
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLithium intercalation mechanisms and critical role of multi-doping in LiFexMn2−x−yTiyO4 as high-capacity cathode material for lithium-ion batteries†
D.
Callegari
 a,
M.
Coduri
a,
M.
Fracchia
a,
P.
Ghigna
a,
L.
Braglia
b,
U.
Anselmi Tamburini
a and
E.
Quartarone
*a
a,
M.
Coduri
a,
M.
Fracchia
a,
P.
Ghigna
a,
L.
Braglia
b,
U.
Anselmi Tamburini
a and
E.
Quartarone
*a
aDepartment of Chemistry and INSTM-GISEL, University of Pavia, Via Taramelli 16, 27100 Pavia, Italy. E-mail: eliana.quartarone@unipv.it
bCNR – Istituto Officina dei Materiali, TASC, I-34149 Trieste, Italy
First published on 10th May 2022
Abstract
The ever-growing demand for Li-ion batteries requires high-capacity electrode materials that should also be environmentally benign, Co-free, secure and durable, to achieve an optimal compromise between sustainability and functional performances. Spinel LiMn2O4 (LMO) is a state-of-the-art material, which, in principle, could satisfy such requirements. However, an undesired cubic–tetragonal phase transition favors Jahn–Teller (J–T) spinel distortion, leading to severe capacity reduction upon cycling below 3 V. Here, we propose a novel dual-doping strategy for LMO, based on the partial substitution of Mn(III) with Fe(III) and Ti(IV) to design new active materials for high-capacity cathodes, namely LiFexMn2−x−yTiyO4 (LFMT), with Li/Mn ratio ranging between 1 and 1.7. The substitution of Mn with Fe and Ti suppresses the J–T distortion, which is often still evident in the case of Ti-doped LMO. This allows cycling in a wider voltage range (4.8–1.5 V), thus resulting in higher capacity and significantly improved stability. The lithiation mechanisms were investigated by combining ex situ X-ray diffraction (XRD) and X-ray absorption spectroscopy (XAS analyses). It demonstrated that the only redox-active metal is Mn, while Fe and Ti are electrochemically inactive. The extensive electrochemical lithiation/delithiation of the LFMT compositions brought unprecedented results, which give evidence of stabilizing cation disorder through the formation of Mn-rich and Mn-poor domains, leading to two spinel phases with different Mn:Ti ratios. These insights into the lithiation mechanism pave the way for a better understanding of the doping chemistry and electrochemistry of Mn-based spinels as cathode materials for Li-ion batteries.
Introduction
The cubic spinel LiMn2O4 is one of the most traditionally studied active materials for cathodes in Lithium-ion batteries (LIBs). This is due to some unique properties, such as its high abundance of manganese and the absence of Co, resulting in a non-toxic, safe, environmentally benign, and thermally stable material.1 The interest in the spinel electrodes is predominantly due to their favorable framework, made of a continuous and energetically accessible 3D framework of face-sharing tetrahedra and octahedra through which Li ions can easily diffuse.2 The electrochemical profile of the Li/LixMn2O4 cell shows two reversible intercalation phenomena, at 4.1 and 3.0 V vs. Li, each of them resulting in a capacity of 120 mA h g−1.1 The first plateau is associated with the removal of one lithium (0 ≤ x ≤ 1), whereas the second plateau corresponds to the insertion of a second Li in the spinel phase (1 ≤ x ≤ 2).1,3 However, it is well known that the spinel cathode can practically use only half of its potential Li capacity and suffers from poor kinetics and a capacity that reduces upon cycling. This problem is more severe at 3 V and slower at 4 V, particularly at elevated temperatures. This is due to a strong synergy between three phenomena: (i) a first-order phase transition (cubic–tetragonal) occurring at 3 V once the spinel is lithiated beyond the LiMn2O4 composition; (ii) a structural rearrangement, causing physical destruction induced by a surface Jahn–Teller (J–T) distortion associated with Mn(III) ions; (iii) a chemical dissolution of Mn into the spinel. The latter process is exacerbated especially in the case of slightly acidic electrolytes (for example in presence of HF-based impurities) and is responsible for a disproportionation reaction of extensively lithiated particles with high Mn(III) concentration to give Mn(IV) (in the solid) and Mn(II) (released in the electrolyte).2 Once dissolved, the manganese ions migrate to the anode where they are reduced to metallic Mn, consuming Lithium and, consequently, decreasing the overall cell capacity.3In order to minimize both the disproportionation step and the impact of the J–T distortion, several attempts to dope the spinel were proposed in literature these last decades.4–13 A first doping strategy concerns the partial Mn(III) substitution with metals such as Al, Mg, Ni, Cr, Co, Fe, Ti. A second approach was to replace some oxygen with fluorine. The cation doping enhances the overall structural stability and decreases the spinel lattice parameter to values lower than 8.23 Å. This seems to be a threshold below which the cycling performances significantly improve.1,5 Moreover, the substitution of Mn(III) ions with other transition metals also results in an effective improvement of the cycling performances at 4 V. On the other hand, the anion doping with fluorine favors the reduction of the Mn average oxidation state, inhibiting the capacity loss.5
One of the most efficient doping strategies in the case of LiMn2O4 is the partial substitution of Mn(III) with Ti(IV) to form LiMn2−xTixO4.6,14,15 The bonding energy of Ti–O is higher than that of Mn–O, therefore an enhancement of the structural stability of the spinel is expected. Moreover, it was demonstrated that when Ti(IV) is present in high content in the spinel structure, the removal and intercalation of Li ions is associated with the increase of Mn(IV) and Mn(II), respectively, that are both non-J–T ions. Due to the inhibition of such distortions, the spinel can store about 2 Li+ occupying the tetragonal 8a and the octahedral 16c sites of the structure, resulting in a very high theoretical capacity (308 mA h g−1 in the specific case of LiMnTiO4) by using the Mn2+/Mn4+ redox couple.6 The concentration reduction of the J–T active Mn(III) can, in principle, also improve the cycling performances in a wider voltage range up to 4.8 V.14 However, it may occur that some Mn(II) ions replace Li+ during the Ti substitution for Mn, with the consequent degradation of the rate capability. For this reason, the Ti doping level needs to be properly modulated and optimized.14
In this work, a new family of dual-doped LiFexMn2−x−yTiyO4 spinel was synthesized via sol–gel reaction by modulating the stoichiometry ratio of the transition metals. These systems were investigated as novel active materials for high-energy cathodes in Co-free Li ion batteries more suitable over a wide voltage range, between 1.5 V and 4.8 V. The effect of the partial dual Mn(III) substitution with Fe and Ti and the potentially synergetic role of such transition metals on the stability of the spinel structure and on the modulation in the amount of Mn(II) ions were investigated by means of electron microscopy, X-ray diffraction and electrochemical characterizations. Ex situ X-ray spectroscopy (XAS) was also performed to understand the chemistry of each element in the spinel and to address the lithiation/delithiation mechanism as well as the possible presence of oxygen redox activity in such complex systems.
Materials and methods
Synthesis of LFMT spinel active materials
LFMT powders were synthesized by means of a sol−gel route starting from a mixture of LiCH3COO·2H2O, Mn(NO3)2·4H2O, and Fe(NO3)·9H2O (Sigma Aldrich) in the proper molar ratios, dissolved in distilled H2O under magnetic stirring at room temperature. A solution of Ti(i-OPr)4 and acetylacetone (acac) (Ti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acac molar ratio of 1:1.1) was then added under vigorous stirring to the dissolved precursors. Citric acid was finally added as a gelling agent. The solution was heated to 80 °C and the resulting gel was dried at 120 °C for 2 hours and finally calcined in air at 600 °C (temperature ramp of 5 °C min−1) for other 2 hours. Several spinels were prepared by changing the stoichiometry of the transition metals, whose compositions are reported in Table 1.
:acac molar ratio of 1:1.1) was then added under vigorous stirring to the dissolved precursors. Citric acid was finally added as a gelling agent. The solution was heated to 80 °C and the resulting gel was dried at 120 °C for 2 hours and finally calcined in air at 600 °C (temperature ramp of 5 °C min−1) for other 2 hours. Several spinels were prepared by changing the stoichiometry of the transition metals, whose compositions are reported in Table 1.
| Composition | Li/Mn | Mn/Fe | Mn/Ti | Fe/Ti | |
|---|---|---|---|---|---|
| LFMTa | LiFe0.53Mn1.00Ti0.47O4 | 1.0 | 1.9 | 2.1 | 1.1 |
| LFMTb | LiFe0.56Mn0.92Ti0.52O4 | 1.1 | 1.6 | 1.8 | 1.1 |
| LFMTc | LiFe0.6Mn0.85Ti0.55O4 | 1.2 | 1.4 | 1.5 | 1.1 |
| LFMTd | LiFe0.74Mn0.58Ti0.68O4 | 1.7 | 0.8 | 0.8 | 1.1 |
Cathode preparation and cell assembly
The cathode slurry was prepared by using 70 wt% of active material (LFMT), 20% of Conductive Carbon black (Imerys, Ensaco 350P), and 10% of a binder (polyvinylidene fluoride, PVdF). The solid content of all slurries was kept between 26 wt% and 28 wt%. LFMT and carbon powders were mixed in zirconia jars by a planetary ball mill at 150 rpm for 10 min twice, with a rest period of 5 min. Subsequently, the polymeric binder was added and mixed with a similar procedure. The as-prepared mixture was dispersed in N-methylpyrrolidone (NMP) to obtain the slurry, which was cast on a carbon-coated Al foil using a doctor blade with a wet thickness of 300 μm. The casted slurry was dried under vacuum at 80 °C to avoid moisture and oxygen contamination. The cathode (active mass ∼2 mg cm−2) was finally cut into 2 cm2 disks and stored in a glove box (MBraum, O2, H2O < 0.5 ppm) before the electrochemical measurements. All the functional tests were performed using a coin cell type (CR2032 – MTI Corp.) assembled in an Ar-filled glove box (H2O and O2 < 0.5 ppm). Metallic Li was used as counter electrode. Electrodes were separated with a Whatman glass fiber separator, imbibed by 120–150 μL of liquid electrolyte, consisting in a solution 1 M LiPF6 in ethylene carbonate:dimethyl carbonate (LP30, EC:DMC, 50:50 vol%) (Sigma-Aldrich).
