
Optical absorption by design in a ferroelectric: co-doping in BaTiO3
Shenglan
Hao
*a,
Minghai
Yao
a,
Gaëlle
Vitali-Derrien
a,
Pascale
Gemeiner
a,
Mojca
Otoničar
b,
Pascal
Ruello
c,
Housnny
Bouyanfif
d,
Pierre-Eymeric
Janolin
a,
Brahim
Dkhil
 a and
Charles
Paillard
a
a and
Charles
Paillard
a
aLaboratoire Structures, Propriétés et Modélisation des Solides, CentraleSupélec, CNRS-UMR8580, Université Paris-Saclay, Gif-sur-Yvette 91190, Paris, France. E-mail: shenglan.hao@centralesupelec.fr
bElectronic Ceramics Department, Jozef Stefan Institute, 1000 Ljubljana, Slovenia
cInstitut des Molécules et Matériaux du Mans, UMR6283 CNRS, Le Mans Université, Le Mans 72085, France
dLPMC UR2081, Université de Picardie Jules Verne, 33 Rue Saint Leu, Amiens 80039, France
First published on 30th November 2021
Abstract
The combination of significant polarization and large light absorption is a challenge in ferroelectric materials. Here we report the optical absorption of BaTiO3 codoped with trivalent and pentavalent transition metal ions replacing the Ti4+ cation, i.e., BaTi1−x(X1/2Y1/2)xO3 (X = Sc, Mn, Fe, Co; Y = Nb, Ta; with x = 0.075). We investigated the optical absorption behavior as a function of different pairs of co-dopants as well as physical properties such as ferroelectricity and piezoelectricity. X-Ray diffraction and Raman spectroscopy measurements show that the tetragonality and polar coherence length decrease after co-doping, which affects the ferroelectric properties. It was found that the onset of absorption can decrease down to 1.5 eV, especially for the (Co3+, Nb5+) co-doped samples, which simultaneously exhibit higher polarization than with the other co-doped ions. The measured photoconductivity confirms that the (Co3+, Nb5+) doping has higher absorption than others. Our density functional theory calculations show that codoping inserts intragap levels which are responsible for lowering the energy of optical absorption, which is in full agreement with our experimental observations. This work opens new perspectives for the use of ferroelectrics in optoelectronic devices.
Introduction
The lack of inversion symmetry inherent to ferroelectric materials, whose electrical polarization can be controlled by external electric fields, has attracted a large and wide interest for their potential optical and photonic applications, e.g., in photovoltaic solar cells, photocatalysts, or innovative elasto-optic devices.1–4 Unlike conventional semiconductor p–n junctions, their so-called bulk photovoltaic (PV) effect can generate voltages larger than the bandgap.5–7 Moreover, new functionalities related to the coupling between their polarization and the PV effect are conceivable. Unfortunately, most ferroelectric materials suffer from a large bandgap, typically larger than 3 eV. This results in poor absorption of the solar spectrum, causing low energy conversion efficiency in most ferroelectric-based solar cells. Lowering the bandgap, while maintaining decent ferroelectric properties (large remnant polarization, low losses upon switching) is therefore a crucial step towards applications of ferroelectrics in photovoltaic-based devices and beyond.Many ferroelectrics crystallize in the perovskite ABO3 structure. A typical example is the model ferroelectric material BaTiO3 (BTO), whose bandgap 3.3 eV. Note that calculations8 have shown that while BTO has an indirect bandgap between Γ–R Brillouin zone points, the direct band gap (Γ–Γ) is very close in energy (less than 0.05 eV whatever the hybrid functional). As a result in the following we assume a direct bandgap. The wide bandgap in BaTiO3 is electronically characterized by a valence band (VB) derived from O 2p states and a conduction band (CB) originating from the B-site transition metal 3d states (3d0 for Ti4+).2 The strong hybridization between the aforementioned orbitals (especially 3d0 states) causes symmetry breaking and leads to polar displacements at the origin of ferroelectric polarization. Therefore, altering the bandgap may also significantly impact the polarization and the related ferroelectric properties. A compromise between having a narrower bandgap while keeping a significant polarization amplitude is thus necessary.
Bandgap engineering strategies have already been devised in an attempt to increase optical absorption. Most studies have focused on tuning the oxygen vacancy concentration, alloying solutions, or chemical substitutions. Control of the oxygen vacancy concentration has been carried out in works on (1 − x)BaTiO3–xBaNi1/2Nb1/2O3−δ and (1 − x)BaTiO3–xBaCo1/2Nb1/2O3−δ, which exhibit a reduced bandgap of about 1.3 eV compared to 3.3 eV obtained in BTO.9,10 In Wu et al.'s work,11 by changing the ratio of Ni and Nb to modify the concentration of oxygen vacancies, the optical bandgap of doped BaTiO3 was significantly reduced down to 1.5 eV. Next, alloying was envisioned as a possible route to bandgap reduction in ferroelectrics, for instance, alloys based on the classical ferroelectric KNbO3 (KNO). Grinberg et al. mixed KNO with BaNi1/2Nb1/2O3−δ to introduce Ni2+ on the B-site and oxygen vacancies, creating an additional electronic band within the bandgap region. The obtained solid solution still exhibited ferroelectricity, while the bandgap was reduced to the range of 1.1–2 eV.12 Similar reports of solid solutions of KNO with BaFe1/2Nb1/2O3, BaCo1/2Nb1/2O3−δ, BiYbO3, and LaFeO3 showed that the bandgap could be shifted down to 1.7 eV, 2.4 eV and 2.2 eV, 2.6 eV, respectively.13–16
At last, several studies have focused on bandgap engineering by chemical substitution. Among those, reports on substitutions on the A-site of the perovskite showed a minor effect on the bandgap value. For instance, Bi/Li co-doped BTO (Ba1−x(Bi0.5Li0.5)xTiO3) exhibits nearly the same bandgap (3.2 eV17) as pure BTO. The inefficiency of this approach can be understood in light of the nature of the states forming the VB and CB described earlier. It is worth noting that a large voltage for this polycrystalline system was explained by the covalent bonding and the nature of the Bi-based orbitals, favoring the shift current of the PV effect.17 Similar observations were made with doping on the B-site, e.g. in Ba(Ti1−xSnx)O3 and [Ba(Zr0.2Ti0.8)O3]0.5[(Ba0.7Ca0.3)TiO3]0.5.18,19 In these compounds the proximity of phase transitions is believed to increase the open-circuit photovoltage. When the substitution on the B-site is made with transition metal acceptors like Fe3+, Mn3+, Co3+, Cr3+ or Ni2+, the bandgap tends to decrease. For example, the substitution of Ti4+ by Cr3+ in BTO showed a redshift of the absorption edge due to d–d band transitions between the Cr3+ center and the Ti4+ sites.20 A similar strategy has been adopted in BiFeO3 compounds doped with Mn3+ (3d4). It was found that defect states develop within the bandgap that act as a scaffold for photogeneration.21 The photogenerated power can be further enhanced by introducing donor and acceptor levels within the bandgap, as done in Fe-doped BaTiO3, through Fe2+ and Fe3+, respectively, allowing to activate a PV effect that is mediated through successive Fe3+/Fe2+ redox cycles.22 However, doping with Cr3+, Mn3+ or Fe3+ usually deteriorates the ferroelectric properties. This can be understood in the framework of the simple tight-binding model of Khomskii as a consequence of the partially occupied nature of the 3d-orbitals of these ions.23 In addition, the use of Fe3+ and Fe2+ as substitutes for Ti4+ ions leads to a charge imbalance, which results in leakage currents that prevent proper polarization measurements.
