
Repurposing pinacol esters of boronic acids for tuning viscoelastic properties of glucose-responsive polymer hydrogels: effects on insulin release kinetics†
Akbar
Ali
a,
Shaista
Nouseen
a,
Saroj
Saroj
b,
Meenakshi
Shegane
c,
Priyankar
Majumder
a,
Aarti
Puri
a,
Tatini
Rakshit
 b,
Debasish
Manna
c and
Suchetan
Pal
*a
b,
Debasish
Manna
c and
Suchetan
Pal
*a
aDepartment of Chemistry, Indian Institute of Technology, Bhilai, Raipur 492015, India. E-mail: suchetanp@iitbhilai.ac.in; suchetan.pal@gmail.com; Web: https://suchetanpal.wixsite.com/bionanolab Tel: +916266274962
bDepartment of Chemistry, Shiv Nadar University, Greater Noida, 201314, UP, India
cDepartment of Chemistry, Indian Institute of Science Education and Research, Bhopal, 462066 MP, India
First published on 6th May 2022
Abstract
In the era of the diabetes pandemic, injectable hydrogels (HGs) capable of releasing the desired amount of insulin under hyperglycemic conditions will significantly advance smart insulin development. Several smart boronic acid-based polymer HGs release insulin under high-glucose conditions. However, the correlation of insulin release characteristics with rheological properties is not well understood yet. Herein, we report a generalized and facile fabrication strategy of a new class of glucose-responsive hydrogels by crosslinking a biocompatible polymer, poly(vinyl alcohol) with pinacol esters of bisboronic acids via transesterification reactions. We show the versatility of the method by fabricating four hydrogels with diverse rheological properties. The HGs embody more than 70% water amenable for hosting insulin in the matrix. HG with high storage modulus, derived from 1,4-benzenediboronic acid bis(pinacol) ester releases ∼3 fold less insulin compared to softer HGs derived from acetylene-1,2-diyl bis(boronic acid pinacol ester) and bis[(pinacolato)boryl]methane under hyperglycemic conditions. We find that HG derived from the bis[(pinacolato)boryl]methane crosslinker exhibits superior insulin release properties due to the softness of the hydrogel matrix. We further show that the newly formulated gel is injectable without any structural change in the released insulin molecules and does not cause cytotoxicity. We believe that glucose-responsive hydrogels with tunable viscoelastic properties will pave the way for developing a variety of hydrogels with programmable insulin release properties.
 Suchetan Pal | Dr Suchetan Pal obtained his BSc and MSc in Chemistry from Jadavpur University and Indian Institute of Technology, Kanpur respectively. Then, he joined Arizona State University, for his doctoral studies. After completing the thesis work on “DNA directed self-assembly of plasmonic nanoparticles,” Dr Pal moved to a joint post-doctoral position at Columbia University and Brookhaven National Laboratory. He completed another postdoctoral stint at the Memorial Sloan Kettering Cancer Center, before joining Indian Institute of Technology, Bhilai as an assistant professor. His primary research focus includes biomolecular nanotechnology for drug delivery, biosensing and bioimaging. |
1. Introduction
Diabetes is an alarming health issue, with 537 million diagnosed adults in 2021.1 Subcutaneous insulin infusion (CSII) or multiple daily injections (MDI) is the primary line of treatment for type 1 and advanced type 2 diabetes.2 The major challenges of these methods are to administer the proper dose of insulin to patients before a meal, so that blood glucose concentration remains close to the desired level (4–7 mM); at the same time, fatal drops in blood glucose level or hypoglycemic events can be avoided.3,4 The inconvenience of frequent subcutaneous injection and the risk of insulin-induced hyperglycemia are significant hindrances in insulin-dependent diabetes treatment for many patients.5 Therefore, active research is focused on developing closed-loop insulin delivery systems that mimic biological glucose-dependent insulin secretion.6–8 These glucose-responsive insulin delivery systems are designed to release insulin according to the blood glucose levels (BGL) of an individual without any adverse consequences. In this direction, a plethora of glucose-responsive hydrogels (HGs) that encapsulate insulin in the matrix and release under hyperglycemic conditions have emerged.9 Glucose oxidase (GOx), glucose-binding lectin proteins, and phenylboronic acid (PBA) are leveraged as glucose-sensing moieties in these HGs.10–21 Nonetheless, enzyme/protein-based glucose-responsive HGs are less likely to be clinically translated due to their inherent instability, immunogenicity, and cytotoxicity.22–24In this regard, PBA-containing HGs have shown enormous promise for glucose-responsive insulin release.25,26 The impressive glucose responsiveness results from reversible boronate ester formation of PBA with 1,2 and 1,3-diol moieties of glucose, the most abundant simple carbohydrates present in the human body.27 PBA's chemical stability makes it suitable for long-term use for glucose-induced insulin release under ambient conditions without any toxicity. Flexibility in incorporating PBA in the HGs also improves durability and reduces the cost. Due to these advantages, several design ideas for PBA incorporation in the HGs have been achieved. In the first design, polymer HGs are synthesized by polymerizing PBA-containing monomers via radical polymerization reactions.28–32 Under hyperglycemic conditions, there is a change in the hydrophilicity in these HGs that leads to swelling and subsequent release of insulin. However, these macromolecular gels are not biodegradable and therefore are used in the form of needles or catheters.33In situ polymer hydrogel microneedle patches loaded with insulin is used to control the BGL in mice and minipigs.34,35 In another work, charge switchable PBA containing polymers showed promising results in mice and pigs.36 Another design strategy uses a PBA-grafted polymer and poly 1,3-diol/carbohydrate-containing polymers to form HGs via glucose-responsive boronate ester bonds.37–41 These HGs are disintegrated under high glucose environments due to the competitive binding of glucose with PBA moieties. However, extensive chemical modifications are required to fabricate such HGs that increase processing time and cost. Recently, glucose-responsive HGs with poly(vinyl alcohol) (PVA) crosslinked with bis-boronic acid crosslinkers have been developed.42,43 This strategy offers a relatively straightforward crosslinking of a biocompatible polymer, PVA, with crosslinkers containing two boronic acid moieties. Nonetheless, the insulin release properties of all these hydrogels are not predictable. Insulin release kinetics depends on the breakage of boronate ester bonds which varies in number across glucose-responsive HGs. However, HGs with preprogrammed release kinetics would be an attractive attribute for closed-loop insulin-dependent diabetes treatment but not yet achieved.
In this work, we aim to expand the bis-boronic acid crosslinked glucose-responsive HG library by developing a general fabrication strategy. A plethora of pinacol ester of bisboronic acid are readily available for Pd-mediated cross-coupling reactions.44 We seek to repurpose these molecules for the fabrication of glucose-responsive HG. We postulate that pinacolato ester of bisboronic acids would crosslink PVA via a thermodynamically favorable transesterification reaction at room temperature and encapsulate insulin during the gelation (Fig. 1). These crosslinks can be of two types: interchain and intrachain. Interchain crosslinks are more likely to create elastically active bonds compared to intrachain bonds.45 We postulated that by changing the structure of the crosslinker, the proportion of interchain and intrachain crosslinks can be altered.45 Further, different crosslinkers may possess different interchain bond strengths due to their structural differences.46,47 Therefore, HGs with different stiffness would be achievable by merely changing the crosslinker. Due to the difference in the number of interchain crosslink, these gels would show different responses under hyperglycemic environment and release entrapped insulin accordingly.
