
Effect of liposome surface modification with water-soluble phospholipid polymer chain-conjugated lipids on interaction with human plasma proteins†
Anna
Adler
 a,
Yuuki
Inoue
b,
Kristina N.
Ekdahl
ac,
Teruhiko
Baba
d,
Kazuhiko
Ishihara
b,
Bo
Nilsson
a and
Yuji
Teramura
*ad
a,
Yuuki
Inoue
b,
Kristina N.
Ekdahl
ac,
Teruhiko
Baba
d,
Kazuhiko
Ishihara
b,
Bo
Nilsson
a and
Yuji
Teramura
*ad
aDepartment of Immunology, Genetics and Pathology (IGP), Uppsala University, Dag Hammarskjölds väg 20, SE-751 85 Uppsala, Sweden
bDepartment of Materials Engineering, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
cLinnaeus Center of Biomaterials Chemistry, Linnaeus University, SE-391 82 Kalmar, Sweden
dCellular and Molecular Biotechnology Research Institute (CMB), National Institute of Advanced Industrial Science and Technology (AIST), AIST Tsukuba Central 5, 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan. E-mail: y.teramura@aist.go.jp; Tel: +81-29-861-6582
First published on 20th September 2021
Abstract
Alternative liposome surface coatings for PEGylation to evade the immune system, particularly the complement system, have garnered significant interest. We previously reported poly(2-methacryloyloxyethyl phosphorylcholine) (MPC)-based lipids (PMPC–lipids) and investigated the surface modification of liposomes. In this study, we synthesize PMPC–lipids with polymerization degrees of 10 (MPC10–lipid), 20 (MPC20–lipid), 50 (MPC50–lipid), and 100 (MPC100–lipid), and coated liposomes with 1, 5, or 10 mol% PMPC–lipids (PMPC–liposomes). Non-modified and PEGylated liposomes are used as controls. We investigate the liposome size, surface charge, polydispersity index, and adsorption of plasma proteins to the liposomes post incubation in human plasma containing N,N,N′,N′-ethylenediamine tetraacetic acid (EDTA) or lepirudin by some methods such as sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE), western blotting, and automated capillary western blot, with emphasis on the binding of complement protein C3. It is shown that the coating of liposome PMPC–lipids can suppress protein adsorption more effectively with an increase in the molecular weight and molar ratio (1–10 mol%). Apolipoprotein A-I is detected on PMPC–liposomes with a higher molecular weight and higher molar ratio of PMPC–lipids, whereas α2-macroglobulin is detected on non-modified, PEGylated, and PMPC–liposomes with a shorter polymer chain. In addition, a correlation is shown among the PMPC molecular weight, molar ratio, and C3 binding. The MPC10–lipid cannot inhibit C3 binding efficiently, whereas surface modifications with 10 mol% MPC20–lipid and 5 mol% and 10 mol% MPC50–lipid suppress both total protein and C3 binding. Hence, liposome modification with PMPC–lipids can be a possible strategy for avoiding complement activation.
1. Introduction
Liposomes are artificial vesicles comprising phospholipids and cholesterol that form one or more concentric bilayers.1 In recent years, liposomes have garnered substantial interest as a drug delivery system because of their ability to encapsulate hydrophilic drugs into the aqueous core and embed hydrophobic drugs into the lipid bilayer. Drug encapsulation provides a physical barrier between the drug and biological environment; hence, the drug is protected from degradation.2 However, a protein corona forms rapidly on the liposome surface upon its injection into blood. The formed protein corona provides a new “biological identity” to the liposome such that its surface chemistry, size, shape, and charge will change, depending on the profile proteins that have adsorbed onto the surface.3 Non-modified liposomes are rapidly cleared from circulation and degraded by the innate immune system, particularly by the complement system, where complement protein C3 is crucial.4,5 Therefore, several liposome-coating strategies have emerged, e.g., modifying the surface with polymers to avoid liposome recognition by the immune system.2 The gold standard liposome coating is to conjugate poly(ethylene glycol) (PEG) to the surface, i.e., PEGylation. PEGylation prevents non-specific protein adsorption and prevents phagocytosis, thereby prolonging the blood residence time. However, PEGylation cannot fully inhibit protein adsorption or protein corona formation on liposomes.6,7 PEGylated liposomes are rapidly cleared from the circulation upon repeated doses through a phenomenon known as accelerated blood clearance (the ABC effect).8 The ABC effect is attributed to the induction of anti-PEG antibodies, primarily IgM, upon the first dose of PEGylated liposomes. Hence, a subsequent dose of PEGylated liposomes is recognized by the anti-PEG antibodies and rapidly eliminated from circulation, as facilitated by the complement system, leading to a loss of therapeutic efficacy.9 The generation of anti-PEG antibodies has been reported in several animal models as well as in humans.10–13 Additionally, anti-PEG antibodies have been reported in the general population where there are people that have never received any PEGylated therapeutics.14 Pre-existing anti-PEG antibodies are attributable to increased exposure to PEG-containing products, including common products such as toothpaste and cosmetics.15 However, natural anti-phosphocholine antibodies have been shown to contribute to the ABC effect observed in lipid nanoparticles.16–18 Therefore, finding alternative liposome surface coatings to PEGylation is of great interest to evade the immune system, especially the complement system.A 2-methacryloyloxyethyl phosphorylcholine (MPC)-based polymer has been successfully used to coat medical devices, such as cardiovascular stents (Endeavor®), left ventricular assist devices (EVAHEART®), oxygenators (PrimO2x®), artificial hip joint systems (Aquala®), and soft contact lenses (Proclear®).19 MPC-based polymers are highly hydrophilic and can retain water, thereby suppressing protein adsorption and inhibiting cell adhesion to the poly(MPC) (PMPC)-coated substrate surface in contact with plasma or whole blood. PMPC is a polymethacrylate bearing a phosphorylcholine group in the side chain. This structure is similar to the phospholipid polar groups in biological membranes, and the MPC unit is electrically neutral under physiological conditions.20 The polymerization of MPC to phospholipid and the use of PMPC–lipid for liposomal coating were first described by Lin et al.21 We have also tried the PMPC polymerization at lipid (1,2-distearoyl-sn-glycerol). After we have reported the synthesis of PMPC–lipids with different polymerization degrees for coating liposomes and revealed that the liposome surface was modifiable using PMPC–lipids to suppress protein adsorption, and that the modification remained stable for a long duration. In addition, the PMPC–lipid-modified surface was not recognized by the anti-PEG antibody.22
In this study, we modified liposomes with PMPC–lipids having different molecular weights of PMPC (from 3 to 30 kDa) and different molar ratios of PMPC–lipids (1, 5, and 10 mol%). Subsequently, we investigated the interaction between human plasma proteins and the modified liposomes to evaluate the protein profile and its relationship with the length of the PMPC-chain, mol% of the PMPC–lipid, and protein adsorption, with emphasis on the binding of C3 to analyze the complement activation. In addition, we investigated the characteristics of the modified liposome, i.e., the size, surface charge, and polydispersity index (PDI), before and after incubation in human plasma.
2. Materials and methods
2.1. Materials
Alpha-2-bromoisobutyryl bromide (BIBB), tris(2-pyridylmethyl) amine (TPMA), cholesterol and bovine serum albumin (BSA) were purchased from Sigma-Aldrich (Munich, Germany); 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) was purchased from Avanti Polar Lipids (Alabaster, AL, USA); 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[monomethoxy poly(ethylene glycol) (5000)] and MPC were purchased from NOF Co. (Tokyo, Japan). Copper(II) bromide (CuBr2), L(+)-ascorbic acid (Asc), a phospholipids C-test Wako kit and a cholesterol quantification kit (T-Cho E) were purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). 1,2-Distearoyl-sn-glycerol (lipid(C18)) was purchased from Bachem Holding AG (Bubendorf, Switzerland). Phosphate buffered saline (PBS) tablets (0.14 M NaCl, 2.7 mM KCl, 10 mM phosphate buffer, pH 7.4) were obtained from Medicago AB (Uppsala, Sweden). LabAssay™ Cholesterol was purchased from Fujifilm (Tokyo, Japan), and a Micro BCA™ Protein Assay Kit, dithiothreitol (DTT) and a 3,3′-diaminobenzidine (DAB) substrate kit were obtained from Thermo Scientific (Rockford, IL, USA). Polyclonal rabbit anti-human antibodies, i.e., apolipoprotein A-I (A0469) and α2-macroglubulin (A033), and polyclonal swine anti-rabbit Ig horseradish peroxidase (HRP) (P0217) were purchased from Dako (Glostrup, Denmark). Thrombin inhibitor lepirudin (Refludan®) was purchased from Celgene (Windsor, UK). N,N,N′,N′-Ethylenediamine tetraacetic acid (EDTA) and sodium n-dodecyl sulfate (SDS) were obtained from Merck KGaA (Darmstadt, Germany). Native C3 was purified in-house from human plasma, based on the procedure reported by Hammer et al.,23 and mAb 169.7 (mouse anti-human complement C3 β-chain) was prepared based on the procedure reported by Nilsson et al.24 and then affinity purified using protein A-Sepharose. Laemmli sample buffer, Coomassie brilliant blue R-250, and destaining solutions were obtained from BioRad (Hercules, CA, USA).PMPC–lipids with polymerization degrees of 10, 20, 50, and 100 (3.7, 6.7, 16, and 30 kDa respectively) were synthesized based on the procedure reported by Adler et al.22 (Scheme 1). These lipids are hereinafter referred to as MPC10–lipid, MPC20–lipid, MPC50–lipid, and MPC100–lipid, respectively. Briefly, we synthesized an atom-transfer radical polymerization initiator composed of 1,2-distearoyl-sn-glycerol (lipid(C18)) and alpha-2-bromoisobutyryl bromide (BIBB). Lipid(C18) and triethylamine were dissolved in dichloromethane, and then BIBB was added to the solution at room temperature. After 24 h, hexane was added to the solution, which was then incubated at room temperature for 1 h. The precipitates were filtered, and the solvents were evaporated from the solution. After this procedure was repeated once, the hexane phase was washed with HCl and saturated aqueous NaCl solution. After dehydrating the organic phase using magnesium sulfate, the solvent was completely removed under reduced pressure. The obtained white powder (Br–lipid(C18), yield: 80%) was analyzed using proton nuclear magnetic resonance (1H NMR) spectroscopy (α-400, JEOL Co., Tokyo, Japan). MPC was polymerized at the end of the lipid initiator (Br–lipid(C18)) to synthesize PMPC–lipids, where the degree of the MPC unit ranged from 10 to 100. Here, the PMPC–lipids were synthesized using an activator regenerated using the electron transfer atom transfer radical polymerization method, as follows: Cu(II)Br2, TPMA, Asc, and MPC were placed in a glass tube at a particular molar ratio and dissolved in methanol. Subsequently, methanol and 1,4-dioxane solutions of Br–lipid(C18) were added to the solutions. The concentration of the lipid initiator was maintained at [MPC]/[lipid initiator] ratios of 10, 20, 50, and 100. After the glass tube was sealed, polymerization was performed by stirring at 40 °C for 24 h.
