
A tunable phosphorescence supramolecular switch by an anthracene photoreaction in aqueous solution†‡
Ting
Su
,
Yao-Hua
Liu
,
Yong
Chen
and
Yu
Liu
 *
*
College of Chemistry, State Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, P. R. China. E-mail: yuliu@nankai.edu.cn
First published on 25th October 2021
Abstract
Purely organic room-temperature phosphorescence (RTP) has attracted tremendous attention, but is rarely reported to possess stimulus responsiveness. Herein, we report a supramolecular RTP switch based on a dicationic guest molecule (BPA) and cucurbituril. BPA possesses a photosensitive 9,10-diphenylanthracene core and terminal 4-(4-bromophenyl)pyridium, which can form 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 supramolecular assembly with cucurbit[7]uril (CB[7]) and an n:n linear supramolecular polymer with cucurbit[8]uril (CB[8]). The assemblies can exhibit tunable fluorescence emission after undergoing a photoreaction of the anthracene unit in BPA. It is interesting that CB[8] can bind two bromophenylpyridinium units to form a green phosphorescent switch. Notably, the RTP of the assemblies can be reversibly switched on/off by UV irradiation or heating through a tunable Förster resonance energy transfer from 4-(4-bromophenyl)pyridium to anthracene. Moreover, the addition of cucurbituril can significantly accelerate the photoreaction rate of anthracene and enable the assembly to respond quickly to UV irradiation, which expands the application potential of this strategy in stimulus responsive RTP materials.
:4 supramolecular assembly with cucurbit[7]uril (CB[7]) and an n:n linear supramolecular polymer with cucurbit[8]uril (CB[8]). The assemblies can exhibit tunable fluorescence emission after undergoing a photoreaction of the anthracene unit in BPA. It is interesting that CB[8] can bind two bromophenylpyridinium units to form a green phosphorescent switch. Notably, the RTP of the assemblies can be reversibly switched on/off by UV irradiation or heating through a tunable Förster resonance energy transfer from 4-(4-bromophenyl)pyridium to anthracene. Moreover, the addition of cucurbituril can significantly accelerate the photoreaction rate of anthracene and enable the assembly to respond quickly to UV irradiation, which expands the application potential of this strategy in stimulus responsive RTP materials.
Introduction
In recent years, purely organic room-temperature phosphorescence (RTP) has gained increasing research interest because of its inherent advantages including a triplet state, longer lifetime and larger Stokes shift.1–4 Therefore, RTP materials have been widely explored for potential applications in light-emitting devices,5–7 biological imaging,8–12 information encryption,13–16 chemical sensing,17–21etc. However, purely organic molecules also have some disadvantages, such as low efficiency intersystem crossing (ISC), weak spin–orbit coupling, easy non-radiative transition, and collision quenching by oxygen or impurities, leading to hardly effective phosphorescence emission at room temperature.22–25 In order to solve the above problems, many strategies to achieve RTP have been proposed, including the introduction of heavy atoms,26,27 crystalline packing,28–30 polymerization modification,31–33 embedding into a matrix,34–38 molecular aggregation39,40 and so on.Among them, a supramolecular method based on macrocyclic compounds has recently attracted great attention in the construction of RTP systems,41–44 because macrocyclic compounds with hydrophobic cavities can specifically bind guest molecules and induce their RTP effect by virtue of a variety of noncovalent interactions, including hydrophobic effect, electrostatic interactions, hydrogen bonding, and van der Waals interactions.45–47 The combination of macrocyclic hosts and modified guests can bring the following benefits to purely organic RTP: (1) the rigid environment of macrocyclic compounds can tightly bind the guest, limit its molecular vibration, and reduce the nonradiative transition of the triplet state;48,49 (2) the hydrophobic cavity can protect the wrapped phosphor from the quenching effect of the external oxygen, water and other impurities.50,51 Based on the above advantages, many efforts have contributed to the application of supramolecular macrocycles in purely organic RTP systems.52–54 Tian and co-workers reported an approach to achieve amorphous purely organic RTP small molecules by modifying different phosphors onto β-cyclodextrin, which could suppress the non-radiative relaxation via intermolecular hydrogen bonding.41 Recently, we reported that by modifying bromophenylpyridinium on hyaluronic acid and co-assembling with cucurbit[8]uril (CB[8]), a type of macrocycle formed by the polymerization of glycoluril and formaldehyde,55,56 the resultant supramolecular assembly exhibited good RTP behavior in aqueous solution, which was successfully applied to mitochondrial-targeted imaging of cancer cells.57 Ma et al. constructed a water-soluble tunable RTP system excited by visible light via 2:2 supramolecular assembly of triazine/bromophenylpyridinium with CB[8] in aqueous solution.58 These outstanding studies inspire the application and development of supramolecular macrocyclic compounds in the construction of purely organic RTP systems. However, there are few reports on purely organic RTP molecular switches with stimulus responsiveness.59–61 Ma and Ding et al. realized photo-controllable purely organic RTP based on the photochromic polymer by synthesizing two polymers containing hexaaryldiimidazole and using their reversible homolysis to form triphenylimidazole radicals under UV irradiation.62
Herein, we wish to report a photoreaction-driven RTP supramolecular switch based on cucurbituril and a dicationic guest molecule possessing a photosensitive 9,10-diphenylanthracene core and terminal 4-(4-bromophenyl)pyridium (BPA, Scheme 1). Interestingly, this 9,10-diphenylanthracene derivative BPA can form not only 1:4 supramolecular assembly with cucurbit[7]uril (CB[7]) but also 1:1 supramolecular linear assembly with CB[8], where 4-(4-bromophenyl)pyridium moieties are encapsulated by a CB[8] cavity in a head-to-tail manner to form biaxial pseudorotaxane. However, 9,10-diphenylanthracene can directly absorb energy from the first excited singlet state of 4-(4-bromophenyl)pyridium by the mechanism of Förster resonance energy transfer (FRET) and block the ISC process, leading to the inhibition of the production of RTP.63,64 In response to UV irradiation, BPA can promote the production of singlet oxygen and combine with it to form endoperoxides (BPAEPO). This combination can be broken reversibly after being heated, so that the molecule can return to its initial reduced state. After the conjugated structure of 9,10-diphenylanthracene is destroyed by a photoreaction, the fluorescence and green phosphorescence of the 4-(4-bromophenyl)pyridium group appear. These results are attributed to the strong supramolecular interaction between CB[8] and BPA, which promotes the ISC pathway, limits molecular motion to suppress the nonradiative transition, and reduces the collision of phosphorescent triplets with triplet oxygen and other impurities. Significantly, the addition of supramolecular macrocycles can accelerate the photoreaction process and shorten the time for guest molecules to combine with singlet oxygen in aqueous solution to form endoperoxides. Such acceleration behavior is helpful to realize the sensitive response to UV irradiation in practical applications and provides a more reliable application prospect for this strategy.
