
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptically controlled large-coercivity room-temperature thin-film magnets†
Anish
Philip
a,
Yifan
Zhou
b,
Girish C.
Tewari
a,
Sebastiaan
van Dijken
b and
Maarit
Karppinen
 *a
*a
aDepartment of Chemistry and Materials Science, Aalto University, FI-00076 Aalto, Finland. E-mail: maarit.karppinen@aalto.fi
bNanoSpin, Department of Applied Physics, Aalto University, FI-00076 Aalto, Finland
First published on 25th November 2021
Abstract
Photo-controlled room-temperature hard magnets could open new horizons for high-density information storage. For this, the material should be fabricated as device-integrable (conformal, stretchable, transparent, etc.) thin films and preferably from readily available and innocuous chemical constituents. Here we report a viable material candidate to address all these criteria. The material basis is the ferrimagnetic ε-Fe2O3 polymorph of trivalent iron oxide with an exceptionally high room-temperature magnetic coercivity. To bring the photo-controllability, azobenzene moieties with trans–cis isomerization tendency are embedded into this matrix as regular monomolecular layers. The strongly emerging atomic/molecular layer deposition (ALD/MLD) technique offers us a scientifically elegant yet industrially feasible tool to fabricate these superlattice thin films with nanoscale precision. We demonstrate reversible changes in both coercivity and magnetization values with alternating irradiations of the films with UV and visible light. Interestingly, the azobenzene layers not only add this switching functionality but also enhance the overall magnetic performance of the ε-Fe2O3 matrix.
1. Introduction
In the big data era, the magnetic data storage density is rapidly gaining importance. To increase the storage density, the number of magnetic grains per unit area should be increased.1 This implies the use of smaller grains, which poses increasingly strict requirements for the magnetic material itself, in particular for its coercivity field. The harder the magnetic material is, the stronger it is against losing the magnetization (due to the ferromagnetic-to-superparamagnetic transition) when the grain size is decreased. In this respect, the rarest of the trivalent iron oxide polymorphs, i.e. ε-Fe2O3, could open new horizons for high-density magnetic storage2–4 owing to its room-temperature (RT) ferrimagnetism with a giant coercive field (up to ∼20 kOe).5–7 On top of that, this material is non-toxic, biocompatible and composed of non-critical elements.The ε-Fe2O3 phase was discovered by Forestier and Guiot-Guillain in 1934,8 but only in trace amounts. However, due to the difficulties in synthesis, it remained a curiosity rather than a viable material candidate up until the first single-phase ε-Fe2O3 nanocrystal samples were grown in 2004,7 followed by the successes in high-quality thin-film fabrication by pulsed laser deposition (PLD)6,9,10 and more recently by atomic layer deposition (ALD).11 The ALD fabrication yielded single-phase polycrystalline ε-Fe2O3 films even on non-epitaxial substrates.
From another perspective, optically controlled molecular magnets have been investigated as enablers of ultra-fast recording at a high density; here the idea is that with light irradiation the magnetic properties could be tuned locally at individual grains. This research avenue was opened along the emergence of molecular magnetism in the 1980s, and the pioneering studies by Sato and Hashimoto in the 1990s on photo-controlled molecular magnets.12 Such multifunctional materials have been challenged by combining magnetic metal species and photo-active organic ligands.13–16 The stimuli-control over the magnetic properties could be achieved through, e.g., light-induced excited spin-state trapping or light-induced metal-to-metal charge transfer.17 The state-of-the-art photomagnetic material variety comprises, e.g., Prussian blue type metal complexes,12,18,19 as well as magnetic nanoparticles14,20,21 and layered inorganics16 modified with photo-responsive organic molecules. However, regarding the magnetic characteristics these inorganic–organic hybrids have remained far inferior to their inorganic counterparts. Moreover, these materials are typically synthesized through multi-step solution-based routes, which are incompatible with the current microfabrication technologies.
We have selected an entirely new strategy to synthesize these materials from gaseous precursors using the state-of-the-art ALD technique. Notably, the ALD technique has been the fastest growing thin-film fabrication technology in microelectronics for years22–25 and is currently rapidly expanding to many other application fields as well owing to its fundamental benefits derived from the unique way of separating the different precursor pulses.24 The chosen strategy has its basis in our recent success in developing a facile ALD process for amazingly stable ε-Fe2O3 thin films exhibiting unique magnetic coercivity characteristics.11 Another important recent advancement in this field is the possibility to combine molecular layer deposition (MLD) cycles for organic molecules with the ALD cycles for the inorganic layers. Indeed, the combined ALD/MLD technique23,26,27 provides us with an ideal route for carefully layer-engineered inorganic–organic superlattices, as it allows the ALD and MLD precursor pulses to be mixed with digital accuracy for essentially any predetermined pattern, regular or irregular.28 We have already demonstrated the insertion of both monomolecular organic layers and thicker Fe-organic layer blocks within the ε-Fe2O3 matrix to enhance the mechanical flexibility of the films; in these experiments terephthalic acid (HOOC–C6H4–COOH) was used as the organic precursor.29,30
In the present work we now fuse together the intrinsic photo-functionality of the organic moiety and the unique magnetic properties of ε-Fe2O3 in a synergistic manner. Our choice for the photochromic organic precursor is the azobenzene dicarboxylic acid (HOOC–C6H4–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–C6H4–COOH) that upon ultraviolet (UV) irradiation undergoes a reversible trans–cis isomerization around the NN bond, accompanied by a considerable structural change.15,31–34 The back-switching is driven by visible (vis) light illumination or thermal relaxation. The azobenzene moiety has been investigated in the context of metal–organic framework (MOF) materials either as an intrinsic linker or as a kind of surface additive.34–36 Most related to the present study, we recently demonstrated a photo-switched water molecule capture/release behavior for ALD/MLD-grown MOF-like Fe-azobenzene thin films.34 In the present work we now embed separate azobenzene layers in the ferrimagnetic ε-Fe2O3 matrix for well-defined superlattice (SL) structures to challenge the photo-control of the unique magnetic properties of the ε-Fe2O3 phase. We believe that these results are a clear step ahead towards the dream of switchable on-and-off type flexible hard magnets operating at room temperature.
