
Morphology, size, and defect engineering in CeOHCO3 hierarchical structures for ultra-wide band microwave absorption†
Haiyan
Wei
,
Xinxin
Wang
,
Guoxiu
Tong
 *,
BaoXin
Fan
,
Xiaojuan
Wang
and
Wenhua
Wu
*,
BaoXin
Fan
,
Xiaojuan
Wang
and
Wenhua
Wu
College of Chemistry and Life Sciences, Key Laboratory of the Ministry of Education for Advanced Catalysis Materials, Zhejiang Normal University, Jinhua 321004, P. R. China. E-mail: tonggx@zjnu.cn; Fax: +86-579-82282269; Tel: +86-579-82282269
First published on 24th November 2021
Abstract
The growing electromagnetic interference problem triggers higher requirements for exploration into new electromagnetic wave (EMW) absorbers and new absorption mechanisms. Herein, we developed a simple and rapid microwave-assisted hydrothermal approach for the controllable synthesis of a butterfly-shaped Ce(OH)CO3 hierarchical structure as an efficient EMW absorber for the first time. Kinetic factors, including the CO(NH2)2/Ce3+ molar ratio (β), reaction temperature (T), and reaction time, can be expediently utilized to tune the size, morphology, and defects of the products, which is helpful for tailoring the EMW absorption capability. The cooperation of selective adsorption and minimum surface free energy results in the evolution of the morphology. The appropriate defects, oxygen vacancies, and surface functional groups along with hierarchical structure can synergistically benefit the superior EMW absorbing capacity. Strong damping and perfect impedance matching can be achieved by tuning the balance between conductivity loss and polarization loss. Excellent comprehensive properties are reached at T = 140 °C and β = 2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with a maximal absorption bandwidth of 9.52 GHz, which is significantly broader than those of the previously reported cerium-based compounds/composites. Our current work not only sheds light on the precisely controllable synthesis of hierarchical structures but also opens up a novel methodology for designing new efficient EMW absorbers.
:1 with a maximal absorption bandwidth of 9.52 GHz, which is significantly broader than those of the previously reported cerium-based compounds/composites. Our current work not only sheds light on the precisely controllable synthesis of hierarchical structures but also opens up a novel methodology for designing new efficient EMW absorbers.
Introduction
The superiority of rare earth compounds, embodied in many unique physicochemical characteristics resulting from the effect of lanthanide contraction and 4f electrons, is generally acknowledged. Cerium is the most abundant rare earth element in the Earth's crust at about 0.0046 wt%.1 Its compounds have received considerable attention from material scientists. Previous studies have indicated that cerium-based compounds have abundant intrinsic structural defects (i.e., oxygen vacancies2,3), and are widely applied in catalysis,4 oxygen sensors,5 water gas shift reactions, fuel cells,6 and biomedicine.7As electromagnetic wave (EMW) absorbents, ceria (CeO2) and its composites have great potential owing to their inherent defects (i.e., oxygen vacancies and Ce interstitial atoms).8 Owing to the similar energy and low potential energy barrier between the 4f and 5d electron states, cerium can vary in its oxidation states between Ce6+, Ce4+, and Ce3+.5 Such changes produce free electrons and more oxygen vacancies,9 which can enhance the polarization loss and conductive loss, and further improve the EMW shielding and absorbing capabilities of materials.10 Meanwhile, oxygen vacancies are capable of inducing ferromagnetic behavior in the surroundings and then endow cerium-based compounds with magnetism,11,12 so as to achieve a perfect matching of materials. Therefore, cerium-based compounds are expected to be potential EMW absorbers.2,13–15 For example, CeO2@Fe core–shell composites prepared by Chaoqun Ge2 showed a wide effective absorption bandwidth (EAB, 90% damping) at 4.24 GHz and a minimum reflection loss (RLmin) of −17 dB. CeO2/MWCNT composites reported by Huan Wang et al.13 reached an EAB of 2.88 GHz and an RLmin of −34.64 dB. Core–shell-structured CeO2/Fe3O4 studied by Jiaheng Wang et al.14 exhibited an RLmin of −28.9 dB and an EAB range of 14.2–18 GHz over a thickness range of 6–10 mm for RL ≤ −5 dB. Co-doped CeO2/RGO nanocomposites developed by Ziyao Shen et al.15 exhibited an EAB of 4 GHz (RL ≤ −10 dB). Much effort has been put into cerium-based compounds for the improvement of EMW absorption properties (EMWAPs). However, most of the existing studies are limited to the composition of CeO2 and other materials, and these composites can do nothing to help satisfy the requirements of a wide EAB and high absorption simultaneously. Furthermore, the effects of oxygen vacancy and morphology on the EMW absorption of pure cerium-based compounds are still unclear.
Cerium hydroxycarbonate (CeOHCO3), a member of the cerium-based compound family, has orthorhombic and hexagonal phases, and is usually utilized to synthesize cerium oxides as a precursor.16,17 But it has rarely been studied as an EMW absorber. Current studies mainly aim at its crystal structures, morphology modulation, valence characteristics, involved hydrothermal equilibria, and optical properties. The novel properties exhibited by nanostructured CeO2 and cerium-based compounds originate from shape, size, and defect effects. So far, plenty of attempts have been made to synthesize polymorphous CeOHCO3, including rods,18 films,19 microplates,20 dendrites,21 spindle-like morphology,22 and so forth. Among various morphologies, hierarchical nanostructures with fine structures and large specific surface areas can bring about many novel optical, electric, and magnetic properties, thus attracting considerable research interest.23–26 Currently, many researchers are devoted to the development of new synthetic routes for preparing hierarchical nanostructures. For example, threefold shape CeOHCO3 dendrites were formed via a hydrothermal reaction at 150 °C for 12 h with the coexistence of cerium(III) nitrate hexahydrate and urea in a water–triethanolamine mixed solution.23 Single-crystal hexagonal CeOHCO3 triangular microplates were synthesized via a cetyltrimethyl ammonium bromide-assisted hydrothermal reaction at 150 °C for 16 h using cerium(III) nitrate hexahydrate as a cerium source, and urea as a source of alkali and carbon.20 Under the same conditions of cerium, alkaline, and carbon sources, the polyvinylpyrrolidone-assisted hydrothermal route was followed. Orthorhombic CeOHCO3 dendrite microstructures were prepared at 180 °C for 12 h,24 and single-crystalline hexagonal CeOHCO3 dendrites at 150 °C for 24 h.25 Besides, CeOHCO3 three-fold dendrites were produced via a complexing agent assisted reaction at 200 °C for 24 h.26 The above synthetic routes, however, have some shortcomings, particularly in the utilization of surfactants, long reaction times, multiple solvents, and expensive raw materials. To deal with these problems, more explorations should be conducted to develop simple, quick, and efficient routes for the mass production of hierarchical nanostructures. Additionally, the synthesis and EMW absorption of butterfly-shaped CeOHCO3 hierarchical structures with tunable size have rarely been reported.
Herein, polymorphous CeOHCO3 with an orthorhombic structure is firstly prepared by a rapid microwave-assisted hydrothermal reaction for ultra-wide band EMW absorption. The samples are formed in an aqueous solution containing Ce(NO3)3 and urea at 120–180 °C for just 20 min. This method is short-cycle, high-yield, and independent of surfactants, organic solvents, and expensive source materials, compared with the reported methods.23–26 We control the size, morphology, and defects of the structure to modulate the balance between conductivity loss and polarization loss, and improve its attenuation and matching properties. On this basis, the EMWAC of the butterfly-shaped CeOHCO3 hierarchical structures is significantly enhanced. In this work, the size and morphology of the structure are continuously modulated by just changing reaction temperature, reaction time, and CO(NH2)2/Ce(NO3)3·6H2O molar ratio. Time-dependent morphology analysis is then performed to decipher the morphological evolution mechanism from rods and dodecahedra to 2-fold butterfly-shaped hierarchical structures. Finally, we systematically study the EMWAC of the butterfly-shaped CeOHCO3 hierarchical structures, and clarified their absorption mechanism for exploring new and efficacious EMW absorbents.
Experimental
Specimen preparation
The source materials for CeOHCO3 were carbamide (CO(NH2)2) and cerium(III) nitrate hexahydrate (Ce(NO3)3·6H2O), both of which were analytical reagents. The butterfly-shaped CeOHCO3 samples were prepared by a rapid microwave-assisted hydrothermal reaction. Briefly, Ce(NO3)3·6H2O (0.7816 g) and CO(NH2)2 (0.2703 g) were added to deionized water (30 mL). After 20 min of strong agitation, the mixed solution was transferred to a microwave digestion tank, which was sealed and kept at 160 °C for 20 min in a Preekem WX-6000 microwave digestion system. Finally, the butterfly-shaped CeOHCO3 specimens were obtained after centrifuging, washing, and drying at 40 °C overnight. Furthermore, the morphology and dimension control of butterfly-shaped CeOHCO3 was achieved by modulating CO(NH2)2/Ce3+ molar ratios [β = 1.5:1 (S1), 2.0:1 (S2), 2.5:1 (S0), 3:1 (S3), and 4:1 (S4)] and reaction temperatures [T = 120 °C (S5), 140 °C (S6), 160 °C (S2), and 180 °C (S7)] (see Table S1, ESI†).
Characterization
The morphology, dimension, composition, and structure analyses of CeOHCO3 specimens were performed on a ZEISS GeminiSEM 300 scanning electron microscope (SEM, 10 kV), an EX-250 energy dispersive X-ray spectrometer (EDX), and a JEM-2100F transmission electron microscope (TEM, 200 kV), respectively. A D/MAX-IIIA X-ray diffraction analyzer (XRD) was utilized to collect information about the crystalline structure (XRD; λ = 0.15406 nm, Cu-Kα radiation) at a scanning rate of 5° min−1. The FTIR spectra of CeOHCO3 specimens were measured on a Model 6700 spectrophotometer (Nicolet) with the help of a thin KBr disk. The XPS spectra were tested on an X-ray photoelectron spectrometer (XPS, ESCALAB 250Xi).Property measurements
The conductivity of the specimens was tested under an RTS-9 four-point probe resistivity measurement system. The measured disc pellets with a thickness of about 1 mm and a diameter of 13 mm were formed by pressing the CeOHCO3 powders in a mold. To measure EM parameters, the specimens (60 wt%) were mixed evenly with paraffin. Then the mixtures were transferred into an abrasive tool and pressed into cylindrical rings. The outer diameter of the cylindrical rings was 7.0 mm and their inner diameter was 3.04 mm. Lastly, the permittivity of the specimens was tested at 2–18 GHz on a Keysight N5230A vector network analyzer based on the coaxial line method.Results and discussion
In general, the oxygen vacancy defects of cerium-based compounds may vary spontaneously with temperature, oxygen partial pressure, doped ions, electric field, or surface stress.5 In our studies, the synthesis of butterfly-shaped CeOHCO3 specimens was realized via a rapid microwave-assisted hydrothermal reaction. The morphology, size, and defects could be modulated in an exquisitely designed and continuously adjusted process, which may be achieved by controlling the kinetic factors (i.e., the CO(NH2)2/Ce3+ molar ratio, reaction temperature, and reaction time). The morphology evolution mechanism of the final products was uncovered with the help of XRD, FTIR, SEM, TEM, and SAED characterization techniques.The CO(NH2)2 to Ce3+ molar ratio is a crucial factor for tailoring the morphology, size, and defects of CeOHCO3 samples. The crystal structure of the specimens is characterized by the XRD patterns in Fig. 1a. As for the specimens formed at β = 4:1, all the diffraction peaks fit perfectly with pure orthorhombic cerium carbonate hydroxide (CeOHCO3) according to the standard JCPDS card (41-0013; Cell = 5.015 × 8.565 × 7.337 Å).24 When β decreases from 3:1 to 1.5:1, a new peak begins to appear at about 40.2° relative to the (032) facet and enhances gradually; another peak around 24.0° relative to the (002) facet overlaps the peaks from the (111) and (021) crystallite facets. Such changes indicate the appearance of a new phase. Compared with the standard JCPDS card (44-0602), all peaks can be accurately calibrated by the standard patterns of cerium oxide carbonate hydrate (Ce2O(CO3)2·H2O) with an orthorhombic structure [Cell = 5.0192 × 8.5675 × 7.3323 Å; Pmcn (62)]. As β increases, these diffraction peaks show the same peak position but different intensities. An increase in the β from 1.5:1 to 3:1 causes the increased peak intensity in the (011) and (012) facets, and the decreased peak intensity in the (110), (111), and (031) facets. When the β further increased to 4:1, the intensities of all diffraction peaks fall significantly. The changes above are generally associated with crystallinity, crystallite size, and element content. The mean crystallite size and inner stress of the CeOHCO3 specimens were calculated using the Hall–Williamson equation, as summarized in Table S1 (ESI†). As β increases from 1.5:1 to 4:1, the crystallite size decreases from 110.1 nm to 53.7 nm, and the internal stress representing the defects increases from 0.086% to 0.176%. This demonstrates that controlling β can tailor the defects in CeOHCO3 and a high β is helpful for the formation of defects and dielectric loss.