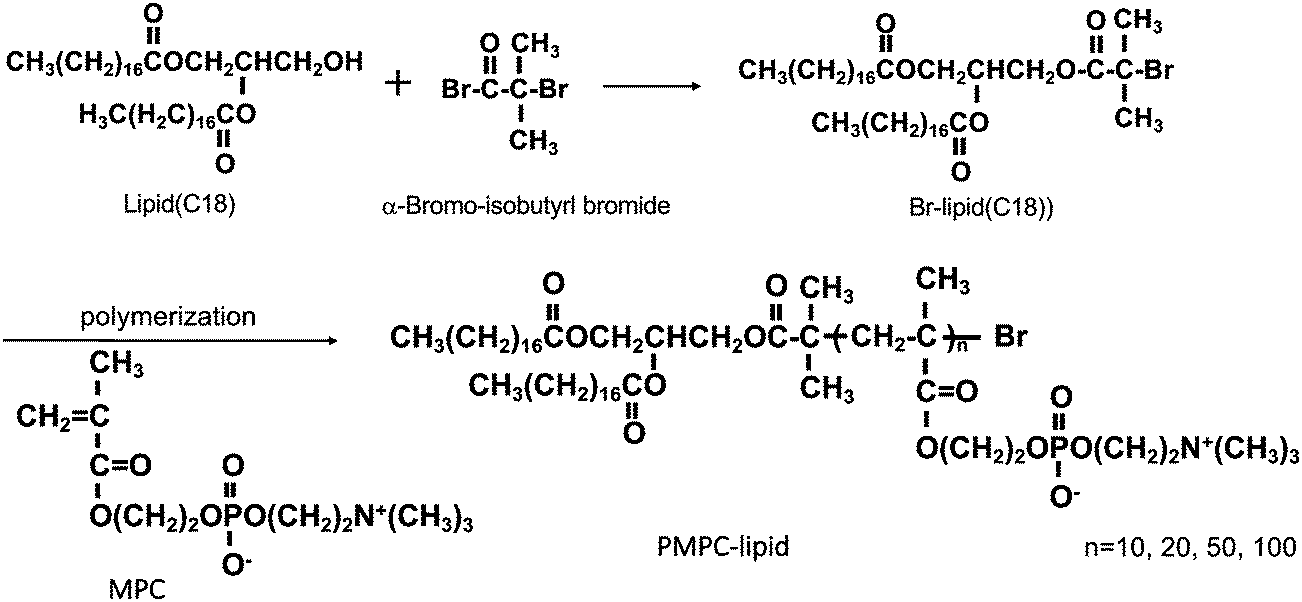 | ||
| Scheme 1 Schematic overview of the synthesis of Br–lipid initiator (C18). MPC was polymerized at Br–lipid(C18) by an ARGET-ATRP method with different degrees of polymerization. (C18); alkyl stearoyl chain, MPC; 2-methacryloyloxyethyl phosphorylcholine, ARGET-ATRP; activator regenerated by electron transfer-atom transfer radical polymerization. | ||
The polymerization solution was poured into an excess of acetone to allow for precipitation, and the precipitates were subsequently dissolved in pure water. The solution was transferred to a dialysis tube (molecular weight cutoff: 3.5 kDa), and impurities were removed by dialysis against pure water for three days Finally, a white powder was obtained by lyophilization. The conversion of MPC to PMPC in the polymerization solution was analyzed using 1H NMR spectroscopy (JEOL, Tokyo, Japan).
2.2. Liposome preparation
Liposomes were prepared using the thin-film hydration method, followed by hand extrusion. Non-modified liposomes containing cholesterol and DPPC at a 1/1 molar ratio were used as controls. Liposomes containing 1, 5, and 10 mol% of polymer–lipids, PEG–lipid, MPC10–lipid, MPC20–lipid, MPC50–lipid, or MPC100–lipid were prepared using cholesterol/DPPC/polymer–lipids at molar ratios of 1/0.98/0.02, 1/0.9/0.1, and 1/0.8/0.2. The amounts used to prepare 1 mL of non-modified liposomes were 5.3 mg cholesterol and 10.1 mg DPPC. For 1 mL of liposomes modified with polymer–lipids, the following amounts were used: 5.3 mg of cholesterol; 9.9, 8.4, or 8.1 mg of DPPC; 1.62, 8.4, or 16.2 mg of PEG–lipid; 0.81, 4.2, or 8.1 mg of MPC10–lipid; 1.62, 8.4, or 16.2 mg of MPC20–lipid; 4.05, 21.0, or 40.5 mg of MPC50–lipid; and 7.02, 36.4, or 70.2 mg of MPC100–lipid for modification with 1, 5, or 10 mol% polymer–lipids, respectively. DPPC, cholesterol, PEG–lipid, or PMPC–lipid were dissolved in 99.9% ethanol at the desired molar ratio. A rotary evaporator was used to evaporate the ethanol and form a uniform lipid film. The lipid films were dried in vacuo for 24 h at 25 °C to ensure the complete removal of ethanol. After the resultant lipid film was hydrated in PBS for 2 h at room temperature (RT), the liposome suspension was extruded through polycarbonate membranes (pore sizes: 1000, 400, 200, and 100 nm; nuclear track-etch membrane; Whatman Ltd., UK) using an Avanti mini extruder (Avanti Polar Lipids). The lipid suspension was passed through each filter 15 times.2.3. Characteristics of modified liposomes
The average particle size, PDI, and zeta potential of the modified liposomes were determined by dynamic light scattering (DLS, Zetasizer Nano ZS, Malvern Instruments, Worcestershire, UK). The liposome samples were diluted in PBS (1/100) to perform size and PDI measurements. For the zeta potential measurements, the liposomes were diluted (1/100) in a 1 mM NaCl aqueous solution. All the measurements were performed in triplicates at 25 °C. The data were analyzed using the software Malvern, version 7.03.2.4. Determination of liposome concentration
To quantify the lipid concentration in the liposome solutions, a chromogenic cholesterol kit (LabAssay™ Cholesterol, Fujifilm) was used according to the manufacturer's instructions to determine the total cholesterol concentration in the samples. The liposomes were treated with 10% SDS at 95 °C for 15 min for solubilization. The lipid concentration was estimated from the total cholesterol concentration based on a cholesterol/lipid molar ratio of 1/1.2.5. Blood collection
Blood was obtained from healthy donors who had not received any medication for a minimum of 10 d prior to donation. To prepare the EDTA plasma, human whole blood was collected in Vacuette® tubes (Greiner Bio-One GmbH, Kremsmünster, Austria) containing K2E K2EDTA (4 mM final concentration) and centrifuged at RT for 15 min at 2000× g. The EDTA plasma was obtained and stored at −80 °C prior to use. To prepare human lepirudin plasma, whole blood was collected in Vacutest® urine collection tubes (Vacutest Kima, Azergrande, Italy) containing 50 μg mL−1 hirudin (final concentration) and centrifuged at RT for 15 min at 2000× g. Lepirudin plasma was obtained immediately prior to use. The anticoagulant lepirudin inhibits thrombin, thereby preserving clotting factors that are absent in serum but does not affect the complement system, unlike other anticoagulants. Ethical approval for the use of human blood was obtained from the regional ethics board of Uppsala (diary no 2008/264).2.6. Incubation of liposomes in human plasma
The modified liposomes were incubated in EDTA or lepirudin plasma at a final lipid concentration of 1 mM for 60 min at 37 °C and 20 rpm on a circulating wheel. The reaction was terminated using 10 mM EDTA, and the liposomes were separated from unbound proteins via ultracentrifugation. Briefly, the plasma containing liposomes was transferred into OptiSeal™ centrifuge tubes (8.9 mL, Beckman Coulter, California, USA) and diluted with PBS to fill the tubes. Subsequently, the samples were centrifuged by ultracentrifugation at 100.000× g for 60 min at 4 °C (Beckman, Optima™ LE-80K Ultracentrifuge with Type 70.1 Ti rotor, Beckman Coulter, Bromma, Sweden). This washing step was repeated thrice. The supernatant was removed by decantation, and the liposome pellet was resuspended in PBS to a final lipid concentration of 4 mM. A small fraction of liposomes from one donor was removed for DLS analysis (see Section 2.3) after incubation in EDTA plasma, and the remaining samples were stored at −80 °C for further post-processing analyses. All 16 liposome compositions were incubated in EDTA and lepirudin plasma from three different donors. As a negative control, PBS containing no liposomes was added to the EDTA or lepirudin plasma in the same volume as the liposomes. The sample were subjected to the same centrifugation protocol. For the SDS–PAGE and western blot, this sample was referred to as “PBS”, whereas in the Micro BCA and automated capillary western blot (Wes) experiments, the values from the PBS background samples were subtracted.2.7. Determination of protein concentration on liposomes using a Micro BCA assay