 | ||
| Scheme 1 Illustration diagram of purely organic RTP activated by a photoreaction. | ||
Results and discussion
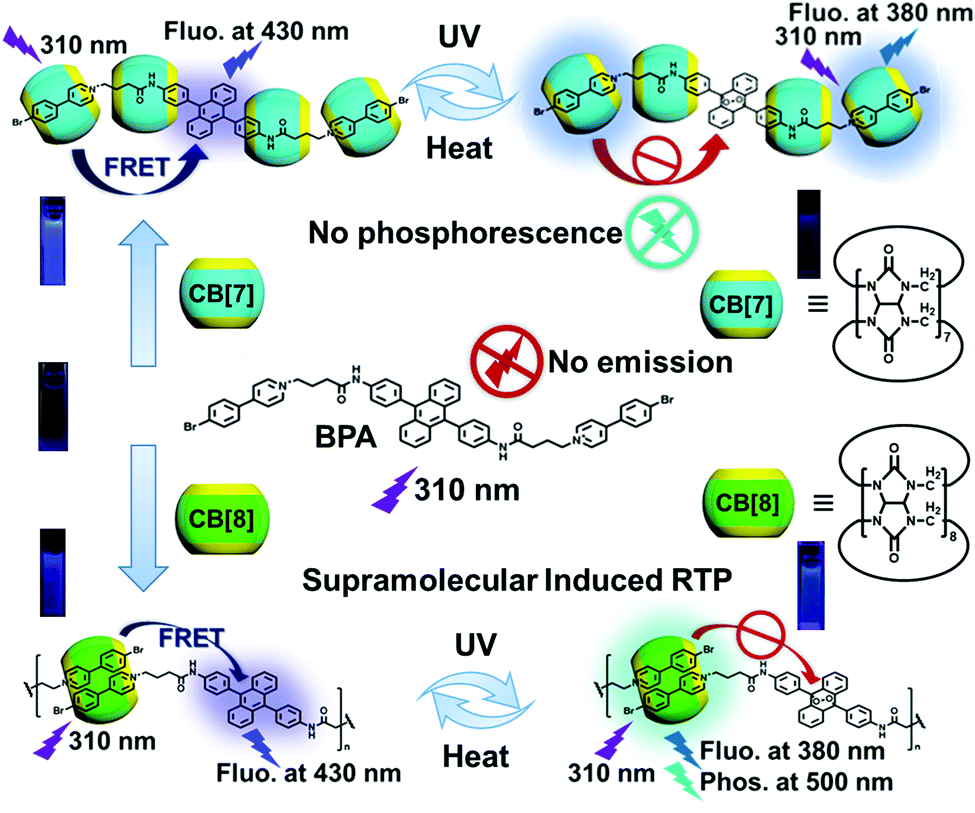
Synthetic routes of the guest molecules, i.e. a phosphor group 4-(4-bromophenyl)pyridium modified 9,10-diphenylanthracene derivative and its photoreaction product endoperoxide, are summarized in Scheme S1 (ESI‡). The dicationic guest molecule BPA was obtained in a total yield of >52% after the salinization between the 9,10-diphenylanthracene derivative 2 and 4-(4-bromophenyl)pyridium and characterized by NMR and mass spectra (Fig. S1–S6, ESI†). According to previous reports, cucurbituril can be used to bind the phosphorescent group 4-(4-bromophenyl)pyridium containing positive charges, enhance its solid phosphorescence intensity and induce visible solution phosphorescence.57,65 In order to explore the influence of the host–guest interaction on the optical properties of BPA, we select two cucurbituril macrocycles CB[7] and CB[8] for subsequent experiments. Job's plot measured by UV-vis absorption and fluorescence emission demonstrates that the stoichiometric ratios of BPA:CB[7] and BPA:CB[8] are 1:4 and 1:1, respectively (Fig. S13, ESI‡). The two-step binding constants of BPA with CB[7] measured by UV-vis absorption titration are Ka1 = 1.53 × 106 M−1 and Ka2 = 5.69 × 105 M−1, and the one-step binding constant of BPA with CB[8] is Ka = 9.39 × 106 M−1 (Fig. S14, ESI‡). Moreover, 1H NMR experiments were carried out to further investigate the host–guest binding mode between BPA and CB[7]/CB[8] (Fig. S15, ESI‡). As shown in Fig. S16 (ESI‡), the addition of CB[7] can passivate and weaken the Hd, He, Hf and Hg signals of 4-(4-bromophenyl)pyridinium and create new peaks in a higher field, indicating that the bromophenylpyridinium moieties of BPA are incorporated into the cavity of CB[7]. With the addition of >1 equivalent CB[7], the hydrogen peak assigned to Ha and Hc protons shifted to a lower field, indicating that the alkyl chain of BPA is also partly included in the CB[7] cavity. In the case of CB[8]/BPA assembly (Fig. S17, ESI‡), significant 1H NMR signal passivation of BPA protons is observed, whose degree gradually deepens with an increase of the CB[8] concentration. Meanwhile, when fixing the host–guest ratio at 1:1 and reducing the total concentration (Fig. S18, ESI‡), the changes in the peak shape and position are observed, indicating that supramolecular polymers may be formed. In the 2D ROESY (rotating-frame Overhauser effect spectroscopy) spectrum of BPA in the presence of 1 equiv. of CB[8] (Fig. S19, ESI‡), a strong ROE cross-peak between He and Hf is observed. According to our previous studies on bromophenylpyridinium derivatives, it is speculated that the two bromophenylpyridinium parts may be wrapped into the cavity of CB[8] in a head to tail orientation.66 In the 2D DOSY spectra (Fig. S20, ESI‡), the self-diffusion coefficient of BPA decreases from 1.255 × 10−10 to 7.896 × 10−11 m2 s−1 after adding CB[8], indicating the formation of a large assembly. In addition, we synthesize a model molecule BPB (Scheme S2 and Fig. S10–12, ESI‡) to simulate the interaction between the two arms of BPA and cucurbituril. Job's plot of the CB[7]/BPB system measured by the UV-vis absorption method confirms the optimal binding ratio of CB[7] to BPB as 2:1, and the two-step binding constants are K1 = 6.93 × 106 M−1 and K2 = 3.70 × 106 M−1, respectively (Fig. S21, ESI‡), which are in the same order of magnitude as the interaction between BPA and CB[7]. By comparing the 1H NMR spectra of BPB and its host–guest complex (Fig. S22, ESI‡), it is found that the hydrogen peak of the bromophenylpyridinium moiety is seriously passivated, and a new set of peaks appear at a higher field with the addition of CB[7]. This phenomenon confirms the existence of the host–guest interaction in a slow exchange manner. With the addition of a gradient concentration of CB[7] (Fig. S23, ESI‡), the peak of BPB decreases gradually, and a new peak at a higher field is observed. When 2 equivalents of CB[7] are added, the original peak disappears completely, indicating that the binding ratio of BPB to CB[7] is 1:2. Moreover, the MALDI-TOF mass spectrum also shows the peak assigned to the 1:2 inclusion complex between BPB and CB[7] at m/z = 2722.764 (Fig. S24, ESI‡). The addition of CB[8] can also make the peak of the aromatic hydrogen of BPB shift to a higher field (Fig. S25, ESI‡). Then, the morphology of the assembly is characterized using a scanning electron microscope (SEM, Fig. S26, ESI‡), where BPA forms a folded film structure on the silicon wafer. BPA and CB[7] are prepared in assembly solution at a ratio of 1:4, the morphologies of which are irregular protrusion after drying on the silicon wafer. While in the 1:1 assembly solution formed by BPA and CB[8], rhombic particles tend to be formed. Through the measurement of dynamic light scattering (Fig. 1a), the average particle size distribution of the assembly formed by BPA and CB[8] is about 1380 nm, which is consistent with the data obtained by SEM photos. The zeta potential of the assembly solution is measured as positive (Fig. 1b), indicating that the surface of particles formed by BPA and cucurbituril has cationic properties.