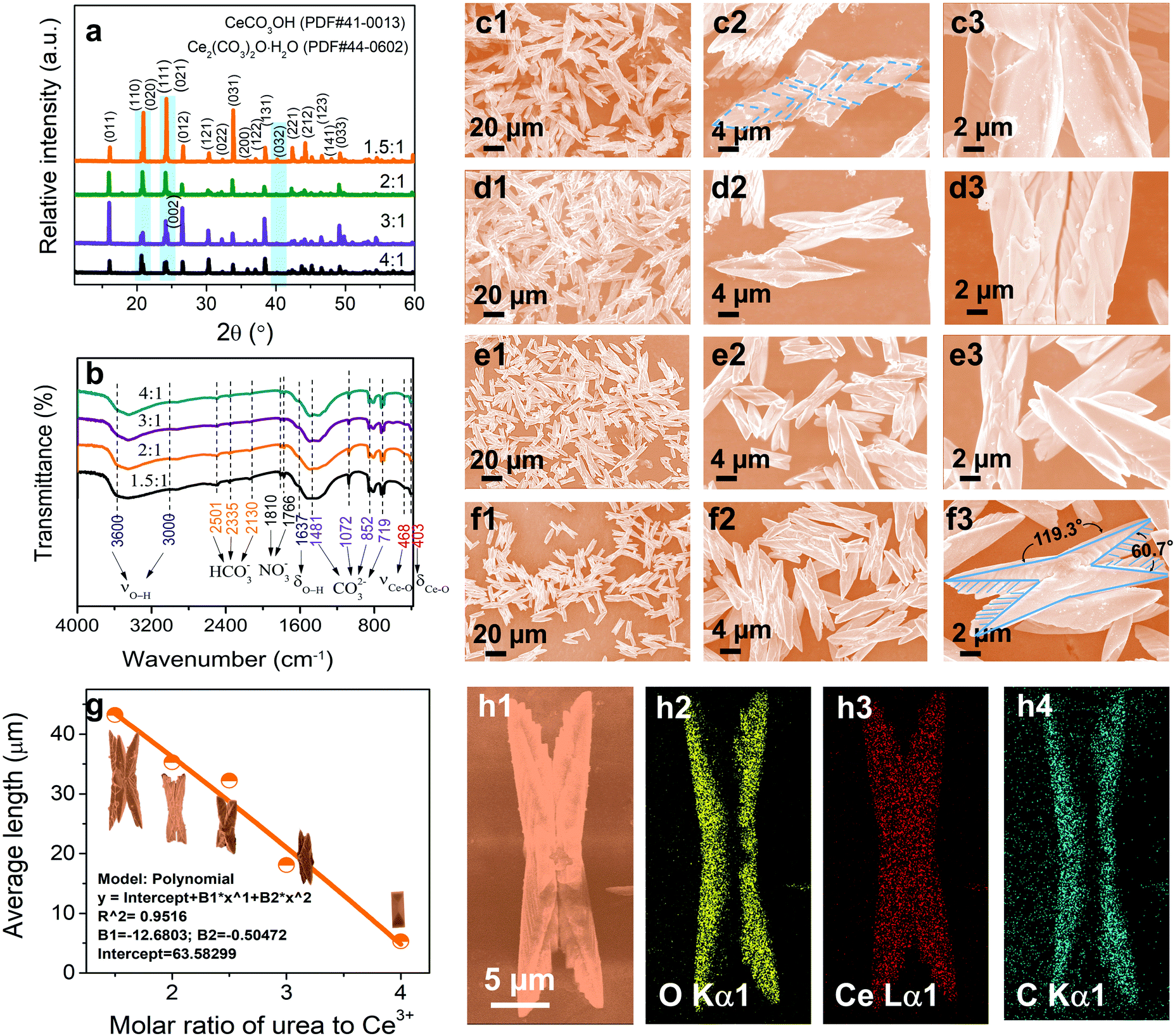 | ||
| Fig. 1 (a) XRD patterns, (b) FTIR spectra, (c1–c3, d1–d3, e1–e3, f1–f3) SEM images, (g) average lengths as a function of β, and (h1–h4) mapping images for the CeOHCO3 specimens yielded under different β: (c1–c3) 1.5:1, (d1–d3) 2:1, (e1–e3) 2.5:1, and (f1–f3, h1–h4) 3:1. | ||
FTIR spectra further confirm the formation of CeOHCO3 and/or Ce2O(CO3)2·H2O. The specimens formed at β = 1.5:1–4:1 take on similar FTIR spectra (Fig. 1b). The wide absorption bands from 3000 cm−1 to 3600 cm−1 and the one at 1637 cm−1 come from the stretching and bending vibrations of the OH− group, respectively, indicating the presence of crystalline water and/or OH−. The wide and intense absorption bands at 1481, 1072, 852, and 719 cm−1 refer to the vibrations of CO32− groups.27 The intense bands at 468 cm−1 and 403 cm−1 come from the Ce–O stretching and bending vibrations,28,29 respectively. Besides, the weak absorption peaks at 2501, 2335, 2130, 1810, and 1766 cm−1 indicate that HCO3− and NO−3 ions are adsorbed on the specimen surfaces.
The β also contributes greatly to the morphological evolution of the cerium-based compounds. Fig. 1c1–f3 illustrate the SEM images of the specimens formed at β = 1.5:1–3:1. The specimens formed at β = 1.5:1–3:1 exhibit a perfect butterfly-shaped configuration with a 2-fold symmetric and hierarchical structure, and the angles between these folds are 119.3° and 60.7° (Fig. 1f3). Their lengths and draw ratios mainly range from 20 μm to 45 μm and from 2 to 4 (Fig. 1g), respectively. Each 2-fold hierarchical structure is a kind of bilayer. A 2-fold symmetric structure looks like a butterfly-shaped configuration. As β varies from 1.5:1 to 3:1, the length of the butterfly-shaped symmetric structure gradually decreases from 40–50 μm to 15–20 μm (Fig. 1g). In general, the growth and nucleation rates increase rapidly with concentration. The decrease in size is owing to the fact that the growth rate is larger than the nucleation rate. The representative EDX spectrum of Ce-based compounds is given in Fig. S1 (ESI†). O, Ce, and C elements exist together in the specimens. As seen from the mapping images in Fig. 1h1–h4, these elements are evenly distributed in each butterfly-like symmetrical structure. The EDX, XRD, and FTIR results indicate that the stoichiometry of Ce-based compounds produced by the urea-assisted hydrothermal method is CeOHCO3 and/or Ce2O(CO3)2·H2O.
As the β further increased to 4:1, the butterfly-shaped symmetric structure evolved into a unique dodecahedron with a symmetric structure and smooth surfaces (Fig. 2a–e). The dodecahedron is approximately 5–8 μm in length and 1.5–2.0 μm in width. The length–width ratio is about 3–4. The crystallinity of CeOHCO3 dodecahedra is tested via SAED patterns, since the crystallinity affects the half-thickness of the CeOHCO3 diffraction peaks. As shown in Fig. 2f, six distinct diffraction rings are found, verifying the polycrystalline features of the CeOHCO3 dodecahedra. The interplanar spacings of 0.433, 0.428, 0.373, 0.337, 0.296, and 0.235 nm are assigned perfectly to the (110), (020), (111), (012), (102), and (131) crystal planes, respectively. The polycrystalline features of the CeOHCO3 dodecahedra can also be verified by the lattice fringes in the short-range order (Fig. 2g). Additionally, the high-density domains with unordered lattice fringes circled by yellow dotted lines are observed in the HRTEM images of CeOHCO3 dodecahedra (Fig. 2g), which correspond to the oxygen vacancy defect (OVD) sites and other lattice defects. The defects can also be confirmed by the foregoing internal stress in XRD data and the subsequent XPS results. The element content and distribution analyses reveal that Ce, C, and O elements are detected and distributed along the dodecahedra (Fig. 2h1–h5). The O/Ce atom ratio is lower than 4, meaning the presence of the OVD. From the XRD, SAED, and HR-TEM analyses, we may deduce that dodecahedra are surrounded by (110), (011), and (111) crystallographic planes because (111), (110), and (100) surfaces are most thermodynamically stable, as illustrated in Fig. 2d and e. Experimental and theoretical studies have confirmed that the generation of oxygen vacancies obeys the following order: {111} < {100} < {110}.4 Therefore, the CeOHCO3 dodecahedra surrounded by {110} facets will possess abundant oxygen vacancy defects, which can improve the conductivity and dielectric loss. Our data demonstrate that changing the CO(NH2)2 to Ce3+ molar ratio can easily adjust the morphology, crystallite size, and defects of the obtained monodisperse hierarchical structure; the higher the CO(NH2)2 to Ce3+ molar ratio, the smaller the size and the more the defects.