The total protein concentration on liposomes incubated in EDTA or lepirudin plasma from three different donors was determined using a Micro BCA™ Protein Assay Kit (Thermo Scientific). Samples were diluted to 1/10 in PBS to a final lipid concentration of 0.4 mM, and the background values obtained for the untreated liposomes of each composition were subtracted. This was performed in duplicate for all samples, and the assay was performed according to the manufacturer's instructions for microtiter plates.2.8. Analysis of adsorbed plasma proteins onto liposomes by SDS–PAGE
The binding of plasma proteins to the surface of liposomes incubated in EDTA plasma was analyzed by SDS–PAGE electrophoresis on a 4–20% gradient gel (Mini-PROTEAN® TXG™ Precast Gels, Hercules, CA, USA, BioRad) under both non-reduced and reduced conditions. After incubation in human plasma and washing by ultracentrifugation, the liposomes were boiled at a concentration of 4 mM lipids in the presence of Laemmli sample buffer (BioRad) and DTT (25 mM) under reduced conditions. The SDS–PAGE electrophoresis was performed at 200 V for 30 min. The proteins were visualized on the gel using Coomassie brilliant blue staining and destaining solution (BioRad) according to the manufacturer's instructions.2.9. LC-MS/MS analysis of gel bands
LC-MS/MS was performed on specific gel bands based on SDS–PAGE analysis, as described in Section 2.8, to determine the specific proteins within each band that had been removed from the gel. Briefly, samples were in-gel digested following the standard operating procedure, followed by desalting using a ZipTip® C18 (Millipore). Subsequently, the dried samples were resolved in 30 μL of 0.1% formic acid and then diluted four times prior to nano-LC-MS/MS. The resulting samples were separated in reversed phase on a C18-column and electro-sprayed on-line to a Q-Exactive Plus Orbitrap mass spectrometer (Thermo Finnigan) with a 35 min gradient. Tandem mass spectrometry was performed applying HCD. Database searches were performed using Proteome Discoverer software (version 1.4). Proteins were identified by searching against Homo sapiens (Human) acquired from Uniprot (release 2019). The search criteria for protein identification were set to at least two matching peptides with a 95% confidence level per protein. The fixed modification was carbamidomethyl, and the variable modifications were oxidation and deamidation. The highest scoring proteins in the samples against Homo sapiens proteome using Proteome Discoverer software are presented.2.10. Western blot
Traditional western blots were performed against apolipoprotein A-I and α2-macroglobulin with 10 mol% liposomes incubated in EDTA plasma from a representative donor and SDS–PAGE, as described in Section 2.8, under reduced conditions. EDTA plasma from the same donor, diluted to 1/100 in PBS, was used as a positive control. The gels were transferred onto a polyvinylidene difluoride transfer membrane (GE Healthcare, Uppsala, Sweden) at 250 mA for 60 min. The membranes were blocked with 1% BSA (Sigma–Aldrich) in PBS (blocking buffer) for 60 min at room temperature (RT). After each incubation step, the membranes were washed three times with 0.1% Tween20-PBS (wash buffer) for 5 min. Polyclonal rabbit anti-human apolipoprotein A-I (Dako) diluted to 12.5 μg mL−1 and polyclonal rabbit anti-human α2-macroglubulin (Dako) antibodies diluted in blocking buffer to 7 μg mL−1 were used for detection. The membranes were incubated with primary antibodies for 60 min at RT and then washed. Subsequently, the membranes were incubated for 60 min at RT with secondary polyclonal swine anti-rabbit Ig HRP (Dako) diluted in blocking buffer to a final concentration of 1.3 μg mL−1, followed by three washes. An additional washing step using PBS for 2 × 5 min was performed to remove Tween20. The bound antibodies were visualized using DAB (Thermo Scientific).2.11. Analysis of adsorbed C3 onto liposome using Wes immunoassay
Simple Western 12–230 kDa assays under reduced conditions were performed using a Wes™ analyzer (Protein Simple, Santa Clara, CA, USA) according to the manufacturer's instructions. In brief, liposomes incubated in EDTA or lepirudin plasma and purified using ultracentrifugation were diluted in 0.1× Wes sample buffer to a final concentration of 1 mM lipids. A serial diluted native C3 (20–0.1 μg mL−1) in 0.1 × Wes sample buffer was used as the standard curve. Mouse anti-human complement C3 β-chain mAb 169.7, at a final concentration of 70 μg mL−1, was used as the primary antibody together with the Wes anti-mouse detection module. Electrophoretic protein separation and immunodetection were performed using the default Simple Western™ settings. The immunodetected signal, i.e., the area under the curve, was analyzed using Compass software (version 4.0.0, ProteinSimple™), which converts electropherograms into virtual blots. The concentration of C3 on the surface of the liposomes was quantified by comparing the peak area against a calibration curve of native C3 with a concentration that was calculated using linear regression analysis.2.12. Phospholipase D-catalyzed hydrolysis of PMPC–lipids
Phospholipase D (PLD)-catalyzed hydrolytic susceptibility of phosphorylcholine moieties of PMPC–lipids was examined by using a phospholipid C-test kit, of which the principle is based on the colorimetric assay of H2O2 co-produced by choline oxidase (COX) catalyzing the oxidation of the liberated choline molecule.25 This kit contains 3,5-dimethoxy-N-ethyl-N-(2-hydroxy-3-sulfopropyl)aniline (DAOS)26 as a color developing reagent instead of phenol originally employed in reference 25 to enhance the absorbance of the final product. PMPC–lipids (MPC10–lipid, MPC100–lipid) were dissolved in pure water and their concentrations as total inorganic phosphorus (Pi) were determined using the Bartlett method.27 Subsequently, the reagent mixture (PLD, COX, horseradish peroxidase, 4-aminoantipyrine, DAOS, ascorbate oxidase) was dissolved in Good's buffer solution in the kit according to its protocol, the reagent solution (3 mL) was added to MPC–lipids solution (20 mL), incubated at 37 °C for 10 min and the absorbance at 600 nm was recorded on a UV/VIS spectrophotometer (V-550, Jasco, Tokyo, Japan). The calibration curve (choline concentration vs. A600) was prepared by using a choline standard solution in the kit.2.13. Statistics