Characterization
SEM and Energy Dispersive X-ray Spectroscopy (EDS) were performed using a Tescan Mira3XMU microscope operated at 20 kV and equipped with an EDAX EDS analysis system. The samples were coated with a carbon thin film using a Cressington 208 carbon coater.Powder X-rays diffraction was carried out by using a Bruker D8 Advance diffractometer in Bragg–Brentano geometry equipped with Ni-filtered Cu Kα radiation. The diffraction patterns of the powder samples as well as the composite cathodes, stuck onto a glass holder, were collected in the 15–65 2θ range, with 0.04 deg. step, for a total acquisition time of 2 hours. Structural analysis was performed via Rietveld refinements using the GSAS package.16 The peak broadening was estimated using WinPLOTR, accounting explicitly for Kα1–Kα2 wavelengths.17
The quality of least squares refinement in this case is also indicated by residual functions Rp and Rwp, defined as:
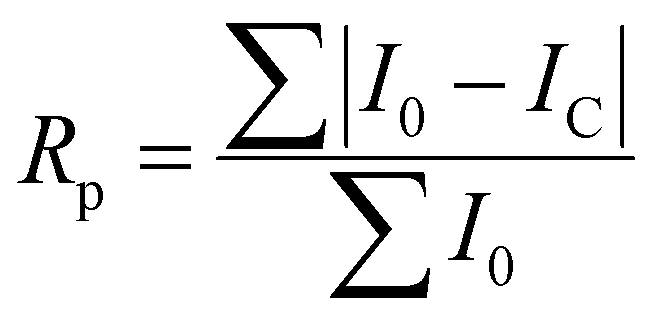
where I0 is the experimental intensity and IC the calculated one.
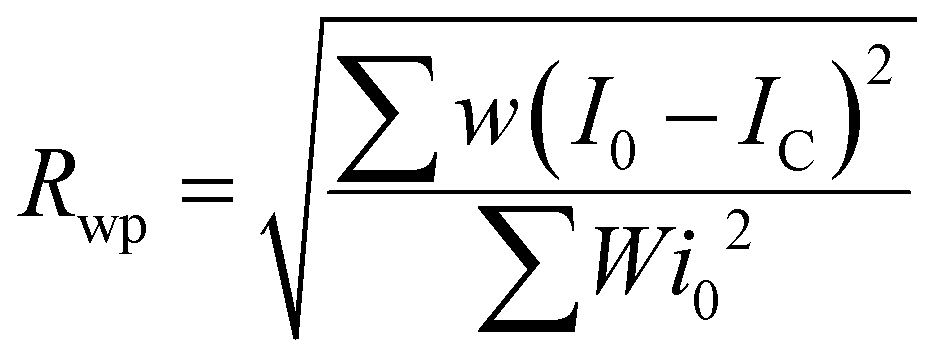
Ex situ XAS spectra were acquired on LFMT electrodes quenched at four different electrochemical conditions, namely at OCV, at 4.8 V vs. Li (in correspondence of the first and the second delithiation), and at 1.5 V vs. Li after 60 cycles. The XAS spectra were measured at the Mn, Fe, and Ti L2,3 edges and at the O K-edge at the APE-HE beamline at the Elettra synchrotron radiation facility in Trieste.18,19 All the electrodes were first mounted in the sample holder in an N2-filled glove box (H2O and O2 < 0.5 ppm), and then moved to the ultrahigh-vacuum chamber of the APE-HE beamline, where the spectra were acquired in total electron yield (TEY) mode. In this configuration, the X-rays beam passes through a Si3N4 membrane and reaches the sample; the secondary emission is then collected with a picoammeter connected to the sample, measuring the drain current. The collected data were calibrated using a reference for each investigated edge (Mn3O4, Fe2O3, and TiO2). All spectra were background-subtracted, flattening the baseline of the spectra, and normalized to peak integrated area.
Potentio-Electrochemical Impedance Spectroscopy (PEIS), Galvanostatic Cycling with Potential Limitation (GCPL), and Potentiodynamic Cycling with Galvanostatic Acceleration (PCGA) were performed on Li/LiMxMnyO4 cells in a wide voltage range between 1.5 and 4.8 V at room temperature and different C rates by means of battery tester (Bio-Logic BCS-810). A theoretical capacity of 280 mA h g−1 was calculated by taking into account the composition and the redox couple actually involved in the electrochemical process.
Electrochemical Impedance Spectroscopy (EIS) was used to measure the bulk conductivity (both ionic and electronic) as well as the transference numbers of ionic and electronic species in the active materials.20 To this aim, Nyquist plots were collected on a symmetric cell including Platinum-sputtered pellets of the spinel powders (∼1.8 mm thick), sandwiched between two Pt plates. Frequency sweeps between 500 and 0.1 Hz were imposed at 100–150 mV as voltage amplitude by means of a frequency response analyzer (Solartron 1255), coupled with an electrochemical interface (Solartron 1287) in the temperature range 25–300 °C.
EIS scans between 100 and 0.1 Hz were also collected at 50 mV during the galvanostatic cycling to investigate Li/LFMT cell impedance before and after the functional tests.
Results
LFMT active materials: morphology, structure, and transport properties
Sol–gel syntheses were attempted using annealing conditions similar to the one used for the synthesis of the spinel-based active materials. Specifically, a greener modified-Pechini route was carried out by using citric acid as complexing agent and nitrates as transition metal (TM) precursors, instead of the conventional chloride, whose removal as impurities is typically more difficult. As summarized in Table 1, four compositions were selected, differing in the Li/Mn molar ratio, while the Fe/Ti ratio was kept constant at 1.1.The pristine spinels were first investigated by XRD, SEM, and electrochemical impedance spectroscopy in order to understand the role played by the TMs dual doping on the morphology, crystal structure, and transport properties of each material.
Fig. S1, ESI† compares the electron microscopy images (both in backscattering and secondary electrons mode) at different magnifications for all the synthesized samples. The LFMT systems exhibit a lamellar- and flake-like morphology consisting of loose agglomerates showing a large distribution in dimension and connected to each other through a porous network, likely formed during the sol–gel process. The undoped LMO prepared by more conventional solid state reactions presents, in fact, a morphology characterized by closely packed particles, whose size strictly depends on the annealing conditions.21
Fig. 1(I) reports the XRD patterns of the pristine powder samples, whose diffraction peaks can all be indexed according to a single cubic LiMn2O4Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. The corresponding lattice constant, a, computed by Rietveld refinements, is reported in Fig. 1(II), as a function of the average ionic radius (i.r.) of the cations and is compared to the one of the undoped Li1+xMn2-xO4 at different degrees of (de)lithiation, as taken from ref. 6.
m space group. The corresponding lattice constant, a, computed by Rietveld refinements, is reported in Fig. 1(II), as a function of the average ionic radius (i.r.) of the cations and is compared to the one of the undoped Li1+xMn2-xO4 at different degrees of (de)lithiation, as taken from ref. 6.
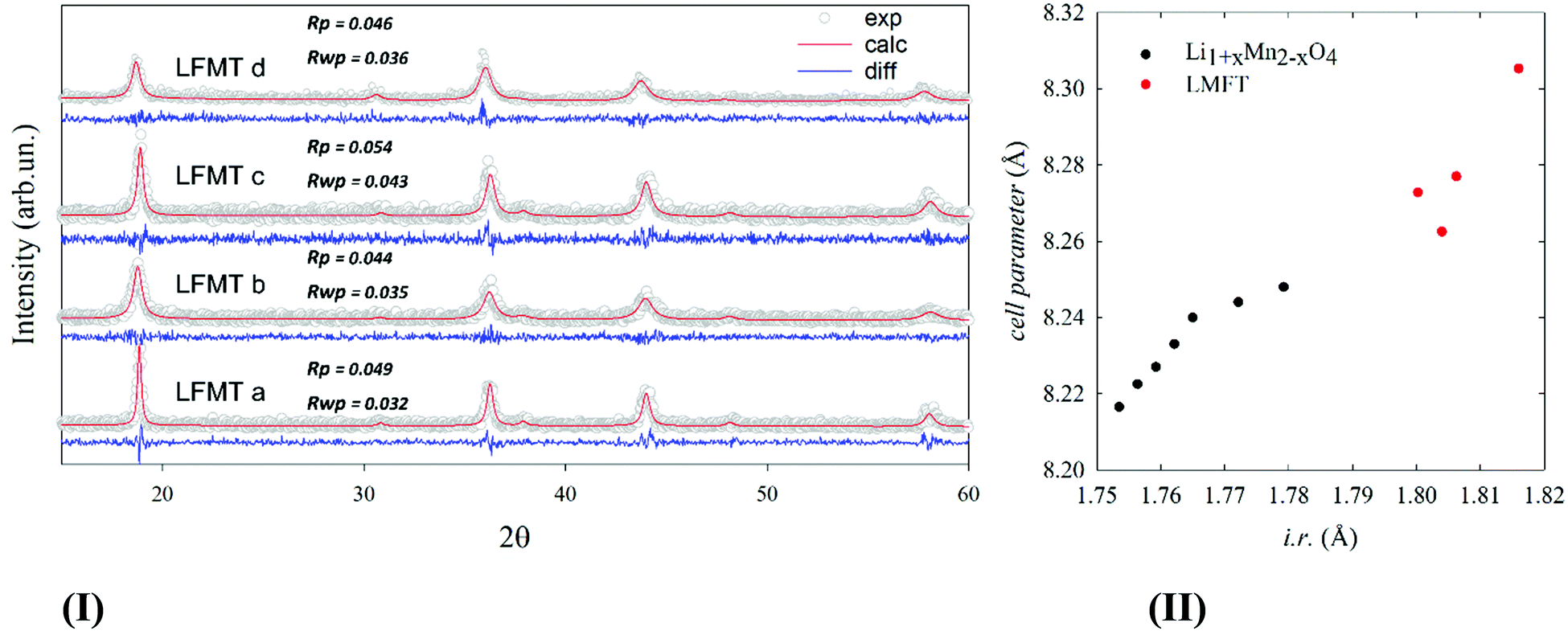 | ||
| Fig. 1 (I) experimental patterns of powdered specimens collected at room temperature (empty gray circles) and corresponding calculated profile computed by Rietveld refinement (red solid line) based on cubic spinel structural model. The difference curve (blue solid line) is shown below each pattern.; (II) cell parameter of LFMT (red) compared to undoped Li1+xMn2−xO4 from ref. 5 plotted as a function of the average ionic radius, i.r., of the cations. | ||
The ionic radii are calculated based on reference values for tetrahedral and octahedral coordination, as reported by Shannon (see Table S1, ESI†), assuming that Li ions lie in the tetrahedral sites and the other cations in the octahedral ones (no site inversion).22 The unit cell expands increasing the Li/Mn molar ratio. As to the undoped Li1+xMn2−xO4, such expansion is attributed to the formation of Mn(IV) cations taking place to compensate the charge when Li ions substitute for Mn(III).5 In particular, the cell expansion up to 8.31 Å gives indirect evidence of the partial Ti/Mn substitution. Indeed, Ti(IV) is bigger than Mn(IV) in octahedral coordination, whereas Fe(III) has negligible effect, having the same size as Mn(III).