N–C6H4–COOH) that upon ultraviolet (UV) irradiation undergoes a reversible trans–cis isomerization around the NN bond, accompanied by a considerable structural change.15,31–34 The back-switching is driven by visible (vis) light illumination or thermal relaxation. The azobenzene moiety has been investigated in the context of metal–organic framework (MOF) materials either as an intrinsic linker or as a kind of surface additive.34–36 Most related to the present study, we recently demonstrated a photo-switched water molecule capture/release behavior for ALD/MLD-grown MOF-like Fe-azobenzene thin films.34 In the present work we now embed separate azobenzene layers in the ferrimagnetic ε-Fe2O3 matrix for well-defined superlattice (SL) structures to challenge the photo-control of the unique magnetic properties of the ε-Fe2O3 phase. We believe that these results are a clear step ahead towards the dream of switchable on-and-off type flexible hard magnets operating at room temperature.
2. Experimental section
A commercial flow-type hot-wall ALD reactor (F-120 by ASM Ltd) was used for depositing the ε-Fe2O3:azobenzene SL thin films from iron chloride (FeCl3, Merck, 95%), deionized water, and azobenzene-4,4′-dicarboxylic acid (AZO; TCI, 95%) precursors. Both FeCl3 and AZO were placed inside the reactor in open boats and heated at 158 and 240 °C, respectively, while the deionized water cylinder was kept outside the reactor. Nitrogen (N2, 99.999%, flow rate at 300 SCCM) was used as both the precursor carrier gas and the purge gas between the precursor pulses. The reactor pressure was kept at 3–5 mbar.The films were deposited at 280 °C on both Si(100) and quartz plates, of which the latter was used for the UV–vis absorption studies. The precursor pulse/purge lengths were adopted from our previous studies on the same precursors, as follows: 2 s FeCl3/4 s N2/1 s H2O/3 s N2 for the ε-Fe2O3 layers and 2.5 s FeCl3/10 s N2/20 s AZO/50 s N2 for the azobenzene layers. The overall pulsing sequence for the SL films was [(FeCl3 + H2O)m + (FeCl3 + AZO)]n + (FeCl3 + H2O)m, where the individual ε-Fe2O3 layer thickness is indicated with m, and n controls the total number of azobenzene layers within the ε-Fe2O3 matrix. The total number of ALD or MLD cycles (controlling the total film thickness) is thus expressed as [n(m + 1) + m].
Grazing incidence X-ray diffraction (GIXRD; X’Pert MPD PRO Alfa 1, PANalytical; CuKα) was used to characterize the crystallinity of the films. The same equipment was used in the X-ray reflectivity (XRR) mode for verifying the SL pattern and the overall film thickness; the data were fitted with X’Pert Reflectivity software by PANalytical. The chemical bonding structure was investigated using Fourier transform infrared (FTIR; Bruker Alpha II) spectroscopy. The spectrum of the bare silicon substrate was subtracted from the FTIR spectra of the samples.
Scanning electron microscopy (SEM; Hitachi S-4700) was used to analyze the surface morphology of the films. For the SEM measurement, the sample specimen was mounted on a carbon tape and analyzed at a current of 15 μA and a voltage of 10 kV. The UV–vis absorption spectra were recorded for the films deposited on quartz plates in the wavelength range of 200–800 nm (Shimadzu UV-2600 spectrometer).
For the trans-to-cis isomerization studies, the films were irradiated with UV light (365 nm) using a 200 W xenon-doped mercury lamp (Hamamatsu Lightning cure LC8) equipped with a cut-off filter (300–480 nm). The reverse cis-to-trans isomerization was achieved with vis-light irradiation. In both cases, the working distance was 10 mm. For the vis-light illumination, total-reflection-type light with the wavelength range from 300 to 800 nm was used. To reduce the power of the Xe-doped Hg lamp and thereby obtain cold light, an optical fiber was utilized in directing the irradiation beam to the thin film. In order to dissipate the heat generated during the irradiation, the sample was placed on a copper plate mounted over cooling blocks.
Magnetic properties were investigated using a vibrating sample magnetometer (VSM; Quantum Design PPMS). A 3 × 4 mm2 sample specimen was glued with GE varnish on a quartz plate and was set parallel to the applied magnetic field. Magnetization versus magnetic field (M–H) isotherms were collected by sweeping the magnetic field from −50 to 50 kOe. The diamagnetic contribution from the substrate was subtracted from the measured data, unless otherwise stated.