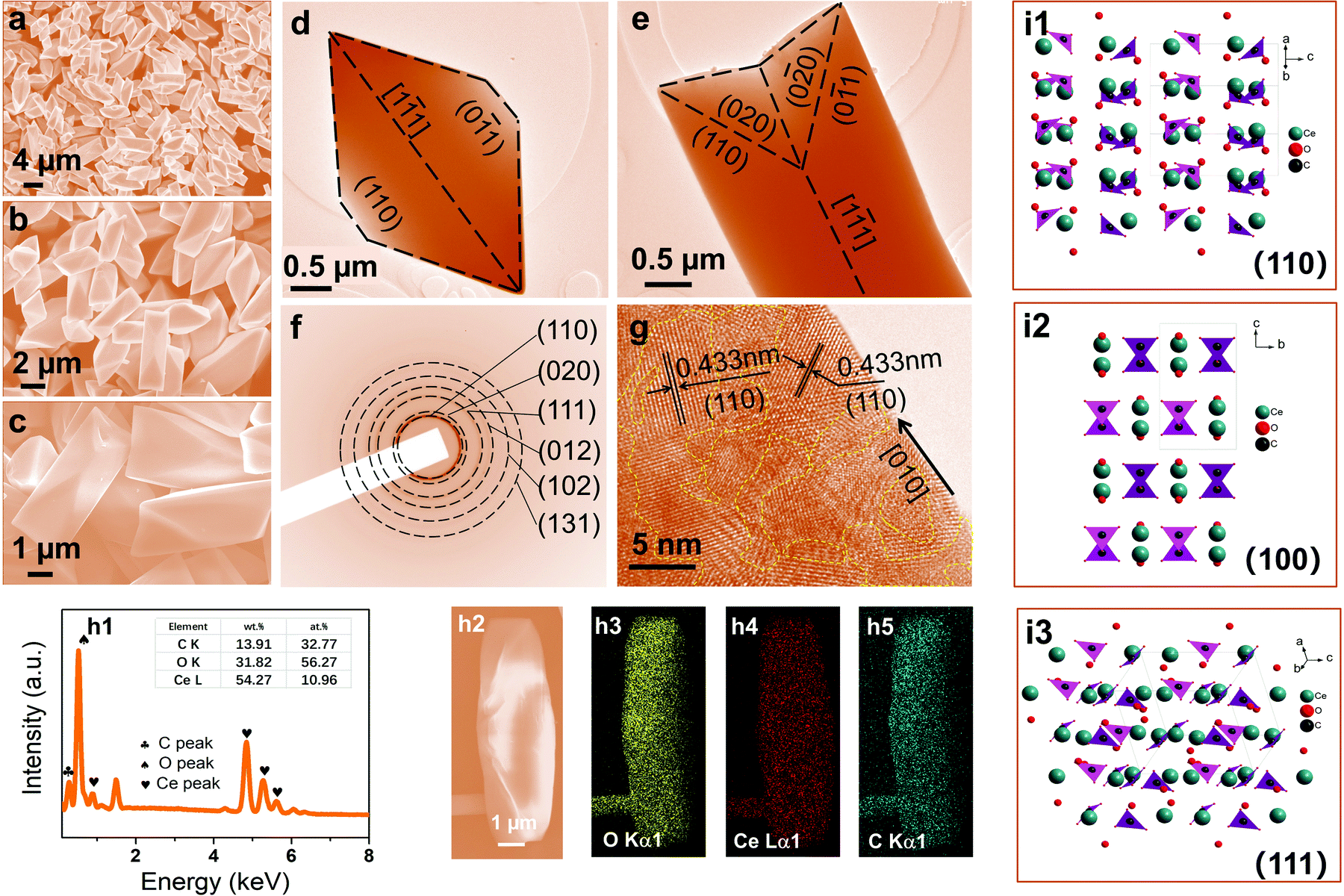 | ||
| Fig. 2 (a–c) SEM images, (d and e) typical TEM images, (f) SAED pattern, (g) HRTEM images, and (h1) EDX spectrum, (h2–h5) the element mapping images, and (i1–i3) the crystal structures of the CeOHCO3 specimens formed under β = 4:1. | ||
In addition to the molar ratio of CO(NH2)2 to Ce3+, reaction temperate can also be used to adjust the size, morphology, and defects of the CeOHCO3 specimens. According to the standard cards (41-0013 and 44-0602), the specimens formed at a low T (i.e., 120 °C) are pure CeOHCO3 and the ones formed at a high T (i.e., 140–180 °C) are a mixture of CeOHCO3 and Ce2O(CO3)2·H2O (Fig. 3a). The peak intensity related to the (032) facet is enhanced with T, meaning an enhanced content and crystallinity of Ce2O(CO3)2·H2O. The peak intensities related to the (011), (110), (111), (031), (221), and (212) facets are significantly enhanced, indicating a preferential growth along these facets. As seen from Table S1 (ESI†), the crystallite size takes on a reversed V-type variation trend with the elevated T; the internal stress exhibits a contrasting trend with a maximum at T = 180 °C. This phenomenon is owing to the oxidation state conversion from Ce3+ to Ce4+ and the higher nucleation rate than the growth rate at T = 180 °C.
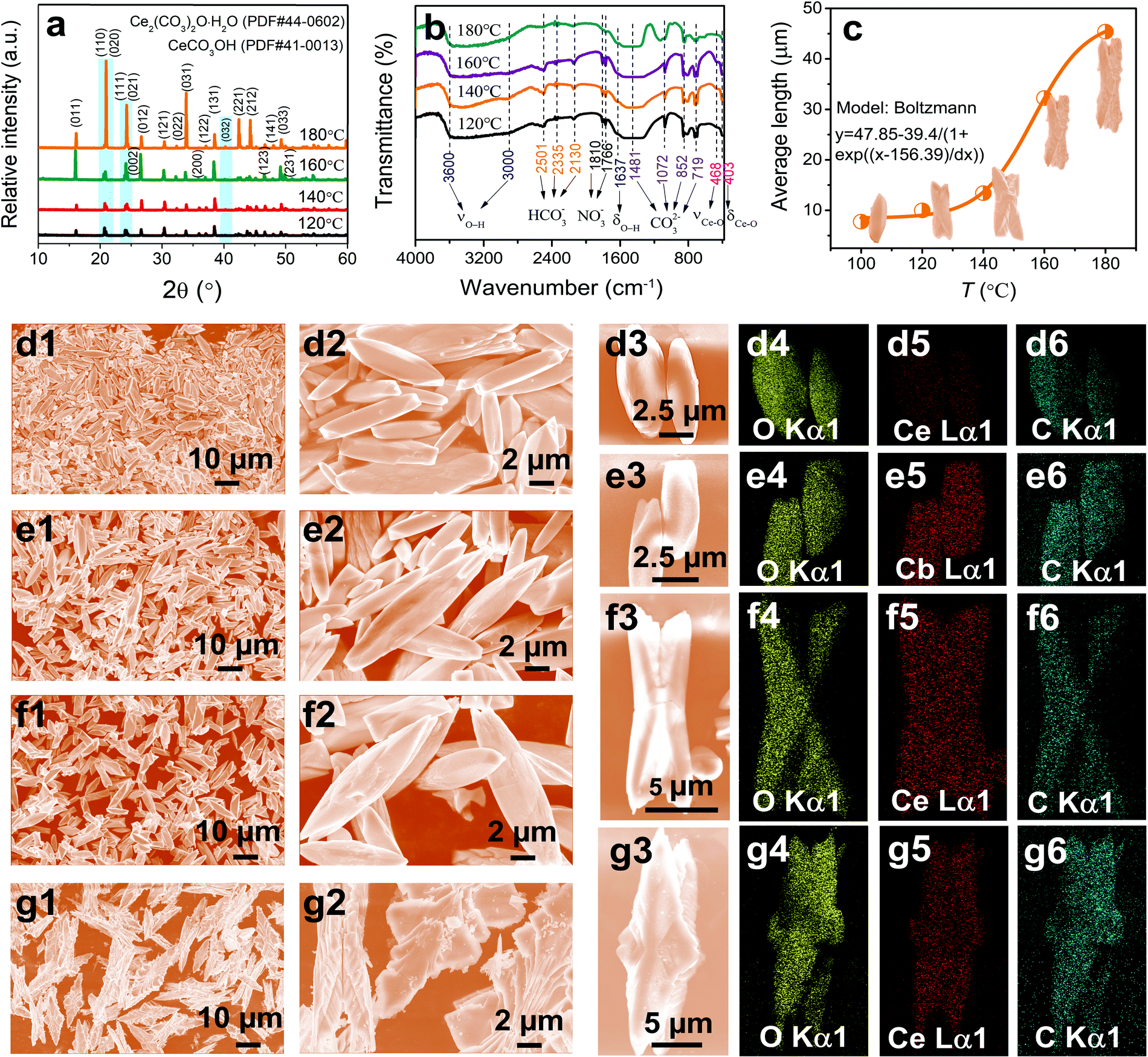 | ||
| Fig. 3 (a) XRD patterns, (b) FTIR spectra, (c) average length as a function of T, (d1–d2, e1–e2, f1–f2, g1–g2) SEM images, and (d3–d6, e3–e6, f3–f6, g3–g6) element mapping images of the CeOHCO3 specimens formed at different T. (d1–d6) 100 °C; (e1–e6) 120 °C; (f1–f6) 140 °C; and (g1–g6) 180 °C. | ||
The FTIR spectra in Fig. 3b further verify the presence of CeOHCO3 and/or Ce2O(CO3)2·H2O. The specimens formed at 120–160 °C take on similar FTIR spectra. The bands at 3000–3600 cm−1 and 1637 cm−1 confirm the existence of crystalline water and/or OH−; the ones at 1481, 1072, 852, and 719 cm−1 refer to the carbonate groups;27 and the ones at 468 cm−1 and 403 cm−1 are associated with the Ce–O stretching and bending vibrations,28,29 respectively. Besides, the weak bands at 2501, 2335, 2130, 1810, and 1766 cm−1 indicate the adsorption of HCO3− and NO−3 on the specimen surfaces. In contrast, the specimens formed at 180 °C show narrower and weaker peaks at 1481–719 cm−1 and a wider peak at about 1072 cm−1. Such changes are related to the conversion from Ce3+ to Ce4+, indicating the appearance of a new phase (Ce(OH)2CO3 or CeOCO3·xH2O).
Changing T can also adjust the specimen morphology. At a relatively low T (100 °C–140 °C), uniform fusiform microrods with two sharp ends are yielded (Fig. 3d1–f3). A V-type indentation appears on the two ends of rods at T = 120 °C (Fig. 3e1–e3). The indentation gradually expands to the middle of rods and splits the rod into two halves at T = 140 °C (Fig. 3f1–f3). At T = 160 °C, a hierarchical structure forms on the surface of indentation (Fig. 1f1–f3). At T = 180 °C, two hierarchically fusiform microflakes assemble symmetrically into a butterfly-shaped configuration with 2-fold symmetry (Fig. 3g1–g3). Fig. 3c shows that the diameter and draw ratio of the specimens are 6–8 μm and 3–4 for 100 °C, 8–10 μm and 4–5 for 120 °C, 12–16 μm and 5–6 for 140 °C, and 40–50 μm and 4–5 for 180 °C, respectively. This is because temperature provides various driving forces for the growth of various lattice planes. Based on the XRD, FTIR, and the mapping analyses in Fig. 3d4–g6, the stoichiometry of Ce-based compounds is found to be CeOHCO3 and/or Ce2O(CO3)2·H2O. These data indicate that a high T favors the conversion of oxidation state and morphology evolution from rods to butterfly-shaped hierarchical structures with large sizes.