 | ||
| Fig. 1 (a) Dynamic light scattering of CB[8]/BPA, and the peak particle size of the normal distribution is about 1380 nm. (b) Zeta potential of the CB[8]/BPA assembly, the peak of which is at +30.8 mV. (c) UV-vis absorption spectra (illustration: partial enlarged view). (d) Prompt photoluminescence spectra (Ex. slit = 2.5 nm, Em. slit = 2.5 nm) ([BPA] = 0.025 mM, [CB[7]] = 0.050 mM and [CB[8]] = 0.025 mM) in water (containing 2.5% of DMSO) at 298 K (λex = 310 nm). | ||
In order to explore the optical properties of BPA and its host–guest complexes in aqueous solution, the UV-vis absorption and photoluminescence emission experiments are performed, where 2.5% dimethyl sulfoxide (DMSO) is added to improve the solubility of BPA in water. The UV-vis absorption spectra of BPA show a series of characteristic peaks of anthracene at 359 nm, 378 nm, and 398 nm and two strong absorptions at 260 nm and 308 nm. The absorption at 308 nm shows either an intensity increase or a bathochromic shift after the addition of CB[7]/CB[8] (Fig. 1c). When excited at 310 nm, the BPA solution presents two weak fluorescence peaks at 380 nm and 430 nm assigned to the characteristic emission of 4-(4-bromophenyl)-pyridium and 9,10-diphenylanthracene, respectively (Fig. 1d), probably due to the nonradiative transition of the aggregation state between BPA molecules caused by the strong tendency for the accumulation of the planar extended π-system of anthracene in solution.67 When CB[7] or CB[8] is added to the BPA solution, these two peaks obviously enhance (Fig. S27, ESI‡); for example, the fluorescence of BPA at 430 nm enhances 26.5 times with the addition of 4 equivalents of CB[7], probably because the binding of CB[7] with the 4-(4-bromophenyl)pyridinium moiety effectively disassembles the aggregated state of BPA. In addition, the inclusion of multiple CB[7] macrocycles on BPA molecules also increases the steric hindrance of the molecules to some extent, reduces the vibration and rotation in the fluorophore, and inhibits the non-radiative transition. These two factors jointly lead to enhanced photoluminescence. On the other hand, the larger cavity of CB[8] can bind two bromophenylpyridinium units at the same time, which can only partially reduce the accumulation of BPA molecules and slightly inhibit the non-radiative transition in molecules, leading to little enhancement in the fluorescence of guest BPA. Two equivalents of CB[7] and then one equivalent of CB[8] were added to BPA solution to explore the synergistic effects of two different sizes of cucurbituril macrocycles (CB[7,8]/BPA) on the photophysical behavior of BPA. We suspect that the addition of CB[8] can squeeze 2 equivalents of CB[7] into the alkyl chain of BPA, so as to further weaken the intermolecular stacking of BPA and weaken the non-radiative relaxation of its excited state. It is noteworthy that there exists intramolecular FRET from the phosphorescent 4-(4-bromophenyl)pyridinium group to the fluorophore 9,10-diphenylanthracene, in which the former jumps to the single excited state (S1) under the excitation of ultraviolet light, and the excitation energy of the fluorophore 9,10-diphenylanthracene matches that of the S1 state of 4-(4-bromophenyl)pyridinium, and the space distance between them is also conducive to the occurrence of energy transfer. Therefore, the system crossing of bromophenylpyridinium phosphorescent groups in the S1 state is blocked, and no phosphorescence is generated through the triplet excited state (T1). The functional group cannot make effective ISC and enter into T1. The result is that the obvious S1 luminescence of 9,10-diphenylanthracene and the relatively weak excited singlet luminescence of bromophenylpyridinium can be observed in the photoluminescence spectrum, and the characteristic phosphorescence emission of 4-(4-bromophenyl)-pyridium at 500 nm cannot be detected using a time-gated fluorescence spectrometer.