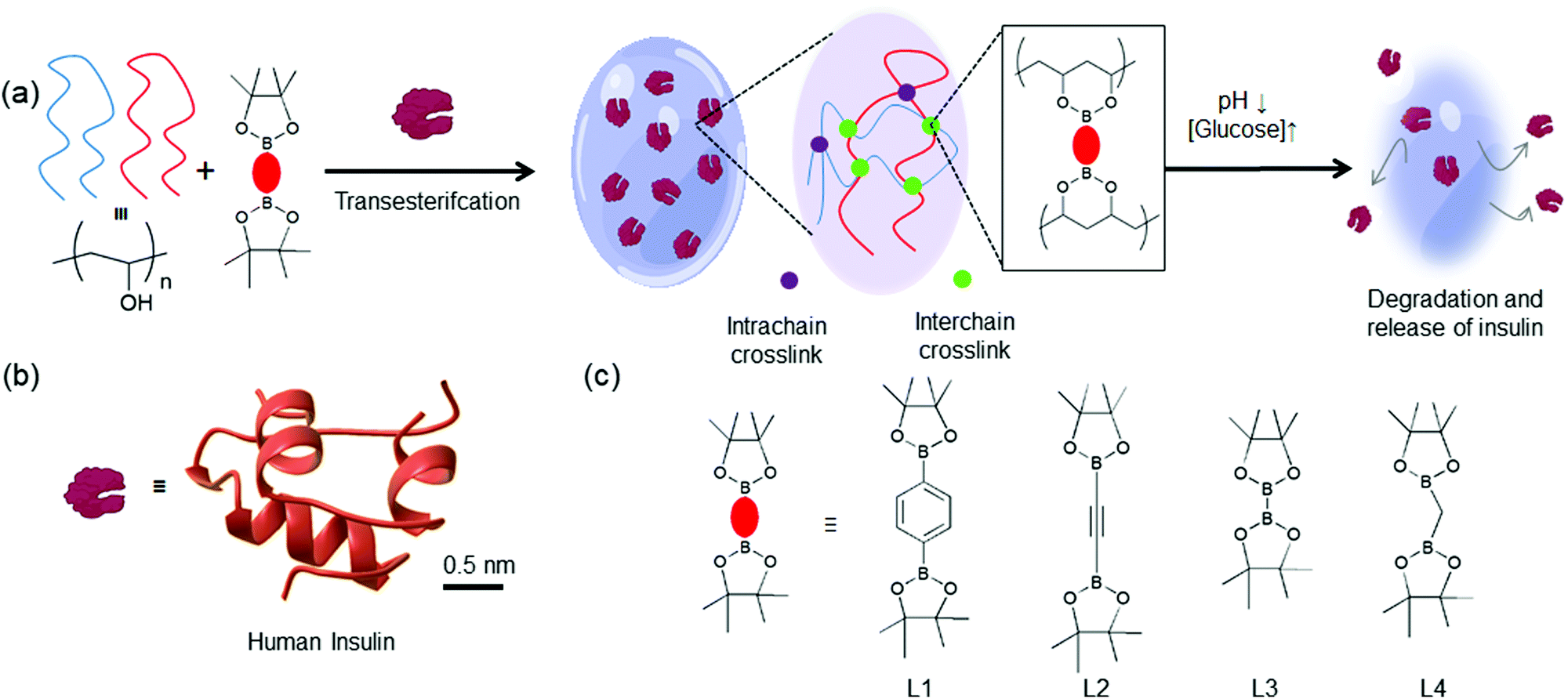 | ||
| Fig. 1 (a) Schematic representation of the glucose-responsive hydrogels developed in this study. PVA chains are crosslinked with pinacol esters of bisboronic acid via a thermodynamically favorable transesterification reaction and entrap molecular guests. There are two different crosslinks: interchain (green) and intrachain (purple). These HGs are expected to undergo structural degradation under low pH and high glucose environments releasing (b) The three-dimensional structure of insulin (PDB: 3I40). (c) Chemical structures of four pinacol ester of bisboronic acid used in this study: L1: 1,4-benzenediboronic acid bis(pinacol) ester, L2: 1,2-diyl bis(boronic acid pinacol ester), L3: bis(pinacolato) diboron, L4: bis(pinacolato)borylmethane. | ||
2. Materials and methods
2.1 Materials
Poly(vinyl alcohol) (PVA, Mw 89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–98000), phosphate buffered saline(PBS), potassium carbonate, glucose, fructose, mannose, galactose, glucosamine, ATTO 495 NHS ester, and acetylene-1,2-diyl bis(boronic acid pinacol ester) were purchased from Sigma Aldrich. Insulin was purchased from HiMedia. 1,4-Benzenediboronic acid bis(pinacol) ester was purchased from TCI Chemicals. Bis(pinacolato) diboron was purchased from Avra Synthesis. Bis(pinacolato)borylmethane was purchased from BLD Pharma. G25 columns were purchased from GE Healthcare. All the experiments were performed with nuclease-free water.
000–98000), phosphate buffered saline(PBS), potassium carbonate, glucose, fructose, mannose, galactose, glucosamine, ATTO 495 NHS ester, and acetylene-1,2-diyl bis(boronic acid pinacol ester) were purchased from Sigma Aldrich. Insulin was purchased from HiMedia. 1,4-Benzenediboronic acid bis(pinacol) ester was purchased from TCI Chemicals. Bis(pinacolato) diboron was purchased from Avra Synthesis. Bis(pinacolato)borylmethane was purchased from BLD Pharma. G25 columns were purchased from GE Healthcare. All the experiments were performed with nuclease-free water.
2.2 Preparation of HGs
PVA was dissolved in 10 mM PBS solution at 40 mg ml−1 concentration under mild heating. Crosslinkers (L1–L4) with 100 mM concentrations were made in an ethanol-100 mM K2CO3(1:1 volume ratio) buffer. Next, equal volumes of PVA and crosslinker solutions were mixed thoroughly and allowed to form gels at room temperature. Depending on the crosslinker, gels were formed in between 1–4 hours. These HGs are exposed to 10 mM PBS solutions with different pH and glucose concentrations for rheological and insulin release studies. Different pH solutions were prepared by adjusting the pH of 10 mM PBS (pH 7.5). pH was measured using a pH meter. Glucose solutions were prepared in 10 mM PBS. The pH values did not change after glucose solution preparation.