3. Results and discussion
We fabricated a series of ε-Fe2O3: azobenzene SL samples using the precursor pulsing sequence of [(FeCl3 + H2O)m + (FeCl3 + AZO)]n + (FeCl3 + H2O)m; see Fig. 1a. The values, n (0–100) and m (20–300), were chosen so that the overall film thickness was in the range of 50 to 250 nm in all samples (Table S1, ESI†). The intended SL structure was verified by XRR analysis (Fig. 2a), while the GIXRD data confirmed that our n = 0 reference sample is of the pure ε-Fe2O3 phase (Fig. 2b),5,9 and the ε-Fe2O3:azobenzene SL films maintain the crystallinity/phase purity upon the insertion of organic layers; in Fig. 2b we present the GIXRD patterns up to n = 20, but even beyond that the films exhibited clear GIXRD peaks due to the ε-Fe2O3 phase. Actually, the degree of crystallinity depended rather on m than on n, which decreases with decreasing m (individual ε-Fe2O3-layer thickness). The SEM images of representative samples indicated an increase in both grain size and polydispersity with increasing n (Fig. S1, ESI†).29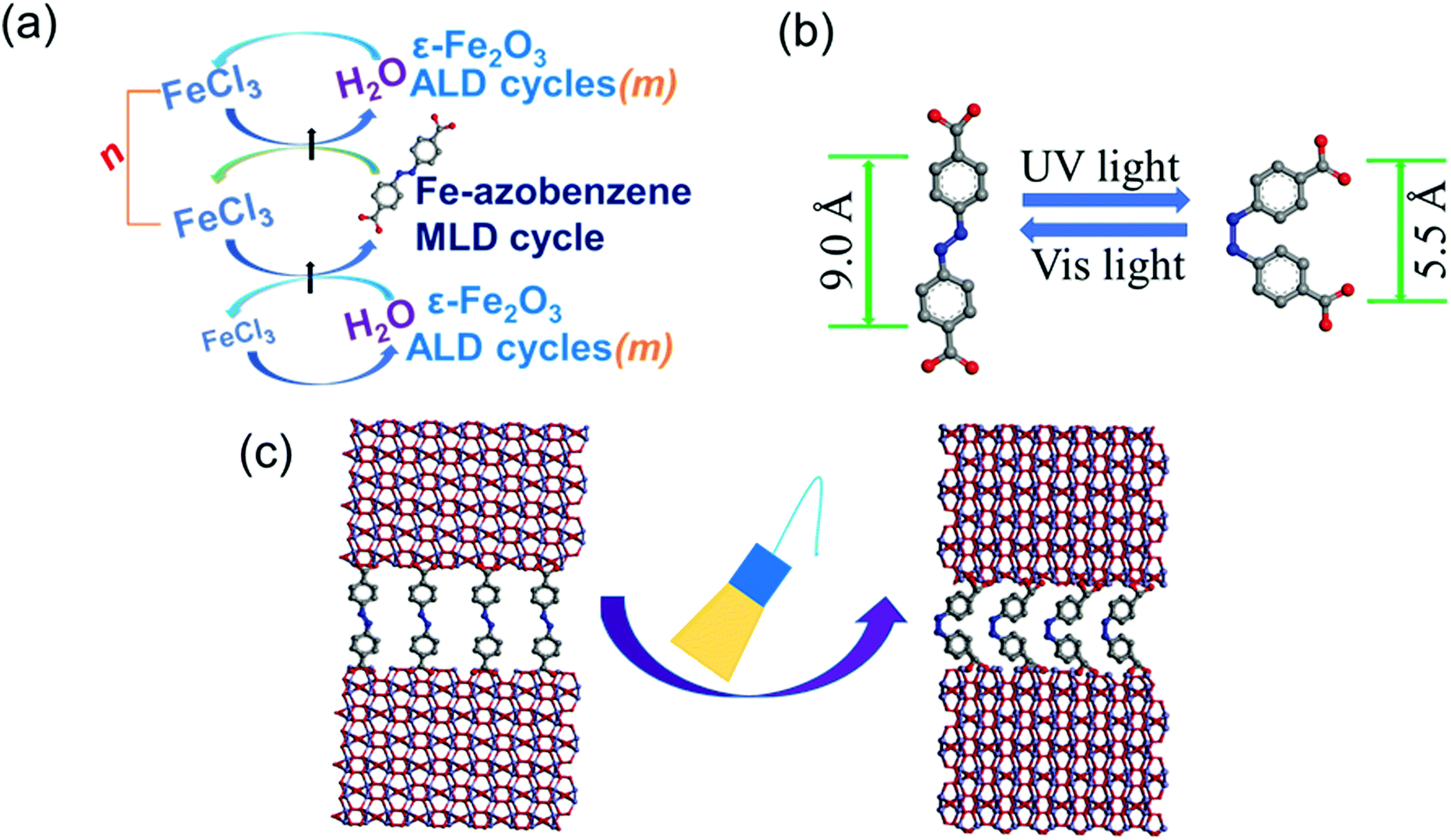 | ||
| Fig. 1 Present research scheme: (a) ALD/MLD process for the ε-Fe2O3:azobenzene SL films; m is the number of (FeCl3 + H2O) ALD cycles applied to deposit each ε-Fe2O3 layer and n stands for the total number of azobenzene layers grown with single (FeCl3 + AZO) MLD cycles into the SL structure. (b) Trans and cis isomers of azobenzene; the molecule lengths indicated are the distances between the carbon atoms at the para positions of the two rings. (c) Azobenzene isomerization within the ε-Fe2O3:azobenzene SL structure, bringing adjacent ε-Fe2O3 layers closer to each other upon UV-light irradiation. | ||
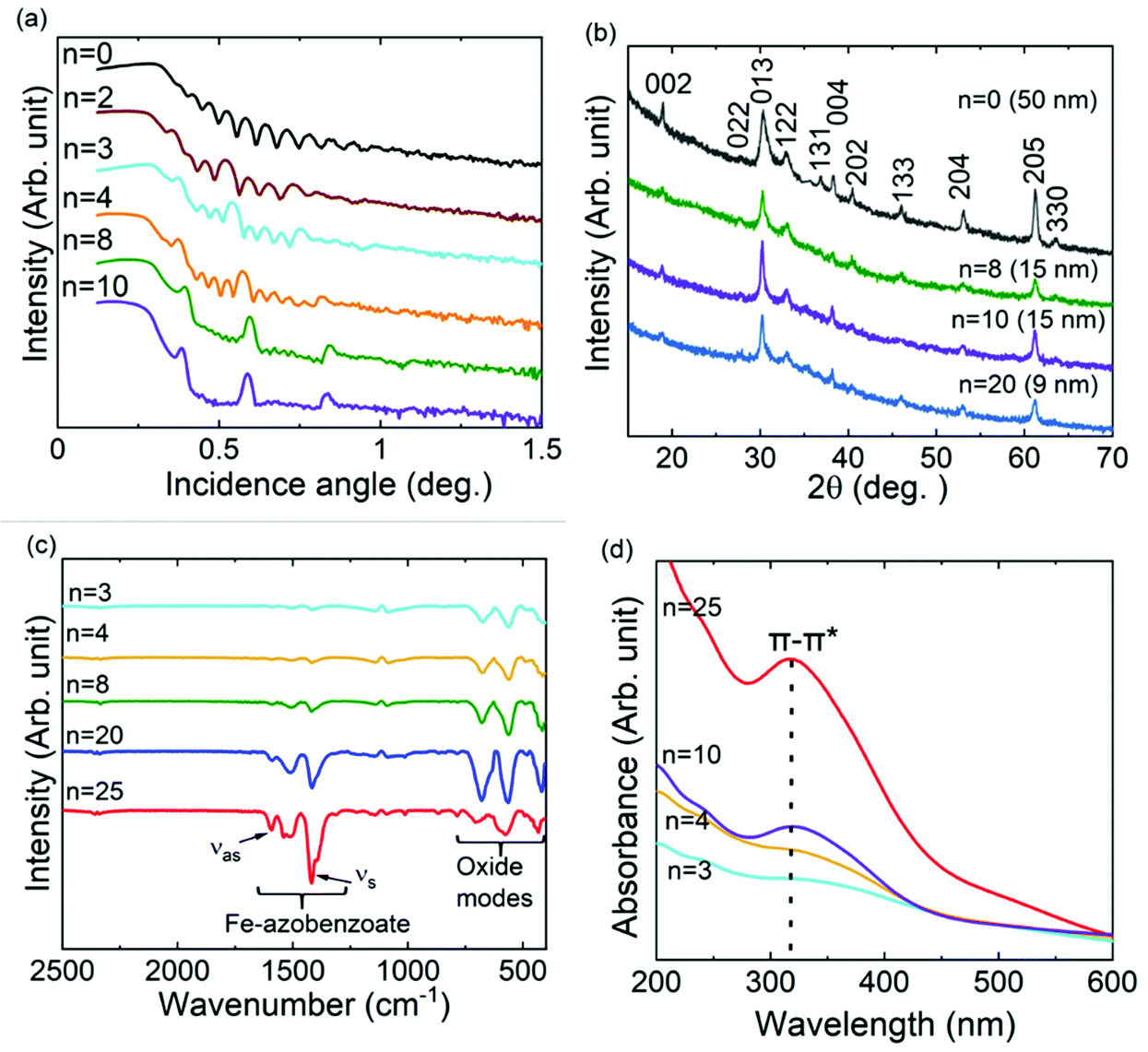 | ||
