org. Chem., 2014, 79, 11316–11329 CrossRef CAS PubMed.
- J.-t Stenhouse, Justus Liebigs Ann. Chem., 1850, 74, 278–297 CrossRef.
- M. M. Lerch, W. Szymański and B. L. Feringa, Chem. Soc. Rev., 2018, 47, 1910–1937 RSC.
- H. Zulfikri, M. A. Koenis, M. M. Lerch, M. Di Donato, W. Szymański, C. Filippi, B. L. Feringa and W. J. Buma, J. Am. Chem. Soc., 2019, 141, 7376–7384 CrossRef CAS PubMed.
- M. M. Lerch, S. J. Wezenberg, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2016, 138, 6344–6347 CrossRef CAS PubMed.
- C. Xiong, G. Xue, L. Mao, L. Gu, C. He, Y. Zheng and D. Wang, Langmuir, 2021, 37, 802–809 CrossRef CAS PubMed.
- Y. Duan, H. Zhao, G. Xue, F. Sun, F. Stricker, Z. Wang, L. Mao, C. He, J. R. de Alaniz, Y. Zheng and D. Wang, J. Phys. Chem. B, 2022, 126, 3347–3354 CrossRef CAS PubMed.
- N. Mallo, P. T. Brown, H. Iranmanesh, T. S. MacDonald, M. J. Teusner, J. B. Harper, G. E. Ball and J. E. Beves, Chem. Commun., 2016, 52, 13576–13579 RSC.
- L. Zhang, Y. Deng, Y. Tang, C. Xie and Z. Wu, Mater. Chem. Front., 2021, 5, 3119–3124 RSC.
- J. Moreno, M. Gerecke, L. Grubert, S. A. Kovalenko and S. Hecht, Angew. Chem., Int. Ed., 2016, 55, 1544–1547 CrossRef CAS PubMed.
- P. Weis, D. Wang and S. Wu, Macromolecules, 2016, 49, 6368–6373 CrossRef CAS.
- R. Yang, Y. Jiao, B. Wang, B. Xu and W. Tian, J. Phys. Chem. Lett., 2021, 12, 1290–1294 CrossRef CAS PubMed.
- R. Yang, X. Ren, L. Mei, G. Pan, X. Z. Li, Z. Wu, S. Zhang, W. Ma, W. Yu, H. H. Fang, C. Li, M. Q. Zhu, Z. Hu, T. Sun, B. Xu and W. Tian, Angew. Chem., 2022, 134, e202117158 Search PubMed.
- S. Peng, J. Wen, M. Hai, Z. Yang, X. Yuan, D. Wang, H. Cao and W. He, New J. Chem., 2019, 43, 617–621 RSC.
- Y. Jiao, R. Yang, Y. Luo, L. Liu, B. Xu and W. Tian, CCS Chem., 2022, 4, 132–140 CrossRef CAS.
- P. Q. Nhien, T. T.-K. Cuc, T. M. Khang, C. H. Wu, B. T.-B. Hue, J. I. Wu, B. W. Mansel, H. L. Chen and H. C. Lin, ACS Appl. Mater. Interfaces, 2020, 12, 47921–47938 CrossRef CAS PubMed.
- H. Suzui, K. Uchiyama, K. Takase, R. Nakagomi, L. Kono, K. Uchida, N. Chauvet, R. Horisaki, H. Hori and M. Naruse, Appl. Phys. Lett., 2022, 120, 071105 CrossRef CAS.
- N. B. Arndt, F. Schlüter, M. Böckmann, T. Adolphs, H. F. Arlinghaus, N. L. Doltsinis and B. J. Ravoo, Langmuir, 2022, 38, 735–742 CrossRef CAS PubMed.
- M. Min, G. S. Bang, H. Lee and B. C. Yu, Chem. Commun., 2010, 46, 5232–5234 RSC.