2.3 Turbidimetric measurements of gel formation kinetics
500 μl of PVA (40 mg ml−1) and 500 μl of crosslinkers (100 mM) were added in the 1 ml quartz cuvette and the absorbance spectra were recorded in 5 minutes interval in a UV-Vis spectrophotometer (Shimadzu, UV-2700). The gel formation led to an increase in the absorbance throughout the 200–800 nm optical window. The absorbance at 800 nm was plotted against the time to determine the gel formation kinetics.2.4 Infrared (IR) spectroscopy
IR spectra of hydrated HGs were recorded using ATR mode in FT-IR spectrometer (Spectrum Two, PerkinElmer). The spectra were recorded in the frequency range of 400–4000 cm−1.2.5 Thermogravimetric analysis
To determine the water content of the HGs, systematic thermogravimetric analysis (TGA) was performed in NETZSCH TG 209F3 Tarsus. TGA measurements were carried out under a nitrogen gas atmosphere at a 20 ml min−1 flow rate. Freshly prepared HGs, weighing between 5 and 50 mg, was heated from 25 °C to 600 °C at a constant rate of 10 °C minute−1. The weight loss in percent was plotted against the temperature.2.6 Scanning electron microscopy
The morphological properties of HGs were examined using a field emission scanning electron microscope (FESEM) (Gemini 500, Zeiss, Germany). The examination was carried out at magnifications of 10000× and 5000× at an accelerated voltage of 3 kV. The freeze-dried hydrogels were prepared and carefully mounted on circular aluminum stubs with double-sided adhesive tape and coated with gold before the imaging.
2.7 Rheological measurements
Rheological properties of the HGs were measured using the Anton Paar MCR102 Modular Compact Rheometer. HGs were placed in between the 25 mm diameter parallel plates and a gap distance of 1 mm. The pressure was 30 PSI for rheometer operation. The test method employed a frequency sweep test using angular frequency 0.1–100 rad s−1 at the constant strain of 0.5% at 25 °C.2.8 Insulin release assay
For the insulin release assay, human recombinant insulin was labeled with ATTO 495 NHS ester and purified with a G25 spin filtration column (GE healthcare). During the preparation of the insulin-loaded HGs, desired amount of insulin (10 μl, 10 mg ml−1 in 1 ml hydrogel) was incorporated. In vitro pH and glucose-responsive insulin release studies were carried out by incubating 1 ml insulin-loaded HGs with 10 ml, 10 mM PBS without or with glucose (100 mM) maintained with different pH environments (pH 4, 7.5, and 10) in an orbital shaking incubator at 37 °C. The samples were collected every hour for up to 4 hours. The aliquots were measured for fluorescence emission using Microplate Reader (Molecular devices, Spectramax i3x, at the excitation of 450 nm and emission of 510 nm). Then the percentage of released insulin calculated from the control insulin solution plotted with the standard calibration curves. All the measurements were done in triplicates.2.9 Circular dichroism spectroscopy
Circular dichroism (CD) spectra of the native insulin and released insulin were recorded using a JASCO J-1500 spectrophotometer. Insulin from the HGs were released using 100 mM glucose solution at room temperature. The cuvettes were kept under a constant temperature of 25 °C using a JASCO PTC-423S Peltier-type thermocouple. All measurements were performed in quartz cuvettes with 2 mm path length.2.10 MALDI-MS analysis
For MALDI-MS analysis, intact samples were reconstituted in 0.1% of TFA:acetonitrile 70:30 (v/v) and 1 μl of sample were spotted with 1 μl of matrix (sinapinic acid) on the MTP 384 ground steel target plate (Make: Bruker). Once samples were dried, the target plate was loaded into a MALDI-TOF MS for the analysis. All samples were analyzed using an Autoflex max MALDI TOF/TOF (Bruker). Measurements were carried out under positive linear mode with 20 kV accelerating voltage. Instrumental parameters were set as mass range m/z 5000–20000 and 70% laser intensity. Following the manufacturer's instructions, the instrument was externally calibrated using a standard protein mix I kit (Make: Bruker).
2.11 Cell viability assay
HeLa, HEK293T, HepG2 cell lines were procured from National Centre for Cell Science (NCCS), Pune, India. All the cell lines were maintained in Dulbecco's modified Eagle's medium (HiMedia), containing 10% Fetal Bovine serum (HiMedia) and 1% Penicillin-Streptomycin (HiMedia). Both the cell lines were incubated at 37 °C with 5% CO2 in a humid environment.Cell viability assay was performed to determine the toxicity of hydrogels against HeLa, HEK293T, HepG2 cells. 10000 cells per well in complete media were seeded in a 96-well plate and incubated overnight for growth and attachment to the surface. 20 mg ml−1 solution of all six samples of hydrogels were prepared by dissolving 200 mg of HG in 10 ml of DMEM media containing 0.5 M glucose. Samples were serially diluted to attain a final concentration of 2 mg ml−1 and 0.2 mg ml−1. Cells were treated with HGs for 24 h along with all experimental controls. After 24 h of incubation, the used media was removed and replaced with 100 μl fresh medium. MTT [3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (SRL) was dissolved at a concentration of 5 mg ml−1 in phosphate buffer saline and 10 μl of MTT solution was added to each well. After 2.5 h incubation at 37 °C, 5% CO2, the formed formazan crystals were dissolved in 100 μl of dimethyl sulfoxide for 20 minutes at shaker. The absorbance values were recorded at 540 nm wavelength in the BioTek Cytation 1 plate reader and the percentage of cell viability was calculated with respect to the untreated control.
3. Results and discussion
3.1 Preparation and characterizations of glucose-responsive HGs
Glucose-responsive HGs were prepared by adding pinacol esters of bisboronic acids with PVA (MW 89000–98000 Da). A collection of such molecules is readily available due to their use in the Pd-mediated cross-coupling reactions. We selected four bis-pinacolato esters of bis-boronic acids: 1,4-benzenediboronic acid bis(pinacol) ester (L1), acetylene-1,2-diyl bis(boronic acid pinacol ester) (L2), bis(Pinacolato)diboron (L3), bis[(pinacolato)boryl]methane (L4), as shown in Fig. 1(c). We carried out extensive gel formation studies to understand the phase behavior by mixing different concentrations of crosslinkers (5, 10, 20, 50 mM) with PVA (5, 10, 15, 20 mg ml−1) at room temperature (Fig. 2 and Fig. S1 ESI†). While L1 produced bulk gels at all concentrations, L2 and L3 did not produce HGs in 5 mg ml−1 PVA and 5, 10, 20 mg ml−1 crosslinker concentrations. For L4, the gel did not form even up to 50 mM crosslinker concentrations. We hypothesized that various crosslinkers inculcated different interchain and intrachain crosslinks. L1, L2, and L3 provided better interchain crosslinks than intrachain crosslinks than L4. Further, we used turbidimetric measurements to understand the kinetics of the gel formation. We measured the absorbance at 800 nm up to 3 hours after mixing PVA (20 mg ml−1) and the crosslinkers (50 mM) (Fig. S2, ESI†). The absorbance immediately increased and attained a plateau for L1, L2, and L3 within one hour, indicating rapid gel formation. For L3, the rise is slower and attained a plateau in 3 hours. These results suggested faster interchain crosslink formation with L1, L2, and L3 leading to a quicker gelation. We would abbreviate the HGs derived from 20 mg ml−1 PVA with 50 mM Ln as HGn (n = 1–4).