The LFMT transport properties were evaluated by means of electrochemical impedance spectroscopy using ionically blocking electrodes. LiMn2O4 is a well-known mixed conductor with predominant Li-ion conduction, whose electronic and ionic impedances are not very different.21 For this reason, it is, in principle, easy to recognize the single components and to extrapolate the ionic and electronic resistances (and consequently the transference numbers, te and tLi+) from the relative values of the intercepts upon the real impedance axis at higher and lower frequencies, respectively.20 Fig. S2(I), ESI† shows, as an example, a typical impedance spectrum of a LFMT pellet as collected using a 2-electrode cell. The Nyquist plot typically shows two semicircles. The first represents a parallel combination of the electronic resistance, Re, and geometrical capacitance, giving clear evidence of a significant electronic leakage through the sample. This is confirmed by the absence of the typical capacitive tail at lower frequencies. The second semicircle is due to the parallel combination of the ionic resistance, Ri, and the geometrical capacitance at high frequency. In some cases, as for the example reported in Fig. S2(I), ESI† also the contribution of the grain boundary was recognized in the intermediate frequency domains, that was readily isolated. The ionic and electronic resistances were used to calculate the conductivity and transference numbers, as clearly described in detail in ref. 19.
Both transport components were studied as a function of the Li/Mn molar ratio and of the temperature. Fig. 2(I) shows the behavior of σi and σe for the LFMT systems versus the Li content in the spinel phase. The Li+ conduction decreases almost linearly by increasing the Li/Mn ratio. This is an expected result, since fewer lattice sites are gradually available for the lithium ionic diffusion when more Li cations are present.23 The conductivity values are in fair agreement with what is usually reported for the LiMn2O4 systems (Li/Mn = 0.5) (e.g. σI,100°C ∼ 10−5 S cm−1), as well as the activation energy calculated by the Arrhenius plots (Fig. 2(II)), ranging between 0.32 and 0.49 eV.21
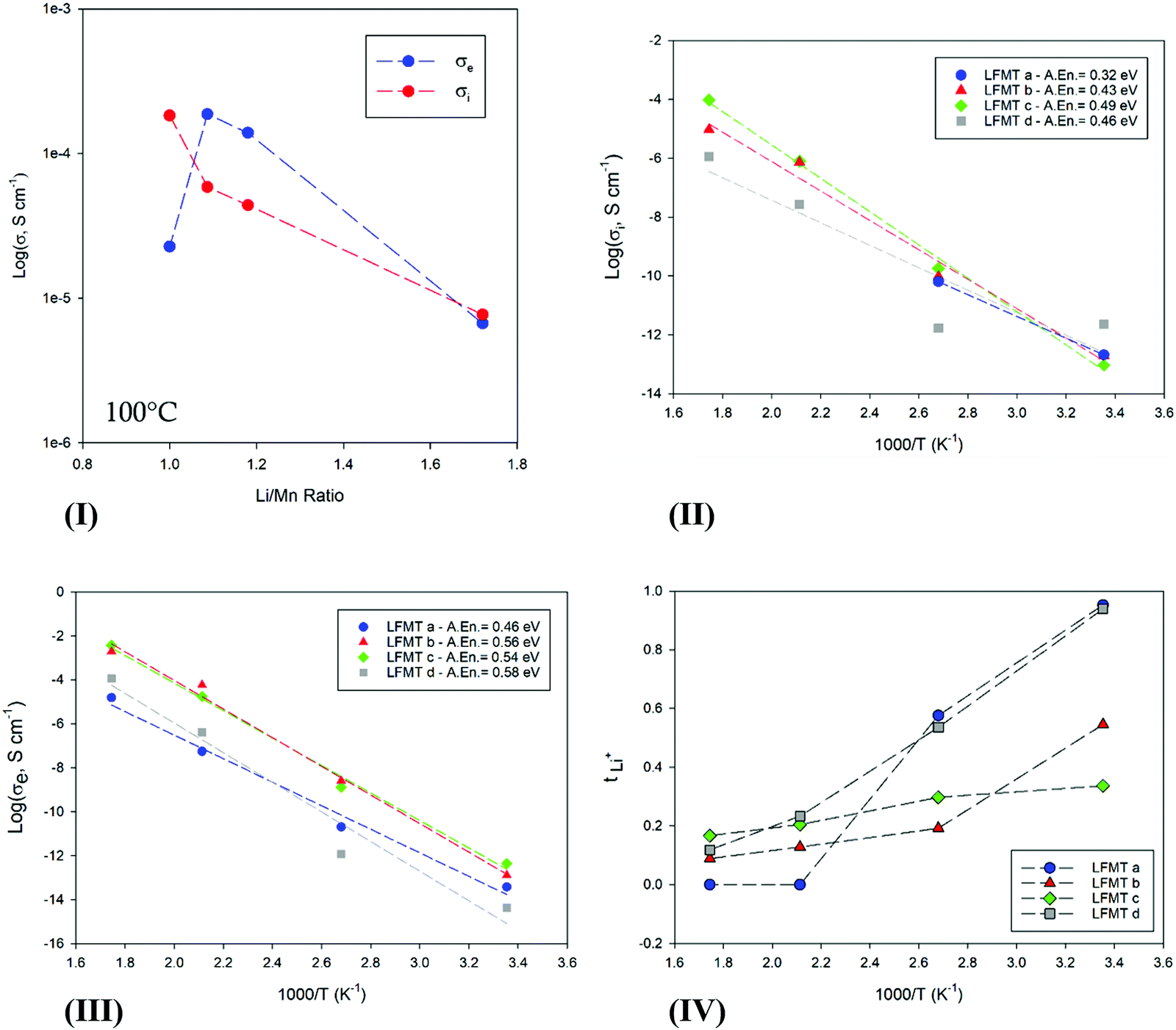 | ||
| Fig. 2 (I) Ionic (red) and electronic (blue) conductivity behavior at 100 °C vs. the Li/Mn ratio; (II) Ionic and (IIIIII) electronic conductivity behavior against temperature; (IV) Li ion transport number, tLi+, vs. T, for the investigated LFMTa–d active materials. In Fig. 2(II), σi values up to 100 °C are only reported for LFMTa, since the conductivity is fully electronic at higher temperature. | ||
The electronic conductivity changes as a function of the Li/Mn ratio with a more complex trend (see Fig. 2(III)). σe,100°C increases with the Li/Mn ratio, achieving the maximum value of ∼0.2 mS cm−1 for the sample LFMTb for which the ratio is 1.1. This behavior is in very good agreement with what is reported in the literature for the electronic transport of LiMn2O4.21,24 The spinel exhibits semiconducting characteristics related to the electron hopping between mixed-valence Mn, being 2.73 Å the calculated critical cation–cation separation in the octahedral configuration.21 Smaller lattice constant allows a more extended overlapping of the wave functions of neighboring Mn with different oxidation states, which consequently results in an enhanced electronic conductivity. In general, the obtained value for the electronic conductivity, exceeding 0.1 mS cm−1 around the curve maximum, are very satisfactory and significantly higher than LiMn2O4. The improved transport is likely due to a sound compromise between an optimal structure for the electron hopping and a good charge carrier concentration related to a proper composition of the dopant TMs. Electronic activation energy, Eae, higher than the ionic one, was determined from the Arrhenius plots reported in Fig. 2(III), with values in the range 0.46–0.58 eV between 25 and 300 °C.
Fig. 2(IV) and Fig. S2(II), ESI† finally show the Li+ and electron transference numbers, respectively tLi+ and te, vs. T, as calculated by the respective Nyquist plots. The curve clearly evidences as the semiconductors character of the material remarkably increase with the temperature for each composition, reaching an almost fully electronic transport at 300 °C. At room temperature, only spinels with the smaller lattice constant (LFMTb and LFMTc) exhibit a significant faction of the electronic component, with transference numbers, te, of 0.7 and 0.5, respectively.
LFMT cathodes: electrochemical performances
All LFMT spinels were investigated as cathode materials and tested in terms of their functional properties, as described in the Experimental section. Fig. S3(I)–(II), ESI† reports the SEM images and the XRD patterns, respectively, obtained for all the cathodes. The structural and morphological features of the aforementioned electrodes are quite similar to those of the pristine active materials.The LFMT electrochemical performances were investigated against Li as counter-electrode between 1.5 and 4.8 V at different current rates, starting from the OCV (∼3.1 V). The spinels exhibit qualitatively similar voltage profiles (Fig. S4, ESI†). Two main plateaus are clearly evident even upon long cycling, one sharper at 2.8–3.0 V and one broader, characteristic of the Li+ extraction from the tetrahedral 8a sites, at around 4 V. One exception is represented by LFMTd, whose first charge does not reach the cut-off voltage of 4.8 V, probably because of unfavored kinetics as a consequence of low content of Mn (Li/Mn = 1.7) and therefore also of Mn(IV). The electrochemical phenomena of the doped spinels are consequently very different from those ones expected for the undoped spinel. They are better evidenced in the dQ/dV curves shown in Fig. 3(I) for all the compositions, where the lithiation/delithiation reactions at ∼3.0 V and 4.0 V are associated with two peaks, describing the redox processes involving Mn. The first process is ascribed to Mn(III)/Mn(II) transition and the second to Mn(IV)/Mn(III) step. As already stated above, the availability of the whole Mn(IV)/Mn(II) redox couple is responsible for the significantly higher specific capacity observed in the doped spinels over LMO, achieving in some cases values higher than 280 mA h g−1 (0.08 C) and high energy density >0.9 W h kg−1, when cycled in a wide voltage range (4.8–1.5 V) (see Fig. S5(I), ESI†).
 | ||
| Fig. 3 (I) dQ/dV plots of LFMTa–d-based cathodes collected in the voltage range between 4.8 and 1.5 V by means of PCGA experiments at C/20; (II) rate performances of LFMTa–d cathodes cycled from 4.8 to 1.5 V at different current densities. | ||
The peak at 3 V is narrower than the corresponding signal observed for the undoped LiMn2O4 spinel, suggesting the hindering of the cubic–tetragonal phase transition typical of the LMO system, as confirmed by the ex situ XRD discussed in the following. The Mn(III)/Mn(IV) occurring around 4 V is a single broad signal, indicating a one-step reaction, contrary to what was observed for the undoped spinel, where two couples of sharp redox peaks are obtained between 3.5 and 4.8 V, These peaks can be theoretically attributed to the 2-step transition: one from LiMn2O4 to Li0.5MnIII0.5MnIV1.5O4 and the other from Li0.5MnIII0.5MnIV1.5O4 to a fully delithiated system (Mn2O4).4 The intensities of both peaks, around 3 and 4 V, decrease coherently with the Mn content in the spinel, as better shown in Fig. S5(I), ESI† reporting the behavior of the 1st cycle-specific capacity as a function of the Li/Mn ratio.
The rate capability is reported in Fig. 3(II) and Fig. S5(II), ESI† for all the compositions. The best functional behavior was obtained for LFMTa and LFMTb, exhibiting discharge capacities between 230–250 mA h g−1 and 130 mA h g−1 in the current density range 21–270 mA g−1, respectively. Lower performances were in contrast achieved by cells including LFMTd, especially at higher current densities where Cdisch values <60 mA h g−1 were delivered at 1C. However, in each composition, the charge/discharge processes are almost reversible. The coulombic efficiency is very close to 1 after the initial 3 cycles and mostly at higher C rates, suggesting that the doped spinels have a stable structure during lithium-ion intercalation/deintercalation.