| Fig. 2 Chemical and structural characterization data for ε-Fe2O3 (n = 0) reference and representative ε-Fe2O3:azobenzene SL samples referred to by the n value (number of organic layers): (a) XRR patterns – the intended SL structure is seen as the appearance of intense SL peaks, between which the number of smaller oscillations equals n (with increasing n these peaks start to overlap). (b) GIXRD patterns – reflections in the n = 0 pattern indexed for the ε-Fe2O3 phase; these peaks are also seen for the SL films, where the intensity decreases with increasing n and decreasing individual ε-Fe2O3-layer thickness (value shown in parentheses). (c) FTIR spectra – the carboxylate group features increase and those due to the Fe–O bonds decrease in intensity with increasing n and decrease in overall ε-Fe2O3 content. (d) UV–vis spectra – the band due to the π–π* transition of trans-azobenzene at ca. 317 nm increases with increasing n. | ||
Chemical bonding at the ε-Fe2O3/azobenzene interfaces was addressed based on the FTIR spectra, shown in Fig. 2c for representative samples. In particular, the presence of the carboxylate groups is evident from the strong symmetric (νs) and asymmetric (νas) stretching bands around 1417 and 1593 cm−1, respectively.33 These carboxylate groups originate from the reaction between Fe–OH surface groups (after the latest FeCl3 + H2O ALD cycle) and the HOOC–C6H4–NN–C6H4–COOH precursor pulse, to form the Fe–OOC–C6H4–NN–C6H4–COO–Fe layers in the SL films, as expected. From the splitting between these bands, i.e. (1593–1417) cm−1 = 176 cm−1, it can be concluded that the azobenzene moieties are bonded to the iron atoms via the carboxylate COO− groups according to a bridging-type mode.33,37 The increase in the organic content is seen as a continuous increase of the peak intensities in the FTIR spectra. The bands observed at 680, 565 and 418 cm−1 are due to ε-Fe2O3, and with increasing number of (FeCl3 + H2O) ALD cycles applied these peaks gain intensity.