An XPS can be utilized to reveal the surface elemental chemistry state of the specimens. In Fig. 4, the binding energies of all elements have been rectified according to a C 1s value of 284.8 eV. The Ce, O, and C elements can be found in the survey spectra (Fig. 4a). The O 1s spectra (Fig. 4b) are fitted into four peaks at 529.3, 530.6, 531.8, and 532.8 eV, which are related to Ce4+–O,30 Ce3+–O,30 –OH,31 and CO32−,32 respectively. The fitted signals at 531.8 eV originate from the –OH groups and oxygen vacancies.33 Due to their same positions, the exact amount of oxygen vacancies is obtained based on the XPS data. The C 1s spectra (Fig. 4c) can be divided into five peaks at 284.8, 286.5, 288.5, 289.9, and 292.0 eV. The peaks at 284.8 eV are of the non-oxygenated ring C.34 The ones at 286.5 eV and 288.5 eV are of C–O–C and O–C–O,35 respectively. The strong ones at 289.9 eV are of O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O.35 These results are in accordance with the FTIR and XRD data. The new peak appearing at 292 eV comes from the n–π* transition of CO bound. It suggests that Ce is bound to the CO32− groups, so the electron density is reduced in the CO direction.36Fig. 4d shows the XPS spectra from Ce 3d electrons, which are fitted into several peaks. The peaks at 886.2 and 904.2 eV are related to Ce3+,37 and the ones at 917, 907, 900.6, 898.5, and 882.9 eV are ascribed to Ce4+.38 The conversion from Ce3+ to Ce4+ is ascribed to NO3− ions and high T. The oxygen vacancy defects (OVD) are usually expressed by cerium(III) content (Ce3+%). To quantify the Ce3+% on the CeOHCO3 surfaces, the relative XPS peak area of Ce3+ is calculated by the expression
O.35 These results are in accordance with the FTIR and XRD data. The new peak appearing at 292 eV comes from the n–π* transition of CO bound. It suggests that Ce is bound to the CO32− groups, so the electron density is reduced in the CO direction.36Fig. 4d shows the XPS spectra from Ce 3d electrons, which are fitted into several peaks. The peaks at 886.2 and 904.2 eV are related to Ce3+,37 and the ones at 917, 907, 900.6, 898.5, and 882.9 eV are ascribed to Ce4+.38 The conversion from Ce3+ to Ce4+ is ascribed to NO3− ions and high T. The oxygen vacancy defects (OVD) are usually expressed by cerium(III) content (Ce3+%). To quantify the Ce3+% on the CeOHCO3 surfaces, the relative XPS peak area of Ce3+ is calculated by the expression 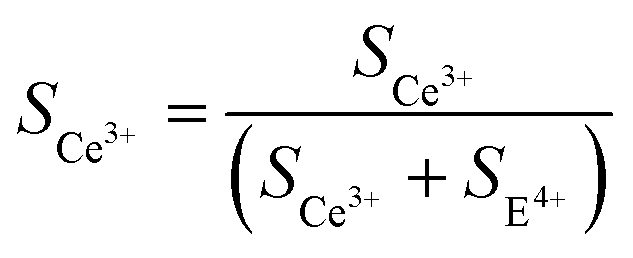 .3,4 The calculated Ce3+% in these materials fluctuates sharply between 66.21% and 64.80% as T rises from 120 °C to 160 °C, and then drops precipitously to 47.86% at 180 °C. The reaction, expressed by the Kröger–Vink notation5
.3,4 The calculated Ce3+% in these materials fluctuates sharply between 66.21% and 64.80% as T rises from 120 °C to 160 °C, and then drops precipitously to 47.86% at 180 °C. The reaction, expressed by the Kröger–Vink notation5
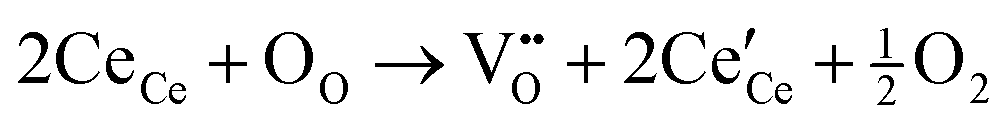
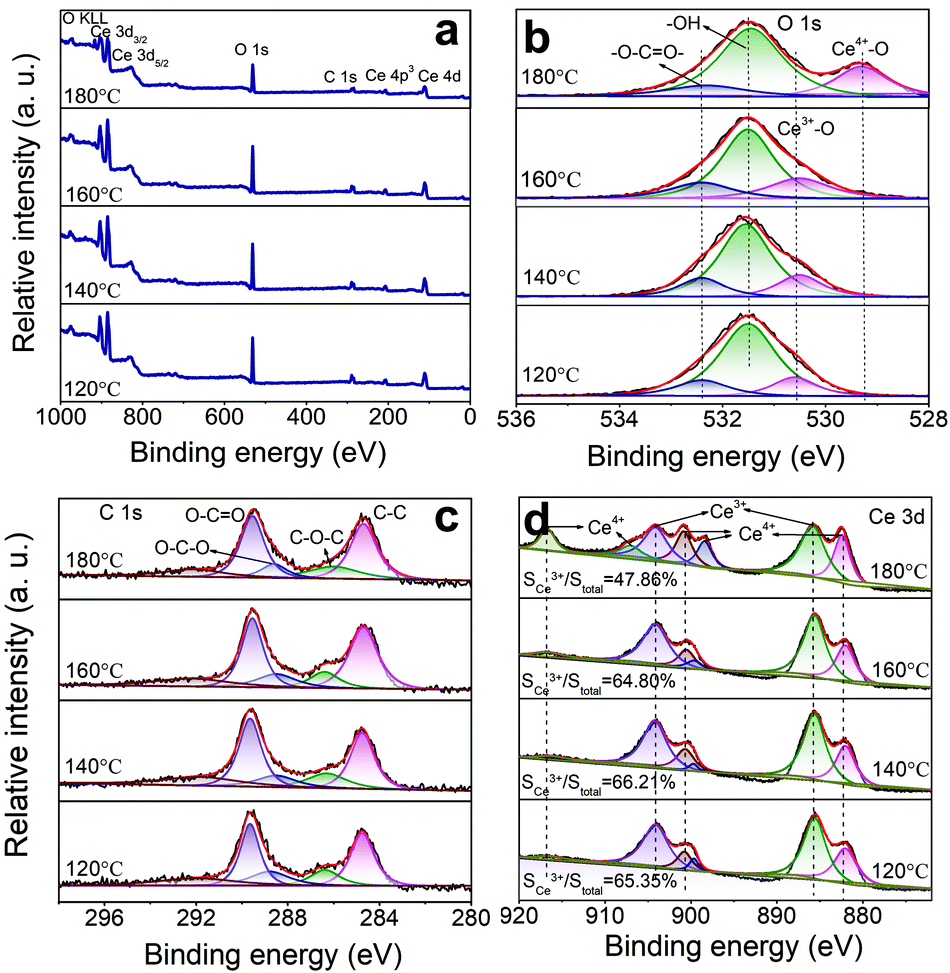 | ||
| Fig. 4 Temperature-dependent XPS spectra: (a) survey spectra, (b) O 1s, (c) C 1s, and (d) Ce 3d. | ||
The basis for elucidating the growth mechanism of the polymorphous cerium-based compounds is laid by the SEM and XRD results concerning the phase and morphology evolution with reaction time, CO(NH2)2 to Ce3+ molar ratio, and reaction temperature. As seen from Fig. 5a, the specimens collected at 1 min are microrods with V-type indentations at the ends. Hierarchical structures begin to form on the indentation surfaces at 5 min (Fig. 5b). With the time further prolonged to 10 min or more, the microrods evolve into hierarchically fusiform microflakes with a 2-fold symmetric structure (Fig. 5c and d). Currently, polymorphic CeOHCO3 micro/nanostructures, such as rods,18 films,19 microplates,20 and dendrites,21 have been synthesized with the help of mixed solvents,23 CTAB,20 PVP,24,25 complexing agents,26 or a long reaction cycle. However, a butterfly-shaped hierarchical structure with 2-fold symmetry, which is obtained in a very short reaction cycle without surfactants, has been rarely explored.
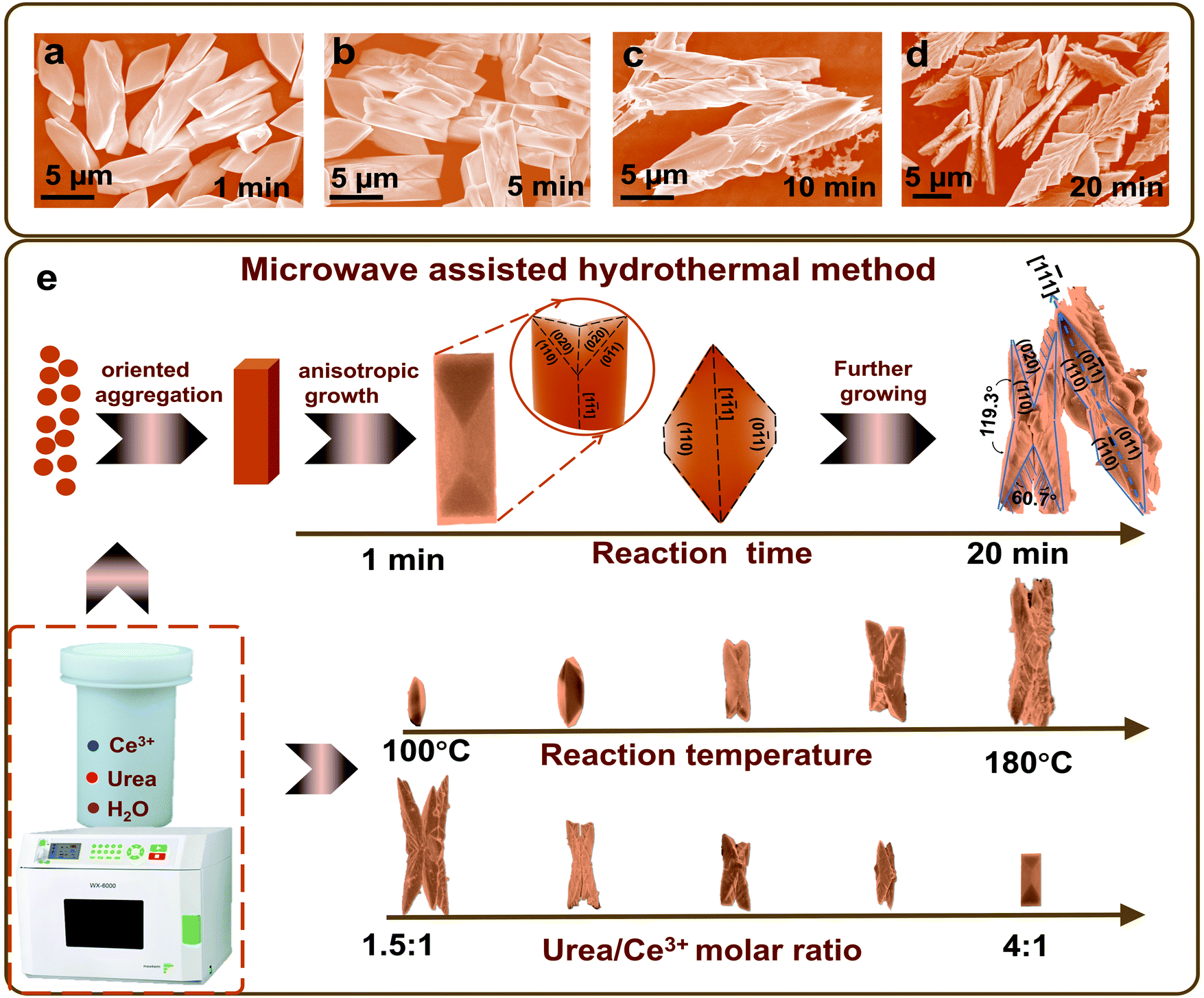 | ||
| Fig. 5 (a–d) Reaction time dependent SEM images of the CeOHCO3 specimens. (e) Schematic illustration of the morphology evolution mechanism of polymorphous CeOHCO3 specimens. | ||
Our results highlight a rapid facile microwave-assisted hydrothermal approach for the mass production of a uniform butterfly-shaped structure, as illustrated in Fig. 5e. The CeOHCO3 hierarchical structure is the result of decomposition, hydrolysis, coordination, reduction, and nucleation in the current Ce(NO3)3–CO(NH2)2 system. The following chemical reactions are involved:40
| CO(NH2)2 → NCO− + NH4+ | (1) |
| NCO− → 3H2O → HCO3− + NH4+ OH− | (2) |
| HCO3− → H+ + CO32− | (3) |
| Ce3+ + CO32− + OH− → CeOHCO3 ↓ | (4) |
| 2CeOHCO3 → Ce2O(CO3)2·H2O | (5) |
It can be seen that the formation of the butterfly-shaped Ce-based compound structure is achieved via the following several steps: first, the slow decomposition of CO(NH2)2 generates NCO− and NH4+ ions (Reaction (1)). Then the NCO− ions hydrolyze to produce HCO3−, and OH− (Reaction (2)). The HCO3− ions hydrolyze further to generate CO32− (Reaction (3)). Finally, the coordination of Ce3+ with CO32− and OH− forms CeOHCO3 sediment (Reaction (4)). Under certain conditions (i.e., high temperature), CeOHCO3 will convert into Ce2O(CO3)2·H2O via the condensation of two OH− ions (Reaction (5)).