The amount of Li ions that de/intercalate from/into the spinels was estimated by the voltage profile at low current density (C/20), as reported in Fig. 4(I). Here the redox plateaus at about 4 V and 2.8–3.0 V are clearly evident. During the lithiation step, the Li+ equivalents progressively inserted through the reduction of the Mn redox couple are strictly dependent on the type of spinel. In particular, they change linearly by increasing the Li/Mn ratio (e.g. nominally from 1.0 to 2.3 for LFMTa), as better shown in Fig. S5(I), ESI.† The plot also shows that, except for the first cycle, the mechanism involved upon lithiation/delithiation is almost symmetric, suggesting very good coulombic efficiency. After the first cycle, the amount of exchanged Li is always x = 1, both on charge and discharge (see Fig. 4(I)).
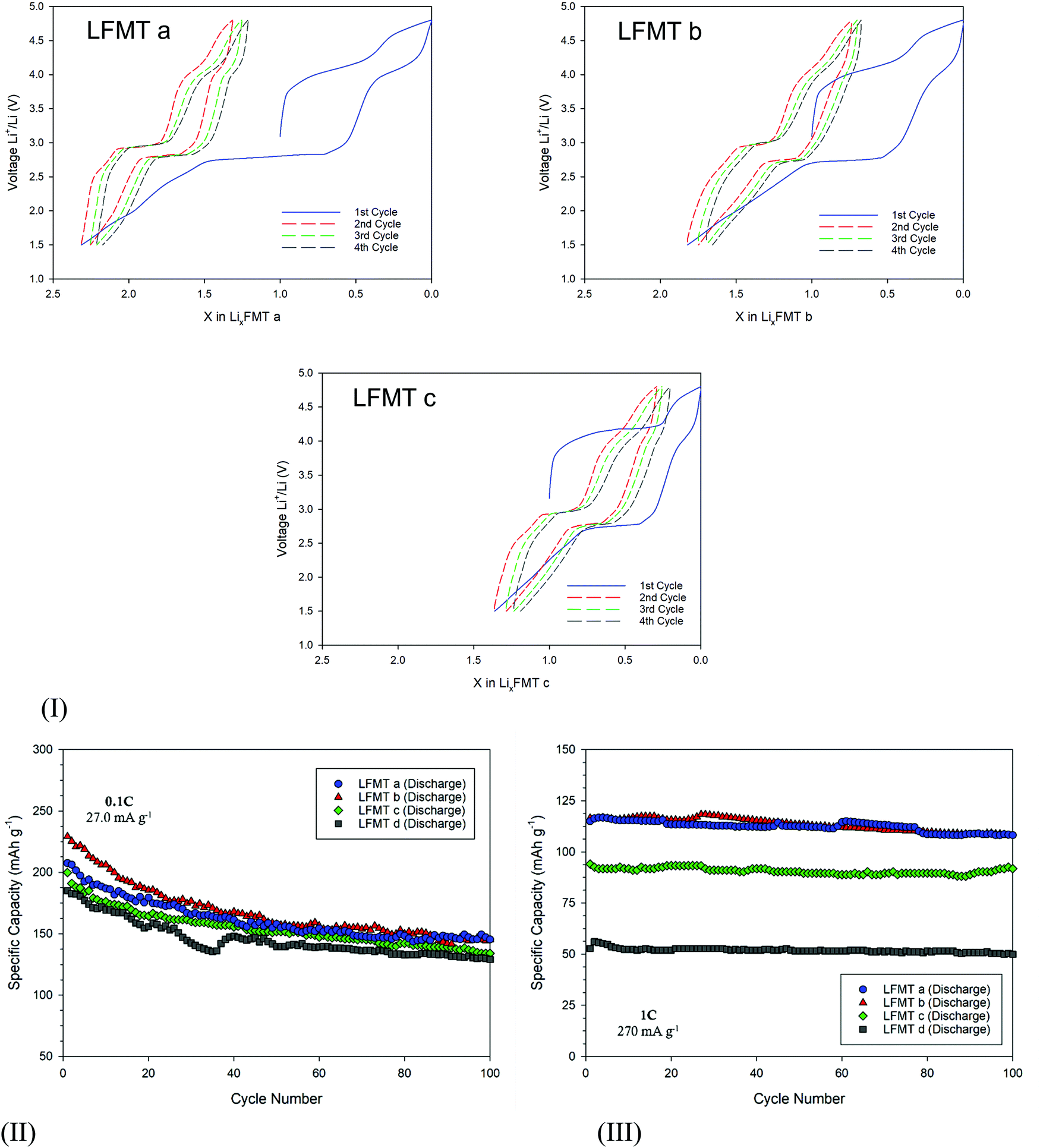 | ||
| Fig. 4 (I) Voltage-composition profiles of the LFMTa–d cathodes during the first charge–discharge cycle with a voltage window 1.5–4.8 V vs. Li/Li+ at C/20 rate; (II) discharge capacity upon 100 cycles at a current density of 27 mA g−1 and (III) 270 mA g−1. | ||
The voltage profiles and the dQ/dV plots also reveal the presence of a plateau at around 4.3–4.4 V during the first oxidation step, which gradually becomes more intense as the Mn content in the spinel decreases (red star in Fig. 3(I)). This signal may be ascribed to an irreversible anionic redox activity that leads to O2 loss, as a consequence of the oxidation of structural O2−, possibly at surface sites, as it is often observed in literature both in layered and spinel-like cathodes.13,25,26 Despite the fact that O2 evolution is in some cases valuable, this process does not contribute, if not marginally, to the delithiation capacity, which in contrast stems from the Mn(IV)–Mn(II) reduction. On the other hand, the anionic redox activity may affect to some extent the cathode capacity retention upon prolonged cycling, inducing potential structural changes. Fig. 4(II) shows the cell stability upon cycling for all the spinels at 27 mA g−1 for 100 cycles. All the compositions show an initial capacity very close or even exceeding 200 mA h g−1 (LFMTa: 207 mA h g−1, LFMTb = 229 mA h g−1; LFMTc = 199 mA h g−1; LFMTd = 185 mA h g−1). However, after 20 cycles, the capacity loss increases with the entity of the irreversible O2 release. Indeed, capacity retention higher than 78% is obtained in the case of LFMTa where the irreversible phenomenon is absent or negligible, whereas more severe losses are observed when the oxygen evolution is more pronounced, such as for LFMTc and LFMTd (capacity retention of about 68%).
At higher current densities (270 mA g−1), the spinel-based electrodes show higher stability with negligible capacity reduction and coulombic efficiency higher than 98% (Fig. 4(III)). However, very different values of specific capacity are obtained, which remarkably decrease when the irreversible anionic redox phenomena are more intense and when the Mn stoichiometry (Mn2−x−y) in the spinel significantly deviates from unity. In particular, capacity higher than 125 mA h g−1 is delivered by the cells including the LFMTa and LFMTb spinels, whose cycling behavior is only marginally affected by O2 loss at high voltage. However, despite capacity reductions are still evident at low C rates, the Mn multi-substitution with transition metals appear to be a very good strategy in order to significantly enhance the long-term stability of the spinel-based cathodes. As well known, in fact, undoped LMO shows severe capacity reduction upon cycling, that might exceed 49% even at very lower current densities, because of the J–T distortion.
The O2 release may be somehow suppressed or limited by using proper additives in the liquid electrolytes. Different amounts of succinic anhydride (4% and 6% vol) were added to the selected electrolyte and used in the LFMTa-based cathode in order to evaluate its possible role on the cycling performances. From the Fig. S6(I), ESI† showing the dQ/dV plot for the cell including a 4% of succinic anhydride, there is no evidence of the peak at high voltage, ascribed to the O2 formation. Furthermore, a significant reduction of the capacity fade is observed particularly at low C rate, when slightly higher amounts of additive are added (e.g. 6 vol%) (Fig. S6(II), ESI†).
LFMT cathodes: ex situ XAS investigation
The chemical environment and TM oxidation states were studied by means of XAS through ex situ experiments carried out on the LFMTc cathode (selected as case study), whose cycling was stopped at specific voltages, namely at OCV, delithiated up to 4.8 V, and lithiated up to 1.5 V (see Fig. 3(I)). Fig. 5 shows the corresponding XAS spectra at the Ti, Mn, and Fe L2,3-edges and at the O K-edge for these samples.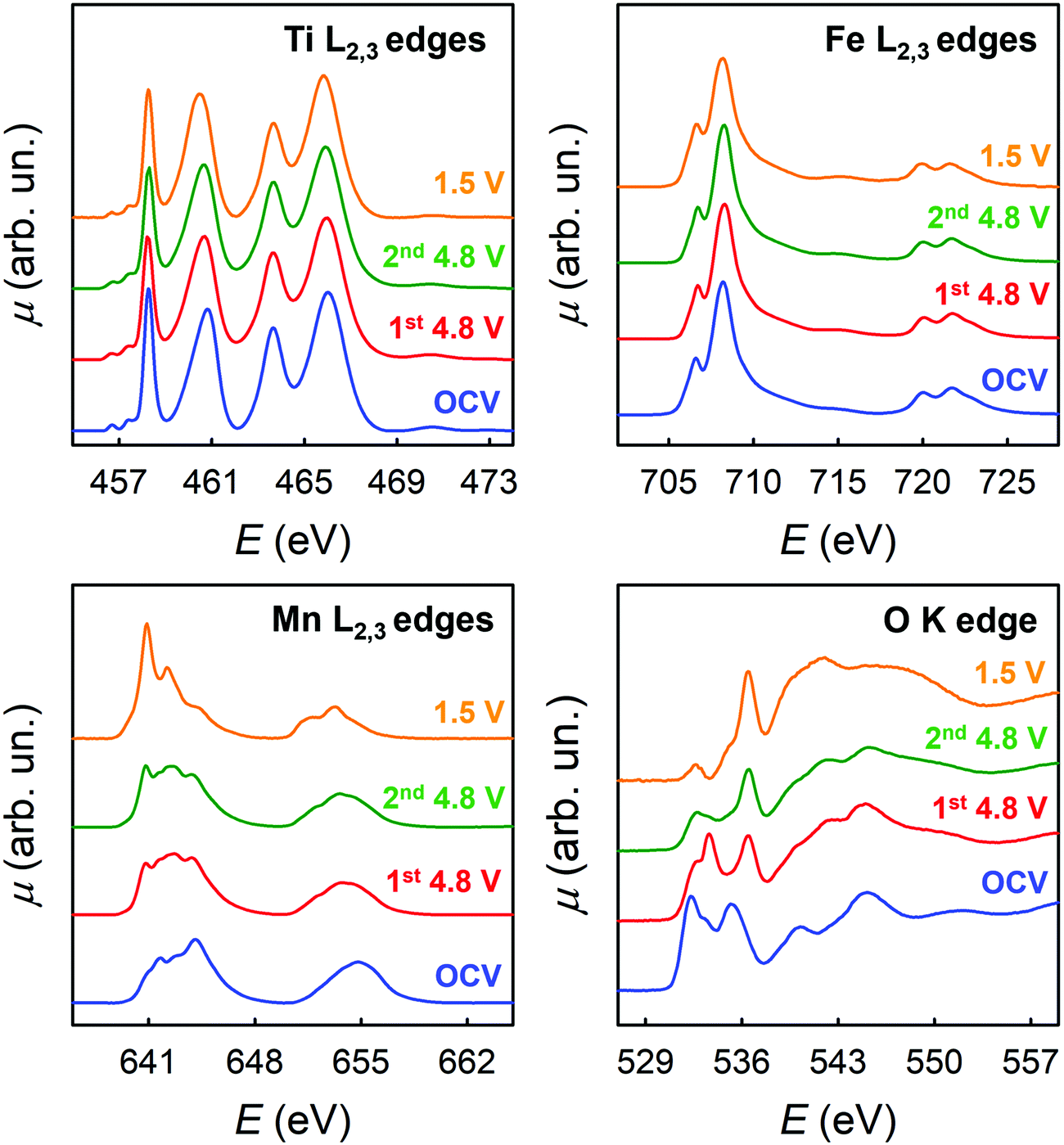 | ||
| Fig. 5 X-ray absorption spectra at the Fe, Mn and Ti L2,3-edges and at the O K-edge on LFMTc cathode at different potentials. | ||
At the L2,3 edges of the TM, the peaks in the XAS spectra arise from dipole allowed electronic transitions from 2p to 3d states. At the O K-edge, the initial states are of 1s character, and therefore dipole allowed electronic transitions probe final states with p character. Therefore, the spectra of Fig. 5 probe in principle all the valence states of the material and can detect their modifications in the lithiation process. However, the detailed interpretation of the L2,3 edges of TMs is complicated by the presence of multiplet interactions. As a general consideration, it is possible to state that the 3d metal L2,3 edges are sensitive to orbital occupancy (oxidation state) and local geometry, while the O K-edge gives a more complete picture of the valence states. In addition, concerning the 3d TMs, the analysis will be here limited to the L3 edge, as the states involved at the L2 edge change only for the different values of the J quantum number.