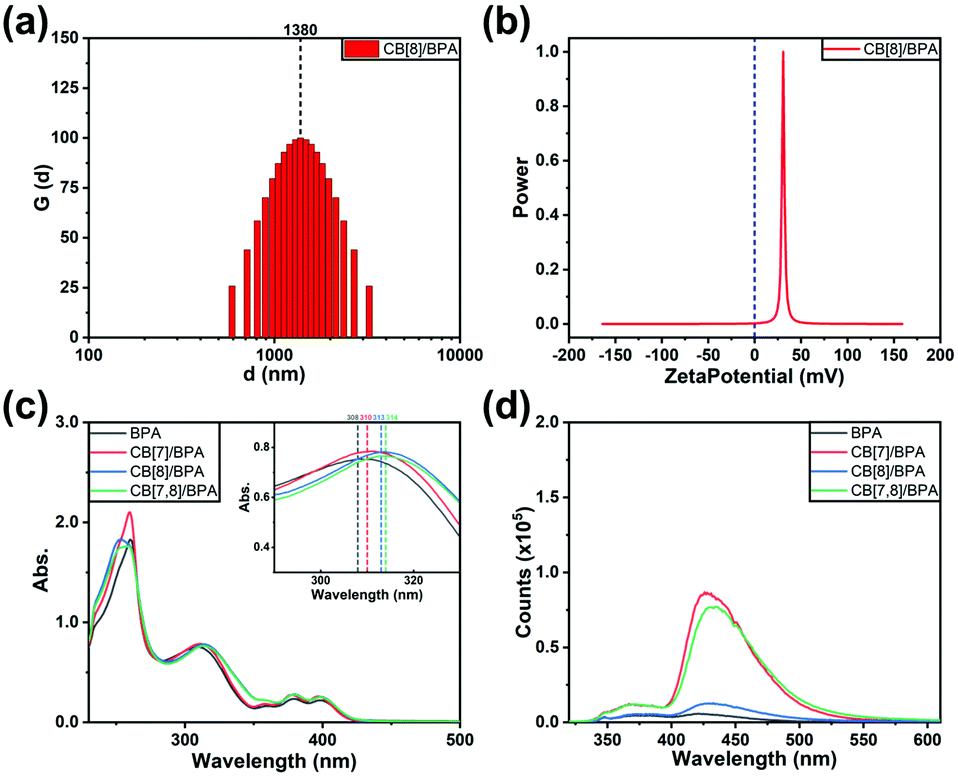
In order to investigate the photoreaction process of cucurbituril/BPA assemblies, the 365 nm UV LED flashlight is used to irradiate the experimental solution. The UV-vis absorption spectra of the solution are monitored at fixed time intervals, and a series of absorption spectra with various photoreaction times are obtained (Fig. S28, ESI‡). In these spectra, we can qualitatively and quantitatively analyze the photo-induced internal peroxycyclization of BPA (Fig. S29, ESI‡). The absorption intensities of three characteristic peaks of anthracene in BPA at 358, 376 and 395 nm gradually weaken with the extension of illumination time and are completely quenched within 50 minutes, probably because of the conversion of BPA to the internal peroxycyclization product BPAEPO. The kinetic study demonstrates that the peroxycyclization of BPA follows a first-order kinetic equation process, and the reaction rate constant is 0.07728 min−1. Significantly, the addition of cucurbituril can greatly accelerate the photoreaction process of BPA. The reaction rate constants of CB[7]/BPA, CB[8]/BPA and CB[7,8]/BPA are measured as 0.13751 min−1, 0.22591 min−1 and 0.42746 min−1, which are 1.8–5.5 times higher than that of free BPA, respectively. One possible reason is that the existence of the host cucurbituril can split the accumulation of BPA in water from the π–π interaction and improve the solubility to a certain extent, so as to reduce the energy dissipation of the thermal form when BPA is excited. This can also be explained by the enhancement of the luminescence of the solution due to the addition of cucurbituril. The molecules are stimulated to jump to S1, some of which release energy in the form of fluorescence and fall back to the ground state, and the other molecules cross to T1 and collide with the oxygen molecules dissolved in water to produce singlet oxygen, followed by the peroxidation process of anthracene. In addition, reduced stacking can also increase the collision probability between BPA and oxygen species, so as to accelerate the generation and combination of singlet oxygen, which is manifested by the increase of the apparent rate constant and the decrease of the BPA half-life. From the change in the photoluminescence spectrum with illumination time shown in Fig. 2a, we can also find that the characteristic emission of anthracene at 430 nm gradually weakens, and the characteristic fluorescence and phosphorescence of bromophenylpyridinium gradually appear (Fig. S30, ESI‡), which also proves the existence of FRET from one point of view.
 | ||
| Fig. 2 The spectra of BPA and its host–guest complexes after photo-induced internal peroxycyclization. (a) Changes in the photoluminescence spectra of CB[7,8]/BPA with 365 nm UV LED flashlight irradiation time. (b) UV-vis absorption spectra (illustration: partial enlarged view). (c) Prompt photoluminescence spectra (Ex. slit = 2.5 nm, Em. slit = 2.5 nm). (d) Phosphorescence spectra (delayed by 0.2 ms, Ex. slit = 20 nm, Em. slit = 10 nm) of BPA (black), CB[7]/BPA (red), CB[8]/BPA (blue), and CB[7,8]/BPA (green) ([BPA] = 0.025 mM, [CB[7]] = 0.050 mM and [CB[8]] = 0.025 mM) in water (containing 2.5% of DMSO) at 298 K (λex = 310 nm). | ||
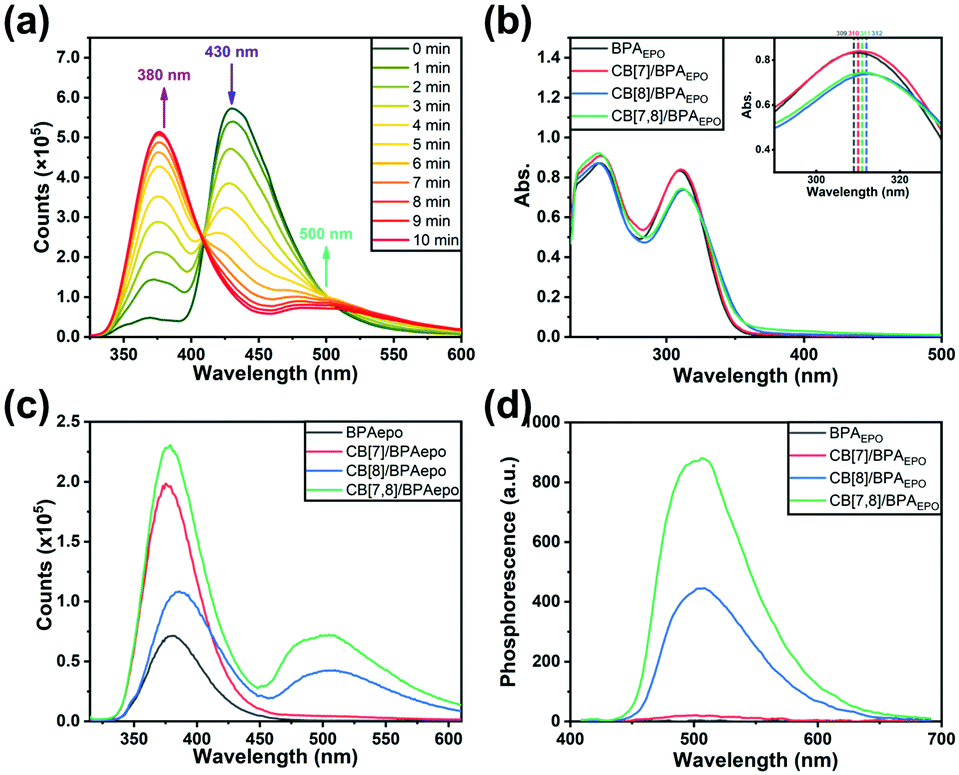
For exploring the molecular properties of BPAEPO after internal peroxide cyclization in more detail and reliably, we tried to directly synthesize BPAEPO and compare the properties with the BPA sample after the photoreaction. By adding a catalytic amount of the tetraphenylporphyrin photosensitizer to the DMSO solution of BPA for promoting the generation of singlet oxygen, under the irradiation of a 365 nm ultraviolet flashlight, a single-step yield of BPAEPO of more than 30% can be achieved. By the comparison of NMR, MS, and FT-IR (Fig. S7–S9 and S31, ESI‡), it can be confirmed that the product of the photoreaction of BPA is the same as the synthesized BPAEPO. The high-resolution mass spectrometry (HR-MS) signal of the oxidation species (Fig. S32a, ESI‡) confirmed the formation of endoperoxide. The UV-vis absorption and photoluminescence spectra of CB[7,8]/BPA after irradiation were very similar to those of CB[7,8]/BPAEPO (Fig. S32b and c, ESI‡). In addition, the decrease of the photoreaction rate under deoxygenated conditions also confirmed that the dissolved oxygen participated in the photoreaction (Fig. S32d, ESI‡). Job's curves are measured by fluorescence spectra and UV-vis absorption spectra, confirming that the optimal bonding ratios of BPAEPO to CB[7] and CB[8] are 1:4 and 1:1, respectively (Fig. S33, ESI‡). These data are consistent with the values measured from BPA, indicating that the photoinduced internal peroxide cyclization has no destructive effect on the molecular skeleton of BPAEPO. Therefore, in the subsequent photophysical property tests, we mostly use the method of preparing solutions for the substances before and after oxidation for comparative experiments. To our surprise, different assemblies also produce different photoluminescence properties after the photo-induced internal peroxidation. After UV irradiation, the fluorescence emissions of BPA, CB[7]/BPA, CB[8]/BPA, and CB[7,8]/BPA at 430 nm disappear (Fig. 2b), while the fluorescence emissions at 380 