All experiments were repeated at least three times, except for the DLS data of liposomes before and after incubation in EDTA plasma (n = 1). Data are presented as mean values ± SD or as representative images. Statistical calculations, one-way ANOVA, followed by Dunnett's multiple comparison tests with a single pooled variance were performed using GraphPad Prism 9 for MacOS version 9.0.0 (GraphPad Software, La Jolla, CA, USA). All liposomes were compared with the PEGylated liposomes. The calculated p-values were defined as follows: *p < 0.05, **p < 0.001, ***p < 0.001, and ****p < 0.0001.3. Results and discussion
3.1. Evaluation of liposome in human EDTA plasma
PMPC–lipids with polymerization degrees of 10, 20, 50, and 100 were synthesized to an alkyl stearoyl (C18) chain (Scheme 1). Liposomes were prepared with 1, 5, and 10 mol% polymer lipids using the thin-film hydration method followed by hand extrusion. The size, PDI, and zeta potential of the liposomes were characterized before and after incubation in EDTA plasma using DLS. Regardless of the lipid composition (i.e., with or without surface modification), the size of all liposomes increased (6–21%) after plasma incubation (Fig. 1A–C). Liposomes with PDI values <0.3 were considered monodisperse.28 All liposome formulations were monodisperse before and after incubation in plasma PDI < 0.3 (Fig. 1D–F). The PEGylated and PMPC-coated liposomes indicated PDI values < 0.09 before incubation in plasma, indicating that the modified liposomes were highly monodisperse. We have previously demonstrated the long-term stability of 1 mol% PMPC–liposomes, which were stable over 98 days.22 This indicates that the surface coverage and molecular weight of PMPC are important for obtaining the optimal shielding effect from protein adsorption in human blood. The zeta potential of all analyzed liposomes became more negative after plasma incubation (Fig. 1G–I), indicating the adsorption of proteins. Furthermore, it indicates that some protein adsorption occurred on the surface of liposomes with and without modification with polymers.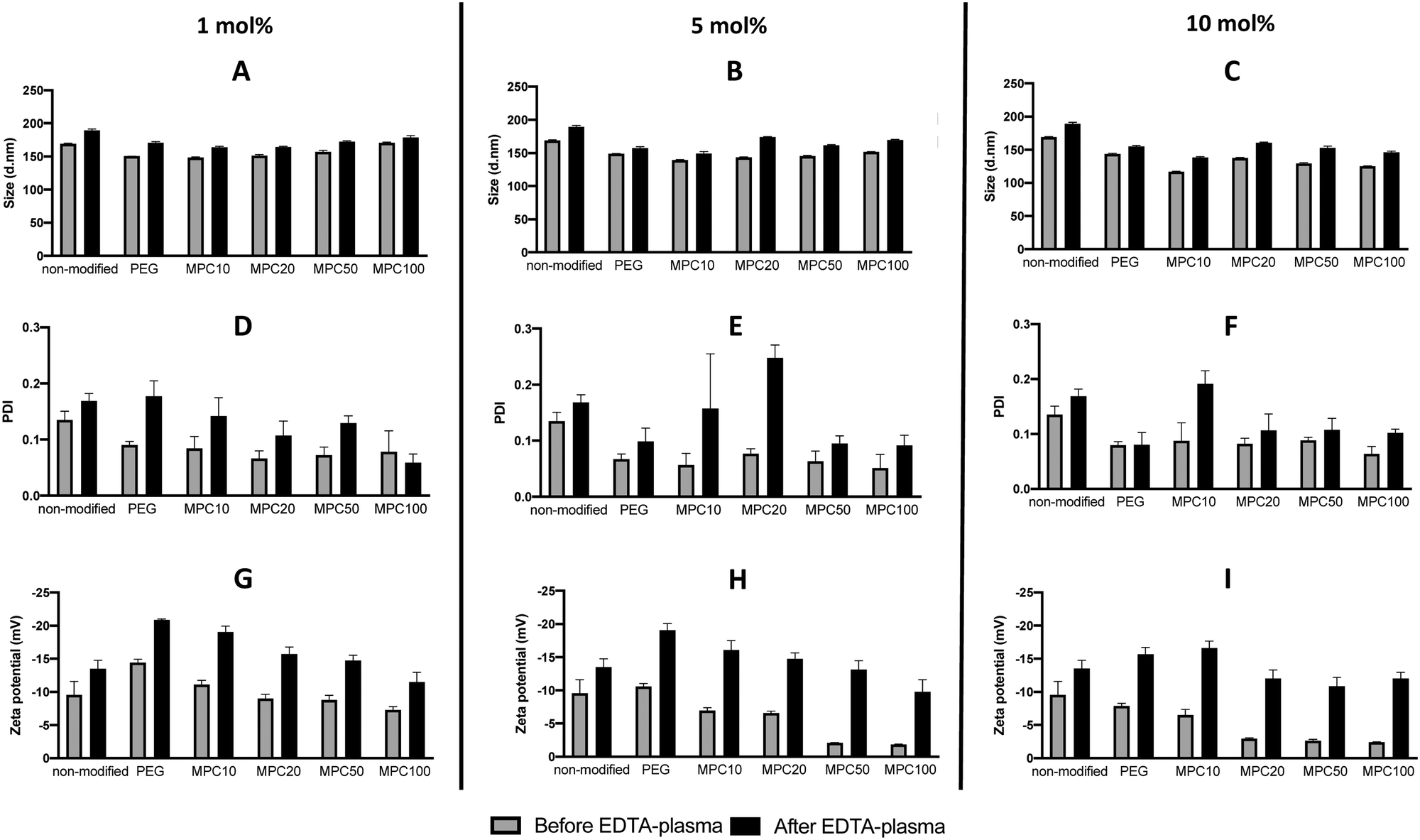 | ||
| Fig. 1 DLS analysis of non-modified, PEGylated, and PMPC–lipid-modified liposomes (1, 5 and 10 mol% polymer–lipids) by measuring the liposome diameters (A, B, and C), PDI (D, E, and F) and zeta potential (G, H, and I) before and after incubation in EDTA-plasma. DLS: dynamic light scattering, PDI: polydispersity index. | ||
This also occurs in vivo when liposomes are injected into the circulation, thereby being exposed to blood plasma proteins which rapidly adsorb to the liposome surface to form a protein corona.3 The zeta potential of nanoparticles with a protein corona, irrespective of their physiochemistry, is generally in the range of −10 to −20 mV,3,29,30 which is the range for our liposomes. This change in the zeta potential is attributable to the fact that the net charge of plasma proteins in a physiological pH is typically negative, indicating that proteins bound to all the liposomal surfaces irrespective of the polymer modification.
3.2. Protein adsorption to modified liposomes in human blood
A Micro BCA kit™ (Thermo Scientific) was used to determine the amount of protein bound to the liposomes after incubation in EDTA or lepirudin plasma. EDTA plasma was used to investigate the passive adsorption of proteins onto the liposomes because no coagulation or complement activation occurred in the presence of EDTA. Human plasma supplemented with lepirudin, a thrombin inhibitor, was used to investigate protein adsorption while the complement system remained active. For non-modified and liposomes modified with 1 and 5 mol% PMPC–lipids and incubated in EDTA plasma, no difference in protein adsorption was observed when compared with 1 and 5 mol% PEGylated liposomes (Fig. 2A and B). Whereas non-modified (300.6 ± 50.4 μg mL−1, p-value = 0.0005) and liposomes modified with 10 mol% MPC10–lipid (215.9 ± 91.2 μg mL−1, p-value = 0.0073) bound more proteins when compared to liposomes modified with 10 mol% PEG–lipids (no proteins were detected) (Fig. 2C). No difference was observed between the control liposomes modified with 10 mol% PEG–lipids and 10% MPC20–lipid (60.3 ± 69.9 μg mL−1), 10% MPC50–lipid (28.8 ± 40.7 μg mL−1) or 10% MPC100–lipid (8.2 ± 12.2 μg mL−1) after incubation in EDTA plasma.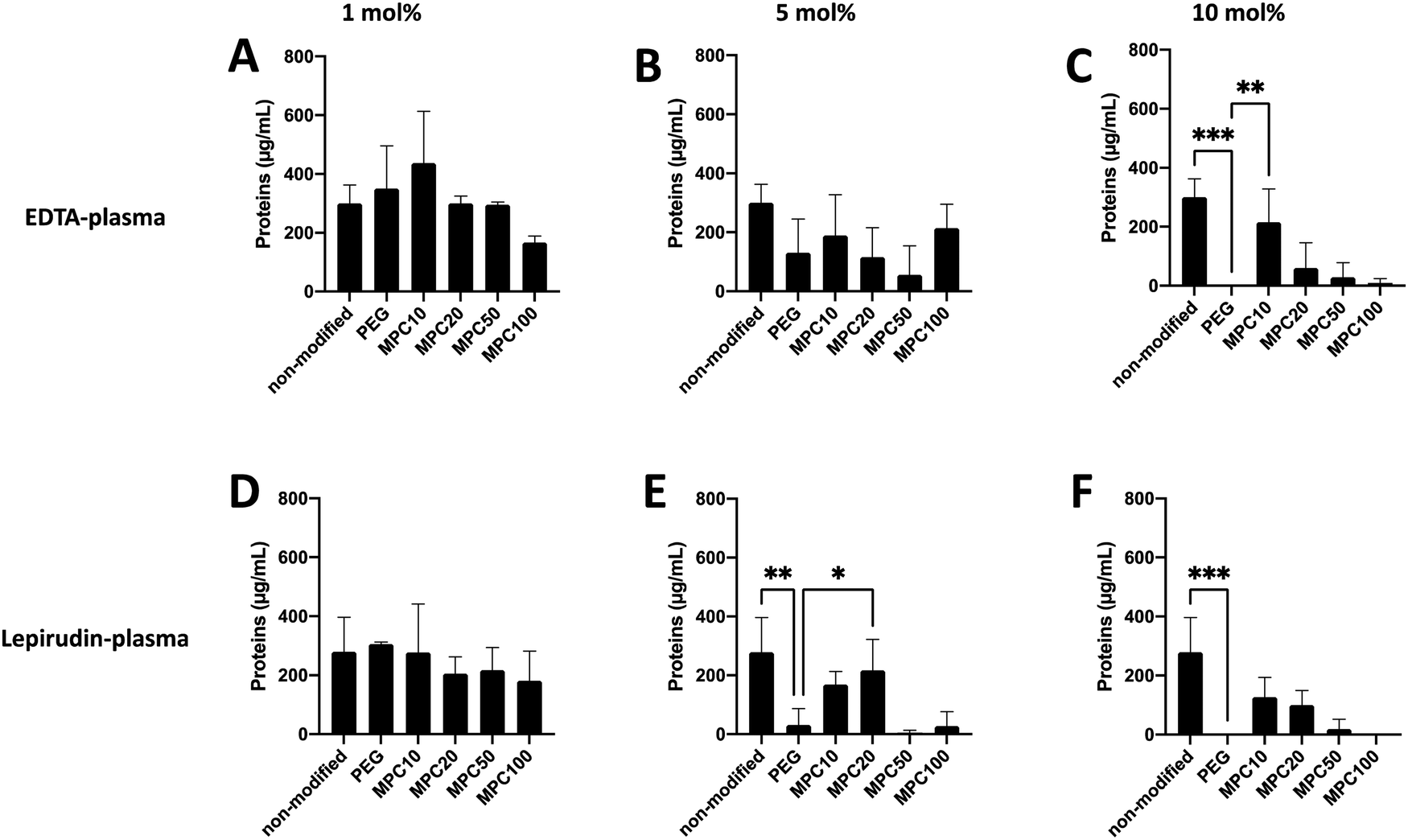 | ||
| Fig. 2 Protein adsorption to non-modified, PEGylated, and PMPC–lipid-modified liposomes (1, 5 and 10 mol% polymer–lipids) at a concentration of 4 mM lipids after incubation in EDTA-plasma (A–C) or lepirudin plasma (D–F) from three different donors (n = 3). Quantified using a Micro BCA kit. | ||
For non-modified and liposomes modified with 1 mol% PMPC–lipids and incubated in lepirudin plasma, no difference in protein adsorption was observed when compared with the liposomes modified with 1 mol% PEG–lipid (Fig. 2D). Non-modified (278.8 ± 96.2 μg mL−1, p-value = 0.0058) and liposomes modified with 5 mol% MPC20–lipid (217.9 ± 85.7, p-value = 0.0058) bound more proteins than liposomes modified with 5 mol% PEG–lipid (31.8 ± 44.9 μg mL−1) whereas there was no difference in protein adsorption to liposome modified PMPC–lipids with a longer polymer chain as observed for liposomes with 5 mol% MPC50–lipid (4.7 ± 6.7 μg mL−1) and 5 mol% MPC100–lipid (27.9 ± 39.4 μg mL−1) (Fig. 2E). No difference in protein adsorption was observed between liposomes modified with 10 mol% MPC10–lipids (126.7 ± 54.5 μg mL−1), 10 mol% MPC20–lipid (100.4 ± 39.5 μg mL−1), 10 mol% MPC50–lipid (19.0 ± 26.9 μg mL−1), and 10 mol% MPC100–lipid (no proteins were detected) when compared to liposomes modified with 10 mol% PEG–lipid (no proteins were detected) (Fig. 2F). Non-modified liposomes (278.8 ± 96.2 μg mL−1) bound more proteins than the 10 mol% PEG–lipid liposomes when incubated in lepirudin plasma.