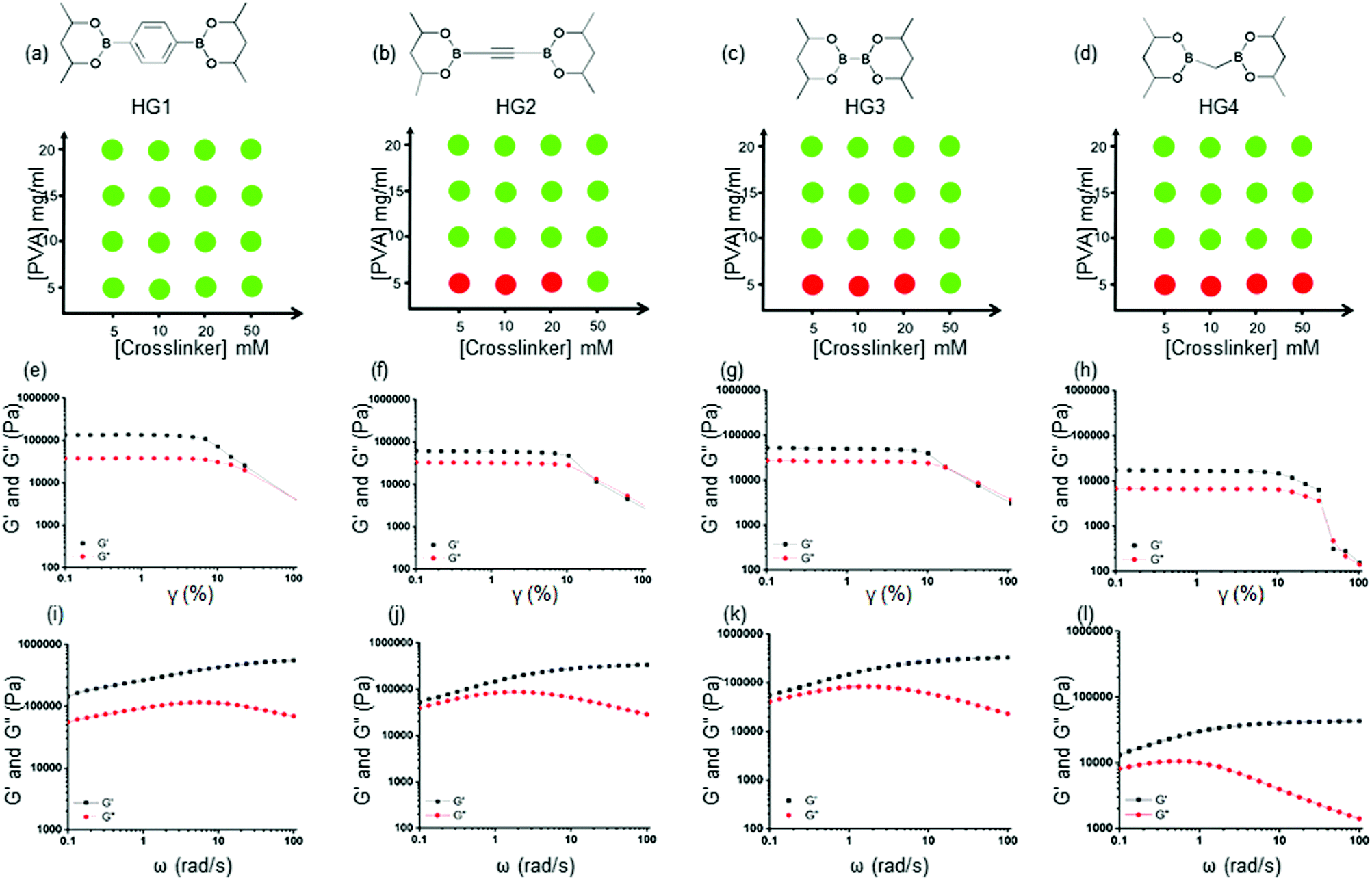 | ||
| Fig. 2 (a)–(d) Phase diagrams of HG1-4. Red and green circles represent solution and gel phase, respectively. (e)–(h) Strain sweep of storage modulus (G′, black) and loss modulus (G′′, red) of HG1-4. (i)–(l) Frequency sweep of storage modulus (G′, black) and loss modulus (G′′, red) of HG1-4. All the gels show elastic solid-like behavior in the frequency range of 0.1–100 rad s−1. HG1 exhibited a significantly higher storage modulus (G′) of 142 kPa (at 0.1 rad s−1) compared to HG2 (52 kPa at 0.1 rad s−1), HG3 (54.5 kPa at 0.1 rad s−1), and HG4(2.8 kPa at 0.1 rad s−1) indicating diverse rheological properties. | ||
We used Fourier transform infrared (FTIR) spectroscopy of the fabricated HGs to obtain information about the interchain bonds. For all the gels, we obtained a peak around 1370 cm−1 corresponding to B–O stretching frequency, indicating the presence of boronate ester bonds (Fig. S3, ESI†). We used scanning electron microscopy (SEM) to assess the microscopic morphologies of the HGs. While HG2-4 micropores were noticed throughout the structures, HG1 did not show any pores visible under the high-resolution of FE-SEM (Fig. S4, ESI†).
3.2 Water content measurement
Next, we performed thermogravimetric analysis (TGA) in a 25–600 °C temperature window to determine the water content in the HG1-4. We observed a steep decline in the HG weights in the temperature range of 70–120 °C due to water evaporation from the HGs (Fig. S5, ESI†). A decrease of weights around 240–360 °C for all the gels was due to PVA decomposition (Fig. S6, ESI†). Careful inspection of TGA data indicated that ∼74.4%, 70.1%, 83.1%, 72.3% water were present in HG1-4, respectively.3.3 Rheological properties
We performed rheological measurements of the HGs to determine their viscoelastic properties. For all the HGs, strain sweep measurements were performed under 0.1–100% strain (γ) as shown in Fig. 2(e)–(h). For all the gels higher storage modulus (G′) than the loss modulus (G′′) and a crossover between moduli at higher strain were observed suggesting structural breakdown. We further carried out angular frequency sweep tests in the angular frequency (ω) range 0.1–100 rad s−1 were carried out under a constant strain of 0.5%. All four HGs exhibited a higher storage modulus (G′) than the loss modulus (G′′), indicating elastic solid-like behaviors for all the gels (Fig. 2(i)–(l)). The storage moduli (G′) increased, and loss moduli (G′′) decreased with the increase in angular frequency, indicating dynamic bond formation and breaking of boronate ester bonds in all the HGs. This observation was consistent with the previous finding for boronate ester crosslinked PVA HGs.48 HG1 possessed a significantly higher storage modulus (G′) of 142 kPa (at 0.1 rad s−1) compared to that of HG2 (52 kPa at 0.1 rad s−1) and HG3 (54.5 kPa at 0.1 rad s−1). We found that the storage modulus (G′) of HG4 was the lowest (2.8 kPa at 0.1 rad s−1) amongst all the gels. These results indicated that the nature of the crosslinker played a pivotal role in determining the rheological properties. We postulated that because of the benzene-based L1 crosslinker formed a more elastically active interchain crosslink compared to L2 and L3. In contrast, borylmethane-based L4 crosslinker produced the softest gel, possibly due to more intrachain crosslinks. The softer nature of the HG4 may be also attributed to the weaker interchain bond strength of the linker. To prove that these gels were formed due to the dynamic boronate ester bonds, we incubated HG4 with 100 mM solutions of tetrahydroxydiboron, benzene-1,4-diboronic acid, and PBA. After 1 hour of incubation, we measured the angular frequency sweep of the HGs (Fig. S7, ESI†). The storage modulus (G′) increased to 16.1 kPa at 0.1 rad s−1 and 22.9 kPa at 0.1 rad s−1 from 2.8 kPa at 0.1 rad s−1 upon the incubation tetrahydroxydiboron and benzene-1,4-diboronic acid, respectively. On the contrary, PBA exposure decreased the storage modulus (G′) to 0.85 kPa. These results indicated that exposure to bis-boronic acid linkers increased elastically active crosslinking, further increasing storage modulus (G′). On the contrary, PBA decreased the interchain crosslinks and rendered the HG soft. The unparalleled flexibility of changing the crosslinkers in situ allowed the dynamic tuning of the rheological properties of the HGs.3.4 Stimuli-responsive degradation of HGs