The spectra at the Ti L2,3-edges are easily discussed, as the spectral shape in all the experimental conditions is unequivocally attributed to Ti(IV) in an octahedral environment (Fig. S7(I), ESI†).27 Ti(IV) has the d0 electronic configuration, and therefore the multiplets arising from the 2p53d1 final state are easily discussed (see the ESI† for additional detail). The octahedral crystal field split the Ti 3d states into threefold degenerate t2g states, and twofold degenerate eg states. Thus, the peak at ca. 460 eV is attributed to the electronic transitions from the Ti 2p to Ti t2g states, while the peak at ca. 463 eV is attributed to Ti 2p to Ti eg states.28,29
The spectra at the Fe L2,3-edges are in principle more difficult to be interpreted, as the electronic configurations of Fe in the different oxidation states give rise to a multiplicity of final states. However, for all the experimental conditions we note a strong resemblance of the Fe L2,3-edges with that of Fe2O3 (see Fig. S7(II), ESI†). It is therefore concluded that in all the samples, Fe is present mainly as Fe(III) in an octahedral environment.
Concerning the spectra at the Mn L2,3 edges, the spectral shape which is found in the different experimental conditions is indicative of a complex behavior. The spectrum at OCV shows a marked peak at ca. 644 eV, which, by comparison with literature data,30 is attributed to Mn(IV). The other spectral features arise from Mn in different oxidation states, indicating that in all the experimental conditions Mn is found in a mixed oxidation state, which renders the interpretation less straightforward than for Fe and Ti, and does not allow a meaningful interpretation in terms of multiplet analysis. Some more specific remarks are however possible. In particular: (i) both spectra at 4.8 V show a clear decrease in the peak at 644 eV; simultaneously, the L2,3 edges develop into a spectral shape which is strongly resembling that of the Mn3O4 spinel.30 Therefore, at this potential, the Mn oxidation state is assigned to a value close to 2.7, even if the presence of residual Mn(IV) cannot be excluded. (ii) at 1.5 V, a strong peak close in energy to 641 eV develops. This peak is attributed to Mn(II) by comparison with the spectrum of MnO.30 This clearly indicates that a large amount of Mn is reduced from Mn(III) to Mn(II) at this potential. Therefore, the results for the TM L2,3 edges show that Ti and Fe are inert during the lithiation process, while Mn participates with both the Mn(IV)/Mn(III) and Mn(III)/Mn(II) redox couples.
The oxygen K-edge spectra which are shown in Fig. 5 show notable differences. This is quite surprising, as the O K-edge XANES of TM oxides, where the TM is in octahedral coordination, usually show impressive similarities due to the hybridization of the TM 4s, 4p, and 3d states with the 2p states of oxygen.31,32 Typically, the first two peaks in the O K-edge XANES are due to electronic transitions from the oxygen 1s orbital to oxygen 2p orbitals hybridized with TM 3d orbitals with t2g and eg symmetry.31,32 Here, we note that there is a marked shift towards higher energies when passing from OCV conditions to 4.8 V. This shift is similar to that observed in the literature comparing the spectra of MnO2 and Mn2O3.30 This is a confirmation of the presence of Mn(IV) in OCV conditions. Additional differences are found by comparing the two spectra at 4.8 V. For the sample after the second de-lithiation (green plot in Fig. 5), the peaks at ca. 533–534 eV show a marked decrease in intensity when compared to 1st de-lithiation (red plot). The intensity of this transition is mainly influenced by two factors: the degree of hybridization between oxygen and TM states (covalence of the TM–O bond) and the filling of the TM 3d states with t2g symmetry.31,32 Recalling the results of the TM L2,3 edges, where almost no changes are found comparing the spectra of these specific samples (green and red plots), a difference in the occupation of TM 3d states seems very unlikely. Thus, we are left with the different hybridization as a cause of this noticeable decrease in intensity.
The decrease is even more evident for the spectrum at 1.5 V. This further decrease cannot be due to the reduction of Mn(III) to Mn(II) which is detected at the Mn L2,3, as the 3d4 and 3d5 electronic configurations do have the same occupancy for the t2g states. Therefore, the degree of the TM–O bond should be involved also in this process.
LFMT cathodes: ex situ XRD investigation
To unravel the mechanism responsible for the spinels cycling performances, the structural evolution was studied during the first charging (delithiation) cycle, the second charge/discharge cycle, and also after extended cycling by means of ex situ XRD experiments. This has been performed selecting the LFMTc composition as a case study, due to its sharp irreversible anion redox activity. Fig. 6 shows the patterns collected after stopping the cycle at a few defined voltages. The pattern of the spinel before electrochemical cycling is also included for the sake of comparison. The Rietveld refinement confirms that the specimen is a single phase with lattice parameter 8.272 Å (see Table S1, ESI†).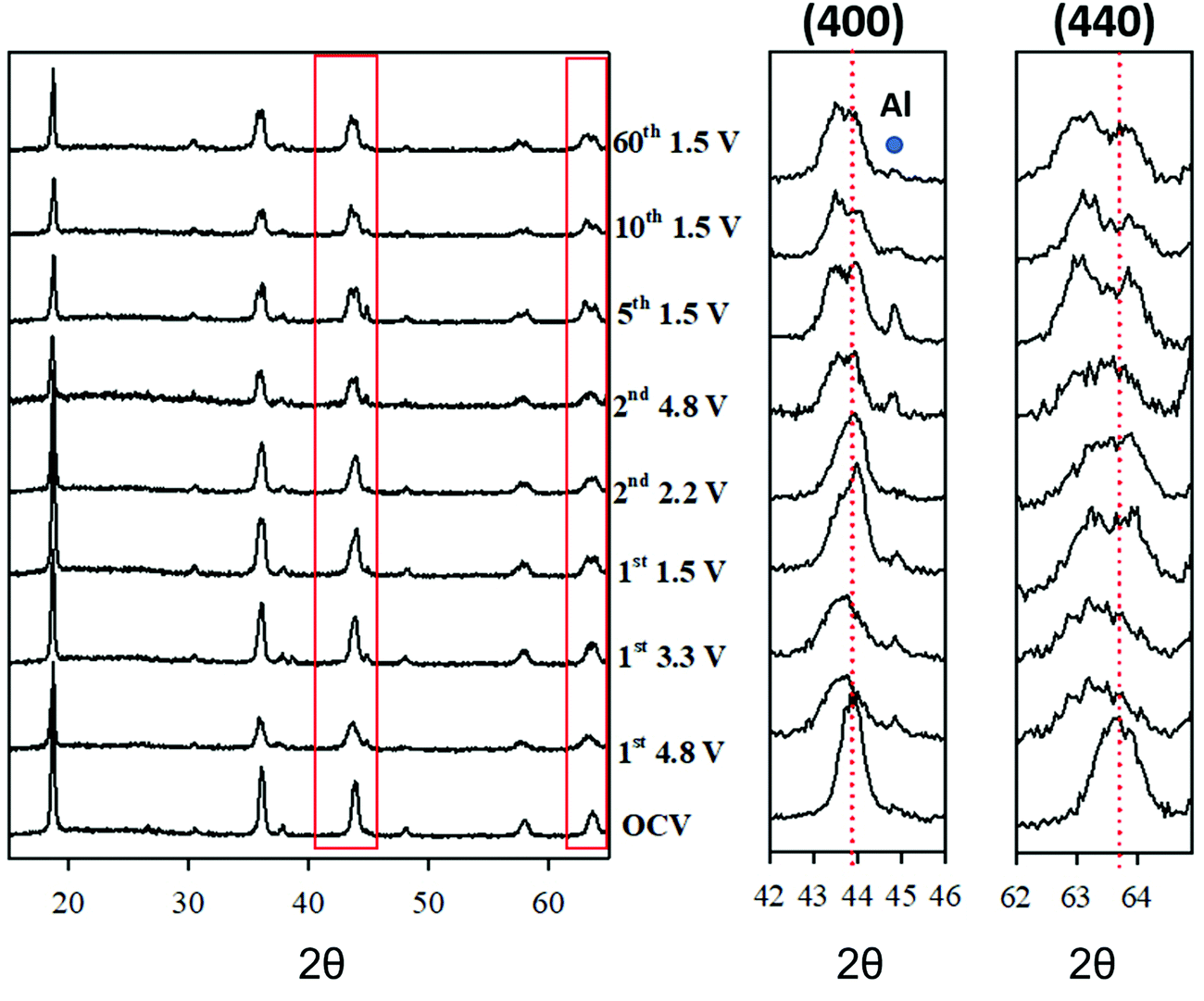 | ||
| Fig. 6 Ex situ experimental XRD patterns during electrochemical cycling of the LFMTc cathode. The 400 and 440 reflections are highlighted in the insets. The red dotted lines refer to the peak position before cycling. | ||
The lattice parameter and the FWHM of the (400) reflection, taken as a reference, are reported in Table 2.