To mitigate this latter flaw of charge imbalance, Das et al. employed a different approach by reducing the bandgap by co-doping with Mn3+ and Nb5+ pairs (replacing two Ti4+ ions). Eventually, a relatively stable polarization was maintained.24 Compared to doping with a single ion,20 the absorption edge of the co-doped BTO presents a more pronounced redshift. In such co-doping strategy, Mn3+ induces intra-gap states, a reduction of the bandgap and a partial loss of polarization which is minimized thanks to strong Jahn–Teller (JT) effect. Nb5+ (d0), on the other hand, acts as a charge compensator, also capable to stabilize ferroelectric ground states in a similar way to ferroelectric niobates.25 It was also shown by the first-principles theory that (1 − x)BaTiO3–xBaMg1/2Mo1/2O3 shows good photovoltaic performance because of the charge balance created by Mg2+ and Mo6+ ions, and increased conduction bandwidth created by the Ti–O–Mo bonds.26
From reports in the literature, it is clear that co-doping could be the best route to (i) decrease the bandgap and (ii) maintain very good ferroelectric properties. There is, however, a need to better understand the effects of different co-dopants on the optical and electrical properties of ferroelectrics. In that endeavour, we employ both experimental and theoretical tools on the model ferroelectric material, BaTiO3. Co-doping with various ion pairs on the B-site is realized to form the BaTi1−x(X1/2Y1/2)xO3 system, and the resulting electrical and optical properties of these compounds are systematically reported herein and compared. We considered several couples of (X3+, Y5+) co-dopants that keep the total average charge of 4+ on the substituted Ti site, i.e. (Sc3+, Nb5+), (Mn3+, Nb5+), (Fe3+, Nb5+), (Co3+, Nb5+) and (Mn3+, Ta5+). For each couple we selected either Nb5+, which has empty d0 orbitals thus favors the polarizability of BTO, or Ta5+ ions, which also have empty d0 orbitals but are less ferroelectrically-active. Indeed, KTaO3 is an incipient ferroelectric that exhibits some polarizability only at low temperature, while a compound like KNbO3 is a room-temperature ferroelectric with similar properties to BaTiO3.27,28 With the exception of Sc3+ which has d0 states, all X3+ ions such as Mn3+, Fe3+, Co3+ have partially filled d-states, i.e., 3d4, 3d5, and 3d6, respectively. We thus expect that only Co3+ will significantly impact the electronic band structure.
Herein, we report the effects of co-doping on the structure, ferroelectric and piezoelectric properties of BTO. The optical absorption behavior is measured and compared to the theoretical one calculated using density functional theory (DFT). We reveal by comparing the results from different pairs of co-dopants that the electronic bandgap is weakly changed, if any, while the unfilled dn≠0 states introduce extra bands within this bandgap that help absorb light below the bandgap by pushing down the onset of absorption to 1.5 eV. Finally, the photoresponse as a function of the wavelength for the best samples is measured, further demonstrating that the co-doping can favor the optical absorption and PV performances.
Experimental
Material preparation
BaTi0.925(X1/2Y1/2)0.075O3 ceramics (abbreviated as XY, where X stands for Sc3+, Mn3+, Fe3+ or Co3+, and Y stands for Nb5+ or Ta5+) were fabricated by the conventional solid-state reaction method. The starting raw materials were: BaCO3 (STREM 99.9%), TiO2 (PROLABO 99%), Sc2O3 (STREM 99%), Mn2O3 (STREM 99%), Fe2O3 (ALFA PRDUKTE 99.9%), Co3O4 (ALFA AESAR 99.7%), Nb2O5 (ALFA AESAR 99.9%) and Ta2O5 (FLUKA 99.9%). Only BaCO3 being very hygroscopic compared to TiO2 was preheated at 500 °C for 5 hours to remove any absorbed moisture. Then, the raw powders were weighed in stoichiometric amounts and mixed by ball-milling in ethanol. After drying, the homogenized powder was pressed into pellets and calcined at 900 °C for 12 hours. After subsequent grinding the calcined powder was again pressed into pellets of about 8 mm diameter and 1 mm thick with polyvinyl alcohol (PVA with 35 wt%) as a binding agent. The pellets were then sintered at 1250–1280 °C for 10 hours. The amounts of dopants was fixed for all ionic pairs to x = 7.5%, which was found to be the optimal concentration for (Mn3+,Nb5+) co-doping.24Characterization
X-Ray diffraction (XRD) patterns were obtained on a Bruker D2 phase diffractometer using Cu-Kα radiation (λ = 1.5406 Å) with 20° ≤ 2θ ≤ 60° and steps of 0.02° with 2 s per step. Raman spectroscopy was recorded at room temperature using a Labram-Horiba spectrometer equipped with a He–Ne, 633 nm laser. The microstructure of the sintered ceramics was analysed by a field-emission scanning electron microscope (FE-SEM; JSM7600, Jeol Ltd, Tokyo, Japan), operated in backscattered electron mode.The sintered pellets were ground into powders for diffuse reflectance spectra. Room-temperature diffuse reflectance spectra were collected in the wavelength range of 200–900 nm using PerkinElmer UV-visible NIR spectroscopy (Lambda 850) equipped with a Harrick Praying Mantis TM diffuse reflectance accessory using the non-absorbing BaSO4 as a reference. The optical band gap was estimated by the Tauc plot of the absorption determined using the Kubelka–Munk formula FKM = k/s = (1 − R)2/(2R) where R is the raw reflectance, k the absorption coefficient, and s the scattering coefficient.29
The polished pellets were coated with silver electrodes on both sides to measure the ferroelectric properties. The polarization–electric-field (P–E) hysteresis loops and piezoelectric strain–electric-field (S–E) loops were recorded by applying a triangular waveform with a frequency of 5 Hz in silicone oil using an AixACCT TF-2000 ferroelectric tester system.