The presence of trans-azobenzene in the SL films was also unambiguously verified by the UV–vis absorption spectra (Fig. 2d). The absorption band due to the π–π* transition of trans-azobenzene is observed at ca. 317 nm; the linear increase of this absorption peak with increasing n indicates that our ALD/MLD process indeed works as intended.32–34
The magnetic properties of the ε-Fe2O3:azobenzene SL films were studied with VSM by applying a magnetic field along the film surface. Fig. 3a displays the 300 K M–H data collected from −50 to 50 kOe for samples with different numbers of azobenzene layers ranging from 10 to 100, while Fig. 3b shows the data for a sample with twenty azobenzene layers measured at different temperatures ranging from 10 to 300 K. The hysteresis loops are typical of a ferro- or ferrimagnetic material.38 The slight but continuous linear increase at high fields is related to flipping of minority spins along the magnetic field direction in a ferrimagnetic system.5
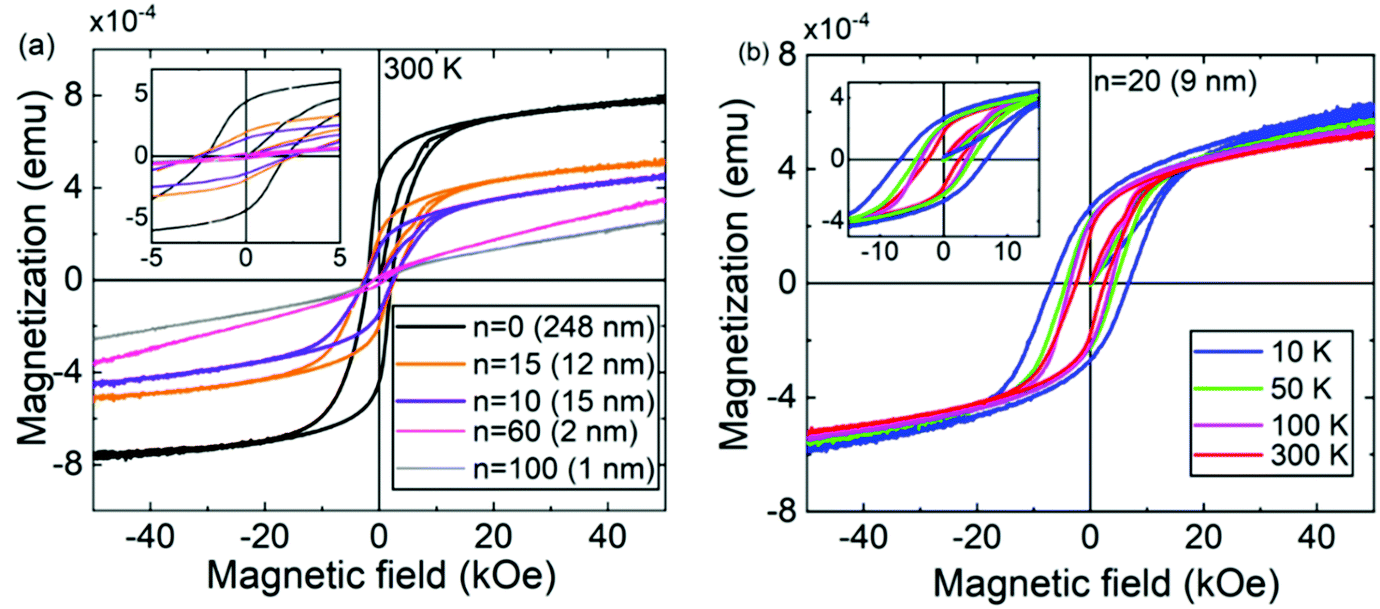 | ||
| Fig. 3 Magnetization versus magnetic field data: (a) M–H curves at 300 K for representative ε-Fe2O3:azobenzene SL films and for a reference ε-Fe2O3 film; introduction of azobenzene decreases the magnetization but enhances the coercivity field; the insets display magnified M–H curves for magnetic fields between −5 and +5 kOe. (b) M–H curves for the n = 20 (9 nm) SL sample at various temperatures; the insets display magnified M–H curves for magnetic fields between −15 and +15 kOe. | ||
Interestingly, the ε-Fe2O3:azobenzene SLs exhibit significantly higher coercivity values than our previously reported ε-Fe2O3:terephthalate SLs and even higher values than the parent ε-Fe2O3 films.11,29 For example, the RT coercivity is 2.6 kOe for the n = 15 SL sample, while it is 2.0 kOe for an ε-Fe2O3 film of the same overall thickness. Notably, even the ε-Fe2O3:azobenzene SLs with ultrathin ε-Fe2O3 layers (down to 1 nm) behave as hard magnets with a RT coercive field as high as ca. 1 kOe. We tentatively attribute the enhanced coercivity of the ε-Fe2O3:azobenzene SL films to the very well persisting crystallinity. Indeed, while the ε-Fe2O3:terephthalate films rapidly lost their crystallinity with increasing number of organic layers,29 the present ε-Fe2O3:azobenzene films remained crystalline even with nanoscale ε-Fe2O3 blocks (Fig. 2b displays the GIXRD patterns of our ε-Fe2O3:azobenzene SL sample series up to n = 20, with ε-Fe2O3 blocks as thin as 9 nm).
Also, the nature of the organic component may play a role in determining the overall magnetic characteristics of the SL films. To investigate the magnetic properties of the Fe-azobenzene moiety alone, we deposited an ca. 200 nm thick Fe-azobenzene film without the ε-Fe2O3 layers by applying (FeCl3 + AZO) cycles only.34 The linear M–H curves measured for this (Fe–OOC–C6H4–NN–C6H4–COO–)n sample at 10 and 300 K (Fig. S2, ESI†) are indicative of a relatively strong paramagnetic nature of the film. From the M–H curves for the ε-Fe2O3:azobenzene SLs (Fig. 3a), it can be seen that while the azobenzene layers enhance the coercive field, they do decrease the film magnetization. This is expected as the iron concentration in the films decreases with increasing n. The ε-Fe2O3:azobenzene SLs were found to be hard magnets even when the number of azobenzene layers increased to 100 with ultrathin (1 nm) ε-Fe2O3 blocks.
We then investigated the photoisomerization characteristics of the ε-Fe2O3:azobenzene SL films. First, the samples were irradiated with UV light (365 nm) for specific time intervals; after all these irradiation periods the corresponding absorbance spectrum was recorded to follow the trans–cis isomerization process, seen as a gradual decrease of the π–π* transition peak at ca. 317 nm; Fig. 4a shows the data for the n = 10 (3 nm) sample in which the individual ε-Fe2O3-layer thickness is 3 nm. The backward (cis–trans) isomerization was confirmed with vis light illumination for 30 min (Fig. 4b). Quite remarkably, the trans–cis–trans isomerization process is essentially reversible.