| Cycle # | Voltage (V) | a spinel1 (Å) | a spinel2 (Å) | FWHM(400) (deg) |
|---|---|---|---|---|
| OCV | 3.1 | 8.2728(7) | 0.605(7) | |
| 1 | 4.8 | 8.302(2) | 1.000(2) | |
| 1 dis | 3.3 | 8.251(4) | 8.310(5) | 0.663(8) |
| 1 | 1.5 | 8.250(3) | 8.330(5) | 0.78(3) |
| 2 ch | 2.2 | 8.250(10) | 8.322(10) | 0.77(2) |
| 2 | 4.8 | 8.253(7) | 8.326(8) | 0.88(2) |
| 5 | 1.5 | 8.250(3) | 8.346(4) | 0.90(2) |
| 10 | 1.5 | 8.248(4) | 8.339(5) | 0.92(2) |
| 60 | 1.5 | 8.251(4) | 8.339(5) | 0.88(2) |
Initially, the spinel was delithiated by charging up to 4.8 from the OCV. The diffraction pattern collected at 4.8 V reveals a clear lattice expansion and a broadening of all reflections. A similar trend was observed in undoped Li1−xMn2+xO4. However, in that case, the change in Li concentration was balanced by adjusting the fraction of Mn. The current behavior, instead, cannot be interpreted as a simple extraction of Li+ ions from the spinel, as this would result in a cell contraction, rather than expansion, as usually reported on Li-based compounds.33–35 The origin of this peculiar behavior can be understood by monitoring the XRD patterns during the electrochemical cycling. Indeed, during the first lithiation, at 3.3 V (see Fig. 6), the spinel peaks split into two separated contributions. The same applies likely at 4.8 V, where the extensive peak broadening (see Table 2), hinting at the presence of significant disorder, hinders the observation of the peak splitting. The following point collected while recharging at 2.2 V after the first full lithiation down to 1.5 V, exhibits still a two-phase system. Since then, a clear phase separation is observed throughout the electrochemical process, the split being less apparent at high voltage, due to important disordering: after 60 cycles, the split is still evident.
A careful analysis of the XRD data excludes that such peak splitting may be ascribed to the cubic/tetragonal phase transition, which is typical of LMO spinel, that was also observed in the case of Ti-doped LMO composition.6 In our case, the split is consistent with the coexistence of two spinel phases, with different composition, and, therefore, different lattice parameter. Here we refer to “spinel 1” and “spinel 2” for the spinel phases with smaller and larger unit cell volume, respectively.
We, here, briefly discuss the character of this phase separation. It is first noted that different phases have been reported to form upon lithiation/delithiation in similar systems. Chen et al. suggested the formation of a rock salt phase, which distorts irreversibly into a tetragonal system.6 Even when the rock salt phase has the same lattice spacing (half unit cell) as spinel, some high intensity reflections observed in the spinel phase are missing. In particular, the splitting of the high angle reflection (333/511, 58° with CuKα radiation) is not consistent with the formation of secondary rock salt phase. Neither are the splits consistent with a tetragonal distortion, since not all the peaks would be affected, as for instance the 111 reflection, which is instead observed to split (see Fig. S3(II), ESI†). Therefore, we can exclude the formation of a rock salt phase or a tetragonal distortion.
Eventually, Levi et al. suggested the occurrence of a layered, rhombohedral phase, presenting diffraction pattern very similar to the spinel, which was observed upon delithiation of LiMn2O4.35 Gummow et al. demonstrated that the spinel and layered phase can be distinguished by the occurrence of few minor peaks (220 and 422 in the spinel setting), which are observed only in the spinel and whose intensity depends on the cation distribution between the two sites.36 The 220 reflection is actually observed only for the spinel 2 phase, while the 422 reflection has a too low intensity.
However, assuming that the Li+ ions occupy only the tetrahedral sites without inversion, the 220 reflection shows negligible intensity. Having no direct proof of the formation of a rhombohedral phase, we consider more reliable the formation of two different spinel phases, with different cation distribution and thus different lattice parameter. More importantly, the combination of the two phases led to reliable fits (see Fig. S8, ESI†).
The two phases exhibited a different behavior upon cycling (see Table 2). The lattice parameter of spinel 1 is constant ∼8.25 Å upon cycling, while spinel 2 expands adding Li, from ∼8.31 Å to ∼8.34 Å. In particular, during the first discharge, the lattice parameter expands from ∼8.31 Å at 3.3 V to ∼8.33 Å at ∼3.1 V, then it contracts to ∼8.32 Å at 2.2 V. The point at 4.8 V is, again, likely biased by the peak broadening. The following lithiations cause the unit cell to expand up to ∼8.34 Å. This suggests that spinel 2 is the phase electrochemically active.
Unfortunately, Fe, Mn, and Ti have very similar scattering factors, hence it is not possible to resolve their distribution between the two phases. However, the presence of Li (or Li vacancies) has a strong impact on the intensity of the XRD patterns of the spinel. In this respect, the spinel 1 phase shows the typical cation arrangement of a normal spinel, with Li (or vacancies) on the tetrahedral sites, and transition metals on the octahedral site. Conversely, the spinel 2 phase shows significantly lower scattering from octahedral sites, thus suggesting the presence of Li+ ions. The presence of large Li+ ions in coordination VI (0.76 Å) is likely the reason for the larger cell volume observed for the spinel 2 phase.22 On the basis of this cation distribution, we estimated that spinel 2 phase is in large excess (roughly 3:1), this ratio only slightly changing upon cycling.
The lithiation/delithiation mechanism
Fig. 7 sketches a possible redox mechanism that may be proposed on the basis of the information coming from the electrochemical, spectroscopic and structural investigations.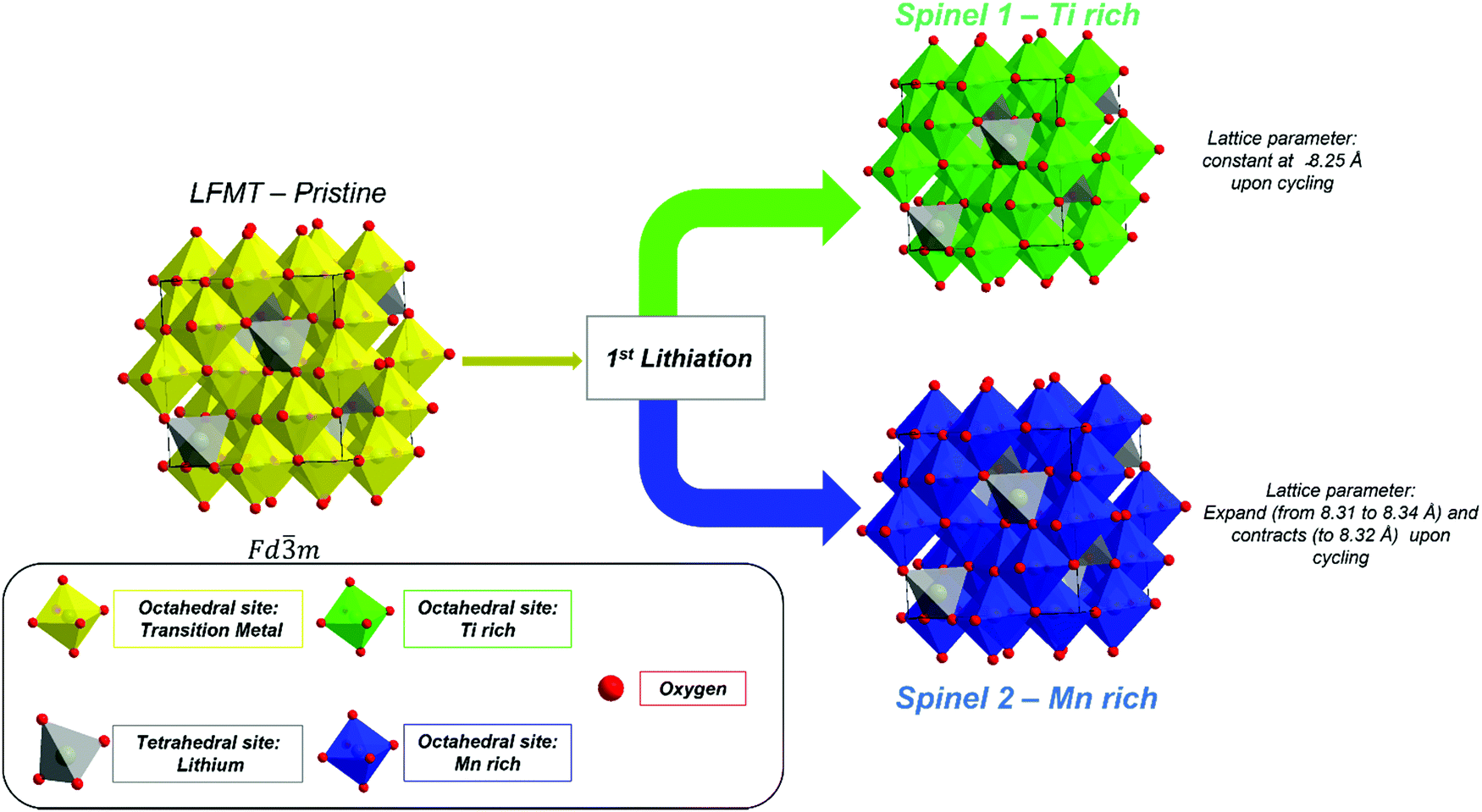 | ||
| Fig. 7 Lithiation mechanism of the LFMTa–d cathodes. | ||
The redox process occurs with the cooperation of a structural modification of the LFMT-based spinels, accompanied by the irreversible phenomena taking place at high voltage during the first charge. By considering the results from the differential capacity and XAS, it is possible to conclude that Mn(IV)/Mn(III) and Mn(III)/Mn(II) are the only redox couple involved in the intercalation/extraction of Li ions from/into the cathode. No further electrochemical process can be observed in the dQ/dV plots and this is confirmed by XAS spectra at the Ti L2,3- and Fe L2,3-edges, which do not evolve upon cycling. Hence, Fe and Ti are electrochemically inactive in these systems, even during the initial cycling step. Therefore, these transition metals act only as stabilizers of the spinel structure in terms of suppression of the J–T distortion and to lowering the J–T-active Mn(III) concentration. In particular, the partial substitution of Mn with Fe(III) contributes to further hinder the cubic to tetragonal phase transition, which is sometimes still observed in the LiMnTiO4-based spinels,6,14 reasonably increasing the overall configurational entropy of the spinel.