nm greatly enhance (15.9–20.7 times), in which CB[7,8]/BPAEPO gives the strongest fluorescence at 380 nm (Fig. 2c). CB[8]/BPAEPO and CB[7,8]/BPAEPO emit the obvious phosphorescence at 500 nm. However, under the same conditions, the phosphorescence of CB[7]/BPAEPO is fairly weak, and the phosphorescence of BPAEPO is negligible (Fig. 2d, delayed 0.2 ms). After nitrogen is introduced into the solution, the phosphorescence lifetime of CB[8]/BPAEPO at 500 nm increases from 133.69 μs to 289.21 μs, while that of CB[7,8]/BPAEPO increases from 405.49 μs to 716.10 μs (Fig. 3a and b), confirming that the photoluminescence at 500 nm is the phosphorescence. The total quantum yields of photoluminescence (Fig. S34, ESI‡) of CB[8]/BPAEPO and CB[7,8]/BPAEPO are 0.78% and 1.29%, respectively. The phosphor group 4-(4-bromophenyl)pyridinium, which originally has no phosphorescence emission in aqueous solution, can form a biaxial pseudorotaxane like structure with CB[8], accompanied by the green phosphorescence emission peak at 500 nm. This phenomenon is promoted by several factors: (1) the encapsulation of the CB[8] cavity provides a hydrophobic environment, which can protect the triplet phosphor from quenching by collision with triplet oxygen or other impurities; (2) the rigid cavity wraps two units of phosphorescent groups through a host–guest interaction, which limits their molecular motion and inhibits nonradiative relaxation; (3) the phosphorescent clusters of two molecules accumulate in the CB[8] cavity, resulting in a more stable π–π interaction, which promotes the occurrence of ISC; and (4) a halogen bond is formed between the C–Br of bromophenylpyridinium and the N atom of adjacent BPA. These factors jointly lead to the strong phosphorescence emission and the long phosphorescence life of CB[8]/BPAEPO and CB[7,8]/BPAEPO, while the CB[7]/BPA system lacks a π–π interaction and a halogen bond to promote ISC between phosphorescent groups. However, in the BPA molecule before the photoreaction, the characteristic absorption of the anthracene moiety just coincides with the fluorescence emission wavelength of the phosphor group, making FRET from the singlet excited state of bromophenylpyridinium to anthracene easy to occur in BPA, which blocks the way of ISC. Therefore, the characteristic fluorescence of bromophenylpyridinium at 380 nm is greatly weakened, and there is no phosphorescence emission. The phosphorescence of the host–guest complex does not reappear until the internal peroxidation process induced by ultraviolet light destroys the structure of anthracene, so as to realize the purely organic RTP emission induced by the photoreaction. Then, we used phosphorescence spectrum monitoring (Fig. 3c) to explore the ability of BPAEPO to decompose into BPA and singlet oxygen under heating conditions. After heating for 20 minutes, it can be observed that the phosphorescence intensity of CB[7,8]/BPAEPO assembly solution decreases by nearly 80%, which can be partially recovered by re-illumination with a 365 nm UV LED flashlight. From the phosphorescence intensity at 500 nm, the oxidation and reduction of the assembly showed good reversible stability (Fig. 3d) under the dual stimulation of UV irradiation and prolonged heating under deoxygenated conditions.
 | ||
| Fig. 3 Phosphorescence lifetime contrast curves for CB[8]/BPAEPO and CB[7,8]/BPAEPO. The phosphorescence decay curves of (a) CB[8]/BPAEPO, CB[8]/BPAEPO-N2 and (b) CB[7,8]/BPAEPO, CB[7,8]/BPAEPO-N2 at 500 nm at 298 K ([BPA] = 0.025 mM, [CB[7]] = 0.050 mM and [CB[8]] = 0.025 mM) in water (containing 2.5% of DMSO) (λex = 310 nm). (c) Phosphorescence intensity change of CB[7,8]/BPA solution after irradiation, heating and re-irradiation. (d) Phosphorescence reversibility for CB[7,8]/BPA detected by the emission intensity change at 500 nm. | ||
In order to explore the potential application of the photoreaction induced phosphorescence system in the material field, we prepared a translucent colorless hydrogel by adding a 3% mass fraction of agarose to the assembly solution after heating dissolution and then cooling. Similar to the solution state, the BPA molecule can undergo a photoreaction under 365 nm UV irradiation to form BPAEPO, which can be excited by a 302 nm light source to produce green phosphorescence. (Fig. 4a). The green phosphorescence cannot be produced without CB[8], which is consistent with the results measured in solution. The mechanical properties of the hydrogel are characterized by the rheology measurement. As shown in Fig. 4b, the linear elastic–viscous area is obtained by the experiments of an amplitude sweep at a strain (g) = 0.01–1000%. Under the strain from 0.01 to 2%, G′ (storage modulus) and G′′ (loss modulus) are almost constants. When the strain is greater than 2%, G′ decreases, but G′′ initially increases and then decreases, indicating that the hydrogel network is destroyed. With an angular frequency ranging from 0.1 to 100 rad per s, neither G′ nor G′′ exhibits obvious changes, and G′ is always larger than G′′, demonstrating good stability of the hydrogels toward the oscillation condition (Fig. 4c). These results provide the possibility for the application of BPA cucurbituril supramolecular assembly in stimulus responsive phosphorescent switching materials.