- A. Gonzalez, E. Kengmana, M. Fonseca and G. Han, Mater. Today Adv., 2020, 6, 100058 CrossRef.
- A. A. Ahmad, Q. M. Al-Bataineh, A. M. Alsaad, D. M. Al-Nawafleh, A. M. Al-Nawafleh and A. D. Telfah, Photochem. Photobiol., 2021 DOI:10.1111/php.13534.
- S. Singh, K. Friedel, M. Himmerlich, Y. Lei, G. Schlingloff and A. Schober, ACS Macro Lett., 2015, 4, 1273–1277 CrossRef CAS.
- M. A. Gerkman, S. Sinha, J. H. Warner and G. G.-D. Han, ACS Nano, 2019, 13, 87–96 CrossRef CAS PubMed.
- J. Fan, B. Bao, Z. Wang, R. Xu, W. Wang and D. Yu, Cellulose, 2020, 27, 493–510 CrossRef CAS.
- T. Li, M. Jevric, J. R. Hauptmann, R. Hviid, Z. Wei, R. Wang, N. E. Reeler, E. Thyrhaug, S. Petersen, J. A.-S. Meyer, N. Bovet, T. Vosch, J. Nygård, X. Qiu, W. Hu, Y. Liu, G. C. Solomon, H. G. Kjaergaard, T. Bjørnholm, M. B. Nielsen, B. W. Laursen and K. Nørgaard, Adv. Mater., 2013, 25, 4164–4170 CrossRef CAS PubMed.
- I. Hnid, D. Frath, F. Lafolet, X. Sun and J. C. Lacroix, J. Am. Chem. Soc., 2020, 142, 7732–7736 CrossRef CAS PubMed.
- C. Honnigfort, L. Topp, N. G. Rey, A. Heuer and B. Braunschweig, J. Am. Chem. Soc., 2022, 144, 4026–4038 CrossRef CAS PubMed.
- L. Q. Zheng, S. Yang, S. Krähenbühl, V. V. Rybkin, J. Lan, I. Aprahamian and R. Zenobi, Mater. Today Chem., 2022, 24, 100797 CrossRef CAS.
- S. Beckford, L. Mathurin, J. Chen, R. A. Fleming and M. Zou, Tribol. Int., 2016, 103, 87–94 CrossRef CAS.
- D. R. Jun, S. K. Moon and S. W. Choi, Colloids Surf., B, 2014, 121, 395–399 CrossRef CAS PubMed.
- W. Yang, C. Liu and Y. Chen, Langmuir, 2018, 34, 3565–3571 CrossRef CAS PubMed.
- H. Zhao, D. Wang, Y. Fan, M. Ren, S. Dong and Y. Zheng, Langmuir, 2018, 34, 15537–15543 CrossRef CAS PubMed.
- K. Ichimura, S. K. Oh and M. Nakagawa, Science, 2000, 288, 1624–1626 CrossRef CAS PubMed.
- Y. Han, J. Li, X. Zhang, F. Xia and Y. Dai, Nanotechnology, 2022, 33, 165601 CrossRef PubMed.
- F. Cardano, M. Frasconi and S. Giordani, Front. Chem., 2018, 6, 102 CrossRef PubMed.
- K. Yadav, S. Mahapatra, T. Halbritter, A. Heckel and T. G. Gopakumar, J. Phys. Chem. Lett., 2018, 9, 6326–6333 CrossRef CAS PubMed.
- D. Wang, Q. Sun, M. J. Hokkanen, C. Zhang, F. Y. Lin, Q. Liu, S. P. Zhu, T. Zhou, Q. Chang, B. He, Q. Zhou, L. Chen, Z. Wang, R. H.-A. Ras and X. Deng, Nature, 2020, 582, 55–59 CrossRef CAS PubMed.
- H. Daglar, H. C. Gulbalkan, G. Avci, G. O. Aksu, O. F. Altundal, C. Altintas, I. Erucar and S. Keskin, Angew. Chem., Int. Ed., 2021, 60, 7828–7837 CrossRef CAS PubMed.