At 4.4 V upon delithiation, an irreversible phenomenon takes places, which is likely related to an irreversible anionic redox process leading to the structural O2− oxidation to O22−n and consequent evolution of O2 in the gas phase. This irreversible step appears to be strictly related to the Li/Mn ratio, increasing its intensity by reducing the Mn amount in the spinel. The oxygen loss is reasonably compensated in terms of charge by the redox processes involving Mn in all its oxidation state and specifically a subsequent reduction Mn(IV)–Mn(III)–Mn(II) occurring at high voltage. This hypothesis is confirmed by the XAS measurements: the ex situ spectrum at the Mn L2,3 edge collected at 4.8 V during the first delithiation reveals the unexpected presence of Mn(II) in addition to Mn(III) and a small fraction of Mn(IV) (Fig. 5), not detected in the spectra obtained at OCV.
The extensive Li insertion/extraction in a wide voltage range (4.8–1.5 V) induces a structural transformation upon cycling, as evidenced by ex situ XRD. As discussed before, all diffraction patterns collected after the first delithiation up to 4.8 V reveal a peak splitting, which is coherent with the formation of two spinel phases with an estimated 25/75 volume ratio (spinel 1/spinel 2). This structural modification is in good agreement with the SEM-EDX analysis. Fig. 8 and Fig. S9, ESI† show the SEM images and the corresponding EDX map for cathodes at OCV, delithiated at 4.8 V, and lithiated at 1.5 V at different selected cycles. The concentration maps show that, while Fe remains well distributed throughout all the samples, independently from the voltage and cycle, the Mn and Ti distribution is inhomogeneous, and two separated domains, one Mn-rich (blue) and the other Ti-rich (green), are clearly noticeable. Similar evidence was reported in the literature in the case of Mg-doped LNixMn2−xO4 spinels, whose initial Ni–Mn ordering is lost during Li insertion due to extensive Ni and Mn migration, achieving Ni-rich and Ni-poor domains in distorted tetragonal phases with different Ni:Mn ratios.37
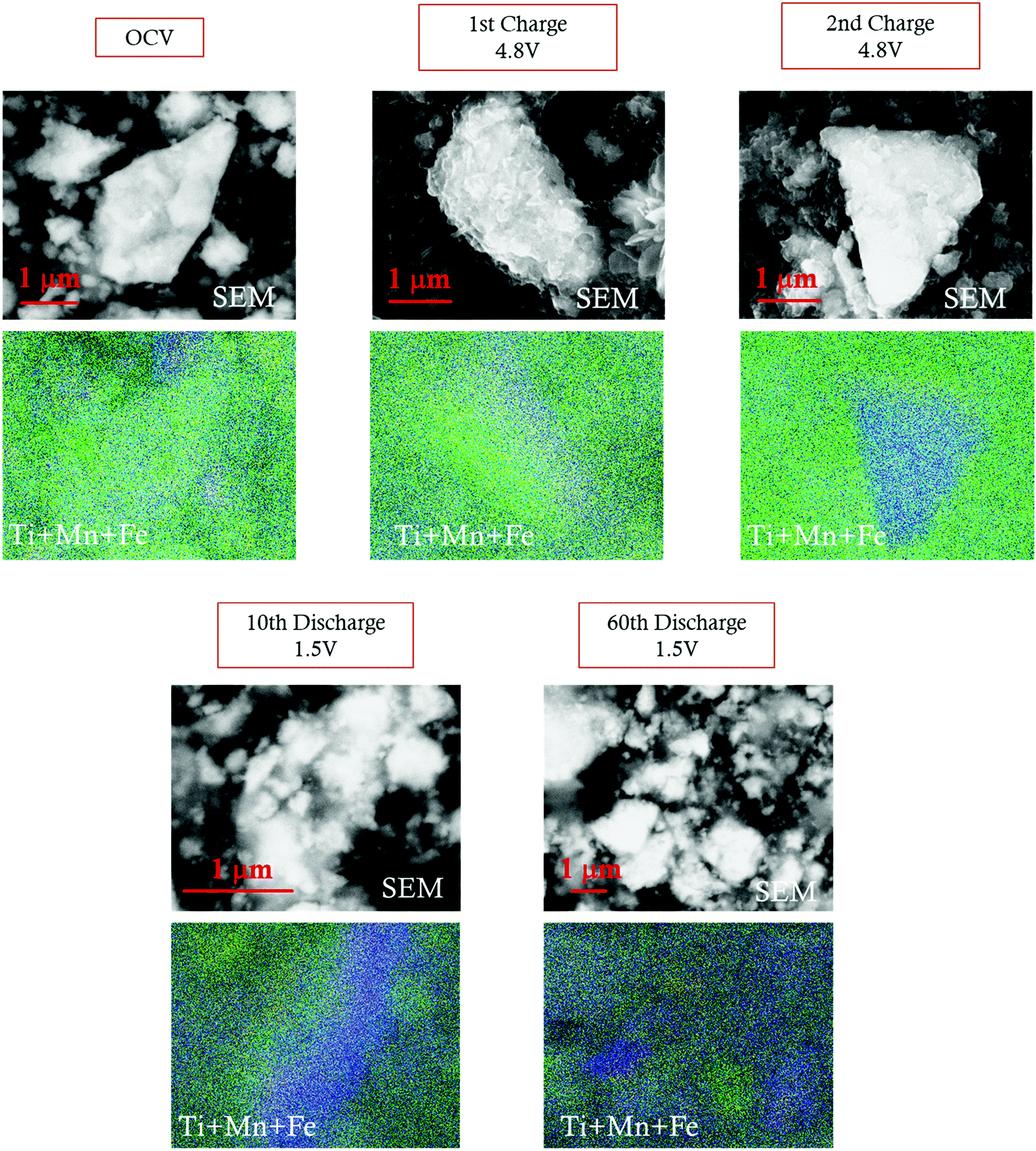 | ||
| Fig. 8 SEM images and SEM-EDX maps of the LFMTc cathode before and after galavanostatic cycling. Fe (yellow); Mn (blue); Ti (green). | ||
While a rough quantification of the volume ratio of the different spinel phases may be attempted, a reliable estimation of their composition is not possible, because of the close scattering factors of the involved TMs. The two spinel phases individuated by XRD were investigated in terms of variations of the lattice parameters upon cycling (Fig. 9). Indeed, the Ti-rich spinel 1 is reasonably electrochemically inactive (or poorly active), since its lattice volume does not change upon Li intercalation/extraction, showing a constant parameter a (8.25 A) during the 1st discharge and 2nd charge. In contrast, the Mn-rich spinel 2 appears to be the active materials. This conclusion is nicely proved by the change of its lattice parameter a upon cycling, as determined by ex situ XRD measurements. Indeed, during the first delithiation (1st charge, Fig. 9) an increase of the lattice parameter with respect to the OCV is noticeable at 4.8 V, thus suggesting a volume expansion of the spinel.
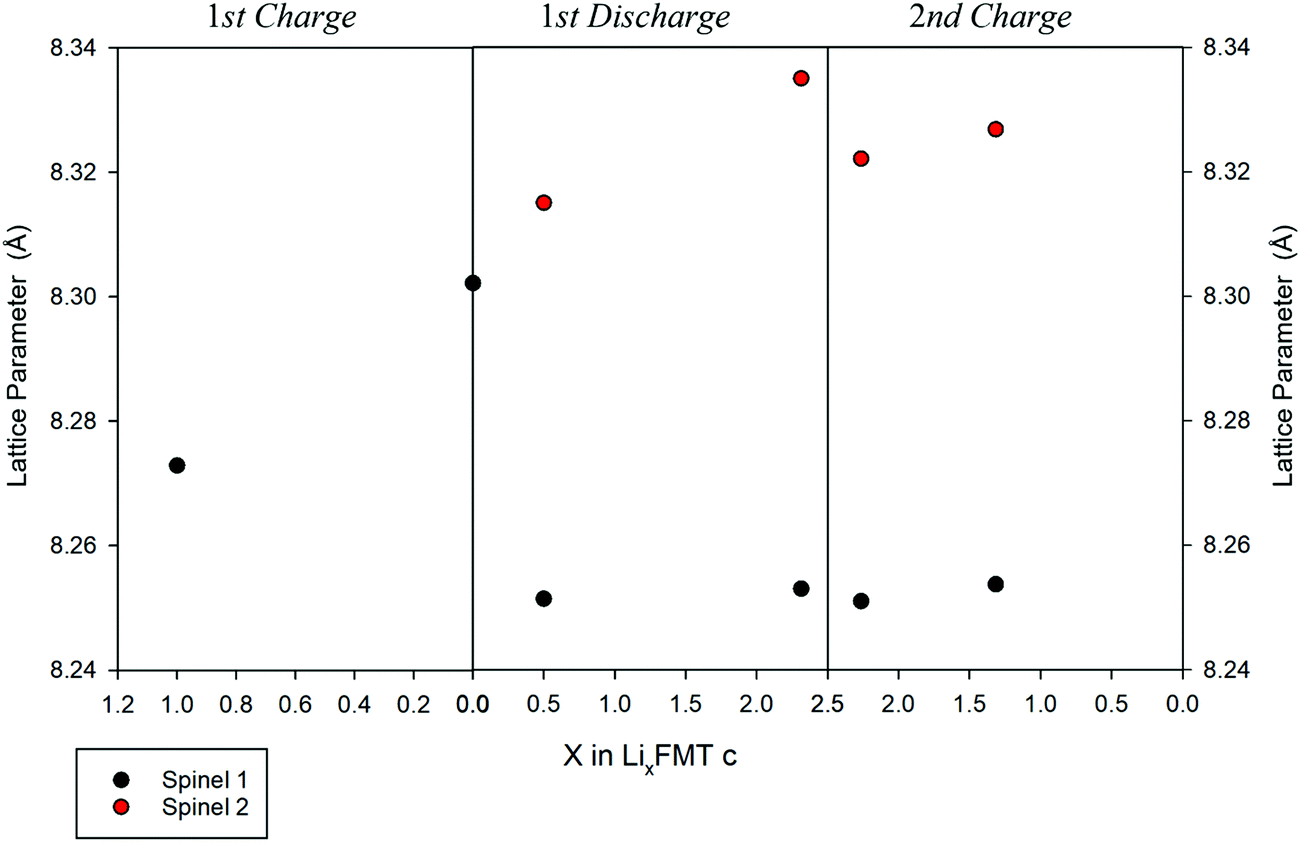 | ||
| Fig. 9 Variation of the lattice parameter, a, upon cycling vs. the Li composition in the LFMTc cathode. | ||
The lattice expansion is even more evident after the 1st lithiation at 1.5 V (Fig. 9, 1st discharge), when the spinel 2 is richer in Li and Mn is almost entirely present as the larger Mn(II) ion, as confirmed by the XAS investigation. During the 2nd charge, the lattice parameter of the spinel 2 decreases with respect the fully lithiated state, indicating a lattice contraction, as expected in case of Li deintercalation.35
The unit cell of spinel 2 is further expanded up to 8.34 Å and contracted upon cycling, demonstrating its electrochemically capability to intercalate and deintercalate Li with excellent reversibility.