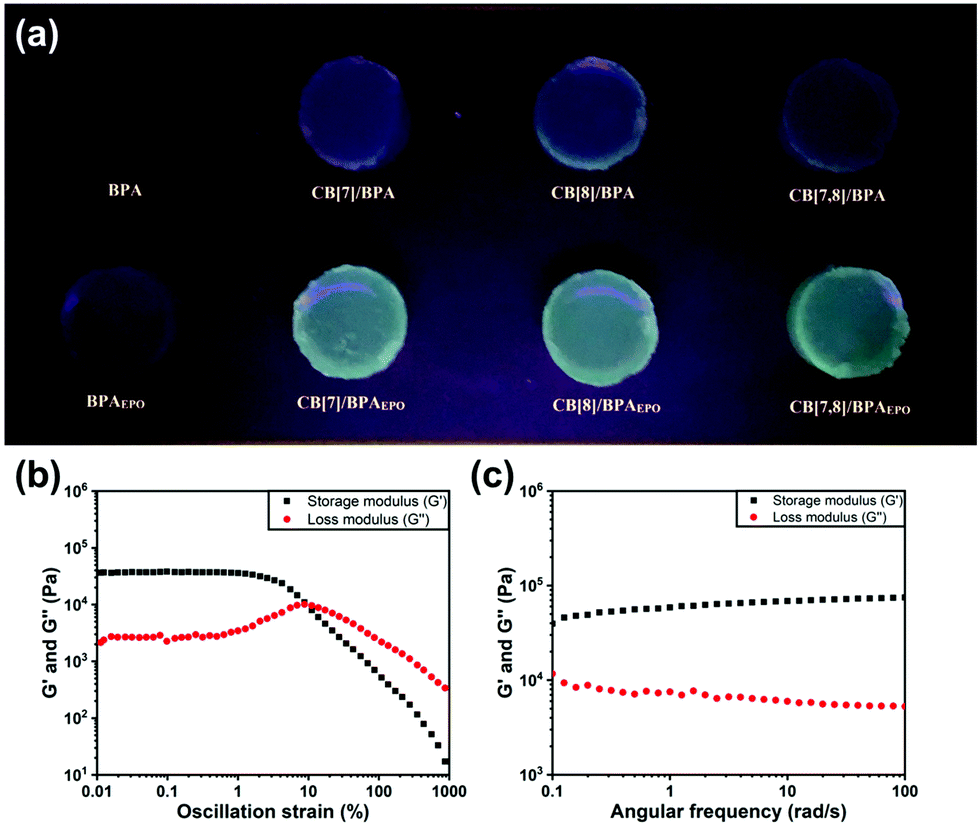 | ||
| Fig. 4 (a) Agarose gel (wt% = 3%) prepared from different assembly solutions under the excitation of 302 nm in a dark environment. (b) G′ (storage modulus) and G′′ (loss modulus) as a function of strain. (c) G′ and G′′ as a function of frequency. | ||
Conclusions
In summary, a photo-sensitive molecule BPA with 9,10-bis(4-aminophenyl)-anthracene and two bromophenylpyridinium arms was synthesized, and its host–guest complexes CB[7]/BPA, CB[8]/BPA and CB[7,8]/BPA were constructed. When BPA has not been oxidized yet, intermolecular FRET from 4-(4-bromophenyl)pyridium to 9,10-diphenylanthracene occurs, resulting in the strong fluorescence emission of supramolecular assembly at 430 nm. The addition of the rigid macrocyclic molecule cucurbituril can effectively reduce the formation of the aggregation state between BPA molecules, inhibit the nonradiative transition and enhance the fluorescence. Irradiation of the assembly solution with a 365 nm UV LED flashlight can induce the anthracene group in BPA to capture singlet oxygen and form a stable internal peroxide, thus prohibiting the FRET pathway. Such an endoperoxide can be reversibly reduced back to BPA by heating, so as to realize the reversible regulation of the supramolecular assembly photoluminescence. Thanks to the 1:2 inclusion of 4-(4-bromophenyl)pyridium by the host molecule, the supramolecular system with CB[8] can emit reversible green phosphorescence at 500 nm. Moreover, the addition of cucurbituril can effectively accelerate the process of the BPA photoreaction by about 1.8 to 5.5 times, giving this supramolecular RTP switch the rapid response capability. Finally, the successful preparation of the agarose based phosphorescent hydrogel also confirmed the application prospect of this RTP supramolecular switch construction strategy based on the anthracene photoreaction for stimulating response materials.
Experimental
Unless otherwise specified, all reagents and solvents were purchased from commercial sources and used without further purification. NMR spectra were recorded using a Bruker AVANCE III HD 400 spectrometer. Chemical shifts (δ) for 1H NMR and 13C NMR spectra are reported in parts per million (ppm) and relative to the solvent residual peak. Low-resolution mass (LR-MS) spectra were performed using a Bruker Autoflex III TOF/TOF200 spectrometer. High-resolution mass (HR-MS) spectra were acquired using a Varian 7.0T FTMS. The UV-vis absorption spectrum was recorded using a Shimadzu UV-3600 spectrometer. The photo-induced endoperoxidation experiments were carried out using a photochemical reaction apparatus with a 365 nm UV LED flashlight (irradiance: 3000 mW cm−2). Fluorescence and phosphorescence spectra were recorded using a Varian Cary Eclipse spectrofluorometer equipped with a plotter unit and a quartz cell (1 cm × 1 cm) at 25 °C. Quantum yields and phosphorescence lifetimes were recorded in a conventional quartz cell (10 × 10 × 45 mm) at 25 °C using an Edinburgh Instruments FS5 employing the single photon counting technique.Solution preparation
The BPA and BPAEPO stock solution was made by dissolving them in DMSO, at a concentration of 1 mM. All the water used in this paper is deionized secondary distilled water. CB[7] and CB[8] stock solutions were dissolved in deionized water at concentrations of 1 mM and 0.25 mM, respectively. All the above stock solution was kept at 25 °C before being used. All samples used for UV-vis absorption and photoluminescence spectroscopy contain DMSO of 2.5% of the solvent volume.Calculation of the apparent rate constant and half-life
By monitoring the UV-vis absorption spectrum of the assembly solution at 375.5 nm, a series of points with decreasing values with an increase of the illumination time were obtained. The data processing software was used to fit the exponential function of this series of points, and the formula is as follows.| y = a + be−kx, |
| t1/2 = ln2/k |
Preparation of supramolecular RTP hydrogels
3 mL of the assembly solution was added at a concentration of 0.025 mm to the cylindrical mold. 90 mg of agarose (3% by mass of the solution) was added to the solution and stirred evenly. The mold was placed in a microwave oven for one minute heating to complete the dissolution of agarose, followed by ice water cooling for 20 minutes, and then the hydrogels can be obtained simply.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant no. 22131008 and 21971127).Notes and references
- Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef CAS PubMed
.