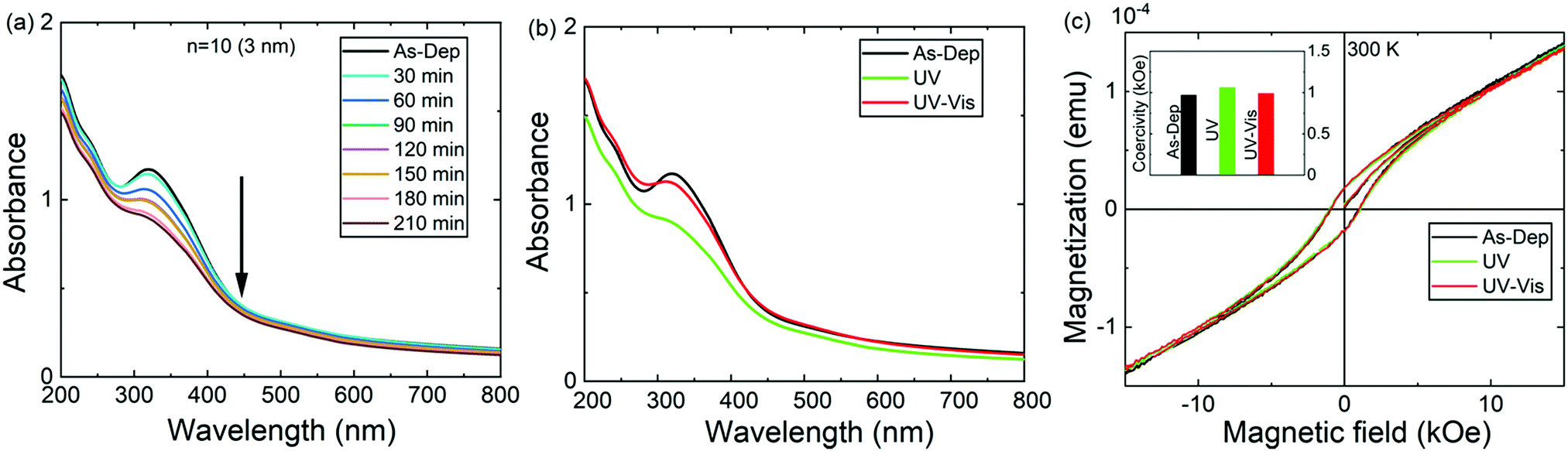 | ||
| Fig. 4 Reversible UV and vis illumination effects: absorption spectra for the n = 10 (3 nm) sample collected (a) after UV irradiation (365 nm) at regular time intervals and (b) after the re-illumination of the 210 min UV-illuminated sample with vis light for 30 min to demonstrate the fully reversible trans–cis–trans isomerization. (c) M–H curves for a representative SL sample, n = 60 (2 nm), before and after consecutive UV (20 min) and vis (20 min) illuminations; the inset shows the changes in coercivity with the irradiation. | ||
The effects of the UV- and vis-irradiation controlled trans–cis and cis–trans azobenzene transitions on the magnetic properties of the ε-Fe2O3:azobenzene films are illustrated in Fig. 4c for the n = 60 (2 nm) sample. The as-deposited sample was first irradiated with UV light for 20 min; this was found to increase the coercivity from 970 to 1060 Oe. To confirm the result, we performed a similar experiment for another sample, n = 100 (1 nm); in this case the coercivity increased from 1140 to 1365 Oe (data shown in Fig. S4, ESI†). These changes are significant and highly promising. The increase in the coercivity is believed to originate from the decreased inter-spacing and thus enhanced interaction between the adjacent ε-Fe2O3 layers upon the trans–cis transformation of the azobenzene moieties which suppresses their size by ca. 3.5 Å,32 and thereby the ε-Fe2O3 inter-layer distance from approx. 9.0 to 5.5 Å, as illustrated in Fig. 1b. Most excitingly, we could also demonstrate for the n = 60 (2 nm) sample the vis-light triggered azobenzene cis-to-trans back-isomerization effect on coercivity, which returned from 1060 to 990 Oe, indicating that the process is essentially reversible (Fig. 4c). Here we like to mention that the aforementioned changes in coercivity (and also in remnant magnetization) were also seen in the measured raw magnetization data before the subtraction of the substrate contribution (see Table S2, ESI†). Finally, we confirmed that no XRD-detectable structural changes occurred during these reversible trans–cis–trans photo-isomerization reactions of the azobenzene layers. From the XRD patterns (shown in Fig. 5 for the n = 10 (15 nm) sample) no indication of any changes in the crystallinity of the ε-Fe2O3 layers was seen, while the XRR data confirmed that the regular SL structure was fully maintained (Fig. S3, ESI†). This is an essential requirement considering the practical applications of our new photo-controlled magnetic coatings. Moreover, by measuring the magnetic properties after one-year storage under ambient conditions (Fig. S5, ESI†), we could confirm that the material is strong against aging too.