Conclusions
We analyzed, for the first time, the microstructure, local environment and electrochemistry of new LiFexMn2−x−yTiyO4 compositions with different Li/Mn ratios. The redox mechanism of these materials was clarified. An easy and fast sol–gel synthesis produced a family of spinels with Li/Mn ratio ranging between 1.0 and 1.7, all presenting the cubic Fdm structure of LiMn2O4 with lattice parameter, a, increasing with the Li/Mn ratio.
Fe/Ti dual-doped spinels show higher electronic conductivity and electronic transport number than undoped LiMn2O4, achieving values that exceed 0.2 mS cm−1 and 0.7 at 90 °C, respectively, in case of compositions presenting smaller lattice parameters.
XAS investigation demonstrated that the only electrochemically active redox couples involves Mn, while both Fe(III) and Ti(IV) do not change their oxidation state upon cycling. Two electrochemical phenomena are clearly observed, one ascribed to Mn(IV)/Mn(III) at around 4 V, and the second one at 3 V, which is assigned to the Mn(III)/Mn(II) transition.
The exchange of more than 2 Li ion through the whole Mn(IV)/Mn(II) redox couple is observed in the LFMT spinels during Li intercalation/deintercalation, providing specific capacity significantly higher than what observed for the undoped spinel (>270 mA h g−1 during the first delithiation step).
Extensive galvanostatic cycling between 1.5 and 4.8 V also demonstrated the presence of irreversible anion redox activity during the first charge, whose entity increases with the Li/Mn ratio in the spinel.
The disorder induced by multiple doping stabilizes the spinel phase, hindering the cubic–tetragonal transition upon prolonged Li intercalation/deintercalation. Such effect results in an unusual behavior through the modification of the LFMT spinel into two different spinel phases with 3:1 mass ratio. The first phase is electrochemically active and shows Mn-rich domains, while the other, including Ti-rich domains, is electrochemically inert, as proved by the lattice parameter variation and SEM-EDX maps upon cycling.
By suppressing the spinel Jahn–Teller distortion, the substitution of Mn(III) with Ti and Fe lowers the concentration of J–T Mn(III), which is responsible for the severe capacity fade in the undoped spinel. Such stabilizing effect, favored by the synergistic presence of Fe and Ti, results in remarkably reduced capacity fade at low current density (<28% after 100 cycles at 27 mA g−1) and to an excellent stability and reversibility in case of higher C rate (270 mA g−1).
The encouraging functional performances of these new LFMT spinels and the resulting structure-property relationship may be an important guide to design an ever-wider range of spinel materials with enhanced stability against J–T distortion as new cathodes for sustainable, greener and Co-free Li-ion batteries.
Author contributions
D. C. prepared the samples and tested the electric and electrochemical properties; M. F., P. G. and L. B. performed XAS measurements; M. C. performed XRD measurements and Rietveld analysis; U. A.-T. performed the SEM characterization; E. Q. conceived the project and wrote the article. All the authors contributed to the paper revision and have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.References
- J. M. Tarascon, W. R. McKinnon, F. Coowar, T. N. Bowmer, G. Amatucci and D. Guyomard, J. Electrochem. Soc., 1994, 141, 1421–1431 CrossRef CAS.
- M. M. Thackeray, Prog. Solid State Chem., 1997, 25, 1–71 CrossRef CAS.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4301 CrossRef CAS.
- X. Zhang, M. Yang, X. Zhao, Y. Wang, M. Wang and L. Ma, J. Mater. Sci.: Mater. Electron., 2015, 26, 6366–6372 CrossRef CAS.
- G. G. Amatucci, N. Pereira, T. Zheng and J.-M. Tarascon, J. Electrochem. Soc., 2001, 148, A171 CrossRef CAS.
- R. Chen, M. Knapp, M. Yavuz, R. Heinzmann, D. Wang, S. Ren, V. Trouillet, S. Lebedkin, S. Doyle, H. Hahn, H. Ehrenberg and S. Indris, J. Phys. Chem. C, 2014, 118, 12608–12616 CrossRef CAS.
- H. C. Wang and C. H. Lu, J. Power Sources, 2003, 119–121, 738–742 CrossRef CAS.
- R. Chen, M. Knapp, M. Yavuz, S. Ren, R. Witte, R. Heinzmann, H. Hahn, H. Ehrenberg and S. Indris, Phys. Chem. Chem. Phys., 2015, 17, 1482–1488 RSC.
- Y. Yu, J. Guo, M. Xiang, C. Su, X. Liu, H. Bai, W. Bai and K. Duan, Sci. Rep., 2019, 9, 1–9 Search PubMed.
- Y. Bao, X. X.-X. Zhang, X. X.-X. Zhang, L. Yang, X. X.-X. Zhang, H. Chen, M. Yang, D. Fang, R. Chen, M. Knapp, M. Yavuz, R. Heinzmann, D. Wang, S. Ren, V. Trouillet, S. Lebedkin, S. Doyle, H. Hahn, H. Ehrenberg, S. Indris, K. Zhou, Y. Li, S. Zheng, M. Zhang, C. Zhang, C. Battaglia, H. Liu, K. Wang, P. Yan, J. Liu, Y. Yang, X. X.-X. Zhang, M. Yang, X. Zhao, Y. Wang, M. Wang, L. Ma, M. Luo, S. Zheng, J. Wu, K. Zhou, W. Zuo, M. Feng, H. He, R. Liu, J. Zhu, G. Zhao, S. Chen, W. Yang, Z. Peng, Q. Wu, Y. Yang, R. Chen, M. Knapp, M. Yavuz, S. Ren, R. Witte, R. Heinzmann, H. Hahn, H. Ehrenberg, S. Indris, K. Zhou, S. Zheng, H. Liu, C. Zhang, H. Gao, M. Luo, N. Xu, Y. Xiang, X. Liu, G. Zhong and Y. Yang, J. Mater. Chem. A, 2015, 43, 120–125 Search PubMed.
- D. Song, H. Ikuta, T. Uchida and M. Wakihara, Solid State Ionics, 1999, 117, 151–156 CrossRef.
- B. Ebin, S. Gürmen, C. Arslan and G. Lindbergh, Electrochim. Acta, 2012, 76, 368–374 CrossRef CAS.
- Z. Cai, H. Ji, Y. Ha, J. Liu, D.-H. Kwon, Y. Zhang, A. Urban, E. E. Foley, R. Giovine, H. Kim, Z. Lun, T.-Y. Huang, G. Zeng, Y. Chen, J. Wang, B. D. McCloskey, M. Balasubramanian, R. J. Clément, W. Yang and G. Ceder, Mater., 2021, 4, 3897–3916 CAS.
- S. Wang, J. Yang, X. Wu, Y. Li, Z. Gong, W. Wen, M. Lin, J. Yang and Y. Yang, J. Power Sources, 2014, 245, 570–578 CrossRef CAS.
- S. Jayapal, R. Mariappan, S. Sundar and S. Piraman, J. Electroanal. Chem., 2014, 720–721, 58–63 CrossRef CAS.
- P. Hervás and R. Millares, CWL Publ. Enterp. Inc., Madison, 2004, vol. 2004, p. 352.
- T. Roisnel and J. Rodríguez-Carvajal, Mater. Sci. Forum, 2001, 378–381, 118–123 CAS.
- G. Panaccione, I. Vobornik, J. Fujii, D. Krizmancic, E. Annese, L. Giovanelli, F. MacCherozzi, F. Salvador, A. De Luisa, D. Benedetti, A. Gruden, P. Bertoch, F. Polack, D. Cocco, G. Sostero, B. Diviacco, M. Hochstrasser, U. Maier, D. Pescia, C. H. Back, T. Greber, J. Osterwalder, M. Galaktionov, M. Sancrotti and G. Rossi, Rev. Sci. Instrum., 2009, 80, 043105 CrossRef CAS.
- C. Castán-Guerrero, D. Krizmancic, V. Bonanni, R. Edla, A. Deluisa, F. Salvador, G. Rossi, G. Panaccione and P. Torelli, Rev. Sci. Instrum., 2018, 89, 054101 CrossRef.
- R. A. Huggins, Ionics, 2002, 8, 300–313 CrossRef CAS.
- J. Guan and M. Liu, Solid State Ionics, 1998, 110, 21–28 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- J. B. Goodenough, Solid State Ionics, 1994, 69, 184–198 CrossRef CAS.
- J. B. Goodenough, Prog. Solid State Chem., 1971, 5, 145–399 CrossRef CAS.
- M. Luo, S. Zheng, J. Wu, K. Zhou, W. Zuo, M. Feng, H. He, R. Liu, J. Zhu, G. Zhao, S. Chen, W. Yang, Z. Peng, Q. Wu and Y. Yang, J. Mater. Chem. A, 2020, 8, 5115–5127 RSC.
- E. Hu, X. Yu, R. Lin, X. Bi, J. Lu, S. Bak, K.-W. Nam, H. L. Xin, C. Jaye, D. A. Fischer, K. Amine and X.-Q. Yang, Nat. Energy, 2018, 3, 690–698 CrossRef CAS.
- R. Brydson, H. Sauer, W. Engel, J. M. Thomass, E. Zeitler, N. Kosugi and H. Kuroda, J. Phys.: Condens. Matter, 1989, 1, 797–812 CrossRef CAS.
- G. Van Der Laan and I. W. Kirkman, J. Phys.: Condens. Matter, 1992, 4, 4189–4204 CrossRef CAS.
- R. Laskowski and P. Blaha, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 1–6 CrossRef.
- B. Gilbert, B. H. Frazer, A. Belz, P. G. Conrad, K. H. Nealson, D. Haskel, J. C. Lang, G. Srajer and G. De Stasio, J. Phys. Chem. A, 2003, 107, 2839–2847 CrossRef CAS.
- F. Frati, M. O.-J. Y. Hunault and F. M.-F. De Groot, Chem. Rev., 2020, 120, 4056–4110 CrossRef CAS PubMed.
- F. M.-F. de Groot, M. Grioni and J. C. Fuggle, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 5715–5723 CrossRef CAS.
- D. Mohanty, S. Kalnaus, R. A. Meisner, A. S. Safat, J. Li, E. A. Payzant, K. Rhodes, D. L. Wood and C. Daniel, RSC Adv., 2013, 3, 7479–7485 RSC.
- W. Zhu, D. Liu, J. Trottier, C. Gagnon, A. Mauger, C. M. Julien and K. Zaghib, J. Power Sources, 2013, 242, 236–243 CrossRef CAS.
- E. Levi, M. D. Levi, G. Salitra, D. Aurbach, R. Oesten, U. Heider and L. Heider, Solid State Ionics, 1999, 126, 109–119 CrossRef CAS.
- R. J. Gummow and M. M. Thackeray, J. Electrochem. Soc., 1993, 140, 3365–3368 CrossRef CAS.
- M. Wagemaker, F. G.-B. Ooms, E. M. Kelder, J. Schoonman, G. J. Kearley and F. M. Mulder, J. Am. Chem. Soc., 2004, 126, 13526–13533 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc00573e |
| This journal is © The Royal Society of Chemistry 2022 |