- Y. Tao, R. Chen, H. Li, J. Yuan, Y. Wan, H. Jiang, C. Chen, Y. Si, C. Zheng, B. Yang, G. Xing and W. Huang, Adv. Mater., 2018, 30, 1803856 CrossRef
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS
- Y. Yu, M. S. Kwon, J. Jung, Y. Zeng, M. Kim, K. Chung, J. Gierschner, J. H. Youk, S. M. Borisov and J. Kim, Angew. Chem., Int. Ed., 2017, 56, 16207–16211 CrossRef CAS PubMed
- W. Ratzke, L. Schmitt, H. Matsuoka, C. Bannwarth, M. Retegan, S. Bange, P. Klemm, F. Neese, S. Grimme, O. Schiemann, J. M. Lupton and S. Höger, J. Phys. Chem. Lett., 2016, 7, 4802–4808 CrossRef CAS PubMed
- R. Kabe, N. Notsuka, K. Yoshida and C. Adachi, Adv. Mater., 2016, 28, 655–660 CrossRef CAS PubMed
- J.-H. Tan, W.-C. Chen, S.-F. Ni, Z. Qiu, Y. Zhan, Z. Yang, J. Xiong, C. Cao, Y. Huo and C.-S. Lee, J. Mater. Chem. C, 2020, 8, 8061–8068 RSC
- S. M. A. Fateminia, Z. Mao, S. Xu, Z. Yang, Z. Chi and B. Liu, Angew. Chem., Int. Ed., 2017, 56, 12160–12164 CrossRef CAS PubMed
- L. M. Hirvonen, M. Fisher-Levine, K. Suhling and A. Nomerotski, Rev. Sci. Instrum., 2017, 88, 013104 CrossRef PubMed
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed
- X. Zhen, C. Xie and K. Pu, Angew. Chem., Int. Ed., 2018, 57, 3938–3942 CrossRef CAS PubMed
- Q. Miao, C. Xie, X. Zhen, Y. Lyu, H. Duan, X. Liu, J. V. Jokerst and K. Pu, Nat. Biotechnol., 2017, 35, 1102–1110 CrossRef CAS
- Y. Su, Y. Zhang, Z. Wang, W. Gao, P. Jia, D. Zhang, C. Yang, Y. Li and Y. Zhao, Angew. Chem., Int. Ed., 2020, 59, 9967–9971 CrossRef CAS
- Z. He, H. Gao, S. Zhang, S. Zheng, Y. Wang, Z. Zhao, D. Ding, B. Yang, Y. Zhang and W. Z. Yuan, Adv. Mater., 2019, 31, 1807222 CrossRef
- Y. Su, S. Z. F. Phua, Y. Li, X. Zhou, D. Jana, G. Liu, W. Q. Lim, W. K. Ong, C. Yang and Y. Zhao, Sci. Adv., 2018, 4, eaas9732 CrossRef PubMed
- G. Chen, S. Guo, H. Feng and Z. Qian, J. Mater. Chem. C, 2019, 7, 14535–14542 RSC
- S. Wang, H. Shu, X. Han, X. Wu, H. Tong and L. Wang, J. Mater. Chem. C, 2021, 9, 9907–9913 RSC
- D.-A. Xu, Q.-Y. Zhou, X. Dai, X.-K. Ma, Y.-M. Zhang, X. Xu and Y. Liu, Chin. Chem. Lett., 2021 DOI:10.1016/j.cclet.2021.08.001
- R. Gao and D. Yan, Chem. Commun., 2017, 53, 5408–5411 RSC
- R. Keruckiene, D. Volyniuk, K. Leitonas and J. V. Grazulevicius, Sens. Actuators, B, 2020, 321, 128533 CrossRef CAS
- X. Yu, W. Liang, Q. Huang, W. Wu, J. J. Chruma and C. Yang, Chem. Commun., 2019, 55, 3156–3159 RSC
- W. Zhao, Z. He, J. W. Y. Lam, Q. Peng, H. Ma, Z. Shuai, G. Bai, J. Hao and B. Z. Tang, Chem, 2016, 1, 592–602 CAS
- Z. Yang, Z. Mao, X. Zhang, D. Ou, Y. Mu, Y. Zhang, C. Zhao, S. Liu, Z. Chi, J. Xu, Y.-C. Wu, P.-Y. Lu, A. Lien and M. R. Bryce, Angew. Chem., Int. Ed., 2016, 55, 2181–2185 CrossRef CAS PubMed
- L. Yang, X. Wang, G. Zhang, X. Chen, G. Zhang and J. Jiang, Nanoscale, 2016, 8, 17422–17426 RSC
- X.-F. Wang, H. Xiao, P.-Z. Chen, Q.-Z. Yang, B. Chen, C.-H. Tung, Y.-Z. Chen and L.-Z. Wu, J. Am. Chem. Soc., 2019, 141, 5045–5050 CrossRef CAS PubMed
- Z. Yang, C. Xu, W. Li, Z. Mao, X. Ge, Q. Huang, H. Deng, J. Zhao, F. L. Gu, Y. Zhang and Z. Chi, Angew. Chem., Int. Ed., 2020, 59, 17451–17455 CrossRef CAS PubMed
- J. Wang, X. Gu, H. Ma, Q. Peng, X. Huang, X. Zheng, S. H. P. Sung, G. Shan, J. W. Y. Lam, Z. Shuai and B. Z. Tang, Nat. Commun., 2018, 9, 2963 CrossRef PubMed
- L. Zhan, Z. Chen, S. Gong, Y. Xiang, F. Ni, X. Zeng, G. Xie and C. Yang, Angew. Chem., Int. Ed., 2019, 58, 17651–17655 CrossRef CAS
- E. Hamzehpoor and D. F. Perepichka, Angew. Chem., Int. Ed., 2020, 59, 9977–9981 CrossRef CAS
- W. Ye, H. Ma, H. Shi, H. Wang, A. Lv, L. Bian, M. Zhang, C. Ma, K. Ling, M. Gu, Y. Mao, X. Yao, C. Gao, K. Shen, W. Jia, J. Zhi, S. Cai, Z. Song, J. Li, Y. Zhang, S. Lu, K. Liu, C. Dong, Q. Wang, Y. Zhou, W. Yao, Y. Zhang, H. Zhang, Z. Zhang, X. Hang, Z. An, X. Liu and W. Huang, Nat. Mater., 2021, 20, 1539–1544 CrossRef CAS PubMed
- F. Gu, B. Ding, X. Ma and H. Tian, Ind. Eng. Chem. Res., 2020, 59, 1578–1583 CrossRef CAS
- Z.-Y. Zhang, W.-W. Xu, W.-S. Xu, J. Niu, X.-H. Sun and Y. Liu, Angew. Chem., Int. Ed., 2020, 59, 18748–18754 CrossRef CAS PubMed
- N. Gan, H. Shi, Z. An and W. Huang, Adv. Funct. Mater., 2018, 28, 1802657 CrossRef