For the photovoltaic measurements, gold electrodes were sputtered on both sides. Before testing, all the samples were pre-poled at 30 kV cm−1 at 150 °C and field-cooled. The photoresponse under dark and different wavelength laser source conditions versus time were measured under a bias of 10 V using a Keithley Model 6430 sub-femtoAmp remote source meter. The laser sources were 325 nm (30 mW, IK Series He-Cd LASER), 405 nm and 532 nm (both with a power 10 mW, Class IIIb Laser), respectively.
Computational methods
We performed density functional theory (DFT) calculations using the Vienna Ab-initio Software Package (VASP).30–34 Structural relaxation was performed on a 3 × 3 × 3 BaTiO3 supercell (135 atoms) using the PBESol exchange functional. A plane wave cut-off of 550 eV was used, alongside a 1 × 1 × 1 Γ-centered k-mesh. Fermi–Dirac occupation with 25 meV smearing was employed. Convergence criteria on the energy and forces were respectively set to 10−6 eV and 10−3 eV A−1. The density of states and optical dielectric constant were subsequently calculated on a 3 × 3 × 3 k-mesh. The optical dielectric constant was calculated in the Independent Particle Approximation (IPA).35Results and discussion
Fig. 1a shows that the room temperature X-ray diffraction patterns of all the samples are very close to that of undoped BTO specimen in its room-temperature tetragonal phase (JCPDS card #05-0626). No parasitic phase could be evidenced on the basis of these X-ray diffractograms. A representative microstructure of the ceramics is shown in Fig. 1b, showing the example of (Mn3+, Nb5+), with grain size typically smaller than 10 μm. As shown in Fig. 1c, the tetragonality, represented by the c/a ratio with a and c being the tetragonal unit cell parameters determined from the diffraction patterns analysis, drops significantly for each pair of co-dopants as compared to the undoped BTO. This is expected as non-ferroelectrically active ions (such as Sc3+, Mn3+, Fe3+, Co3+) are replacing the ferro-active Ti4+ ions. These results also indicate that a lower ferroelectricity is expected as the c/a is, in the case of the tetragonal polar symmetry, directly proportional to the square of the polarization.36 Note that in the case of (Mn3+, Nb5+) co-dopants the decrease of c/a is significantly lower than for the other dopants and might be related to the strong JT distortions about Mn3+ that adequately couple the Mn3+–O6 octahedra to the overall ferroelectric BaTiO3 lattice as recently revealed using X-ray absorption spectroscopy.25 Such an effect, if any, seems to be much weaker in the case of Co3+ dopant which is also a JT active ion. It is also worth mentioning that when (Mn3+, Nb5+) is replaced by (Mn3+, Ta5+), the positive effect of Mn on the polarization is reduced because of the less-polarizable Ta5+ compared to Nb5+. In Table 1, we also reported the volume of the unit cell, determined from the XRD patterns, with respect to the average ionic radius for each (X3+, Nb5+) pairs i.e. rave = (rX + rNb)/2. It can be seen that the volume depends on the substitution elements confirming the dopants have been inserted in BTO unit cell. For instance, because Sc3+ has a larger radius (74.5 pm) and Fe3+ has a smaller one (55 pm) compared to Ti4+ (60.5 pm) the volume of (Sc3+, Nb5+) and (Fe3+, Nb5+) co-doping increases and decreases respectively compared to that of pure BTO.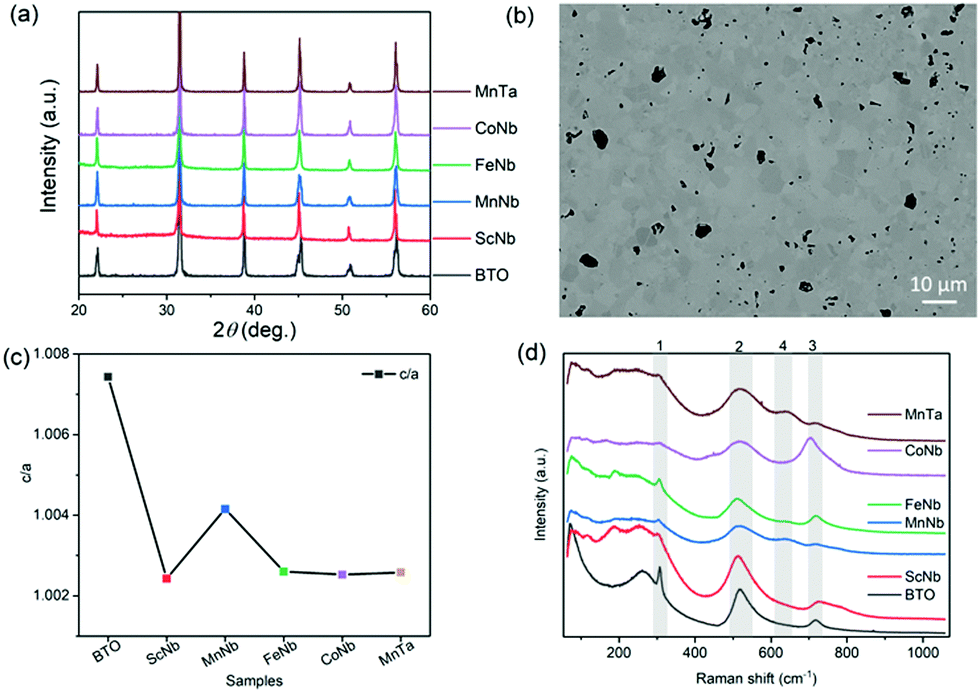 | ||
| Fig. 1 (a) XRD patterns of the pure BTO and co-doped BTO samples measured at room temperature. (b) Typical microstructure of the ceramics (here the case of (Mn3+, Nb5+) co-doping). Black areas are pores and the different shades of grains come from different grain orientations as commonly observed in polycrystalline ceramics (c) tetragonality i.e. c/a ratio depending on the dopants. (d) Raman spectra of undoped BTO and the co-doped samples collected at room temperature. | ||
| Composition | Volume (Å3) | P r (μC cm−2) | E c (kV cm−1) | S max (%) | r ave (Å) |
|---|---|---|---|---|---|
| BTO | 64.54 | 6.384 | 3.632 | 0.164 | 0.61 |
| Sc3+, Nb5+ | 64.75 | 1.632 | 2.255 | 0.0660 | 0.695 |
| Mn3+, Nb5+ | 64.37 | 0.663 | 3.428 | 0.0404 | 0.61 |
| Fe3+, Nb5+ | 64.55 | 0.228 | 3.803 | 0.00884 | 0.595 |
| Co3+, Nb5+ | 64.44 | 8.652 | 5.939 | 0.0737 | 0.595 |
| Mn3+, Ta5+ | 64.41 | 1.055 | 2.292 | 0.0622 | 0.61 |
Fig. 1(d) shows the room temperature Raman spectra for all the samples. The typical Raman spectrum of undoped BTO shows three major groups of Raman-active bands (denoted 1 to 3 in Fig. 1d), which are the fingerprints of the BTO tetragonal phase.37 In the range between 200–400 cm−1, which is usually attributed to vibrations associated to polar BO6 octahedra in ferroelectric perovskite oxides,38 there is a sharp peak at 305 cm−1, corresponding to B1(TO)/E(TO + LO) modes (TO and LO refer to transverse and longitudinal optic modes, respectively), representative of the long-range ferroelectric phase related to the cooperative displacement of Ti4+ in Ti–O chains.38 The two bands at 520 cm−1 and 720 cm−1, assigned to the A1(TO)/E(TO) and A1(LO)/E(LO) modes, respectively, are dominated by oxygen vibrations (bending and stretching) of the TiO6 octahedra. The co-doping of BTO has a clear impact on both the intensity and width of the sharp mode at 305 cm−1 that is related to the polar order. While the intensity of the mode can be related to the decrease of the tetragonality, i.e., polarization amplitude, its broadening is related to the lifetime of the associated phonon. The clear increase in the half-width of the Raman peaks with co-doping in Fig. 1d thus hints that coherence length in the tetragonal phase is shortened when co-dopants are added. In other words, the doping not only reduces the tetragonality and the polarization magnitude, but also induces some clustering through disorder which might originate from chemical defects/dopants or local chemical order, static/dynamic