- C. D. Wu and M. Zhao, Adv. Mater., 2017, 29, 1605446 CrossRef PubMed.
- Y. Wang, J. Yan, N. Wen, H. Xiong, S. Cai, Q. He, Y. Hu, D. Peng, Z. Liu and Y. Liu, Biomaterials, 2020, 230, 119619 CrossRef CAS PubMed.
- H. Y. Li, Y. L. Wei, X. Y. Dong, S. Q. Zang and T. C. Mak, Chem. Mater., 2015, 27, 1327–1331 CrossRef CAS.
- M. Hartmann and X. Kostrov, Chem. Soc. Rev., 2013, 42, 6277–6289 RSC.
- A. M.-M. Jani, D. Losic and N. H. Voelcker, Prog. Mater. Sci., 2013, 58, 636–704 CrossRef.
- L. Y. Long, Y. X. Weng and Y. Z. Wang, Polymers, 2018, 10, 623 CrossRef PubMed.
- P. Hong, Z. Liu, Y. Gao, Y. Chen, M. Zhuang, L. Chen, X. Liu and H. Xiang, Adv. Polym. Tech., 2019, 2019, 9536320 CrossRef.
- X. Fang, X. Zhao, W. Fang, C. Chen and N. Zheng, Nanoscale, 2013, 5, 2205–2218 RSC.
- M. Pera-Titus, Chem. Rev., 2014, 114, 1413–1492 CrossRef CAS PubMed.
- P. Yang, S. Gai and J. Lin, Chem. Soc. Rev., 2012, 41, 3679–3698 RSC.
- J. E. Lofgreen and G. A. Ozin, Chem. Soc. Rev., 2014, 43, 911–933 RSC.
- J. Liu, W. Bu, L. Pan and J. Shi, Angew. Chem., 2013, 125, 4471–4475 CrossRef.
- L. Wang and Q. Li, Chem. Soc. Rev., 2018, 47, 1044–1097 RSC.
- T. Yamaguchi, K. J. Imwiset and M. Ogawa, Langmuir, 2021, 37, 3702–3708 CrossRef CAS PubMed.
- A. Khalil, P. Rostami, G. K. Auernhammer and A. Andrieu-Brunsen, Adv. Mater. Interfaces, 2021, 8, 2100252 CrossRef CAS.
- H. Zhu, S. Yang, D. Chen, N. Li, Q. Xu, H. Li, J. He and J. Lu, Adv. Mater. Interfaces, 2016, 3, 1500683 CrossRef.
- K. Karimipour, J. K. Rad, A. R. Ghomi, H. Salehi-Mobarakeh and A. R. Mahdavian, Dyes Pigm., 2020, 175, 108185 CrossRef CAS.
- H. Sakaino, D. J. Broer, S. C. Meskers, E. Meijer and G. Vantomme, Angew. Chem., Int. Ed., 2022, 61, e202200839 CrossRef CAS PubMed.
- G. Liu, M. Wang, H. Gao, C. Cui and J. Gao, Eur. Polym. J., 2021, 161, 110828 CrossRef CAS.
- J. K. Rad, A. R. Ghomi, K. Karimipour and A. R. Mahdavian, Macromolecules, 2020, 53, 1613–1622 CrossRef.
- J. Meng, X. Liu, C. Niu, Q. Pang, J. Li, F. Liu, Z. Liu and L. Mai, Chem. Soc. Rev., 2020, 49, 3142–3186 RSC.
- R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dincă, A. Bavykina, J. Gascon, A. Ejsmont, J. Goscianska, M. Kalmutzki, U. Lächelt, E. Ploetz, C. S. Diercks and S. Wuttke, Angew. Chem., Int. Ed., 2021, 60, 23975–24001 CrossRef CAS PubMed.