Collectively, these results indicate that the 1 mol% PMPC–lipids were insufficient to modify the surface to suppress protein adsorption in human plasma. Depending on the interaction between adsorbed proteins and liposome surface, a hard and soft protein corona is formed. Tightly bound proteins make up the hard corona, whereas the soft corona consists of proteins loosely associated to the liposome surface.7 Weber et al. reported that the ultracentrifugation performed during washing might disrupt protein corona formation owing to weak interactions.7 We can assume that in our assays, the protein detected was more tightly bound; hence, they should be the hard corona. As described in previous studies, the surface modification of liposome surfaces with PEG cannot fully suppress protein adsorption,3,31 and based on our current data, this appears to be applicable to PMPC-modified liposomes as well.
The total amounts of plasma proteins that adsorbed onto the liposomes did not differ when they were incubated in EDTA or lepirudin plasma; however, we speculated that the composition of the protein corona would differ because the complement system was fully functional in lepirudin plasma, but not in EDTA plasma. This is important because the fate of the liposomes in vivo is determined by the type of proteins adsorbed onto the liposome surface. The effect of the PEG chain length on protein adsorption has been investigated in several studies, and the results indicate that an optimal length of the PEG chain exists that can effectively inhibit protein adsorption.31,32 For example, Doxil®, a PEGylated liposomal drug, is composed of hydrogenated soy phosphatidylcholine, cholesterol, and PEG–lipid (PEG molecular weight: 2 kDa) (56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :39:5, by molar ratio),33,34 which has been used for cancer treatment. By contrast, the optimal MPC–polymer molecular weight and the ratio required for liposome coating may differ from that of PEGylated liposomes because PMPC is a rigid polymer compared with PEG.35 In this study we used liposomes modified with PEG–lipid having a 5 kDa PEG-chain as the negative control since we know PEGylation can suppress protein binding, and non-modified liposomes were used as the positive control. In fact, the PEGylated liposomes showed less binding of proteins in human plasma. The main aim of this study was to compare the different PMPC–lipids with each other for optimization, and therefore all matching PEG–lipids to the PMPC–lipids (based on the molecular weight) were not examined in this study. However, we considered that liposomes modified with PEG–lipid (5 kDa) could be a proper and representative control for PEGylated liposomes. In our experiments, we observed that PMPC–lipids with a higher molecular weight and an increasing ratio of PMPC–lipids can suppress protein adsorption in human plasma. This is consistent with our observations in a previous study, where we investigated the interaction among albumin, C3, and fibrinogen with PMPC–lipids using quartz crystal microbalance with dissipation monitoring (QCM-D). The QCM-D experiments indicated that PMPC–lipids with a higher molecular weight suppressed protein adsorption more effectively.22 Next, we investigated whether the different surface modifications yielded different protein adsorption profiles.
:39:5, by molar ratio),33,34 which has been used for cancer treatment. By contrast, the optimal MPC–polymer molecular weight and the ratio required for liposome coating may differ from that of PEGylated liposomes because PMPC is a rigid polymer compared with PEG.35 In this study we used liposomes modified with PEG–lipid having a 5 kDa PEG-chain as the negative control since we know PEGylation can suppress protein binding, and non-modified liposomes were used as the positive control. In fact, the PEGylated liposomes showed less binding of proteins in human plasma. The main aim of this study was to compare the different PMPC–lipids with each other for optimization, and therefore all matching PEG–lipids to the PMPC–lipids (based on the molecular weight) were not examined in this study. However, we considered that liposomes modified with PEG–lipid (5 kDa) could be a proper and representative control for PEGylated liposomes. In our experiments, we observed that PMPC–lipids with a higher molecular weight and an increasing ratio of PMPC–lipids can suppress protein adsorption in human plasma. This is consistent with our observations in a previous study, where we investigated the interaction among albumin, C3, and fibrinogen with PMPC–lipids using quartz crystal microbalance with dissipation monitoring (QCM-D). The QCM-D experiments indicated that PMPC–lipids with a higher molecular weight suppressed protein adsorption more effectively.22 Next, we investigated whether the different surface modifications yielded different protein adsorption profiles.
3.3. Identification of specific proteins on liposomes
SDS–PAGE was used to investigate the protein corona to observe the difference between the various liposome compositions after incubation in EDTA plasma. The gels used were under reduced and non-reduced conditions (Fig. 3A and B, respectively). All liposomes exhibited similar protein patterns, except two bands, i.e., one at 25 and the other at 180 kDa. The 25 kDa band became more visible with a higher molar ratio of PMPC–lipids and a higher molecular weight of PMPC. It did not appear in any of the liposomes with 1 mol% lipid polymer (Fig. S1, ESI†). The band at approximately 180 kDa was detected only under reduced conditions, indicating that it originated from a protein consisting of several subunits bound together by disulfide bonds, and was observed for non-modified, PEGylated, and PMPC–liposomes with a lower molecular weight. The band almost completely disappeared as the molar ratio of PMPC–lipids and molecular weight of PMPC increased, and this was observed for all liposome compositions at 1 mol% polymer–lipids (Fig. S1 (ESI†), data for 5 mol% is not shown). SDS–PAGE was performed on all liposome compositions incubated in lepirudin plasma from three different donors. They exhibited the same protein pattern as the liposomes incubated in EDTA plasma (Fig. S2 (ESI†), only liposomes modified with 10 mol% polymer–lipid are shown).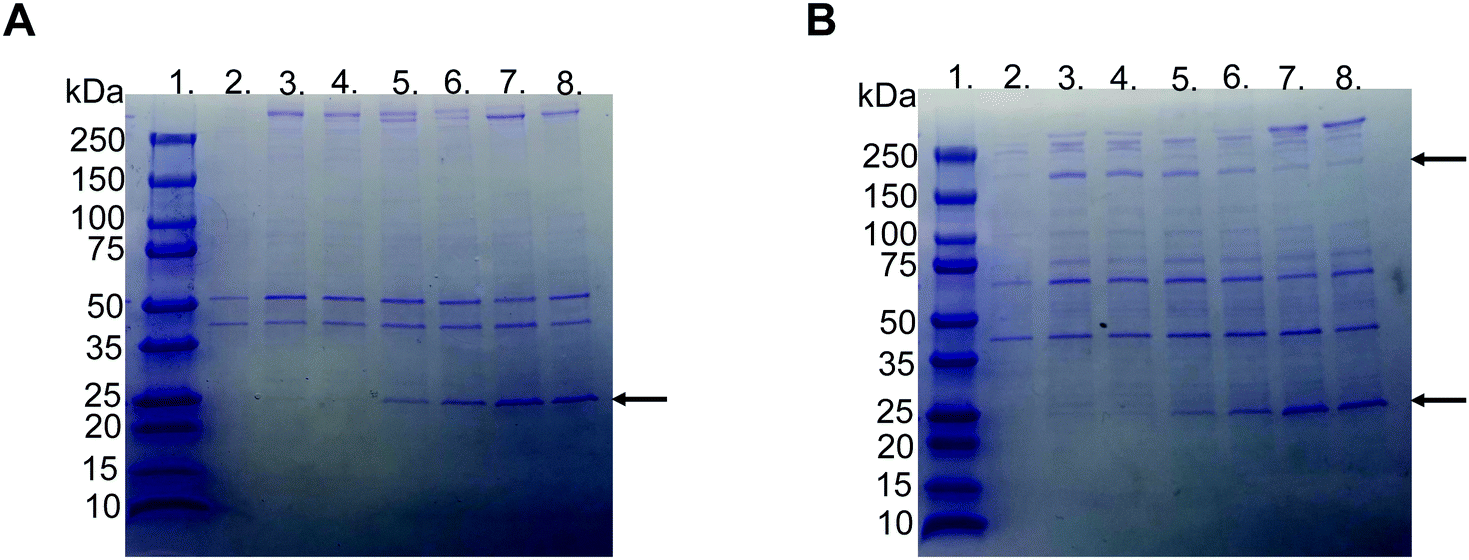 | ||