atomic shifts and oxygen octahedral distortion (e.g. because of JT effect) or local symmetry change.9,25,39 Such a disorder results in a relaxation of the Raman selection rules and may explain the broadening as well as the asymmetries of bands located at 520 cm−1 and 720 cm−1 in Fig. 1d. These latter are also regarded as an indicator of ferroelectricity as they have mixed A and E characters, which further points to the fact that co-doping indeed partially disrupts the long-range ferroelectric order. Note that the disorder is also observed in the low wavenumber region around 200 cm−1, as is observed in the cubic phase of pure BTO (expected to show no active Raman mode in case of strict selection rules) because of reminiscent Ti4+ polar displacements.37 The Raman spectra of samples co-doped with Mn or Fe also exhibit a Raman active mode at 635 cm−1 (denoted 4). As Mn ion is JT active, it may thus lead to extra modes because of local distorsions resulting in selection rules relaxation. However, Fe is not JT active and Co, which is JT active, does not show the 635 cm−1 mode. Rather, it is believed that such an extra band corresponds to the A1g mode of the hexagonal phase of BTO.40–46 Indeed, it is known that partial substitution of Ti4+ for transition metal ions can stabilize the hexagonal polymorph of BTO at room temperature.47 Such phase is believed to be present only in trace amounts as no extra Bragg peaks can be revealed in the diffraction patterns. Note that a few works have also attributed this extra band to the presence of oxygen vacancies.39 As a result, the existence of the hexagonal phase and/or oxygen vacancies may imply the presence of mixed valence transition metal ions (e.g. Mn3+/Mn4+ or Co3+/Co4+).
In order to characterize the ferroelectric and piezoelectric properties of all our samples, the polarization versus electric field (P–E) hysteresis loops and the strain versus electric field (S–E) curves measured at room temperature are shown in Fig. (a) and (b), respectively (see also Table 1). In the case of undoped BTO (black in Fig. 2), a typical ferroelectric P–E hysteresis loop and a butterfly-like S–E curve are obtained, indicative of the ferroelectric and piezoelectric character, respectively. When co-doping with (Fe3+, Nb5+), these aforementioned features are suppressed and the P–E curve shows a linear dielectric behavior, while the S–E curve displays a parabolic-like shape, typical of electrostriction (i.e., S ∝ E2 while piezoelectricity shows a linear dependency, i.e., S ∝ E). Note that electrostriction is a universal feature occurring in all materials, including non-ferroelectric ones, while all ferroelectrics are piezoelectrics. Table 1 also shows that there is a good correlation between the c/a ratio and the strain and polar reponse. The tetragonality decrease observed in Fig. 1c upon doping is indeed consistent with the reduction of the polarization amplitude displayed in Fig. 2a, which, in the extreme case of (Fe3+, Nb5+) doping, drops to roughly zero. Moreover, our Raman investigation shows that doping increases the chemical/polar disorder, e.g., through the broadening of the 305 cm−1 Raman mode. In the case of (Fe3+, Nb5+), the induced disorder appears strong enough to suppress the long range polar order despite the unit cell remaining on average tetragonal. Some uncorrelated local polar regions most likely remain due to the persistence of the 305 cm−1 mode. Comparing with other (X3+, Nb5+) pairs, iron is found here not only to disrupt the long-range polar correlation as evidenced from Raman data, but also to completely destroy the macroscopic polarization character (see Table 1) and to lead to a non-polar tetragonal symmetry.
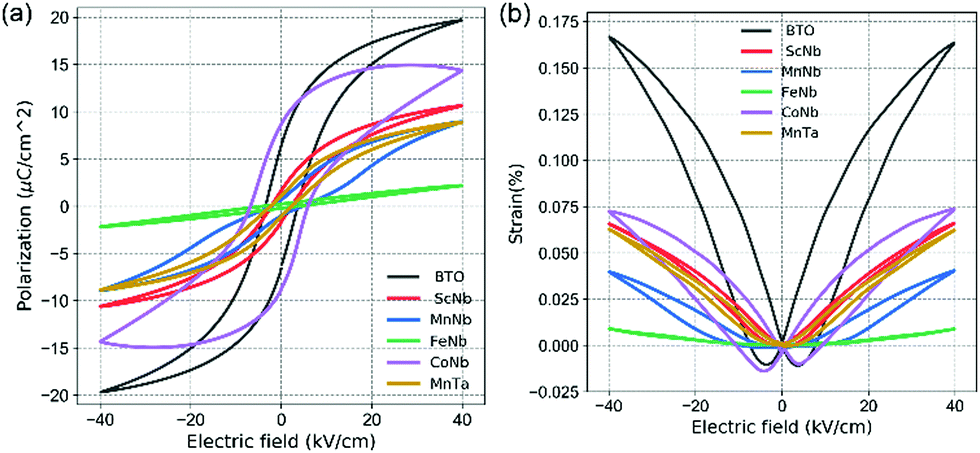 | ||
| Fig. 2 (a) P–E and (b) S–E loops for undoped and co-doped BTO. | ||
In the case of (Mn3+, Nb5+) doping, the P–E curve shows a “pinched” loop with remanent polarization close to zero (blue in Fig. 2) and reduced coercive field EC compared to pure BTO (Table 1). Such “pinching” was attributed to defect dipoles48 where defects associates with oxygen vacancies form, e.g. 2MnTi−–VO2+. The defect complexes can also impact the photovoltaic response.22 In addition to the pinched nature of its P–E loop, (Mn3+, Nb5+) co-doped BTO shows a decreased piezoelectric response (Table 1), with a clear electrostrictive nature observable from the S–E loop, plotted in blue in Fig. 2b. This can be understood as the result of the disruption of the long range polar order and formation of polar clusters. Such clustering is also likely to occur in the case of (Mn3+, Ta5+) and (Sc3+, Nb5+), as both also exhibit a slim P–E loop (without “pinching” like in (Mn3+, Nb5+) sample). Usually, the slim loops are associated to the so-called ferroelectric relaxors known to be characterized by polar clusters.49 Interestingly, doping pairs that show the most degraded ferroelectric properties correlate with those exhibiting a marked Raman peak around 635 cm−1 associated with the parasitic hexagonal phase and/or oxygen vacancies in Fig. 1d. On the contrary, the loop showing strongest ferroelectric and piezoelectric character from the doped samples is observed for (Co3+, Nb5+) co-doping (see purple in Fig. 2a, b and Table 1), and accordingly shows little to no peak around 635 cm−1 in Fig. 1b. Despite retaining good ferroelectric properties, (Co3+, Nb5+) co-doped samples exhibit a 56% lower maximum effective piezoelectric coefficient (deff = Smax/Emax) and about 33% larger coercive field (i.e., 14 kV cm−1). The remnant polarization (2Pr = 17 μC cm−2), on the other hand, is larger than in undoped BTO. These values suggest that the JT distorsion due to Co ions may adequately couple to the overall polarization of BTO. Nevertheless, given the swollen-shape of the P–E loop, some leakage currents are not excluded and may parasitically contribute to the total polarization response.