β and T are key factors affecting the driving forces for CO(NH2)2 decomposition, hydrolysis, coordination, reduction, nucleation, and assembly. Therefore, we can adjust β and T to realize the morphological evolution of specimens. As seen from reactions 1–3, the high β can enhance the concentration of anions (i.e., NCO−, CO32−, OH−, and HCO3−), which promotes the nucleation and growth of CeOHCO3 nanocrystals. At a low β value, the nucleation rate is lower than the growth rate, which helps the CeOHCO3 nanocrystals grow into large-size ones. Moreover, the growth rate in different crystallographic planes varies greatly, causing the anisotropic growth and the formation of a hierarchical structure. Conversely, at a high β value, the nucleation rate is higher than the growth rate, unfavorable for the growth of CeOHCO3 nanocrystals, resulting in the small size. Furthermore, the reaction temperature provides a driving force for CO(NH2)2 decomposition, hydrolysis, coordination, reduction, and nucleation. Obviously, a high T value accelerates the decomposition of CO(NH2)2, resulting in the rapid release of anions (i.e., NCO−, CO32−, OH− and HCO3−). Afterward, a high growth rate promotes the growth of CeOHCO3 nanocrystals, leading to their increased size with T.
The formation of the hierarchical structure is usually associated with the anisotropic growth from a center to a preferred orientation or the oriented attachment assisted by surface-capped agents and ions (OH−).41–43 In this study, the evolution of the hierarchical structure is owing to selective adsorption and minimum surface free energy. On the one hand, cerium is 7-fold coordinated on the {111} surface, but 6-fold coordinated on the {100} and {110} surfaces. In this case, the adsorption energy of ions (i.e., CO32−, OH−, HCO3− and NO−3) decreases in the order {100} > {110} > {111}, which favors the coordination of surface cerium atoms upon these ions.44 Such selective coordination or adsorption promotes the growth of {100}, {110}, and {111} facets. On the other hand, the {111}, {110}, and {100} surfaces are most thermodynamically stable for the cerium-based compounds.12 The surface free energy of the cerium-based compounds will be minimized if {100}, {110}, and {111} facets are used as their surfaces. Moreover, the difference in the growth rate of various crystallographic planes is very large, causing the anisotropic growth and the formation of the hierarchical structure.
The EMWAPs of the cerium-based compounds formed at different β and T are listed in Fig. 6. When T is constant (160 °C), the variation of β affects the EMWAPs of the CeOHCO3 specimens. The CeOHCO3 specimens take on an ultra-wide absorption band over 6–18 GHz, which covers the X and Xu frequency bands. The absorption bands are consistent with the λ/4 of the specimens, meaning the λ/4 damping models. The EABmax (RL ≤ −10 dB, 90% damping) takes on an inverted U-type change trend with a crest value of 9.52 GHz at β = 2:1. The EAB is larger than 6 GHz within the thickness range of 2.5–3.9 mm. The EABmax is 9.52 GHz at 3.4 mm thickness (Fig. 6b1), and the optimal absorption is −44.95 dB under 9.76 GHz (Fig. 6b2). Therefore, β = 2:1–3:1 is the best condition to explore the temperature effect on the EMWAPs of CeOHCO3 specimens. If β keeps unchanged (2.5:1) and T increases from 120 °C to 180 °C, the CeOHCO3 specimens exhibit excellent EMWAPs (Fig. 6e1–e2, f1–f2, and g1–g2). The optimal comprehensive properties are achieved at T = 140 °C and β = 2.5:1 with an RLmin of −47.35 dB at 11.12 GHz, corresponding to a 3.0 mm thickness (Fig. 6f2). The EAB is more than 6 GHz within the thickness range of 2.5–3.7 mm, and the EABmax is 9.52 GHz (Fig. 6f1).
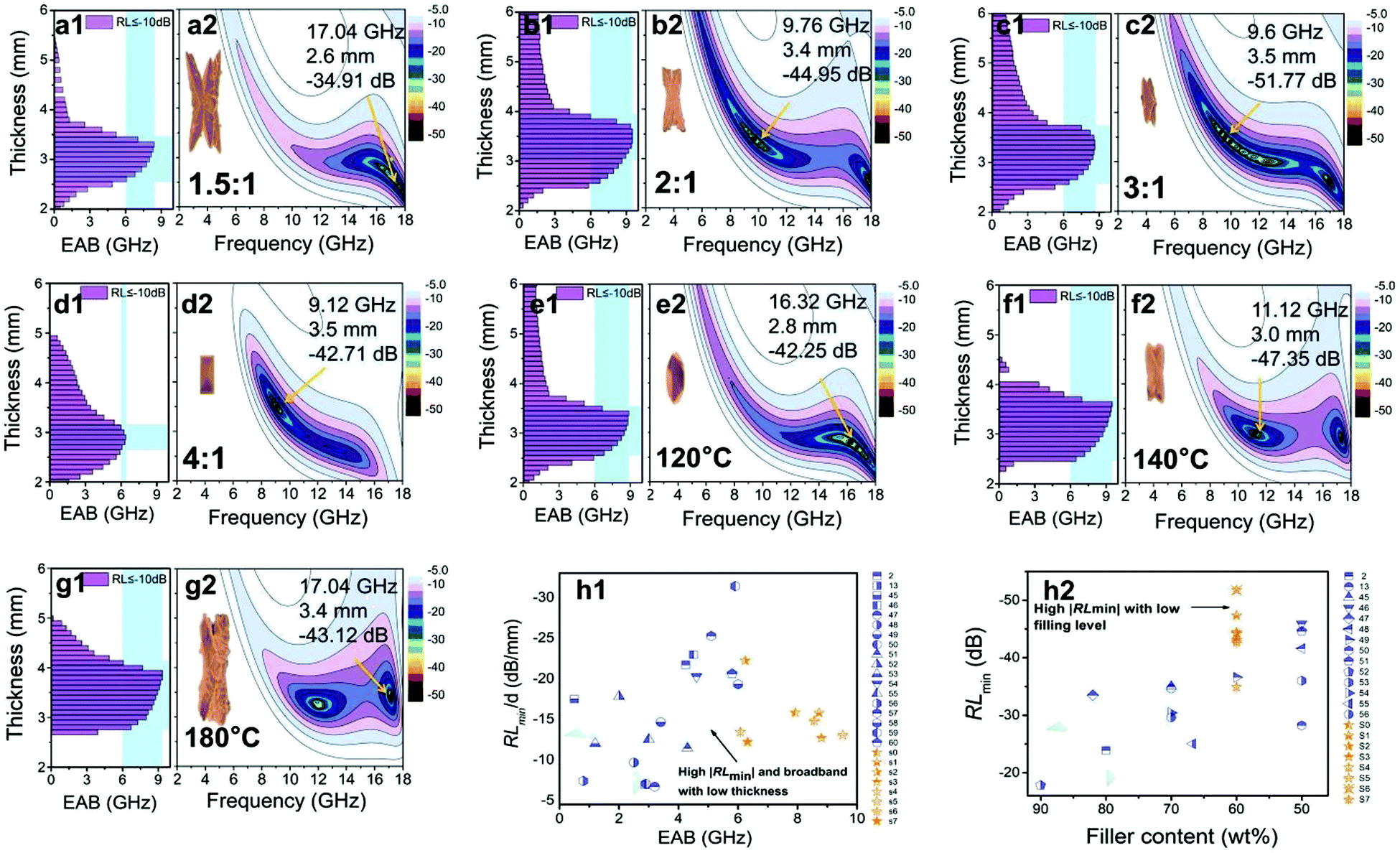 | ||
| Fig. 6 Frequency characteristics: (a1–g1) EAB (RL ≤ −10 dB) and (a2–g2) 2D RL curves of the cerium-based compounds formed at different β and T. (h1–h2) The EMWAP comparisons of the CeOHCO3 specimens with other absorption materials.2,13,45–60 | ||
Fig. 6h1 and Table S2 (ESI†) show that the specimen thickness of 2.5 mm with EAB ≥ 6.0 GHz mm is much smaller than those of CeO2/MWCNT (5.0 mm),13 rGO/α-Fe2O3 hydrogel (5.0 mm),47 Fe3O4 nanorings (4.3 mm),48 and RGO/Fe3O4 (3.9 mm).51 Meanwhile, the EABmax of 9.52 GHz is 2.07–19.0 times that of other cerium-based composites (0.5–4.6 GHz).2,13,45–60 Additionally, the RLmin of −47.35 dB is lower than that of other absorbers;2,13,45,47,49,50 the filling ratio of CeOHCO3 specimens (60 wt%) is lower than those of Zn modified-CeO2 microspheres (70 wt%),45 CeO2/MWCNT (70 wt%),13 and core–shell structured CeO2@Fe (80 wt%)2 (Fig. 6h2). This indicates the light weight of CeOHCO3 specimens. Our data demonstrate that the CeOHCO3 specimens produced at T = 140 °C and β = 2.5:1 are lighter with stronger absorption over a wider absorption frequency range. Moreover, controlling the defects and morphology of CeOHCO3 specimens can achieve prominent absorption performance with strong absorption, light weight, and small matching thickness within a wide frequency range.