- Y. Wei, T. E. Parmentier, K. P. de Jong and J. Zečević, Chem. Soc. Rev., 2015, 44, 7234–7261 RSC.
- M. Tu, H. Reinsch, S. Rodríguez-Hermida, R. Verbeke, T. Stassin, W. Egger, M. Dickmann, B. Dieu, J. Hofkens, I. F. Vankelecom, N. Stock and R. Ameloot, Angew. Chem., 2019, 131, 2445–2449 Search PubMed.
- F. Marlow, K. Hoffmann and J. Caro, Adv. Mater., 1997, 9, 567–570 CrossRef.
- K. Hoffmann, U. Resch-Genger and F. Marlow, Microporous Mesoporous Mater., 2000, 41, 99–106 CrossRef CAS.
- I. Casades, S. Constantine, D. Cardin, H. García, A. Gilbert and F. Márquez, Tetrahedron, 2000, 56, 6951–6956 CrossRef CAS.
- C. Schomburg, M. Wark, Y. Rohlfing, G. Schulz-Ekloff and D. Wöhrle, J. Mater. Chem., 2001, 11, 2014–2021 RSC.
- O. M. Yaghi, G. Li and H. Li, Nature, 1995, 378, 703–706 CrossRef CAS.
- K. Ohara, Y. Inokuma and M. Fujita, Angew. Chem., Int. Ed., 2010, 49, 5507–5509 CrossRef CAS PubMed.
- H. Schwartz, U. Ruschewitz and L. Heinke, Photochem. Photobiol. Sci., 2018, 17, 864–873 CrossRef CAS PubMed.
- A. M. Rice, C. R. Martin, V. A. Galitskiy, A. A. Berseneva, G. A. Leith and N. B. Shustova, Chem. Rev., 2020, 120, 8790–8813 CrossRef CAS PubMed.
- S. Castellanos, F. Kapteijn and J. Gascon, CrystEngComm, 2016, 18, 4006–4012 RSC.
- R. Haldar, L. Heinke and C. Wöll, Adv. Mater., 2020, 32, 1905227 CrossRef CAS PubMed.
- A. L. Leistner and Z. Pianowski, Eur. J. Org. Chem., 2022, e202101271 Search PubMed.
- F. Sun, X. Xiong, A. Gao, Y. Duan, L. Mao, L. Gu, Z. Wang, C. He, X. Deng and Y. Zheng, Chem. Eng. J., 2022, 427, 132037 CrossRef CAS.
- I. M. Walton, J. M. Cox, J. A. Coppin, C. M. Linderman, D. G.-D. Patel and J. B. Benedict, Chem. Commun., 2013, 49, 8012–8014 RSC.
- Z. Li, G. Wang, Y. Ye, B. Li, H. Li and B. Chen, Angew. Chem., 2019, 131, 18193–18199 CrossRef.
- D. N. Dybtsev, H. Chun and K. Kim, Angew. Chem., 2004, 116, 5143–5146 CrossRef.
- D. Hermann, H. Emerich, R. Lepski, D. Schaniel and U. Ruschewitz, Inorg. Chem., 2013, 52, 2744–2749 CrossRef CAS PubMed.
- D. Hermann, H. A. Schwartz, M. Werker, D. Schaniel and U. Ruschewitz, Chem. – Eur. J., 2019, 25, 3606–3616 CrossRef CAS PubMed.
- H. A. Schwartz, S. Olthof, D. Schaniel, K. Meerholz and U. Ruschewitz, Inorg. Chem., 2017, 56, 13100–13110 CrossRef CAS PubMed.
- H. A. Schwartz, M. Werker, C. Tobeck, R. Christoffels, D. Schaniel, S. Olthof, K. Meerholz, H. Kopacka, H. Huppertz and U. Ruschewitz, ChemPhotoChem, 2020, 4, 195–206 CrossRef CAS.