Let us now have a look at the light absorption properties of the co-doped samples. Fig. 3a and the zoom in Fig. 3b present Tauc plots involving the Kubelka–Munk function F (a proxy for the absorption coefficient) versus photon energy as determined from diffuse reflectance spectroscopy experiments. The plots indicate that the optical bandgap of undoped BTO is 3.4 eV (see intersection of the linear fit with the x axis in Fig. 3a), with the onset of absorption at 3.2 eV, as observed in Fig. 3b. With doping, the bandgap as determined using Tauc plots decreases to about 3.3 eV for (Sc3+, Nb5+), to 3.2 eV for (Fe3+, Nb5+), and to 3.0 eV for (Mn3+, Nb5+), (Mn3+, Ta5+) and (Co3+, Nb5+). Note that it is difficult to accurately determine the bandgap using Tauc plots with such absorption curves, as there is some degree of arbitrariness to locate the appropriate energy region for the linear fit. Hence, we found it important to additionally extract the onset of absorption which can be seen as the optical bandgap in contrast to the electronic bandgap separating the conduction band from the valence band.
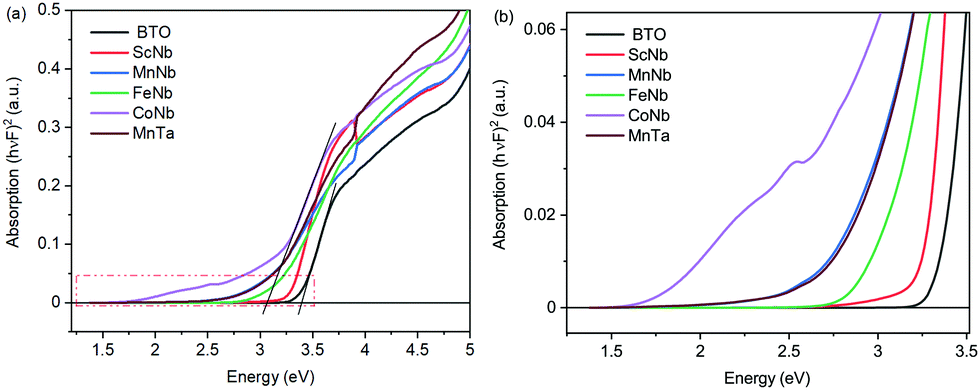 | ||
| Fig. 3 (a) Absorption as a function of incident energy for all the compositions, the intersection of the lines (indicative here for undoped BTO and (Co3+, Nb5+) doped BTO) with the abscisse axis gives the bandgap value according to Tauc plot (b) zoom in of the absorption cuves. | ||
Interestingly, while the bandgap of the doped samples extracted from Tauc plots in Fig. 3a only moderately decreases, a much larger variation is observed when considering the absorption onset (Fig. 3b). Indeed, from 3.2 eV in undoped BTO, the absorption onset drops to 2.75 eV for (Sc3+, Nb5+) and (Fe3+, Nb5+), then to 1.9 eV for (Mn3+, Nb5+) and (Mn3+, Ta5+), finally reaching 1.5 eV for (Co3+, Nb5+). The value of the absorption onset thus strongly depends on the X3+ transition metal element. As an illustration, in the case of Mn-samples, both (Mn3+, Nb5+) and (Mn3+, Ta5+) have almost identical optical absorption curves. Interestingly, despite similar absorption properties, both samples display different ferroelectric-like features, as described above. Among the X3+ dopant ions, Sc ion shows less effect on the absorption than Fe, while the impact of Mn and Co is significantly stronger. This can be understood by the partially filled dn≠0 orbitals of Fe3+, Mn3+ and Co3+ as well as the JT effect of both Mn3+ and Co3+ ions in contrast to the 3d0 orbitals of Sc3+.
In order to get a deeper insight into the band structure and its consequences on the absorption properties, DFT calculations were performed. Fig. displays the density of electronic states (DOS) for undoped BTO, (Sc3+, Nb5+), (Mn3+, Nb5+) and (Co3+, Nb5+) co-doped samples. The electronic bandgap using the GGA-PBESol exchange–correlation functional is underestimated as it is found to be 2.5 eV in undoped BTO (Fig. 4a). As expected, the conduction band (CB) has a strong Ti 3d-character, while the valence band is mainly due to O 2p-orbitals. It is interesting to note that whatever the doping (Fig. 4b–d), these features are kept and the CB and VB remain dominated by Ti- and O-orbitals. Consequently and while underestimated, the bandgap is found almost unchanged whatever the dopants, which is consistent with our experimental findings (see Fig. 3a). Interestingly, the partially filled dn≠0 orbitals of Mn3+ and Co3+ (also for Fe3+, not shown here) bring new intragap electronic bands (Fig. 4c and d), as also previously reported,21,24,25 which is not the case of Sc3+ with 3d0 orbitals (Fig. 4b). These extra bands inevitably impact on the absorption as we found experimentally (see Fig. 3b). Note that the band structure is impacted by the dn≠0 orbitals of the transition metals and thus Sc3+, Nb5+ as Ta5+ have no effects on the band structure; indeed, the density of states of (Mn3+, Nb5+) and (Mn3+, Ta5+) (not shown here) are similar and the experimental absorption curves are identical, as described previously.