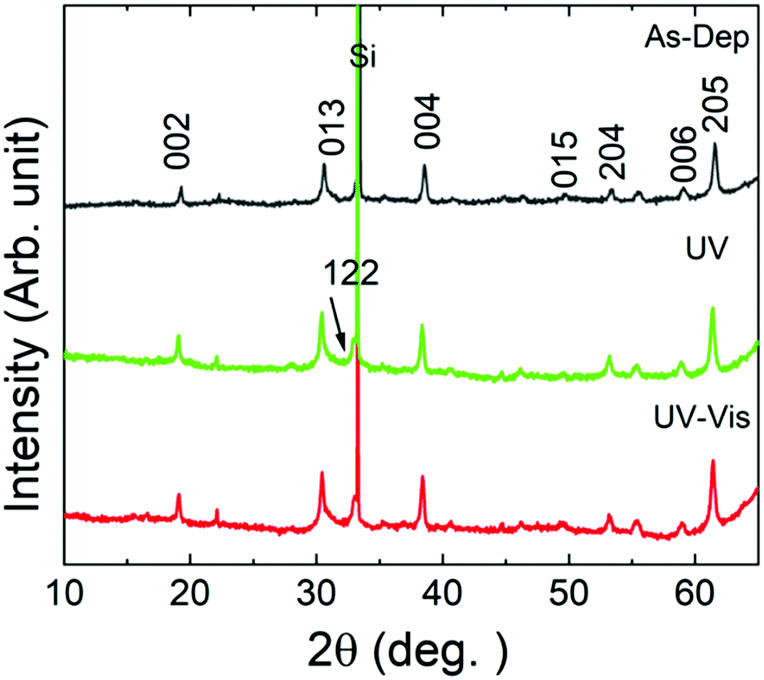 | ||
| Fig. 5 XRD patterns of the n = 10 (15 nm) sample collected after deposition and after consecutive UV and visible light illuminations; the illumination time was 20 min for both UV and visible light illuminations. Indexes are for the ε-Fe2O3 phase. | ||
4. Conclusions
This work has highlighted the exciting possibilities provided by the currently strongly emerging ALD/MLD technique in the field of magnetic thin films, in particular towards novel applications building on stimuli-responsive magnetic thin films. The strong magnetic properties in these thin films emerge from the d electrons of the rather unique iron oxide material ε-Fe2O3, for which the facile ALD process based on simple precursors, FeCl3 and H2O, provides an industry-feasible and sustainable fabrication process. Then, the further tunability of these room-temperature ferrimagnetic films is a kind of designers’ dream and originates from the fact that they can be adorned with ultrathin organic layers with any chosen frequency by simply including MLD organic precursor pulses within the ALD fabrication process. These embedded organic layers can provide the films with additional valuable properties and most straightforwardly improved mechanical properties.In the present study we introduced photoresponsive azobenzene moieties within the ε-Fe2O3 films. This was achieved without compromising the crystallinity and the unique magnetic characteristics of the films. For example, for the ε-Fe2O3:azobenzene superlattice film with 15 monomolecular azobenzene layers embedded within a 200 nm thick film the GIXRD pattern evidenced the high degree of crystallinity of the 12 nm thick individual ε-Fe2O3 layers, and the magnetic measurements verified the high coercivity value of 2630 Oe. Most importantly, we could demonstrate the fully reversible trans–cis–trans photoisomerization reactions in these ε-Fe2O3:azobenzene SL films upon successive UV and visible light illuminations and then finally show that the photo-controlled suppression and expansion of the spacing azobenzene layer between the adjacent ε-Fe2O3 layers upon the azobenzene isomerization resulted in reversible changes in the coercivity value. Even though this switching effect was yet far from complete (being of the order of 10–20% at the best), we positively believe that our work provides new horizons for the future development of mechanically flexible and photo-controllable room-temperature thin-film magnets with the desired high coercivity characteristics.
Author contributions
A. P. and M. K. conceived and planned the study. The samples were synthesized and structurally and chemically characterized by A. P. Magnetic properties were measured by A. P. and Y. Z., and the data were interpreted and discussed by A. P., G. C. T., S. V. D. and M. K. A. P. and M. K. wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the funding from the Academy of Finland (Profi 3 and PREIN). This work made use of the RawMatters Finland infrastructure (RAMI) facilities and OtaNano-Nanomicroscopy Center (Aalto-NMC) at Aalto University.References
- W. Kuch, Nat. Mater., 2003, 2, 505–506 CrossRef CAS PubMed.
- S. I. Ohkoshi, A. Namai, M. Yoshikiyo, K. Imoto, K. Tamazaki, K. Matsuno, O. Inoue, T. Ide, K. Masada, M. Goto, T. Goto, T. Yoshida and T. Miyazaki, Angew. Chem., Int. Ed., 2016, 55, 11403–11406 CrossRef CAS PubMed.
- H. Tokoro, A. Namai and S. I. Ohkoshi, Dalton Trans., 2021, 50, 452–459 RSC.
- S. I. Ohkoshi, K. Imoto, A. Namai, M. Yoshikiyo, S. Miyashita, H. Qiu, S. Kimoto, K. Kato and M. Nakajima, J. Am. Chem. Soc., 2019, 141, 1775–1780 CrossRef CAS PubMed.
- J. Tuček, R. Zbořil, A. Namai and S. I. Ohkoshi, Chem. Mater., 2010, 22, 6483–6505 CrossRef.
- M. Gich, I. Fina, A. Morelli, F. Sánchez, M. Alexe, J. Gàzquez, J. Fontcuberta and A. Roig, Adv. Mater., 2014, 26, 4645–4652 CrossRef CAS.
- J. Jin, S. I. Ohkoshi and K. Hashimoto, Adv. Mater., 2004, 16, 48–51 CrossRef CAS.
- H. Forestier and G. Guiot-Guillain, C. R. Acad. Sci. (Paris), 1934, 199, 720 CAS.
- M. Gich, J. Gazquez, A. Roig, A. Crespi, J. Fontcuberta, J. C. Idrobo, S. J. Pennycook, M. Varela, V. Skumryev and M. Varela, Appl. Phys. Lett., 2010, 96, 112508 CrossRef.