The Debye equation 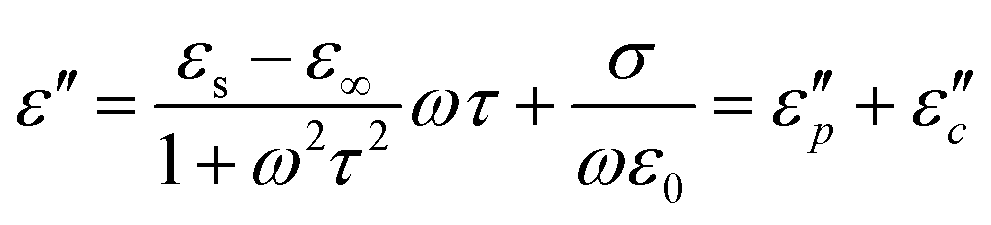 61 reveals that the dielectric loss mainly stems from conductance loss
61 reveals that the dielectric loss mainly stems from conductance loss 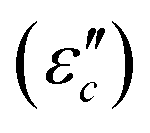 and polarization loss
and polarization loss 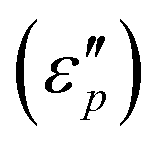 . To ascertain their contributions, we systematically study the variation regularity of
. To ascertain their contributions, we systematically study the variation regularity of 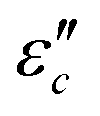 and
and 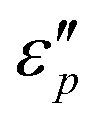 with β and T. The
with β and T. The 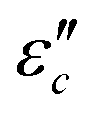 can be obtained based on the free electron theory,
can be obtained based on the free electron theory, 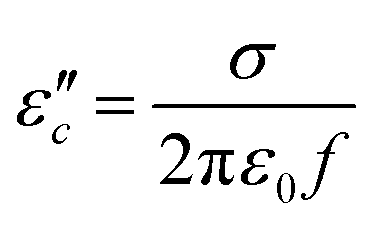 .62 According to the formula, the
.62 According to the formula, the 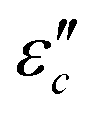 varies directly as conductivity and inversely as frequency. This result, therefore, can explain why the
varies directly as conductivity and inversely as frequency. This result, therefore, can explain why the 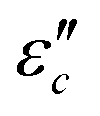 decreases with the increasing f (Fig. 7a1–a2). This demonstrates that conductance loss is significant for low-frequency absorption.
decreases with the increasing f (Fig. 7a1–a2). This demonstrates that conductance loss is significant for low-frequency absorption.
 | ||
Fig. 7 Frequency characteristics: (a1–a2) 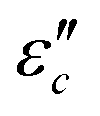 and (d1–d2) and (d1–d2) 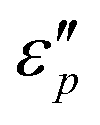 of the CeOHCO3 specimens formed at different β and T. Conductance loss mechanism: (b1–b2) conductivity (σ), (c1–c2) the oxygen vacancy, (c2) defects, (c3) delocalization Π64 bonds in functional groups, and (c4) local conductive network. Polarization loss mechanism: (e1–e2) Cole–Cole curves (ε′ vs. of the CeOHCO3 specimens formed at different β and T. Conductance loss mechanism: (b1–b2) conductivity (σ), (c1–c2) the oxygen vacancy, (c2) defects, (c3) delocalization Π64 bonds in functional groups, and (c4) local conductive network. Polarization loss mechanism: (e1–e2) Cole–Cole curves (ε′ vs.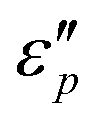 ) (f1) orientation polarization, (f2) interface polarization, and (f3) defect/polar polarization. (f4) Electric-field (E-field) distributions of fusiform CeOHCO3 rods and hierarchical structures simulated by the CST MICROWAVE STUDIO program. ) (f1) orientation polarization, (f2) interface polarization, and (f3) defect/polar polarization. (f4) Electric-field (E-field) distributions of fusiform CeOHCO3 rods and hierarchical structures simulated by the CST MICROWAVE STUDIO program. | ||
By contrast, the conductivity effect is more complicated. The conductivity of the CeOHCO3 specimens is determined by oxygen vacancies, functional groups (i.e., NCO−, CO32−, and HCO3−), defects, and their morphologies. First, the conversion between Ce6+, Ce4+, and Ce3+ can generate more oxygen vacancies and free electrons, enhancing the conductivity.9,10 The oxygen vacancy defect (OVD) sites can be confirmed by the high density of domains circled by yellow dotted lines in the HRTEM images (Fig. 7c1–c2). Oxygen vacancies can also be quantitatively analyzed by the relative XPS peak area of Ce3+ (Ce3+%). As seen from Fig. 4d, Ce3+% fluctuates slightly in the range of 66.21% to 64.80% as T increases from 120 °C to 160 °C and then drops precipitously to 47.86% at 180 °C. A high T (≥180 °C) favors the conversion from cerium(III) to cerium(IV), reducing oxygen vacancies. Second, the internal stress varies between 0.198% and 0.111% with T increasing from 120 °C to 160 °C and reaches a crest value of 0.206% at T = 180 °C (Table S1, ESI†). The high internal stress corresponds to the high amount of defects, which can block the transfer of free electrons (Fig. 7c2) and result in low conductivity and conductive loss. Third, the delocalization Π64 bonds generated from functional groups (i.e., NCO−, CO32−, and HCO3−) can provide free charges. The migration of these free charges can improve the conductivity (Fig. 7c3).63 The FTIR data in Fig. 1b and 3b confirm the presence of functional groups (i.e., NCO−, CO32−, and HCO3−).
Besides, local conductive micro-networks easily generate in the butterfly-shaped hierarchical structures (Fig. 7c4). The induced microcurrents form in the alternate EM field, causing the conductive loss and the induced magnetic field. Fig. 4e reveals that the diameter and draw ratio of the specimens increase as T increases from 120 °C to 180 °C, favorable for the formation of conductive micro-networks. In sum, the conductivity is owing to the combined action of oxygen vacancies, functional groups, defects, and their morphologies. The high conductivity obtained at a low temperature (120 °C) is ascribed to the high OVD and functional groups; the low conductivity obtained at a high temperature (180 °C) is ascribed to the low OVD and functional groups as well as high internal stress defects.
Fig. 7d1–d2 show an uptrend of 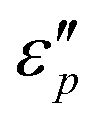 with frequency. With the increase of β, the
with frequency. With the increase of β, the 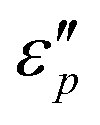 reaches a maximal value at β = 4:1; with the increase of T, the maximal
reaches a maximal value at β = 4:1; with the increase of T, the maximal 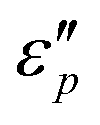 value appears at T = 160 °C. At low frequencies, negative
value appears at T = 160 °C. At low frequencies, negative 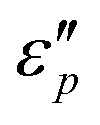 values appear in the low-frequency range (2–11 GHz) due to the high conductive loss and strong resonance. In the presence of the large microcurrents, the alternating magnetic field can induce an electric field and radiate out electric energy. A negative
values appear in the low-frequency range (2–11 GHz) due to the high conductive loss and strong resonance. In the presence of the large microcurrents, the alternating magnetic field can induce an electric field and radiate out electric energy. A negative 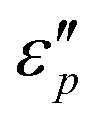 will be generated if the induced electric field is higher than the original electric field. Three resonance peaks appear at low, middle, and high frequencies, corresponding to three relaxation processes. This can be verified by the
will be generated if the induced electric field is higher than the original electric field. Three resonance peaks appear at low, middle, and high frequencies, corresponding to three relaxation processes. This can be verified by the 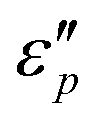 & ε′ plots of the specimens. Fig. 7e1–e2 present three Cole–Cole semicircles, which are related to triple relaxation processes based on the Debye dipole relaxation expression.64 The conductivity loss causes the formation of a contorted semicircle along with linear tails. Compared with conductance loss (Fig. 7a1–a2), the polarization loss possesses higher values in the high-frequency range and lower ones in the low-frequency range. Thus, the dielectric loss mainly stems from conductance loss in the low-frequency range and the polarization loss in the high-frequency range.
& ε′ plots of the specimens. Fig. 7e1–e2 present three Cole–Cole semicircles, which are related to triple relaxation processes based on the Debye dipole relaxation expression.64 The conductivity loss causes the formation of a contorted semicircle along with linear tails. Compared with conductance loss (Fig. 7a1–a2), the polarization loss possesses higher values in the high-frequency range and lower ones in the low-frequency range. Thus, the dielectric loss mainly stems from conductance loss in the low-frequency range and the polarization loss in the high-frequency range.
Generally, the polarization loss originates from multiple polarizations (interface, defect/dipole, and orientation). The interfaces among nanocrystallites lead to the enhanced polarization, which contributes greatly to the polarization loss. High T and low β values promote the formation of the hierarchical structure that can form the extra surfaces. The HR-TEM image in Fig. 2g reveals that many interfaces are formed among the nanocrystallites. As seen from Table S1 (ESI†), crystallite size decreases and increases with the increase of β and T, respectively. The small size corresponds to the large specific surface area. These data demonstrate that high β and low T provide a large specific surface area, generating the strong interfacial polarization.
The defects and large quantities of surface functional groups in/on the CeOHCO3 specimens are desirable for polarization loss. A lot of oxygen vacancies and defects act as a dipole center for the occurrence of dipole/defect polarization (Fig. 7c2).65 The defects vary directly as the internal stress. Table S1 (ESI†) shows that the internal stress exhibits large values at a low or high T and a large β. The higher the internal stress, the larger the dipole/defect polarization. In this case, the low or high T and large β are helpful for the creation of dipole/defect polarization. As seen from Fig. 4d, oxygen vacancies quantitatively analyzed by Ce3+% exhibited large values of 66.21% to 64.80% at T = 120 °C–160 °C, and a small value of 47.86% at 180 °C. A high T (≥180 °C) favors the formation of oxygen vacancies and dipole/defect polarization.
The hierarchical structure with large shape anisotropy can generate strong orientation polarization. The shape anisotropy varies directly as the draw ratio. As seen from Fig. 1g and 3c, the draw ratio increases with the increasing T and the decreasing β. This indicates that high T and low β values are helpful in the enhancement of orientation polarization.
To further approve the aforementioned loss mechanism of CeOHCO3 hierarchical structures, frequency-dependent electric field distributions are simulated by the commercial CST program. Using four CeOHCO3 rods or hierarchical structures as cell units, the electric field is simulated for cell units in response to the alternating applied EM field. As shown in Fig. 7f4, fusiform CeOHCO3 rods exhibit strong electric field distribution at the top of rods, corresponding to the strong orientation polarization. When fusiform rods evolve into hierarchical structures, a strong electric field is distributed not only at the top of the rods but also at the edges. This indicates that the hierarchical structure possesses high orientation polarization and interfacial polarization. The polarization behaviors can be studied by electron holography.66–71 Moreover, a strong electric field is found at the high frequency of 10–18 GHz rather than at the low frequency (2 GHz). These simulated results reveal that polarizations originating from the oxygen vacancies, defects, and surface functional groups are major contributors to high-frequency absorption. Additionally, multiple scattering/reflection, cross polarization, and electric coupling can improve the EMWACs owing to the enhanced EMW transmission path and dielectric loss.