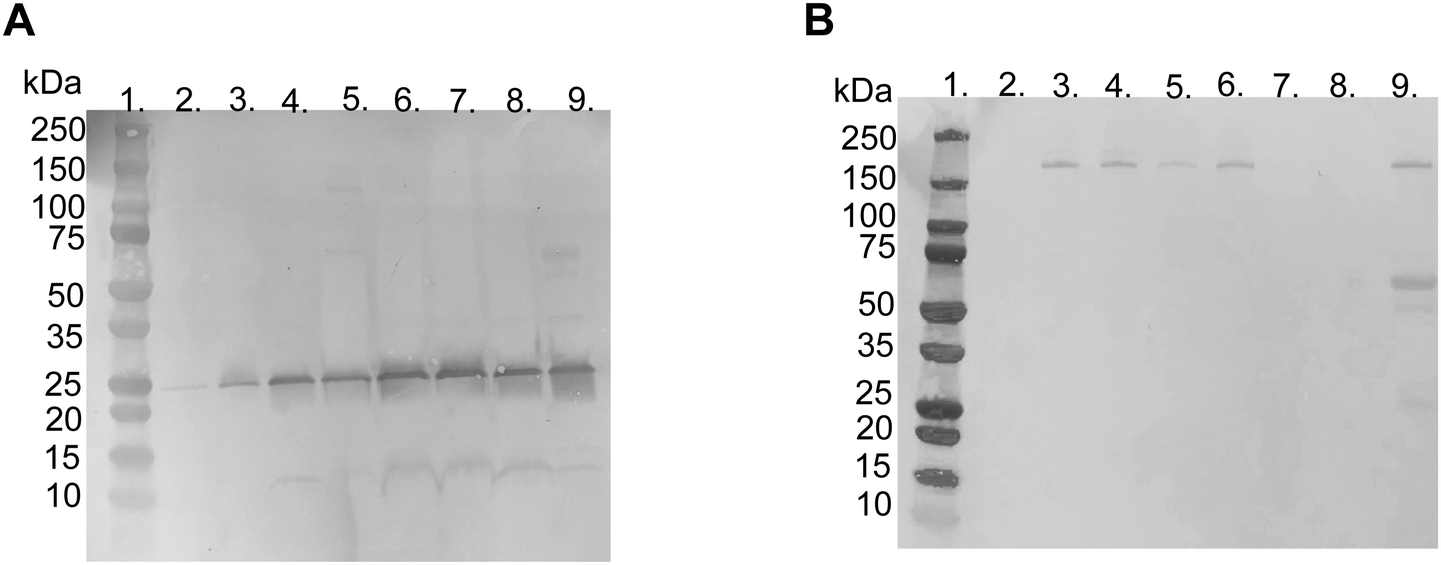
| Fig. 3 Representative picture of SDS-PAGE gels with Coomassie Brilliant Blue staining of proteins adsorbed to liposomes modified with 10 mol% polymer–lipid after incubation in EDTA-plasma. (A) Non-reduced and (B) reduced with DTT. Lane 1; ladder 10-250 kDa, lane 2; PBS, lane 3; non-modified liposomes, lane 4; PEGylated liposomes, lane 5; MPC10-, lane 6; MPC20-, lane 7; MPC50- and lane 8; MPC100–liposomes. The arrows indicate apolipoprotein A-I (25 kDa) and α2-macroglobulin (180 kDa). | ||
LC-MS/MS analysis was performed by extracting the 25 and 180 kDa bands from the gel (Fig. S3A and B, ESI†). The protein in the 25 kDa band was identified as apolipoprotein A-I (score 230), whereas that in the 180 kDa band was identified as α2-macroglobulin (score 761) (Table 1). Subsequently, we performed western blots for liposomes modified with 10 mol% polymer–lipids incubated in EDTA–plasma using antibodies for apolipoprotein A-I and α2-macroglobulin to verify the bands at 25 and 180 kDa. Apolipoprotein A-I was detected in all liposome preparations (Fig. 4A). Apolipoprotein A-I has previously been shown to be associated with non-modified and PEGylated liposomes.6,7 α2-Macroglobulin was detected on non-modified, PEG-, MPC10- and MPC20–liposomes (Fig. 4B). Similarly, the binding of α2-macroglobulin has been demonstrated in non-modified and PEGylated liposomes incubated in fetal bovine serum and citrated human plasma.6,36 Apolipoprotein A-I is a protein involved in the destabilization of lipid vesicles in the circulation37 and α2-macroglobulin is a plasma protease inhibitor with broad specificity.36,38 However, the possible effects of these proteins in the protein corona on the liposome circulation time still need to be investigated.
| Gel band | Highest scoring protein | Score | Sequence coverage (%) |
|---|---|---|---|
| 25 kDa | Apolipoprotein A-I | 230 | 74.9 |
| 180 kDa | α2-Macroglobulin | 761 | 44.9 |
 | ||
| Fig. 4 Western blot, under reduced conditions with DTT, using specific polyclonal antibodies for detection of (A) apolipoprotein A-I (25 kDa) and (B) α2-macroglobulin (180 kDa) binding to liposomes modified with 10 mol% polymer–lipid after incubation in human EDTA-plasma. Lane 1; ladder 10-250 kDa, lane 2; PBS, lane 3; non-modified liposomes, lane 4; PEGylated liposomes, lane 5; MPC10-, lane 6; MPC20-, lane 7; MPC50-, lane 8; MPC100–liposomes and lane 9; EDTA-plasma diluted 1/100. | ||
3.4. Quantification of C3 bound to liposomes
The binding of C3 to the liposomes following incubation in EDTA or lepirudin plasma was investigated using the Wes immunoassay. The opsonization of liposomes by C3 binding is correlated with decreased circulation time owing to the complement activation and uptake of liposomes by phagocytic cells.39,40 The passive binding of C3 to the liposomes was investigated by incubating the liposomes in human EDTA plasma. Insignificant amounts of C3 were passively absorbed into the liposomes (Fig. S4A–F, ESI†). This is expected because the complement system is not active in EDTA plasma.The active binding of C3 to liposomes was investigated using fresh human lepirudin plasma. Non-modified (15.0 ± 1.7 μg mL−1, p-value = 0.02), 1 mol% MPC10 (23.9 ± 3.0 μg mL−1, p-value = 0.0003), and 1 mol% MPC20 (16.7 ± 4.8 μg mL−1, p-value =0.008) liposomes bound more C3 when compared with liposomes containing 1 mol% PEG, inhibiting the binding of C3 (2.6 ± 0.4 μg mL−1). There was no significant difference between liposomes modified with 1 mol% PEG–lipids and 1 mol% MPC50–lipid (10.5 ± 5.3 μg mL−1) or 1 mol% MPC100–lipid (11.8 ± 3.8 μg mL−1) (Fig. 5A and B). Non-modified liposomes (15.0 ± 1.7 μg mL−1, p-value = 0.022) and liposomes containing 5 mol% MPC10–lipid (15.6 ± 9.46 μg mL−1, p-value = 0.017) bound more C3 when compared to liposomes with 5 mol% PEG–lipid (1.3 ± 0.04 μg mL−1). No difference in C3 binding was observed between liposomes modified with 5 mol% MPC20–lipid (2.7 ± 2.0 μg mL−1), MPC50–lipid (2.5 ± 1.6 μg mL−1) and MPC100–lipid (1.2 ± 0.2 μg mL−1) when compared with the liposomes containing 5 mol% PEG–lipid (Fig. 5C and D). Liposomes modified with 10 mol% PEG–lipid (1.7 ± 0.02 μg mL−1) clearly reduced the C3 binding when compared to non-modified (15.0 ± 1.7 μg mL−1, p-value = 0.04) and 10 mol% MPC10–lipid (16.0 ± 7.7 μg mL−1, p-value = 0.03) liposomes (Fig. 5E and F). No difference was observed between liposomes containing 10 mol% PEG–lipid and liposomes modified with 10 mol% MPC20–lipid (4.6 ± 1.8 μg mL−1), 10 mol% MPC50–lipid (2.5 ± 0.4 μg mL−1), and 10 mol% MPC100–lipid (9.7 ± 7.2 μg mL−1). These experiments showed that the proteins, in our case C3, in the liposome's protein corona differed depending of the plasma type used. This must be considered when designing experiments to investigate liposome interactions with plasma proteins, particularly with regard to complement proteins. The lepirudin plasma provided a better understanding of the activation of the complement system via modified liposomes in vivo. In Wes using the anti C3 β-chain, the native C3 and its activated form C3b or its degradation fragments such as iC3b and C3c could not be distinguished; hence, the form detected on the liposome surface could not be determined. Similarly, we observed that the 1 mol% PMPC–lipids could not adequately suppress C3 binding to the liposomes. Moreover, the polymer chain length was correlated with C3 binding. The short MPC10–lipid was insufficient to inhibit C3 binding, whereas both the 10 mol% MPC20- and 5 and 10 mol% MPC50–liposomes suppressed the total protein and C3 binding to liposomes; hence, these liposome compositions should be evaluated further. Additionally, the immunogenicity and ABC effect of the PMPC–liposomes in vivo should be investigated.