Next, we qualitatively investigated the response of the HGs under different pH environments (pH 4, 6, 7.5, 9) at 37 °C (Fig. S8, ESI†). HG1 did not show any visual changes up to 24 hours of incubation (Fig. S8, ESI†). HG2-4 were more transparent when compared to HG1. Hence, the variations were difficult to assess but by inverting the tubes, we observed more degradation of HGs under acidic pH of 4 and 6 compared to pH 7.4 and 9. Then, we evaluated the carbohydrate responsiveness of HG1-4 by exposing the HGs in increasing concentrations (0 mM, 5 mM, 10 mM, 100 mM, and 500 mM) of four simple carbohydrates (D-glucose, D-fructose, D-galactose, D-mannose) and glucosamine. We incubated HGs up to 24 hours at 37 °C and observed visual changes. While HG1 did not show any change, HG 2–4 degraded entirely at higher carbohydrate concentrations (100 mM, 500 mM), as shown in Fig. S9, ESI.† We further measured the dynamic rheological properties of HG4 incubated with 100 mM glucose solution under pH 4, 7.5 and 10. We observed a decrease in the storage moduli (G′), and loss moduli (G′′) indicating structural disintegration as shown in Fig. S10, ESI.†3.5 Glucose responsive insulin release under different pH conditions
Further, we examined the glucose-responsive release of insulin under different pH environments (pH 4, 7.5, and 10) under physiological conditions. We selected three HGs, HG1, HG2, and HG4, with drastically different rheological properties to assess the insulin release kinetics from the HGs. We labelled insulin with a fluorophore, ATTO495, and incorporated the insulin in the HGs to quantify its release. We did not observe any change in the viscoelastic properties of the HGs after insulin incorporation. We incubated the insulin-loaded HGs with 100 mM and 0 mM glucose concentrations at pH 4, 7.5, and 10 under physiological temperature and studied the insulin release up to 4 hours from the incubation (Fig. 3). We observed a background insulin release from all the gels at 0 mM glucose conditions. The release at pH 4 was significantly higher than pH 7.5 and 10. This was due to the hydrolysis of boronate ester bonds at a low pH environment that induced structural disintegration and higher insulin release than pHs 7.5 and 10. The release of insulin at pH 10 was slightly higher than that of pH 7.4, possibly due to a negatively charged interior at higher pH that caused a faster release of negatively charged insulin. This effect was more prominent for HG2 and HG4 than that of HG1. The release of insulin under 0 mM glucose conditions was increased with the decreased hardness of the HGs. In other words, HG1 released the least amount of insulin, followed by HG2 and HG4 under any pH conditions. These results suggested a higher number of interchain crosslinks, leading to higher storage modulus (G′) diminished insulin release from the HGs.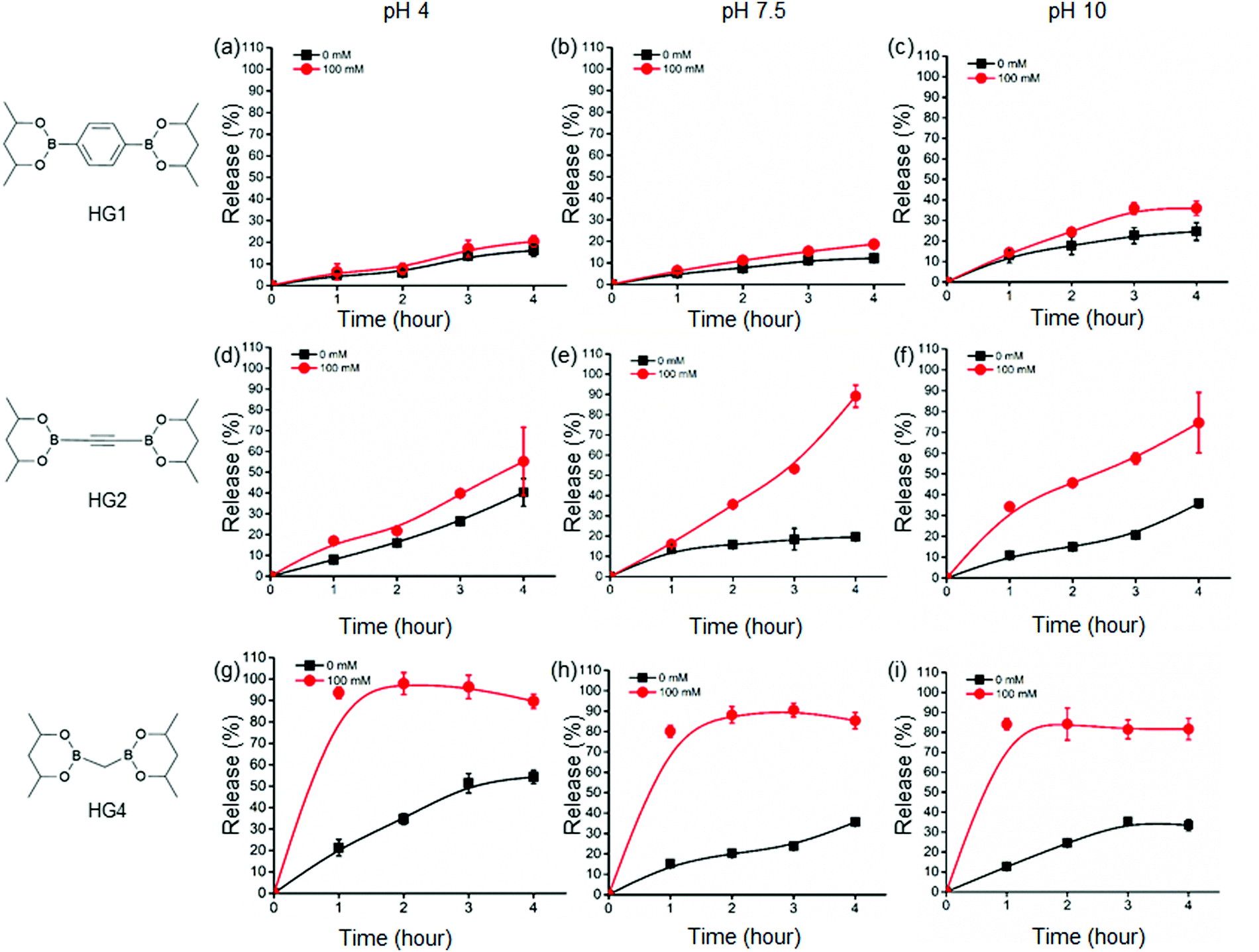 | ||
| Fig. 3 In vitro insulin release profiles of HG1, HG2 and HG3 under 0 mM glucose (black) and 100 mM glucose (red) in different pH conditions (4, 7.5 and 10). HG1 showed slow release of insulin compared to HG2. HG4 exhibited the fastest insulin release kinetics. | ||
Additionally, we observed an increase in insulin release under 100 mM glucose for all the HGs. HG1 exhibited the least glucose response showing 20%, 19%, and 36% release at 4 hour time point under 100 mM glucose in pH 4, 7.5, and 10 environments, respectively (Fig. 3(a)–(c)). On the contrary, HG2 and HG4 showed significantly faster insulin release profiles. At 4 hour time point, 55%, 89%, and 75% of insulin was released from HG2 under 100 mM glucose at pH 4, 7.5, and 10, respectively (Fig. 3(d)–(f)). Interestingly, HG4 attained more than 90% insulin release under 100 mM glucose at pH 4 within the first hour (Fig. 3(g)). More than 75% insulin was released within 3 hours under 100 mM glucose at pH 7.5 and 10 (Fig. 3(h) and (i)).