- R. Liang, J. Samanta, B. Shao, M. Zhang, R. J. Staples, A. D. Chen, M. Tang, Y. Wu, I. Aprahamian and C. Ke, Angew. Chem., Int. Ed., 2021, 60, 23176–23181 CrossRef CAS PubMed.
- G. Zhang, C. Fu, H. Zhang and H. Zhang, CrystEngComm, 2021, 23, 4513–4521 RSC.
- T. Fan, Z. Li, G. Liu, C. Fan and S. Pu, J. Solid State Chem., 2022, 309, 122950 CrossRef CAS.
- T. Fu, Y. L. Wei, C. Zhang, L. K. Li, X. F. Liu, H. Y. Li and S. Q. Zang, Chem. Commun., 2020, 56, 13093–13096 RSC.
- D. E. Williams, C. R. Martin, E. A. Dolgopolova, A. Swifton, D. C. Godfrey, O. A. Ejegbavwo, P. J. Pellechia, M. D. Smith and N. B. Shustova, J. Am. Chem. Soc., 2018, 140, 7611–7622 CrossRef CAS PubMed.
- T. Gong, Q. Sui, P. Li, X. F. Meng, L. J. Zhou, J. Chen, J. Xu, L. Wang and E. Q. Gao, Small, 2019, 15, 1803468 CrossRef PubMed.
- N. Sun, Y. Jin, H. Wang, B. Yu, R. Wang, H. Wu, W. Zhou and J. Jiang, Chem. Mater., 2022, 34, 1956–1964 CrossRef CAS.
- O. Shekhah, J. Liu, R. Fischer and C. Wöll, Chem. Soc. Rev., 2011, 40, 1081–1106 RSC.
- L. Feng, K. Y. Wang, G. S. Day, M. R. Ryder and H. C. Zhou, Chem. Rev., 2020, 120, 13087–13133 CrossRef CAS PubMed.
- C. H. Liu, H. L. Nguyen and O. M. Yaghi, AsiaChem Magazine, 2020, 1(1), 18 Search PubMed.
- S. M.-T. Abtab, D. Alezi, P. M. Bhatt, A. Shkurenko, Y. Belmabkhout, H. Aggarwal, Ł. J. Weseliński, N. Alsadun, U. Samin and M. N. Hedhili, Chem, 2018, 4, 94–105 Search PubMed.
- D. Wang, L. Zhao, H. Zhao, J. Wu, M. Wagner, W. Sun, X. Liu, M. S. Miao and Y. Zheng, Commun. Chem., 2019, 2, 118 CrossRef.
- X. Kong, H. Deng, F. Yan, J. Kim, J. A. Swisher, B. Smit, O. M. Yaghi and J. A. Reimer, Science, 2013, 341, 882–885 CrossRef CAS PubMed.
- A. Meeks, M. M. Lerch, T. B.-H. Schroeder, A. Shastri and J. Aizenberg, J. Am. Chem. Soc., 2022, 144, 219–227 CrossRef CAS PubMed.
- X. Wang, Y. He and J. Leng, Macromol. Mater. Eng., 2022, 307, 2100778 CrossRef CAS.
- F. H. Schacher, P. A. Rupar and I. Manners, Angew. Chem., Int. Ed., 2012, 51, 7898–7921 CrossRef CAS PubMed.
- M. H. Burk, S. Schröder, W. Moormann, D. Langbehn, T. Strunskus, S. Rehders, R. Herges and F. Faupel, Macromolecules, 2020, 53, 1164–1170 CrossRef CAS.
- K. Ghebreyessus, I. Uba, D. Geddis and U. Hömmerich, J. Solid State Chem., 2021, 303, 122519 CrossRef CAS.
- L. Liu, R. Zeng, J. Jiang, T. Wu, P. Zhang, C. Zhang, J. Cui and J. Chen, Dyes Pigm., 2022, 197, 109919 CrossRef CAS.
- V. Graca, C. M. Sousa and P. J. Coelho, Dyes Pigm., 2021, 192, 109388 CrossRef CAS.