The attenuation constant (A) and matching performance are key factors evaluating the EMWAPs of materials,72 which satisfy the formulae 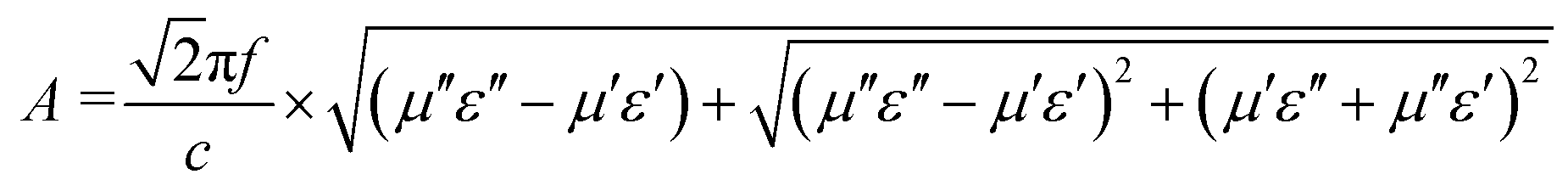 and
and 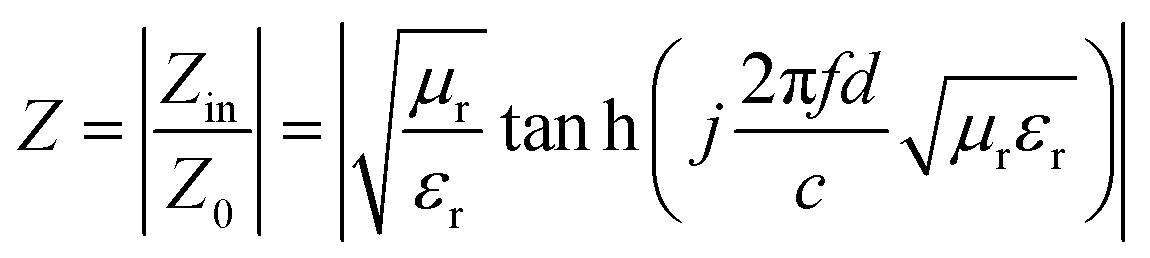 , respectively. Fig. 8a and c show that A is proportional to f, indicating the excellent high-frequency absorption. The variation regulation of A with β and T is as follows: A4:1 > A2:1 > A3:1 > A1.5:1 at 8.5–18 GHz and A2:1 > A3:1 > A1.5:1 > A4:1 at 2–8.5 GHz; A160°C > A140°C > A180°C > A120°C at 9–18 GHz, A120°C > A140°C > A160°C > A180°C at 9–18 GHz, and A180°C ≈ A140°C ≈ A160°C > A120°C at 2–8.5 GHz. According to the data in Fig. 7a1–b1 and d1–e1, the excellent high-frequency absorption is ascribed to the high polarization loss; the low-frequency absorption is related to the combined action of conductive loss and polarization loss. The increased attenuation results from the synergistic effect of the hierarchical structure, oxygen vacancies, defects, and abundant surface functional groups.
, respectively. Fig. 8a and c show that A is proportional to f, indicating the excellent high-frequency absorption. The variation regulation of A with β and T is as follows: A4:1 > A2:1 > A3:1 > A1.5:1 at 8.5–18 GHz and A2:1 > A3:1 > A1.5:1 > A4:1 at 2–8.5 GHz; A160°C > A140°C > A180°C > A120°C at 9–18 GHz, A120°C > A140°C > A160°C > A180°C at 9–18 GHz, and A180°C ≈ A140°C ≈ A160°C > A120°C at 2–8.5 GHz. According to the data in Fig. 7a1–b1 and d1–e1, the excellent high-frequency absorption is ascribed to the high polarization loss; the low-frequency absorption is related to the combined action of conductive loss and polarization loss. The increased attenuation results from the synergistic effect of the hierarchical structure, oxygen vacancies, defects, and abundant surface functional groups.
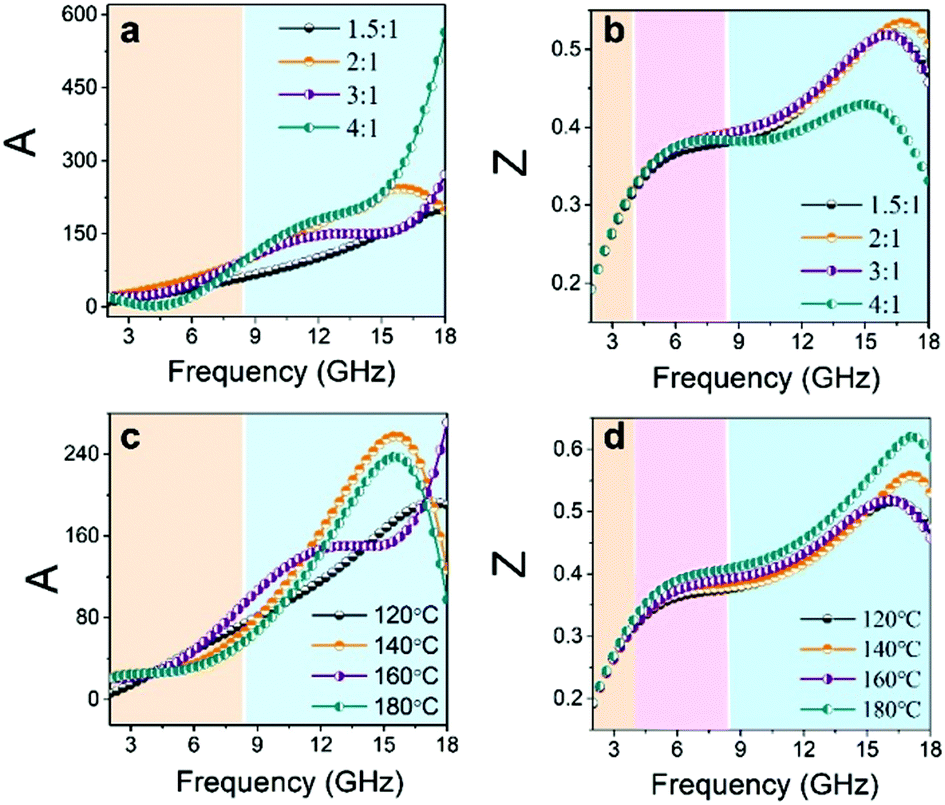 | ||
| Fig. 8 Frequency characteristics: (a and c) attenuation constant (A) and (b and d) impedance matching (expressed by modulus of normalized characteristic impedance Z) of the CeOHCO3 specimens formed at different β and T. | ||
Fig. 8b and d show that all the Z values increase with f and two resonance peaks are found at about 4–7 GHz and 16–18 GHz. These data demonstrate that CeOHCO3 exhibits good matching performances in the high-frequency range. The Z value varies with β and T as follows: Z4:1 < Z2:1 ≈ Z3:1 ≈ Z1.5:1 at 6–18 GHz and A180°C > A160°C > A160°C > A120°C at 4–18 GHz. The outstanding matching performance is ascribed to the balance modulation between conductivity loss and polarization loss caused by controlling the OVD density, size, defects, and morphology. Above all, the superior EMWAPs for CeOHCO3 at β = 2:1–3:1, T = 140 °C and 180 °C are attributed to the combination of good matching and high attenuation.
Conclusions
In summary, the orthorhombic phase Ce(OH)CO3 with continuously tunable morphology and size has been successfully prepared via a rapid microwave-assisted hydrothermal route without any surfactants, organic solvents, or expensive source materials. The morphology, dimension, and defects can be conveniently adjusted by just controlling the reaction temperature, reaction time, and CO(NH2)2/Ce3+ molar ratio. Both the selective adsorption and minimum surface free energy steer the morphology evolution from rods and dodecahedra to 2-fold butterfly-shaped hierarchical structures. Benefitting from the hierarchical structure, oxygen vacancies, and surface functional groups, the as-prepared Ce(OH)CO3 hierarchical structure possesses high conductivity and excellent electromagnetic wave absorption as a result of strong damping and perfect matching. The CeOHCO3 specimens formed at T = 140 °C and 180 °C and β = 2:1–3:1 take on lighter weight and stronger absorption over the X and Xu frequency bands, which obey the λ/4 damping models. Due to its excellent properties, our work suggests that the obtained Ce(OH)CO3 hierarchical structure is a promising EMW absorber. Additionally, the fabrication method adopted in this study is simple, high-yield, and low-cost, opening up an innovative route to fabricate hierarchical structures with controllable defects for EM applications.
Author contributions
Haiyan Wei: methodology, data curation, writing–original draft preparation; Xinxin Wang: calculation, visualization; Guoxiu Tong: conceptualization, writing, reviewing and editing; BaoXin Fan: software, validation; Xiaojuan Wang and Wenhua Wu: validation. All authors discussed the results and assisted during manuscript preparation.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The support of this research by funding from the National Natural Scientific Foundation of China (52073260), the Public Utility Items of Zhejiang Province (LGG21E020002), the Industrial Key Projects of Jinhua City (2019A12238), and the Self-Topic Fund of Zhejiang Normal University (2020ZS04) is appreciated.References
- R. Ma, S. Zhang, G. Liu, T. Wen, P. C. Gu, L. Li, G. X. Zhao, F. L. Niu, Q. F. Huang, Z. W. Tang and X. K. Wang, Catal. Today, 2019, 335, 20 CrossRef CAS.
- C. Q. Ge, L. Y. Wang, G. Liu and H. Q. Chen, J. Magn. Magn. Mater., 2019, 485, 228 CrossRef CAS.
- T. C. Campbell and C. H. F. Peden, Science, 2005, 309, 713 CrossRef PubMed.
- M. Konsolakis and M. Lykaki, Catalysts, 2021, 11, 452 CrossRef CAS.
- C. W. Sun, H. Li and L. Q. Chen, Energy Environ. Sci., 2012, 5, 8475 RSC.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987 CrossRef CAS PubMed.
- A. B. Shcherbakov, V. V. Reukov Cheng, A. V. Yakimansky, E. L. Krasnopeeva, O. S. Ivanova, A. L. Popov and V. K. Ivanov, Polymers, 2021, 13, 924 CrossRef CAS PubMed.
- G. L. Wu, Y. H. Cheng, F. Xiang, Z. R. Jia, Q. Xie, G. Q. Wu and H. J. Wu, Mater. Sci. Semicond. Process., 2016, 41, 6 CrossRef CAS.
- R. K. Singhal, S. Kumar, A. Samanya, M. Dhawan, S. C. Sharma and Y. T. Xing, Mater. Chem. Phys., 2012, 132, 534 CrossRef CAS.
- M. S. Cao, W. Zhou, X. L. Shi and Y. J. Chen, Appl. Phys. Lett., 2007, 91, 491 Search PubMed.
- M. Y. Ge, H. Wang, E. Z. Liu, J. F. Liu, J. Z. Jiang, Y. K. Li, Z. A. Xu and H. Y. Li, Appl. Phys. Lett., 2008, 93, 062505 CrossRef.
- L. N. Wang and F. M. Meng, Mater. Res. Bull., 2013, 48, 3492 CrossRef CAS.