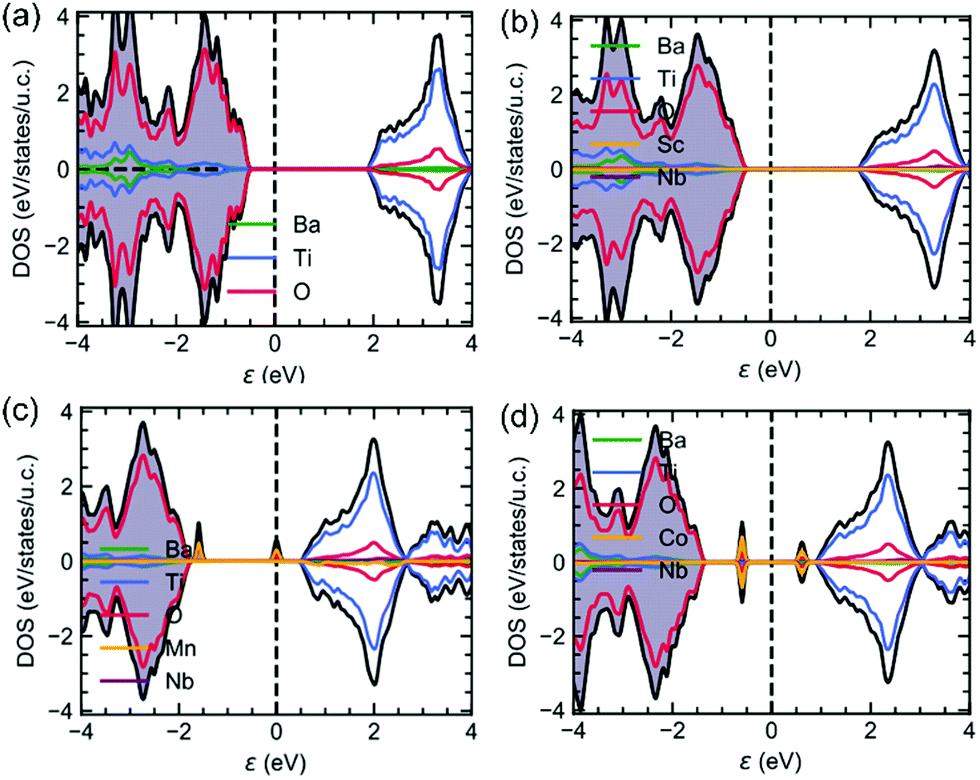 | ||
| Fig. 4 The density of states of (a) undoped BTO, (b) (Sc3+, Nb5+), (c) (Mn3+, Nb5+) and (d) (Co3+, Nb5+) co-doped samples. | ||
Using these calculations results, it is also possible to calculate at 0 K the imaginary part of permittivity ε′′ which is involved in the absorption. Fig. 5a–c display ε′′ (both components, i.e., parallel and perpendicular to the polarization, the latter being along the [111] rhombohedral direction because the calculations are done at 0 K in the rhombohedral phase) for (Sc3+, Nb5+), (Mn3+, Nb5+) and (Co3+, Nb5+), respectively. The absorption of undoped BTO is added for comparison (black lines in Fig. 5). It is interesting to see that the calculated absorption curves are in very good qualitative agreement with the experimental data. Indeed, it is remarkable to see that, e.g. in case of (Co3+, Nb5+), the experimental absorption curve (Fig. 3b) is very similar to the calculated one (Fig. 5c). Interestingly, and as suspected from the DOS plotted in Fig. 4b, the absorption spectrum of the (Sc3+, Nb5+) co-doping pair is similar to that of undoped BTO; this is because none of the doping (Sc or Nb) provide partially filled d states. A similar result was found experimentally in Fig. 3. Both undoped BTO and (Sc3+, Nb5+) co-doped BTO exhibit a steep absorption above the bandgap. In contrast, co-doping strategies involving dn≠0 ions such as Co3+ and Mn3+ show a smoothed absorption that starts to rise below the bandgap, due to the intragap states seen in Fig. 4c and d that provide a springboard for additional absorption e.g. optical transition between either occupied X3+ d-states and the conduction band or between the valence band and the empty X3+ d-state band. It is worth mentioning here that these results also highlight the difficulties one has to face when experimentally determining the bandgap value using the Tauc plot.
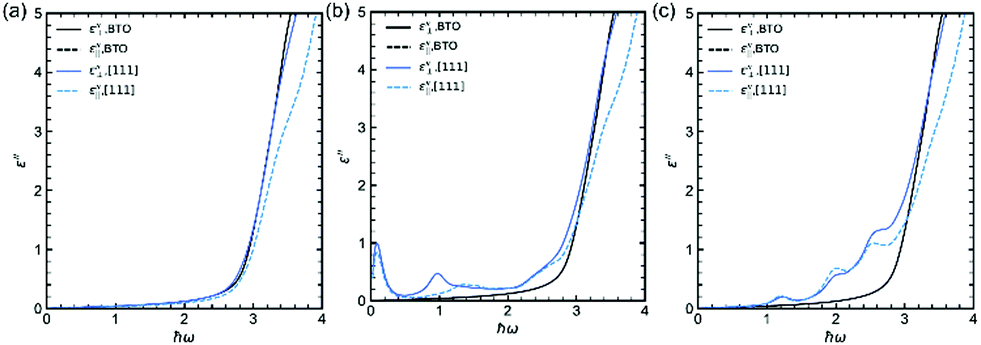 | ||
| Fig. 5 Imaginary part of the permittivity of (a) (Sc3+, Nb5+), (b) (Mn3+, Nb5+) and (c) (Co3+, Nb5+) samples compared to undoped BTO. | ||
Another way to attest of the enhanced absorption efficiency when doping with Mn and Co is to measure the photoresponse when light is illuminating the sample using a perpendicular electrode configuration (see Fig. 6a). Fig. 6b–d show the time dependence of the current under 10 V bias (photoconductivity) with the light switching on/off with laser diodes with wavelength 325 nm, 405 nm and 532 nm, corresponding to an energy of about 3.8 eV, 3.1 eV and 2.3 eV, respectively.