These observations were rationalized based on the binding of D-glucose with the boronate ester-crosslinks. It has been demonstrated that for boronate ester-based hydrogels, the release of entrapped protein cargo depends on the temperature, network design and surface erosion.49 It was well established that drug molecules are released from HGs due to a concentration gradient following a diffusion-controlled release mechanism. Further, dissociation of interchain crosslinks led to the disintegration of the HGs via a dissociation-controlled release (Fig. 4(a) and (b)).50 In this work, degradation-controlled release was dominant at a lower pH and higher glucose concentrations. All the HGs showed an increased release at pH 4 (0 mM glucose) compared to pH 7.5 (0 mM glucose). HG1 having more interchain crosslinks than HG2 and HG4, as evidenced by the rheological measurements, showed slightly higher dissociation-controlled insulin release at 100 mM glucose conditions than 0 mM glucose for all pH environments. HG2 being softer compared to HG1, exhibited better glucose-responsive insulin release. In contrast, HG4, which possessed the lowest storage modulus (G′), exhibited the most facile insulin release under any pH conditions.
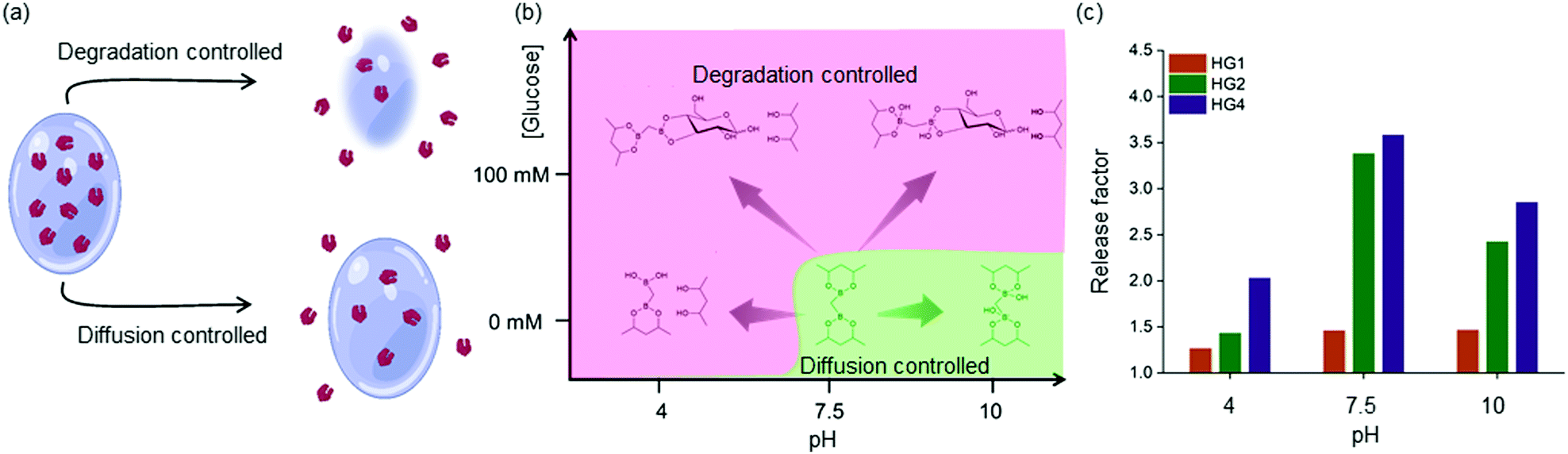 | ||
| Fig. 4 (a) and (b) Two mechanisms for insulin release from HG4: diffusion controlled and dissociation controlled. Dissociation controlled release is predominant under acidic pH and high glucose concentrations, (c) release factors (insulin released under 100 mM glucose over insulin released under without glucose) different pH for HG1,2 and 4. HG4 show the largest release factor under all the pH conditions than HG1 and HG2. | ||
The efficacy of glucose-specific release from an HG was defined by the release factor, a ratio of released insulin under glucose-rich condition (100 mM), and non-glucose condition (0 mM) over time (Fig. 4(c)). The release factor was the highest for HG4 (2.0 at pH 4, 3.6 at pH 7.5, 2.8 at pH 10) followed by HG2 (1.4 at pH 4, 3.4 at pH 7.5, 2.4 at pH 10) and HG1 (1.2 at pH 4, 1.4 at pH 7.5, 1.5 at pH 10). The order of glucose responsiveness was as follows: HG4 > HG2 > HG1. The release factors at pH 7.5 were larger than pH 4 and 10 because of the increased release under no glucose conditions. These results suggested that control of stiffness of the HGs allowed tuning the release kinetics of encapsulated insulin. Softer the gel, the faster was the insulin release under hyperglycemic conditions.