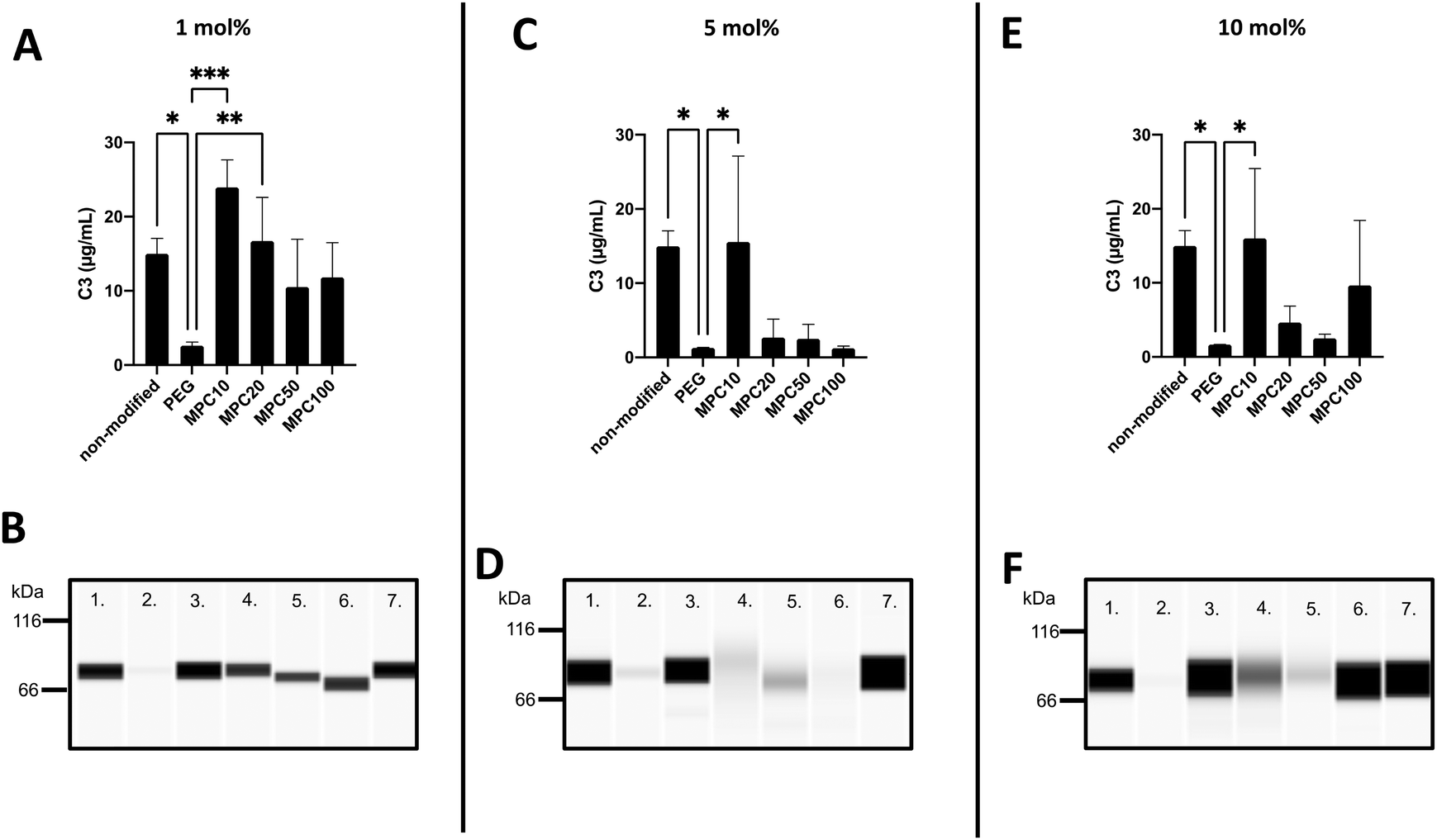 | ||
| Fig. 5 Wes immunoassay, under reduced conditions with DTT, using a specific monoclonal antibody against the C3 β-chain for detection of the active binding of C3 to liposomes modified with 1, 5 and 10 mol% polymer–lipids after incubation in lepirudin plasma (n = 3). (A, C, and E): Quantification of the amount of surface bound C3 to liposomes at a lipid concentration of 4 mM. (B, D, and F): Representative Wes virtual blots of lane 1; non-modified liposomes, lane 2; PEGylated liposomes, lane 3; MPC10-, lane 4; MPC20-, lane 5; MPC50-, lane 6; MPC100–liposomes and lane 7; 5 μg mL−1 native C3 (for reference). | ||
3.5. Phospholipase D-catalyzed hydrolysis of PMPC–lipids
Neither MPC10–lipid (2.30 mM Pi) nor MPC100–lipid (1.91 mM Pi) showed the color development in the presence of PLD. That is, PLD could not catalyze the hydrolysis of the phosphorylcholine moieties of the PMPC–lipids within the time required to hydrolyze the phosphatidylcholines. It is considered that: (i) bulky PLD molecules hardly access the vicinity of phosphorylcholine moieties in long polymer chains due to steric hindrance (e.g., thick hydration layer, overlapping or entangled main chains as well as phosphorylcholine branches, etc.); and/or (ii) phosphorylcholine moieties conjugated to the polymer chain might not be available as a substrate for PLD due to lower specificity.4. Conclusions
We fabricated liposomes modified with 1, 5, and 10 mol% PEG-, MPC10-, MPC20-, MPC50- and MPC100–lipids and then investigated their interactions with human plasma proteins. Apolipoprotein A-I was to a larger extent associated with PMPC–liposomes with a higher molar ratio of PMPC–lipids and with longer polymer chains, whereas the binding of α2-macroglobulin was observed for non-modified, PEGylated, and PMPC–liposomes with a shorter polymer chain. Liposome modification with PMPC–lipids could be a possible strategy to avoid complement activation, in particular 10 mol% MPC20–lipid and 5 mol% and 10 mol% MPC50–lipid, which can suppress protein binding, particularly the binding of C3.Conflicts of interest
The authors declare no conflict of interest.Author contributions
AA and YT designed the research project. AA, YT and TB performed the experiments. AA and YT wrote the manuscript with editorial assistance from YI, KNE, KI and BN. All authors have read and approved the final manuscript.Acknowledgements
This work was supported by the Mass Spectrometry Based Proteomics Facility in Uppsala. This research was supported in part by the Bilateral Joint Research Project (Japan–Sweden) of the Japan Society for the Promotion of Science (JSPS) and STINT; a Grant-in-Aid for Scientific Research (B) (26702017), a Grant-in-Aid for Scientific Research for Fostering Joint International Research (15KK0230, 18KK0305), and a Grant-in-Aid for Challenging Research (Exploratory) (19K22951) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan; StemTherapy; and grants 2018-04199, 2016-2075-5.1, and 2016-04519 from the Swedish Research Council. The research project RELIEF has received funding from the Eurostars-2 joint program (project ID: E! 113670) with co-funding from the European Union Horizon 2020 Research and Innovation Program and further co-funding for the French, German, Dutch, and Swedish partners from BPIfrance, the German Federal Ministry of Education and Research, RVO, VINNOVA.References
- A. Akbarzadeh, R. Rezaei-Sadabady, S. Davaran, S. W. Joo, N. Zarghami and Y. Hanifehpour, et al., Liposome: classification, preparation, and applications, Nanoscale Res. Lett., 2013, 8(1), 102 CrossRef PubMed.
- M. L. Immordino, F. Dosio and L. Cattel, Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential, Int. J. Nanomed., 2006, 1(3), 297–315 CrossRef CAS PubMed.
- C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. Chan, Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake, J. Am. Chem. Soc., 2012, 134(4), 2139–2147 CrossRef CAS PubMed.
- J. Szebeni and S. M. Moghimi, Liposome triggering of innate immune responses: a perspective on benefits and adverse reactions, J. Liposome Res., 2009, 19(2), 85–90 CrossRef CAS.
- T. M. Allen and P. R. Cullis, Liposomal drug delivery systems: from concept to clinical applications, Adv. Drug Delivery Rev., 2013, 65(1), 36–48 CrossRef CAS PubMed.
- S. Palchetti, V. Colapicchioni, L. Digiacomo, G. Caracciolo, D. Pozzi and A. L. Capriotti, et al., The protein corona of circulating PEGylated liposomes, Biochim. Biophys. Acta, Biomembr., 2016, 1858(2), 189–196 CrossRef CAS PubMed.
- C. Weber, M. Voigt, J. Simon, A. K. Danner, H. Frey and V. Mailander, et al., Functionalization of Liposomes with Hydrophilic Polymers Results in Macrophage Uptake Independent of the Protein Corona, Biomacromolecules, 2019, 20(8), 2989–2999 CrossRef CAS PubMed.