- X. Guan, L. Yao, K. Z. Rushchanskii, S. Inkinen, R. Yu, M. Ležaić, F. Sánchez, M. Gich and S. van Dijken, Adv. Electron. Mater., 2020, 6, 1901134 CrossRef CAS.
- A. Tanskanen, O. Mustonen and M. Karppinen, APL Mater., 2017, 5, 056104 CrossRef.
- O. Sato, T. Iyoda, A. Fujishima and K. Hashimoto, Science, 1996, 272, 704–705 CrossRef CAS.
- M. Suda, Bull. Chem. Soc. Jpn., 2018, 91, 19–28 CrossRef CAS.
- T. Yamamoto, E. A. Yurieva, K. Tsuda, T. Hosomi, S. M. Aldoshin and Y. Einaga, Phys. Status Solidi RRL, 2017, 11, 1700161 CrossRef.
- M. Suda, N. Kameyama, A. Lkegami and Y. Einaga, J. Am. Chem. Soc., 2009, 131, 865–870 CrossRef CAS PubMed.
- G. Abellán, C. Martí-Gastaldo, A. Ribera and E. Coronado, Acc. Chem. Res., 2015, 48, 1601–1611 CrossRef.
- M. Reczyński, K. Nakabayashi and S. I. Ohkoshi, Eur. J. Inorg. Chem., 2020, 2669–2678 CrossRef.
- S. I. Ohkoshi and H. Tokoro, Acc. Chem. Res., 2012, 45, 1749–1758 CrossRef CAS PubMed.
- D. M. Pajerqwski, M. J. Andrus, J. E. Gardner, E. S. Knowles, M. W. Melsel and D. R. Talham, J. Am. Chem. Soc., 2010, 132, 4058–4059 CrossRef.
- R. Mikami, M. Taguchi, K. Yamada, K. Suzuki, O. Sato and Y. Einaga, Angew. Chem., Int. Ed., 2004, 43, 6135–6139 CrossRef CAS PubMed.
- M. Suda, M. Nakagawa, T. Iyoda and Y. Einaga, J. Am. Chem. Soc., 2007, 129, 5538–5543 CrossRef CAS PubMed.
- S. M. George, Chem. Rev., 2010, 110, 111–131 CrossRef CAS.
- R. W. Johnson, A. Hultqvist and S. F. Bent, Mater. Today, 2014, 17, 236–246 CrossRef CAS.
- R. K. Ramachandran, J. Dendooven and C. Detavernier, J. Mater. Chem. A, 2014, 2, 10662–10667 RSC.
- H. H. Sønsteby, E. Skaar, Ø. S. Fjellvåg, J. E. Bratvold, H. Fjellvåg and O. Nilsen, Nat. Commun., 2020, 11, 1–7 Search PubMed.
- Y. Zhao, L. Zhang, J. Liu, K. Adair, F. Zhao, Y. Sun, T. Wu, X. Bi, K. Amine, J. Lu and X. Sun, Chem. Soc. Rev., 2021, 50, 3889–3956 RSC.
- X. Meng, J. Mater. Chem. A, 2017, 5, 18326–18378 RSC.
- T. Tynell, A. Giri, J. Gaskins, P. E. Hopkins, P. Mele, K. Miyazaki and M. Karppinen, J. Mater. Chem. A, 2014, 2, 12150–12152 RSC.
- A. Philip, J. P. Niemelä, G. C. Tewari, B. Putz, T. E. J. Edwards, M. Itoh, I. Utke and M. Karppinen, ACS Appl. Mater. Interfaces, 2020, 12, 21912–21921 CrossRef CAS.
- J.-P. Niemelä, A. Philip, N. Rohbeck, M. Karppinen, J. Michler and I. Utke, ACS Appl. Nano Mater., 2021, 4, 1692–1701 CrossRef.
- G. Abellán, E. Coronado, C. Martí-Gastaldo, A. Ribera, J. L. Jordá and H. García, Adv. Mater., 2014, 26, 4156–4162 CrossRef.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- A. Khayyami and M. Karppinen, Chem. Mater., 2018, 30, 5904–5911 CrossRef CAS PubMed.
- A. Khayyami, A. Philip and M. Karppinen, Angew. Chem., Int. Ed., 2019, 58, 13400–13404 CrossRef CAS PubMed.
- A. B. Kanj, K. Müller and L. Heinke, Macromol. Rapid Commun., 2018, 39, 1700239 CrossRef.
- K. Griffiths, N. R. Halcovitch and J. M. Griffin, Chem. Mater., 2020, 32, 9925–9936 CrossRef CAS.
- A. Tanskanen and M. Karppinen, Sci. Rep., 2018, 8, 8976 CrossRef CAS PubMed.
- R. K. Kotnala and J. Shah, Ferrite Materials: Nano to Spintronics Regime, Elsevier, 2015, vol. 23 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1 – Summary of the [(FeCl3 + H2O)m + (FeCl3 + AZO)]n + (FeCl3 + H2O)m films studied. Fig. S1 – Top-view SEM images of representative ε-Fe2O3:azobenzene SL films. Fig. S2 – Magnetization versus magnetic field isotherms for the Fe-azobenzene film measured at various temperatures. Fig. S3 – Effect of UV irradiation on the structural properties of ε-Fe2O3:azobenzene SL films. Fig. S4 – M–H curves for the n = 100 (1 nm) SL film before and after UV (20 min) irradiation. Table S2 – Coercivity and remnant magnetization values for two representative samples: n = 60 (2 nm) and n = 100 (1 nm), taken from the measured raw data without the diamagnetic background (substrate) signal subtraction. Fig. S5 – Effect of aging on the magnetic and structural properties of ε-Fe2O3:azobenzene SL films. See DOI: 10.1039/d1tc05100h |
| This journal is © The Royal Society of Chemistry 2022 |