3.6 Insulin stability, injectability and biocompatibility of HG4
Next, we investigated the insulin stability in the HG4 using far-UV CD spectroscopy and MALDI mass spectrometry. We recorded the far-UV CD spectra of native insulin and insulin released from HG4 after 2,4- and 24 hours incubation at 37 °C (Fig. 5(a)). The minima at 209 and 222 nm and maxima at 195 nm indicated the predominant α-helix structure of insulin. The ratio of 209 and 222 nm peaks indicated the changes in the conformation. The ratio was ∼1.3 for native insulin and released insulin after 2 and 4 hours. However, the ratio was found to be 1.5, and the 195 nm maximum decreased in intensity for the insulin released after 24 hours due to slight structural changes. Nonetheless, when we carried out MALDI mass spectrometry of the released insulin, we did not find any molecular weight change indicating no insulin aggregation (Fig. S10, ESI†). These results collectively indicate that insulin hosted in the hydrogels remained structurally unchanged in the HG environment. Further, we studied the shear thinning properties of the HG4 with a step shear strain recovery test (alternating periods 0.5% and 50% strain, Fig. 5(b)). We observed that under high strain, the storage modulus (G′) decreased than the loss modulus (G′′) due to the reversible breaking of boronate ester bonds. Then, we tested the injectability of the HG4 through a G25 needle (Fig. 5(c)). Due to the shear-thinning properties, HG4 loaded with insulin could be easily injected. Finally, we performed extensive cytotoxicity studies of HG4 using human epithelial kidney cells (HeK293, healthy), HeLa (cervical cancer), and HepG2 (liver cancer), as shown in Fig. 5(d). We did not observe any significant cytotoxicity of the hydrogels in either of the cell lines indicating that the material could be suitable for smart insulin delivery applications in vivo.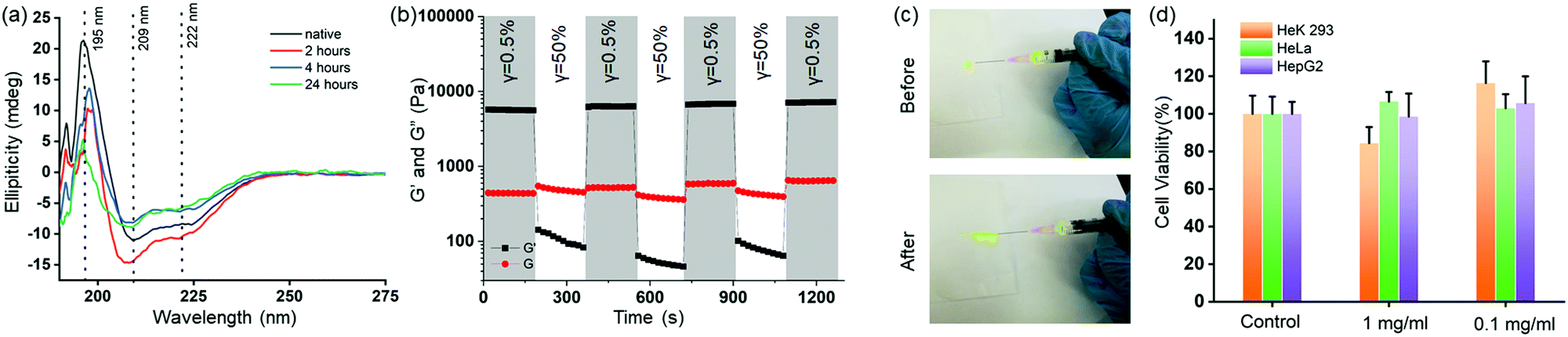 | ||
| Fig. 5 (a) Far-UV CD spectra of native insulin (black) and released insulin from HG4, indicating a minor change in the secondary structure of the released insulin after 24 hours. (b) Strain sweep with alternate low strain (γ = 0.5%) and high strain (γ = 50%) of storage modulus (G′, black) and loss modulus (G′′, red) of insulin loaded HG4 shows shear-thinning properties. (c) Photograph of the insulin-loaded HG4 is being injected from a syringe. (d) Cell viability assay with HGK293, HeLa, and HepG2 cells indicate nontoxic nature of the gels. The gel was added in 1 and 0.1 mg ml−1 concentrations in the growth media. For control, no gel was added. | ||
4. Conclusions
In this work, we developed a facile fabrication strategy of glucose-responsive HGs by crosslinking a biocompatible polymer PVA with bis-boronic acid via boronate ester bond formation. Commercially available pinacol esters, well-known reagents for Pd-catalyzed coupling reactions, were used as precursors for the crosslinkers. All hydrogels contained more than 70% water and demonstrated a 50-fold difference in storage moduli between the softest and the hardest HGs by changing the crosslinker due to the change in the number of elastically active crosslinks. Under physiological temperature, all hydrogels showed glucose-dependent insulin release properties in acidic, neutral, and alkaline pH environments. Interestingly, HG4 derived from bis[(pinacolato)boryl]methane showed significantly faster insulin release kinetics than the other counterparts. The released insulin was structurally similar to the native insulin with no significant denaturation. We further established good injectability and non-cytotoxicity of these newly developed HGs. We have significantly enlarged the library of boronic acid crosslinked glucose-responsive HGs with a new design principle for programmable insulin release properties. We are moving forward for detailed in vivo anti-diabetic properties, and safety studies with these HGs developed in this work, being optimistic for the clinical trials in the near future.Author contributions
Methodology, synthesis, in vitro studies, validation, formal analysis, data interpretation, investigation, writing – original draft, visualization: A. A., S. N. Methodology, in vitro studies, validation, formal analysis, data interpretation: S. S., M. S., P. M., A. P. Conceptualization, data interpretation, writing and editing: D. M., T. R. Conceptualization, methodology, writing and editing, supervision, project administration, funding acquisition: S. P.Conflicts of interest
S. P. is founder and director of CGel07 Biotech private limited that aims to commercialize smart insulin hydrogels.Acknowledgements
S. P. thanks SERB (SRG/2019/000953), DBT (BT/12/IYBA/2019/14) and IIT Bhilai for generous funding. T. R. thanks SERB CRG (CRG/2019/007013) for funding. D. M. thanks SERB CRG (CRG/2019/001667) for funding. S. P. Is thankful to IIT Bhilai for providing research facilities and fellowship to Akbar Ali. We thank Dr Sanjib Banerjee for Rheology measurements.Notes and references
- A. Ali, S. Nouseen, S. Saroj, M. Shegane, P. Majumder, A. Puri, T. Rakshit, D. Manna and S. Pal, I. D. Federation, Brussels, Belgium, 2015 Search PubMed.
- K. Jeitler, K. Horvath, A. Berghold, T. W. Gratzer, K. Neeser, T. R. Pieber and A. Siebenhofer, Diabetologia, 2008, 51, 941–951 CrossRef CAS PubMed.
- Y. Ohkubo, H. Kishikawa, E. Araki, T. Miyata, S. Isami, S. Motoyoshi, Y. Kojima, N. Furuyoshi and M. Shichiri, Diabetes Res. Clin. Pract., 1995, 28, 103–117 CrossRef CAS PubMed.
- J. Yang and Z. Cao, J. Controlled Release, 2017, 263, 231–239 CrossRef CAS.
- P. E. Cryer, J. Clin. Invest., 2007, 117, 868–870 CrossRef CAS PubMed.
- J. Wang, Z. Wang, J. Yu, A. R. Kahkoska, J. B. Buse and Z. Gu, Adv. Mater., 2020, 32, 1902004 CrossRef CAS PubMed.
- K. M. Bratlie, R. L. York, M. A. Invernale, R. Langer and D. G. Anderson, Adv. Healthcare Mater., 2012, 1, 267–284 CrossRef CAS.
- A. N. Zaykov, J. P. Mayer and R. D. DiMarchi, Nat. Rev. Drug Discovery, 2016, 15, 425–439 CrossRef CAS PubMed.
- S. Fuchs, A. U. Ernst, L.-H. Wang, K. Shariati, X. Wang, Q. Liu and M. Ma, Chem. Rev., 2021, 121(18), 11458–11526 CrossRef CAS PubMed.
- C. M. Hassan, F. J. Doyle and N. A. Peppas, Macromolecules, 1997, 30, 6166–6173 CrossRef CAS.
- J. Yu, Y. Zhang, Y. Ye, R. DiSanto, W. Sun, D. Ranson, F. S. Ligler, J. B. Buse and Z. Gu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8260–8265 CrossRef CAS.
- T. Uchiyama, Y. Kiritoshi, J. Watanabe and K. Ishihara, Biomaterials, 2003, 24, 5183–5190 CrossRef CAS.