- E. T. Dams, P. Laverman, W. J. Oyen, G. Storm, G. L. Scherphof and J. W. van Der Meer, et al., Accelerated blood clearance and altered biodistribution of repeated injections of sterically stabilized liposomes, J. Pharmacol. Exp. Ther., 2000, 292(3), 1071–1079 CAS.
- S. M. Moghimi, A. J. Andersen, S. H. Hashemi, B. Lettiero, D. Ahmadvand and A. C. Hunter, et al., Complement activation cascade triggered by PEG-PL engineered nanomedicines and carbon nanotubes: the challenges ahead, J. Controlled Release, 2010, 146(2), 175–181 CrossRef CAS PubMed.
- T. Ishida, R. Maeda, M. Ichihara, K. Irimura and H. Kiwada, Accelerated clearance of PEGylated liposomes in rats after repeated injections, J. Controlled Release, 2003, 88(1), 35–42 CrossRef CAS PubMed.
- T. Ishida, M. Ichihara, X. Wang, K. Yamamoto, J. Kimura and E. Majima, et al., Injection of PEGylated liposomes in rats elicits PEG-specific IgM, which is responsible for rapid elimination of a second dose of PEGylated liposomes, J. Controlled Release, 2006, 112(1), 15–25 CrossRef CAS PubMed.
- T. Ishida and H. Kiwada, Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes, Int. J. Pharm., 2008, 354(1-2), 56–62 CrossRef CAS PubMed.
- P. Laverman, M. G. Carstens, O. C. Boerman, E. T. Dams, W. J. Oyen and N. van Rooijen, et al., Factors affecting the accelerated blood clearance of polyethylene glycol-liposomes upon repeated injection, J. Pharmacol. Exp. Ther., 2001, 298(2), 607–612 CAS.
- A. W. Richter and E. Akerblom, Polyethylene glycol reactive antibodies in man: titer distribution in allergic patients treated with monomethoxy polyethylene glycol modified allergens or placebo, and in healthy blood donors, Int. Arch. Allergy Appl. Immunol., 1984, 74(1), 36–39 CrossRef CAS PubMed.
- Q. Yang and S. K. Lai, Anti-PEG immunity: emergence, characteristics, and unaddressed questions, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7(5), 655–677 CAS.
- G. Besin, J. Milton, S. Sabnis, R. Howell, C. Mihai and K. Burke, et al., Accelerated Blood Clearance of Lipid Nanoparticles Entails a Biphasic Humoral Response of B-1 Followed by B-2 Lymphocytes to Distinct Antigenic Moieties, Immunohorizons, 2019, 3(7), 282–293 CrossRef CAS PubMed.
- R. Fiskesund, J. Steen, K. Amara, F. Murray, A. Szwajda and A. Liu, et al., Naturally occurring human phosphorylcholine antibodies are predominantly products of affinity-matured B cells in the adult, J. Immunol., 2014, 192(10), 4551–4559 CrossRef CAS PubMed.
- Y. Cruz-Leal, A. Lopez-Requena, I. Lopetegui-Gonzalez, Y. Machado, C. Alvarez and R. Perez, et al., Phosphocholine-Specific Antibodies Improve T-Dependent Antibody Responses against OVA Encapsulated into Phosphatidylcholine-Containing Liposomes, Front. Immunol., 2016, 7, 374 Search PubMed.
- K. Ishihara, Revolutionary advances in 2-methacryloyloxyethyl phosphorylcholine polymers as biomaterials, J. Biomed. Mater. Res., Part A, 2019, 107A, 933–943 CrossRef PubMed.
- K. Ishihara, T. Ueda and N. Nakabayashi, Preparation of Phospholipid Polymers and Their Properties as Polymer Hydrogel Membranes, Polym. J., 1990, 22, 355–360 CrossRef CAS.
- W. Lin, N. Kampf, R. Goldberg, M. J. Driver and J. Klein, Poly-phosphocholinated Liposomes Form Stable Superlubrication Vectors, Langmuir, 2019, 35(18), 6048–6054 CrossRef CAS PubMed.
- A. Adler, Y. Inoue, Y. Sato, K. Ishihara, K. N. Ekdahl and B. Nilsson, et al., Synthesis of poly(2-methacryloyloxyethyl phosphorylcholine)-conjugated lipids; its characterization and surface properties of modified liposomes for protein interactions, Biomater. Sci., 2021, 9, 5854–5867 RSC.
- C. H. Hammer, G. H. Wirtz, L. Renfer, H. D. Gresham and B. F. Tack, Large scale isolation of functionally active components of the human complement system, J. Biol. Chem., 1981, 256(8), 3995–4006 CrossRef CAS.
- B. Nilsson, K. E. Svensson, P. Borwell and U. R. Nilsson, Production of mouse monoclonal antibodies that detect distinct neoantigenic epitopes on bound C3b and iC3b but not on the corresponding soluble fragments, Mol. Immunol., 1987, 24(5), 487–494 CrossRef CAS PubMed.
- M. Takayama, S. Itoh, T. Nagasaki and I. Tanimizu, A new enzymatic method for determination of serum choline-containing phospholipids, Clin. Chim. Acta, 1977, 79(1), 93–98 CrossRef CAS.
- K. Tamaoku, K. Ueno, K. Akiura and Y. Ohkura, New water-soluble hydrogen donors for the enzymatic photometric determination of hydrogen peroxide. II. N-ethyl-N-(2-hydroxy-3-sulfopropyl)aniline derivatives, Chem. Pharm. Bull., 1982, 30(7), 2492–2497 CrossRef CAS.
- G. R. Bartlett, Phosphorus assay in column chromatography, J. Biol. Chem., 1959, 234(3), 466–468 CrossRef CAS.
- M. Danaei, M. Dehghankhold, S. Ataei, F. Hasanzadeh Davarani, R. Javanmard and A. Dokhani, et al., Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems, Pharmaceutics, 2018, 10(2), 57 CrossRef PubMed.
- F. Giulimondi, L. Digiacomo, D. Pozzi, S. Palchetti, E. Vulpis and A. L. Capriotti, et al., Interplay of protein corona and immune cells controls blood residency of liposomes, Nat. Commun., 2019, 10(1), 3686 CrossRef CAS PubMed.
- G. Caracciolo, Liposome-protein corona in a physiological environment: challenges and opportunities for targeted delivery of nanomedicines, Nanomedicine, 2015, 11(3), 543–557 CrossRef CAS PubMed.
- D. Pozzi, V. Colapicchioni, G. Caracciolo, S. Piovesana, A. L. Capriotti and S. Palchetti, et al., Effect of polyethyleneglycol (PEG) chain length on the bio-nano-interactions between PEGylated lipid nanoparticles and biological fluids: from nanostructure to uptake in cancer cells, Nanoscale, 2014, 6(5), 2782–2792 RSC.
- Y. Teramura, K. Kuroyama and M. Takai, Influence of molecular weight of PEG chain on interaction between streptavidin and biotin-PEG-conjugated phospholipids studied with QCM-D, Acta Biomater., 2016, 30, 135–143 CrossRef CAS PubMed.
- H. I. Chang and M. K. Yeh, Clinical development of liposome-based drugs: formulation, characterization, and therapeutic efficacy, Int. J. Nanomed., 2012, 7, 49–60 CAS.
- U. Bulbake, S. Doppalapudi, N. Kommineni and W. Khan, Liposomal Formulations in Clinical Use: An Updated Review, Pharmaceutics, 2017, 9(2), 12 CrossRef PubMed.
- K. Yoshimoto, T. Hirase, J. Madsen, S. P. Armes and Y. Nagasaki, Non-Fouling Character of Poly[2-(methacryloyloxy)ethyl Phosphorylcholine]-Modified Gold Surfaces Fabricated by the 'Grafting to' Method: Comparison of its Protein Resistance with Poly(ethylene glycol)-Modified Gold Surfaces, Macromol. Rapid Commun., 2009, 30(24), 2136–2140 CrossRef CAS PubMed.
- M. E. Price, R. M. Cornelius and J. L. Brash, Protein adsorption to polyethylene glycol modified liposomes from fibrinogen solution and from plasma, Biochim. Biophys. Acta, Biomembr., 2001, 1512(2), 191–205 CrossRef CAS.
- P. Sirnio, J. P. Vayrynen, K. Klintrup, J. Makela, M. J. Makinen and T. J. Karttunen, et al., Decreased serum apolipoprotein A1 levels are associated with poor survival and systemic inflammatory response in colorectal cancer, Sci. Rep., 2017, 7(1), 5374 CrossRef PubMed.
- S. Yoshino, K. Fujimoto, T. Takada, S. Kawamura, J. Ogawa and Y. Kamata, et al., Molecular form and concentration of serum alpha2-macroglobulin in diabetes, Sci. Rep., 2019, 9(1), 12927 CrossRef PubMed.
- T. Ishida, H. Kojima, H. Harashima and H. Kiwada, Biodistribution of liposomes and C3 fragments associated with liposomes: evaluation of their relationship, Int. J. Pharm., 2000, 205(1-2), 183–193 CrossRef CAS PubMed.
- Y. Klapper, O. A. Hamad, Y. Teramura, G. Leneweit, G. U. Nienhaus and D. Ricklin, et al., Mediation of a non-proteolytic activation of complement component C3 by phospholipid vesicles, Biomaterials, 2014, 35(11), 3688–3696 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tb01485d |
| This journal is © The Royal Society of Chemistry 2022 |