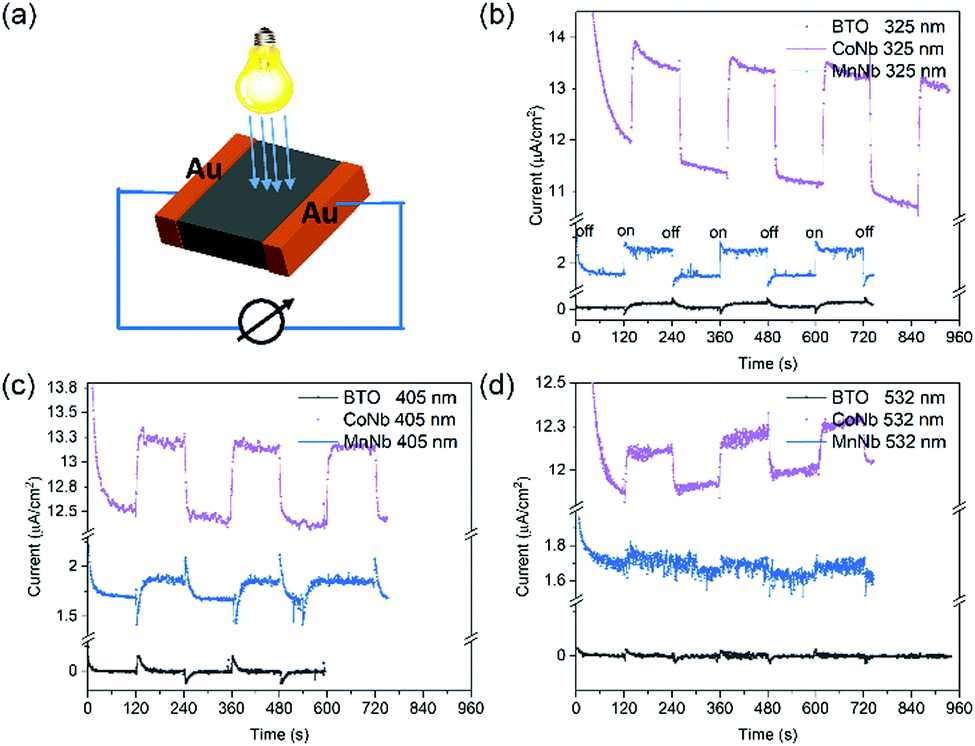 | ||
| Fig. 6 (a) Device configuration for the photoelectric measurement. (b and c) The photoresponse versus time curves with laser (325 nm, 405 nm, 532 nm) of undoped BTO, (Co3+, Nb5+), and (Mn3+, Nb5+) samples. | ||
Despite some residual leakage current, observed in dark conditions and associated to free carriers or defects, when the laser is on there is a clear stable photogenerated current. At times t when the light is on (off) this current rises (or drops) until reaching a plateau that is the steady state. The initial transient current jump is associated to a pyroelectric current due to the sudden heating (cooling) of the sample when the laser diode is on (off) Interestingly, the photoconductivity changes according to the illuminating wavelength value as well as to the type of doping with respect to undoped BTO. For undoped BTO whose experimental electronic bandgap obtained using Tauc plot is 3.4 eV and the optical bandgap, i.e., the real absorption onset is 3.2 eV (see Fig. 3), there is no detectable current (except jumps of pyroelectric current) when the incident energy is lower than the absorption onset (i.e., 2.3 eV in Fig. 6d or 3.1 eV in Fig. 6c). In contrast, it becomes significant when illuminated using above bandgap light of 3.8 eV (325 nm). When co-doping with (Mn3+, Nb5+) and (Co3+, Nb5+), the absorption onset shifts down to 1.9 eV and 1.5 eV, respectively, which explains why we do see photogenerated response even using the lowest energy diode, i.e., 2.3 eV (532 nm). Moreover, as expected from absorption spectra (Fig. 3), the photoresponse of (Co3+, Nb5+) is higher than that of (Mn3+, Nb5+) and (Sc3+, Nb5+), which supports all previous experimental and theoretical results. Interestingly, (Co3+, Nb5+) co-doped samples show also the best ferroelectric properties among the investigated co-doped BTO and thus could be considered as a good potential candidate for photovoltaic-based applications.
Conclusions
In this work, two different transition metal ions are introduced into BTO to substitute the Ti4+ in the B site of the perovskite, while keeping the overall charge balance. The co-doping with 3dn≠0 ions introduces intra-gap levels within the bandgap of BTO and improves the absorption (by lowering the energy). The onset of absorption can be significantly decreased down to 1.5 eV for co-dopants (Co3+, Nb5+) while retaining very good ferroelectric properties. The experimental optical absorption results are well supported by ab initio calculations. Such chemical approach appears as an excellent strategy to decrease the optical bandgap, and increase the optical absorption in BTO and can be largely extended to other ferroelectrics, making it available for the development of the next generation of high-performance ferroelectric-based photovoltaic devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the French National Research Agency (ANR) with the projects UP-DOWN (ANR-18-CE09-0026) and RelaxSolaire (ANR-18-CE92-0025). We also acknowledge a public grant overseen by the ANR as part of the “Investissements d’Avenir” program (ANR-10-LABX-0035, LabexNanoSaclay) and PHC Slovenian-French Proteus project BI-FR/19-20-PROTEUS-009. We also thank GENCI/CINES for computational support through access to High Performance Computing resources on the Occigen machine (Project A0070910952). We would like also thank the Chinese Scholarship Council.References
- P. Lopez-Varo, L. Bertoluzzi, J. Bisquert, M. Alexe, M. Coll, J. Huang, J. A. Jimenez-Tejada, T. Kirchartz, R. Nechache, F. Rosei and Y. Yuan, Phys. Rep., 2016, 653, 1–40 CrossRef CAS.
- C. Paillard, X. Bai, I. C. Infante, M. Guennou, G. Geneste, M. Alexe, J. Kreisel and B. Dkhil, Adv. Mater., 2016, 28, 5153–5168 CrossRef CAS.
- D. Sando, Y. Yang, C. Paillard, B. Dkhil, L. Bellaiche and V. Nagarajan, Appl. Phys. Rev., 2018, 5, 041108 Search PubMed.
- M. Lejman, G. Vaudel, I. C. Infante, I. Chaban, T. Pezeril, M. Edely, G. F. Nataf, M. Guennou, J. Kreisel, V. E. Gusev, B. Dkhil and P. Ruello, Nat. Commun., 2016, 7, 1–10 Search PubMed.
- A. M. Glass, D. Von Der Linde and T. J. Negran, Appl. Phys. Lett., 1974, 25, 233–235 CrossRef CAS.
- S. Y. Yang, J. Seidel, S. J. Byrnes, P. Shafer, C. H. Yang, M. D. Rossell, P. Yu, Y. H. Chu, J. F. Scott, J. W. Ager, L. W. Martin and R. Ramesh, Nat. Nanotechnol., 2010, 5, 143–147 CrossRef CAS.
- A. Bhatnagar, A. Roy Chaudhuri, Y. Heon Kim, D. Hesse and M. Alexe, Nat. Commun., 2013, 4, 2835 CrossRef.