- K. Podual, F. J. Doyle, III and N. A. Peppas, Polymer, 2000, 41, 3975–3983 CrossRef CAS.
- Y. Wu, H. Hu, J. Hu and S. Liu, Macromol. Rapid Commun., 2012, 33, 1852–1860 CrossRef CAS PubMed.
- T. Traitel, Y. Cohen and J. Kost, Biomaterials, 2000, 21, 1679–1687 CrossRef CAS.
- Z. Gu, T. T. Dang, M. Ma, B. C. Tang, H. Cheng, S. Jiang, Y. Dong, Y. Zhang and D. G. Anderson, ACS Nano, 2013, 7, 6758–6766 CrossRef CAS PubMed.
- T. Miyata, A. Jikihara, K. Nakamae and A. S. Hoffman, Macromol. Chem. Phys., 1996, 197, 1135–1146 CrossRef CAS.
- A. A. Obaidat and K. Park, Biomaterials, 1997, 18, 801–806 CrossRef CAS.
- S. Tanna, T. S. Sahota, K. Sawicka and M. J. Taylor, Biomaterials, 2006, 27, 4498–4507 CrossRef CAS PubMed.
- K. Lin, J. Yi, X. Mao, H. Wu, L.-M. Zhang and L. Yang, React. Funct. Polym., 2019, 139, 112–119 CrossRef CAS.
- M. Zhang, C.-C. Song, F.-S. Du and Z.-C. Li, ACS Appl. Mater. Interfaces, 2017, 9, 25905–25914 CrossRef CAS PubMed.
- Q. Wu, L. Wang, H. Yu, J. Wang and Z. Chen, Chem. Rev., 2011, 111, 7855–7875 CrossRef CAS PubMed.
- L. Zhao, C. Xiao, L. Wang, G. Gai and J. Ding, Chem. Commun., 2016, 52, 7633–7652 RSC.
- S. Y. Cheng, I. Constantinidis and A. Sambanis, Biotechnol. Bioeng., 2006, 93, 1079–1088 CrossRef CAS PubMed.
- W. L.-A. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375–1397 CrossRef CAS PubMed.
- R. Ma and L. Shi, Polym. Chem., 2014, 5, 1503–1518 RSC.
- G. Springsteen and B. Wang, Tetrahedron, 2002, 58, 5291–5300 CrossRef CAS.
- A. Matsumoto, S. Ikeda, A. Harada and K. Kataoka, Biomacromolecules, 2003, 4, 1410–1416 CrossRef CAS PubMed.
- A. Matsumoto, T. Ishii, J. Nishida, H. Matsumoto, K. Kataoka and Y. Miyahara, Angew. Chem., Int. Ed., 2012, 51, 2124 CrossRef CAS PubMed.
- A. Matsumoto, K. Yamamoto, R. Yoshida, K. Kataoka, T. Aoyagi and Y. Miyahara, Chem. Commun., 2010, 46, 2203–2205 RSC.
- A. Matsumoto, R. Yoshida and K. Kataoka, Biomacromolecules, 2004, 5, 1038–1045 CrossRef CAS PubMed.
- S. Chen, T. Miyazaki, M. Itoh, H. Matsumoto, Y. Moro-oka, M. Tanaka, Y. Miyahara, T. Suganami and A. Matsumoto, ACS Appl. Polym. Mater., 2020, 2, 2781–2790 CrossRef CAS.
- A. Matsumoto, M. Tanaka, H. Matsumoto, K. Ochi, Y. Moro-oka, H. Kuwata, H. Yamada, I. Shirakawa, T. Miyazawa, H. Ishii, K. Kataoka, Y. Ogawa, Y. Miyahara and T. Suganami, Sci. Adv., 2017, 3, eaaq0723 CrossRef PubMed.
- J. Yu, J. Wang, Y. Zhang, G. Chen, W. Mao, Y. Ye, A. R. Kahkoska, J. B. Buse, R. Langer and Z. Gu, Nat. Biomed. Eng., 2020, 4, 499–506 CrossRef CAS PubMed.
- Z. Wang, J. Wang, H. Li, J. Yu, G. Chen, A. R. Kahkoska, V. Wu, Y. Zeng, D. Wen, J. R. Miedema, J. B. Buse and Z. Gu, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 29512–29517 CrossRef CAS PubMed.
- J. Wang, J. Yu, Y. Zhang, X. Zhang, A. R. Kahkoska, G. Chen, Z. Wang, W. Sun, L. Cai, Z. Chen, C. Qian, Q. Shen, A. Khademhosseini, J. B. Buse and Z. Gu, Sci. Adv., 2019, 5, eaaw4357 CrossRef CAS PubMed.
- J. Lee, J. H. Ko, K. M. Mansfield, P. C. Nauka, E. Bat, H. D. Maynard, J. Lee, J. H. Ko, K. M. Mansfield, P. C. Nauka, E. Bat, H. D. Maynard and E. Bat, Macromol. Biosci., 2018, 18, e1700372 CrossRef PubMed.
- L. Zhao, L. Niu, H. Liang, H. Tan, C. Liu and F. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 37563–37574 CrossRef CAS PubMed.
- B. Cai, Y. Luo, Q. Guo, X. Zhang and Z. Wu, Carbohydr. Res., 2017, 445, 32–39 CrossRef CAS.
- V. Yesilyurt, M. J. Webber, E. A. Appel, C. Godwin, R. Langer and D. G. Anderson, Adv. Mater., 2016, 28, 86–91 CrossRef CAS PubMed.
- Y. Dong, W. Wang, O. Veiseh, E. A. Appel, K. Xue, M. J. Webber, B. C. Tang, X.-W. Yang, G. C. Weir, R. Langer and D. G. Anderson, Langmuir, 2016, 32, 8743–8747 CrossRef CAS PubMed.
- A. Ali, S. P. Nagumantri, T. Rakshit and S. Pal, Macromol. Chem. Phys., 2021, 222, 2100121 CrossRef CAS.
- R. Nishiyabu, Y. Takahashi, T. Yabuki, S. Gommori, Y. Yamamoto, H. Kitagishi and Y. Kubo, RSC Adv., 2020, 10, 86–94 RSC.
- A. J.-J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC.
- E. M. Grad, I. Tunn, D. Voerman, A. S. de Léon, R. Hammink and K. G. Blank, Front. Chem., 2020, 8 DOI:10.3389/fchem.2020.00536.
- B. Marco-Dufort, R. Iten and M. W. Tibbitt, J. Am. Chem. Soc., 2020, 142, 15371–15385 CrossRef CAS PubMed.
- W. L.-A. Brooks, C. C. Deng and B. S. Sumerlin, ACS Omega, 2018, 3, 17863–17870 CrossRef CAS PubMed.
- T. Narita, K. Mayumi, G. Ducouret and P. Hebraud, Macromolecules, 2013, 46, 4174–4183 CrossRef CAS.
- B. Marco-Dufort, J. Willi, F. Vielba-Gomez, F. Gatti and M. W. Tibbitt, Biomacromolecules, 2021, 22, 146–157 CrossRef CAS PubMed.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tb00603k |
| This journal is © The Royal Society of Chemistry 2022 |