- S. Kuila, K. V. Rao, S. Garain, P. K. Samanta, S. Das, S. K. Pati, M. Eswaramoorthy and S. J. George, Angew. Chem., Int. Ed., 2018, 57, 17115–17119 CrossRef CAS PubMed
- W.-J. Fang, J.-J. Zhang, H. Zhao, J. Ni, S.-Q. Liu, Z. Liu, A.-Y. Ni, P.-P. Zhang and H.-H. Wei, Adv. Opt. Mater., 2020, 8, 2000482 CrossRef CAS
- L. Ma, S. Sun, B. Ding, X. Ma and H. Tian, Adv. Funct. Mater., 2021, 31, 2010659 CrossRef CAS
- Y. Zhang, L. Gao, X. Zheng, Z. Wang, C. Yang, H. Tang, L. Qu, Y. Li and Y. Zhao, Nat. Commun., 2021, 12, 2297 CrossRef CAS PubMed
- J. Sun, J. Jia, B. Zhao, J. Yang, M. Singh, Z. An, H. Wang, B. Xu and W. Huang, Chin. Chem. Lett., 2021, 32, 1367–1371 CrossRef CAS
- X.-Q. Liu, K. Zhang, J.-F. Gao, Y.-Z. Chen, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2020, 59, 23456–23460 CrossRef CAS PubMed
- H. Liu, N. Ando, S. Yamaguchi, P. Naumov and H. Zhang, Chin. Chem. Lett., 2021, 32, 1669–1674 CrossRef CAS
- D. Li, F. Lu, J. Wang, W. Hu, X.-M. Cao, X. Ma and H. Tian, J. Am. Chem. Soc., 2018, 140, 1916–1923 CrossRef CAS PubMed
- X.-K. Ma, W. Zhang, Z. Liu, H. Zhang, B. Zhang and Y. Liu, Adv. Mater., 2021, 33, 2007476 CrossRef CAS PubMed
- P. Wei, X. Zhang, J. Liu, G.-G. Shan, H. Zhang, J. Qi, W. Zhao, H. H.-Y. Sung, I. D. Williams, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9293–9298 CrossRef CAS PubMed
- X.-K. Ma and Y. Liu, Acc. Chem. Res., 2021, 54, 3403–3414 CrossRef CAS PubMed
- Y. Zhou, K. Jie, R. Zhao and F. Huang, Adv. Mater., 2020, 32, 1904824 CrossRef CAS PubMed
- Y. Chen, F. Huang, Z.-T. Li and Y. Liu, Sci. China: Chem., 2018, 61, 979–992 CrossRef CAS
- X.-Y. Lou and Y.-W. Yang, Adv. Opt. Mater., 2018, 6, 1800668 CrossRef
- Z.-Y. Zhang, D.-Q. Ye, Q.-Q. Gao, Z.-C. Shi, M. Xie, S.-Z. Zhan, Y.-L. Huang, G.-H. Ning and D. Li, Inorg. Chem. Front., 2021, 8, 2299–2304 RSC
- L. Zuo, G. Qin, Y. Lan, Y. Wei and C. Dong, Sens. Actuators, B, 2019, 289, 100–105 CrossRef CAS
- L. Straub, D. González-Abradelo and C. A. Strassert, Chem. Commun., 2017, 53, 11806–11809 RSC
- T. Li and X. Ma, Dyes Pigm., 2018, 148, 306–312 CrossRef CAS
- X.-K. Ma, Y.-M. Zhang, Q. Yu, H. Zhang, Z. Zhang and Y. Liu, Chem. Commun., 2021, 57, 1214–1217 RSC
- S. Garain, B. C. Garain, M. Eswaramoorthy, S. K. Pati and S. J. George, Angew. Chem., Int. Ed., 2021, 60, 19720–19724 CrossRef CAS PubMed
- C. Li, X. Li and Q. Wang, Chin. Chem. Lett., 2021 DOI:10.1016/j.cclet.2021.08.011
- Z.-Y. Zhang and Y. Liu, Chem. Sci., 2019, 10, 7773–7778 RSC
- Y.-H. Liu, Y.-M. Zhang, H.-J. Yu and Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 3870–3880 CrossRef CAS PubMed
- W.-L. Zhou, Y. Chen, Q. Yu, H. Zhang, Z.-X. Liu, X.-Y. Dai, J.-J. Li and Y. Liu, Nat. Commun., 2020, 11, 4655 CrossRef CAS PubMed
- J. Wang, Z. Huang, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 9928–9933 CrossRef CAS
- H.-J. Yu, Q. Zhou, X. Dai, F.-F. Shen, Y.-M. Zhang, X. Xu and Y. Liu, J. Am. Chem. Soc., 2021, 143, 13887–13894 CrossRef CAS PubMed
- J. Yang, M. Fang and Z. Li, Acc. Mater. Res., 2021, 2, 644–654 CrossRef CAS
- X. Lin, J. Wang, B. Ding, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2021, 60, 3459–3463 CrossRef CAS PubMed
- Y. Li, F. Gu, B. Ding, L. Zou and X. Ma, Sci. China: Chem., 2021, 64, 1297–1301 CrossRef CAS
- Y. Zhou, H.-Y. Zhang, Z.-Y. Zhang and Y. Liu, J. Am. Chem. Soc., 2017, 139, 7168–7171 CrossRef CAS PubMed
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, H. Savoie, K. J. Flanagan, C. Sy, E. Sitte, M. Telitchko, F. Laquai, R. W. Boyle and M. O. Senge, J. Am. Chem. Soc., 2017, 139, 6282–6285 CrossRef CAS PubMed
- Z.-Y. Zhang, Y. Chen and Y. Liu, Angew. Chem., Int. Ed., 2019, 58, 6028–6032 CrossRef CAS PubMed
- F.-F. Shen, Y. Chen, X. Dai, H.-Y. Zhang, B. Zhang, Y. Liu and Y. Liu, Chem. Sci., 2021, 12, 1851–1857 RSC
- D. G. Congrave, B. H. Drummond, V. Gray, A. D. Bond, A. Rao, R. H. Friend and H. Bronstein, Polym. Chem., 2021, 12, 1830–1836 RSC
Footnotes |
| † Dedicated to Professor Daoben Zhu on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, methods for synthesis, relevant characterization details, and supporting photographs. See DOI: 10.1039/d1tc04100b |
| This journal is © The Royal Society of Chemistry 2022 |