- L. Qu, X. Xu, J. Song, D. Wu, L. Wang, W. Zhou, X. Zhou and H. Xiang, Dyes Pigm., 2020, 181, 108597 CrossRef CAS.
- Q. Yan, C. Li, S. Wang, Z. Lin, Q. Yan and D. Cao, Dyes Pigm., 2020, 178, 108352 CrossRef CAS.
- D. Zhong, Z. Cao, B. Wu, Q. Zhang and G. Wang, Sens. Actuators, B, 2018, 254, 385–392 CrossRef CAS.
- Y. Okui and M. Han, Chem. Commun., 2012, 48, 11763–11765 RSC.
- W. C. Xu, S. Sun and S. Wu, Angew. Chem., Int. Ed., 2019, 58, 9712–9740 CrossRef CAS PubMed.
- H. Zhou, C. Xue, P. Weis, Y. Suzuki, S. Huang, K. Koynov, G. K. Auernhammer, R. Berger, H. J. Butt and S. Wu, Nat. Chem., 2017, 9, 145–151 CrossRef CAS PubMed.
- S. Yang, J. D. Harris, A. Lambai, L. L. Jeliazkov, G. Mohanty, H. Zeng, A. Priimagi and I. Aprahamian, J. Am. Chem. Soc., 2021, 143, 16348–16353 CrossRef CAS PubMed.
- Y. Guo, J. Xiao, Y. Sun, B. Song, H. Zhang and B. Dong, J. Mater. Chem. A, 2021, 9, 9364–9370 RSC.
- Z. Y. Zhang, Y. He, Z. Wang, J. Xu, M. Xie, P. Tao, D. Ji, K. Moth-Poulsen and T. Li, J. Am. Chem. Soc., 2020, 142, 12256–12264 CrossRef CAS PubMed.
- M. A. Gerkman, R. S. Gibson, J. Calbo, Y. Shi, M. J. Fuchter and G. G.-D. Han, J. Am. Chem. Soc., 2020, 142, 8688–8695 CrossRef PubMed.
- Y. Zhou, M. Chen, Q. Ban, Z. Zhang, S. Shuang, K. Koynov, H. J. Butt, J. Kong and S. Wu, ACS Macro Lett., 2019, 8, 968–972 CrossRef CAS.
- G. D. Goh, Y. L. Yap, H. Tan, S. L. Sing, G. L. Goh and W. Y. Yeong, Crit. Rev. Solid State Mater. Sci., 2020, 45, 113–133 CrossRef CAS.
- A. Julià-López, J. Hernando, D. Ruiz-Molina, P. González-Monje, J. Sedó and C. Roscini, Angew. Chem., 2016, 128, 15268–15272 CrossRef.
- A. Julià-López, D. Ruiz-Molina, J. Hernando and C. Roscini, ACS Appl. Mater. Interfaces, 2019, 11, 11884–11892 CrossRef PubMed.
- M. Raeesi, Z. Alinejad, V. Hamrang and A. R. Mahdavian, J. Colloid Interface Sci., 2020, 578, 379–389 CrossRef CAS PubMed.
- T. Sheth, S. Seshadri, T. Prileszky and M. E. Helgeson, Nat. Rev. Mater., 2020, 5, 214–228 CrossRef.
- H. Torres-Pierna, D. Ruiz-Molina and C. Roscini, Mater. Horiz., 2020, 7, 2749–2759 RSC.
- S. Santiago, P. Giménez-Gómez, X. Muñoz-Berbel, J. Hernando and G. Guirado, ACS Appl. Mater. Interfaces, 2021, 13, 26461–26471 CrossRef CAS PubMed.
- Z. Wang, X. Wang, S. Cong, F. Geng and Z. Zhao, Mater. Sci. Eng., R, 2020, 140, 100524 CrossRef.
| This journal is © The Royal Society of Chemistry 2022 |