- H. Wang, H. L. Xing, Q. C. Liu, H. X. Jia, A. J. Chen and Y. Liu, J. Mater. Sci.: Mater. Electron., 2018, 29, 19308 CrossRef CAS.
- J. H. Wang, P. F. Zhu, J. Q. Wang, S. W. Or, S. L. Ho and J. Tan, AIP Adv., 2016, 7, 55811 CrossRef.
- Z. Y. Shen, H. L. Xing, H. Wang, H. X. Jia, Y. Liu, A. J. Chen and P. Y. Yang, J. Alloys Compd., 2018, 753, 28 CrossRef CAS.
- M. Y. Cho, K. C. Roh, S. M. Park, H. J. Choi and J. W. Lee, Mater. Lett., 2010, 64, 323 CrossRef CAS.
- Y. G. Wang, F. Y. Zhang, Y. Guo, Y. Q. Wang, J. W. Ren, D. S. Qiao, X. H. Liu and G. Z. Lu, Mater. Chem. Phys., 2010, 120, 23 CrossRef CAS.
- R. J. Qi, Y. J. Zhu, G. F. Cheng and Y. H. Huang, Nanotechnology, 2005, 16, 2502 CrossRef CAS.
- Z. H. Han, Y. T. Qian, K. B. Tang, G. Q. Lu, S. H. Yu and N. Guo, Chem. Commun., 2003, 1117 CAS.
- Z. Y. Guo, F. L. Du, G. C. Li and Z. L. Cui, Inorg. Chem., 2006, 45, 4167 CrossRef CAS PubMed.
- B. Bakiz, F. Guinneton, J. P. Dallas, S. Villain and J. R. Gavarri, J. Cryst. Growth, 2008, 310, 3055 CrossRef CAS.
- D. F. Yan, T. Li, P. Liu, S. P. Mo, J. P. Zhong, Q. M. Ren, Y. H. Sun, H. R. Cheng, M. L. Fu, J. L. Wu, P. R. Chen, H. M. Huang and D. Q. Ye, Chemosphere, 2021, 279, 130658 CrossRef CAS PubMed.
- D. E. Zhang, X. J. Zhang, X. M. Ni, J. M. Song and H. G. Zheng, Chem. Phys. Lett., 2006, 430, 326 CrossRef CAS.
- M. Z. Wu, Q. H. Zhang, Y. M. Liu, Q. Q. Fang and X. S. Liu, Mater. Res. Bull., 2009, 44, 1437 CrossRef CAS.
- K. Li and P. S. Zhao, Mater. Res. Bull., 2010, 45, 243 CrossRef CAS.
- L. W. Qian, X. Wang and H. G. Zheng, Cryst. Growth Des., 2011, 12, 271 CrossRef.
- J. Fujita, A. E. Martell and A. K. Nakamoto, J. Chem. Phys., 1962, 36, 339 CrossRef CAS.
- P. F. Lin and J. Z. Lin, J. Appl. Math. Mech., 2009, 30, 957 CrossRef.
- N. T. McDevitt and W. L. Baun, Spectrochim. Acta, Part A, 1964, 20, 799 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. Smart, Appl. Surf. Sci., 2010, 257, 887 CrossRef CAS.
- J. J. He, Y. H. Xu, W. Wang, B. Hu, Z. J. Wang, X. Yang, Y. Wang and L. W. Yang, Chem. Eng. J., 2020, 379, 122431 CrossRef CAS.
- G. F. Zhang, P. Qin, R. Nasser, S. K. Li, P. Chen and J. M. Song, Chem. Eng. J., 2020, 387, 124029 CrossRef CAS.
- T. K. Sahu, S. Alam, D. Gogoi, N. R. Peela and M. Qureshi, ACS Appl. Energy Mater., 2020, 3, 5610 CrossRef CAS.
- Q. Y. Huang, Y. B. Wang, B. T. Zhou, Y. Z. Wei, F. Gao and T. Fujita, Corros. Sci., 2021, 179, 109165 CrossRef CAS.
- I. V. Plyuto, A. P. Shpak, I. V. Babich, Y. V. Plyuto, L. F. Sharanda, J. Stoch and J. A. Moulijn, Surf. Interface Anal., 1999, 27, 911 CrossRef CAS.
- J. Q. Liu, P. X. Wu, S. S. Li, M. Q. Chen, W. T. Cai, D. H. Zou, N. W. Zhu and Z. Dang, Chemosphere, 2019, 225, 115 CrossRef CAS PubMed.
- Y. Ding and W. L. Chen, Inorg. Chem. Commun., 2020, 117, 107937 CrossRef CAS.
- E. Beche, P. Charvin, D. Perarnau, S. Abanades and G. Flamant, Surf. Interface Anal., 2008, 40, 264 CrossRef CAS.
- Z. X. Yang, T. K. Woo, M. Baudin and K. Hermansson, J. Chem. Phys., 2004, 120, 7741 CrossRef CAS PubMed.
- S. K. Meher and G. R. Rao, J. Colloid Interface Sci., 2012, 373, 46 CrossRef CAS PubMed.
- L. Liu, N. He, T. Wu, P. B. Hu and G. X. Tong, Chem. Eng. J., 2019, 335, 103 CrossRef.
- G. X. Tong, J. H. Yuan, W. H. Wu, Q. Hu, H. S. Qian, L. C. Li and J. P. Shen, CrystEngComm, 2012, 14, 2071 RSC.
- G. X. Tong, F. T. Liu, W. H. Wu, J. P. Shen, X. Hu and Y. Liang, CrystEngComm, 2012, 14, 5963 RSC.
- A. R. Symington, R. M. Harker, M. T. Storr, M. Molinari and S. C. Parker, J. Phys. Chem. C, 2020, 124, 23210 CrossRef CAS.
- H. J. Wu, L. D. Wang, Y. M. Wang and S. L. Guo, Appl. Surf. Sci., 2012, 258, 10047 CrossRef CAS.
- Z. Q. Wang, P. F. Zhao, D. N. He, Y. Cheng, L. S. Liao, S. D. Li, Y. Y. Luo, Z. Peng and P. W. Li, Phys. Chem. Chem. Phys., 2018, 20, 14155 RSC.
- H. Zhang, A. J. Xie, C. P. Wang, H. S. Wang, Y. H. Shen and X. G. Tian, J. Mater. Chem. A, 2013, 1, 8547 RSC.
- Y. Liu, T. T. Cui, W. Tong, Y. N. Li and G. X. Tong, Nanotechnology, 2016, 27, 165707 CrossRef PubMed.
- K. D. Jiang, Y. Liu, Y. F. Pan, R. Wang, P. B. Hu, R. J. He, L. L. Zhang and G. X. Tong, Appl. Surf. Sci., 2017, 404, 40 CrossRef CAS.
- G. X. Tong, F. F. Du, L. J. Xiang, F. T. Liu, L. L. Mao and J. G. Guan, Nanoscale, 2014, 6, 778 RSC.
- M. Zong, Y. Huang, Y. Zhao, X. Sun, C. H. Qu, D. D. Luo and J. B. Zheng, RSC Adv., 2013, 3, 23638 RSC.
- Z. Zou, A. G. Xuan, Z. G. Yan, Y. X. Wu and N. Li, Chem. Eng. Sci., 2010, 65, 160 CrossRef CAS.
- T. Wang, H. D. Wang, X. Chi, R. Li and J. B. Wang, Carbon, 2014, 74, 312 CrossRef CAS.
- J. S. Deng, S. M. Li, Y. Y. Zhou, L. Y. Liang, B. Zhao, X. Zheng and R. Zhang, J. Colloid Interface Sci., 2018, 509, 406 CrossRef CAS PubMed.
- T. Liu, P. H. Zhou, J. L. Xie and L. J. Deng, J. Appl. Phys., 2011, 110, 033918 CrossRef.
- L. Jing, G. Q. Wang, Y. P. Duan and Y. Z. Jiang, J. Alloys Compd., 2009, 475, 862 CrossRef CAS.
- P. B. Liu, S. Gao, G. Z. Zhang, Y. Huang, W. B. You and R. C. Che, Adv. Funct. Mater., 2021, 31, 27 Search PubMed.
- J. J. Zhang, Z. H. Li, X. S. Qi, X. Gong, R. Xie, C. Y. Deng, W. Zhong and Y. W. Du, Composites, Part B, 2021, 222, 109067 CrossRef CAS.
- J. Qiao, X. Zhang, C. Liu, L. F. Lyu, Y. F. Yang, Z. Wang, L. L. Wu, W. Liu, F. L. Wang and J. R. Liu, Nano-Micro Lett., 2021, 13, 1 CrossRef CAS PubMed.
- Y. Wang, X. C. Di, Y. Q. Fu, X. M. Wu and J. T. Cao, J. Colloid Interface Sci., 2021, 587, 561 CrossRef CAS PubMed.
- M. Zhang, C. Han, W. Q. Cao, M. S. Cao, H. J. Yang and J. Yuan, Nano-Micro Lett., 2021, 13, 27 CrossRef PubMed.
- X. F. Yang, M. M. Liu, Y. Q. Lan, L. S. Wu, R. Ji, G. X. Tong, P. J. Gong and W. H. Wu, Chem. Eng. J., 2021, 381, 130779 CrossRef.
- X. F. Yang, B. X. Fan, X. X. Wang, X. Tang, J. L. Wang, G. X. Tong, X. J. Wang and W. F. Tian, J. Environ. Chem. Eng., 2021, 9, 105672 CrossRef CAS.
- M. M. Liu, X. F. Yang, W. Shao, T. Wu, R. Ji, B. X. Fan and G. X. Tong, Carbon, 2021, 174, 625 CrossRef CAS.
- P. C. Watts, W. K. Hsu, A. Barnes and B. Chambers, Adv. Mater., 2010, 15, 7 Search PubMed.
- R. C. Che, L.-M. Peng, X. F. Duan, Q. Chen and X. L. Liang, Adv. Mater., 2004, 16, 401 CrossRef CAS.
- H. Sun, R. C. Che, X. You, Y. S. Jiang, Z. B. Yang, J. Deng, L. B. Qiu and H. S. Peng, Adv. Mater., 2014, 26, 8120 CrossRef CAS PubMed.
- Q. H. Liu, Q. Cao, H. Bi, C. Y. Liang, K. P. Yuan, W. She, Y. J. Yang and R. C. Che, Adv. Mater., 2016, 28, 486 CrossRef CAS PubMed.
- J. W. Liu, R. C. Che, H. J. Chen, F. Zhang, F. Xia, Q. S. Wu and M. Wang, Small, 2012, 8, 1214 CrossRef CAS PubMed.
- R. C. Che, C. Y. Zhi, C. Y. Liang and X. G. Zhou, Appl. Phys. Lett., 2006, 88, 033105 CrossRef.
- Z. C. Wu, K. Pei, L. S. Xing, X. F. Yu, W. B. You and R. C. Che, Adv. Funct. Mater., 2019, 29, 1901448 CrossRef.
- L. Quan, F. X. Qin, D. Estevez, H. Wang and H. X. Peng, Carbon, 2017, 125, 630 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Physical parameters and synthesis conditions of CeOHCO3 specimens and EMWAP summarization of the CeOHCO3 specimens with other absorption materials. See DOI: 10.1039/d1tc04430c |
| This journal is © The Royal Society of Chemistry 2022 |