- S. Piskunov, E. Heifets, R. I. Eglitis and G. Borstel, Comput. Mater. Sci., 2004, 29, 165–178 CrossRef CAS.
- Q. Lou, J. Zeng, Z. Man, L. Zheng, C. Park, A. Kassiba, Y. Liu, X. Chen and G. Li, J. Am. Ceram. Soc., 2019, 102, 7405–7413 CrossRef CAS.
- D. Zheng, H. Deng, S. Si, Y. Pan, Q. Zhang, Y. Guo, P. Yang and J. Chu, Ceram. Int., 2020, 46, 6073–6078 CrossRef CAS.
- L. Wu, A. R. Akbashev, A. A. Podpirka, J. E. Spanier and P. K. Davies, J. Am. Ceram. Soc., 2019, 102, 4188–4199 CrossRef CAS.
- I. Grinberg, D. V. West, M. Torres, G. Gou, D. M. Stein, L. Wu, G. Chen, E. M. Gallo, A. R. Akbashev, P. K. Davies, J. E. Spanier and A. M. Rappe, Nature, 2013, 503, 509–512 CrossRef CAS.
- L. Yu, H. Deng, W. Zhou, P. Yang and J. Chu, Mater. Lett., 2017, 202, 39–43 CrossRef CAS.
- L. Yu, J. Jia, G. Yi, Y. Shan and M. Han, Mater. Lett., 2016, 184, 166–168 CrossRef CAS.
- C. Pascual-Gonzalez, G. Schileo and A. Feteira, Appl. Phys. Lett., 2016, 109, 132902 CrossRef.
- C. Elicker, C. Pascual-González, L. T. Gularte, M. L. Moreira, S. S. Cava and A. Feteira, Mater. Lett., 2018, 221, 326–329 CrossRef CAS.
- S. Pal, A. B. Swain, P. P. Biswas, D. Murali, A. Pal, B. R. K. Nanda and P. Murugavel, Sci. Rep., 2018, 8, 1–7 Search PubMed.
- L. Kola, D. Murali, S. Pal, B. R. K. Nanda and P. Murugavel, Appl. Phys. Lett., 2019, 114, 183901 CrossRef.
- A. Bihari Swain, D. Murali, B. R. K. Nanda and P. Murugavel, Phys. Rev. Appl., 2019, 11, 044007 CrossRef CAS.
- I. C. Amaechi, G. Kolhatkar, A. H. Youssef, D. Rawach, S. Sun and A. Ruediger, RSC Adv., 2019, 9, 20806–20817 RSC.
- H. Matsuo, Y. Noguchi and M. Miyayama, Nat. Commun., 2017, 8, 207 CrossRef.
- Y. Noguchi, Y. Taniguchi, R. Inoue and M. Miyayama, Nat. Commun., 2020, 11, 966 CrossRef CAS.
- D. I. Khomskii, J. Magn. Magn. Mater., 2006, 306, 1–8 CrossRef CAS.
- S. Das, S. Ghara, P. Mahadevan, A. Sundaresan, J. Gopalakrishnan and D. D. Sarma, ACS Energy Lett., 2018, 3, 1176–1182 CrossRef CAS.
- S. Mukherjee, D. Phuyal, C. U. Segre, S. Das, O. Karis, T. Edvinsson and H. Rensmo, J. Phys. Chem. C, 2021, 125, 14910–14923 CrossRef CAS.
- O. Shafir, J. Yang, A. M. Rappe and I. Grinberg, J. Appl. Phys., 2019, 126, 174101 CrossRef.
- C. Ang, A. S. Bhalla and L. E. Cross, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 1841041–1841046 CrossRef.
- G. L. Linyun Liang, Y. L. Li, L. Chen and S. Y. Hu, J. Appl. Phys., 2009, 106, 104118 CrossRef.
- B. Philips-Invernizzi, D. Dupont and C. Caze, Opt. Eng., 2001, 40, 1082–1092 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- M. Gajdoš, K. Hummer, G. Kresse, J. Furthmüller and F. Bechstedt, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 45112 CrossRef.
- T. Qi, I. Grinberg and A. M. Rappe, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 134113 CrossRef.
- U. D. Venkateswaran, V. M. Naik and R. Naik, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 14256–14260 CrossRef CAS.
- J. Pokorný, U. M. Pasha, L. Ben, O. P. Thakur, D. C. Sinclair and I. M. Reaney, J. Appl. Phys., 2011, 109, 114110 CrossRef.
- V. K. Veerapandiyan, S. Khosravi, G. Canu, A. Feteira, V. Buscaglia, K. Reichmann and M. Deluca, J. Eur. Ceram. Soc., 2020, 40, 4684–4688 CrossRef CAS.
- M. C. Maldonado-Orozco, M. T. Ochoa-Lara, J. E. Sosa-Márquez, R. P. Talamantes-Soto, A. Hurtado-Macías, R. López Antón, J. A. González, J. T. Holguín-Momaca, S. F. Olive-Méndez and F. Espinosa-Magaña, J. Am. Ceram. Soc., 2019, 102, 2800–2809 CrossRef CAS.
- N. V. Dang, T.-L. Phan, T. D. Thanh, V. D. Lam and L. V. Hong, J. Appl. Phys., 2012, 111, 113913 CrossRef.
- A. Rani, J. Kolte and P. Gopalan, Ceram. Int., 2015, 41, 14057–14063 CrossRef CAS.
- H. M. Nguyen, N. V. Dang, P.-Y. Chuang, T. D. Thanh, C.-W. Hu, T.-Y. Chen, V. D. Lam, C.-H. Lee and L. V. Hong, Appl. Phys. Lett., 2011, 99, 202501 CrossRef.
- H. Yamaguchi, H. Uwe, T. Sakudo and E. Sawaguchi, J. Phys. Soc. Jpn., 1987, 56, 589–595 CrossRef CAS.
- T.-L. Phan, P. Zhang, D. Grinting, S. C. Yu, N. X. Nghia, N. V. Dang and V. D. Lam, J. Appl. Phys., 2012, 112, 13909 CrossRef.
- A. Kirianov, N. Ozaki, H. Ohsato, N. Kohzu and H. Kishi, Jpn. J. Appl. Phys., 2001, 40, 5619–5623 CrossRef CAS.
- J. Wang, H. Zhang, Y. Li and Z. Li, J. Mater. Sci.: Mater. Electron., 2010, 21, 811–816 CrossRef CAS.
- J. Li, J. Lv, D. Zhang, L. Zhang, X. Hao, M. Wu, B.-X. Xu, M. Otonicar, T. Lookman and B. Dkhil, Phys. Rev. Appl., 2021, 16, 14033 CrossRef CAS.
- C. Zhao, Y. Huang and J. Wu, InfoMat, 2020, 2, 1163–1190 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |