
A tunable phosphorescence supramolecular switch by an anthracene photoreaction in aqueous solution†‡
 *a
*a
Abstract
Purely organic room-temperature phosphorescence (RTP) has attracted tremendous attention, but is rarely reported to possess stimulus responsiveness. Herein, we report a supramolecular RTP switch based on a dicationic guest molecule (BPA) and cucurbituril. BPA possesses a photosensitive 9,10-diphenylanthracene core and terminal 4-(4-bromophenyl)pyridium, which can form 1 : 4 supramolecular assembly with cucurbit[7]uril (CB[7]) and an n : n linear supramolecular polymer with cucurbit[8]uril (CB[8]). The assemblies can exhibit tunable fluorescence emission after undergoing a photoreaction of the anthracene unit in BPA. It is interesting that CB[8] can bind two bromophenylpyridinium units to form a green phosphorescent switch. Notably, the RTP of the assemblies can be reversibly switched on/off by UV irradiation or heating through a tunable Förster resonance energy transfer from 4-(4-bromophenyl)pyridium to anthracene. Moreover, the addition of cucurbituril can significantly accelerate the photoreaction rate of anthracene and enable the assembly to respond quickly to UV irradiation, which expands the application potential of this strategy in stimulus responsive RTP materials.
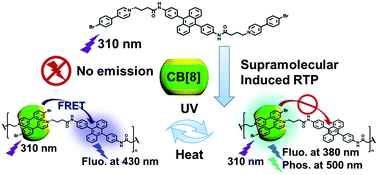
- This article is part of the themed collection: Special issue in honour of Daoben Zhu
 Please wait while we load your content...
Please wait while we load your content...

