
One-step electrochemical deposition of antifouling polymers with pyrogallol for biosensing applications
Shang-Lin
Yeh
a,
Piyush
Deval
 a,
Jhih-Guang
Wu
b,
Shyh-Chyang
Luo
b and
Wei-Bor
Tsai
*a
a,
Jhih-Guang
Wu
b,
Shyh-Chyang
Luo
b and
Wei-Bor
Tsai
*a
aDepartment of Chemical Engineering, National Taiwan University, No. 1, Sec. 4, Roosevelt Rd., Taipei, 10617, Taiwan. E-mail: weibortsai@ntu.edu.tw
bDepartment of Material Science and Engineering, National Taiwan University, No. 1, Sec. 4, Roosevelt Rd., Taipei, 10617, Taiwan
First published on 31st December 2021
Abstract
Electrochemical techniques are highly sensitive and label-free sensing methods for the detection of various biomarkers, toxins, or pathogens. An ideal sensing element should be electroconductive, nonfouling, and readily available for conjugation of ligands. In this work, we have developed a facile, one-step electrodeposition method based on pyrogallol polymerization for preparation of a nonfouling and biotinylated surface on indium tin oxide (ITO). A copolymer of sulfobetaine methacrylate and aminoethyl methacrylate (pSBAE) was synthesized and deposited on ITO in the presence of pyrogallol via cyclic voltammetry. The deposition took less than 15 minutes to sufficiently inhibit cell adhesion. Using biotinylated pSBAE, the modified surface resisted nonspecific protein adsorption from the fetal bovine serum solution and detected added avidin concentrations. The results show an efficient platform to fabricate an electrochemical biosensor for the detection of biomarkers. We expect that this facile one-step technology could be applied to conjugate various biosensing elements for nonfouling biosensors.
Introduction
Electrochemical techniques, such as cyclic voltammetry and electrochemical impedance spectroscopy (EIS), are highly sensitive and label-free sensing methods for the detection of various biomarkers, toxins, or pathogens.1 EIS applies a redox probe, such as Fe(CN)63−/4− to measure the ability of Fe ions to become oxidized and reduced at a working electrode.2 Impedimetric biosensors make use of the interactions of biomolecules with a conductive transducer surface. On a pristine working electrode, Fe ions could be oxidized and reduced without hinderance. When the working electrode is covered with proteins or other biomolecules, Fe ion transport is impeded, so the charge transfer resistance (Rct) of the circuit increases.3 Biomolecule attachment could be determined via Rct. Because of the affordability and availability of impedance instrumentation, the technique has become one of the popular types of electrochemical biosensing platform.For the construction of electrochemical biosensors, ideal biosensing elements should be electroconductive, nonfouling, and readily used for conjugation of ligands that recognize specific target biomolecules. A simple and reliable surface modification technology to meet the requirements of electrochemical biosensors is highly desirable. In recent years, the poly(dopamine) (PDA) deposition technique,4 based on spontaneous self-polymerization of dopamine (DA) under alkaline conditions (pH 8.5), has attracted much attention in the construction of biosensors. DA polymerization forms a thin polydopamine ad-layer that strongly interacts with various organic and inorganic materials, such as polymers, metals, and metal oxides.5,6 The PDA ad-layer contains many functional groups such as quinone, catechol, amine, and imine, which could bind ligands.7 Furthermore, the PDA deposition process cannot only be initiated by self-oxidation at alkaline pH, but also electrochemical stimuli using cyclic voltammetry.8,9 The PDA ad-layer prepared by electrodeposition can also assist immobilization of molecules on the conductive surfaces for further applications. Characterization of PDA makes it a good technique for modification of electrochemical biosensors.10–12
Bioelectrodes generally experience surface biofouling, triggered by nonspecific attachment of proteins, lipids, cells, and microorganisms. Surface modification with antifouling hydrophilic polymers, such as polysaccharides, poly(ethylene glycol) (PEG), and poly(zwitterions), could greatly reduce nonspecific binding of biomolecules. These highly hydrophilic polymers adsorb a great amount of water to form a barrier to reduce nonspecific interactions between surfaces and biological systems.13 A recent study used PDA as a platform to co-immobilize thiolated PEG and thiolated aptamers through the Michael addition reaction as a biosensor, detecting adenosine triphosphate.10 The study demonstrated that in the presence of PEG protection, the aptasensor was capable of sensing adenosine triphosphate in human plasma with a significantly reduced nonspecific adsorption effect. Similarly, a recent study constructed sensitive and antifouling biosensors via copolymerization of PDA and poly(sulfobetaine), followed by attachment of thiolates aptamers to the PDA for detection of carcinoembryonic antigen.14
In addition to dopamine, many polyphenolic molecules possess similar characteristics for surface modifications.15–17 An advantage of polyphenolic compounds is their lower price compared to dopamine. Polyphenolic molecules, such as pyrogallol, tannic acid and gallic acid, containing catecholic or pyrrolic moieties, are prone to oxidization to quinones and further self-polymerize on most materials under oxidized conditions. It has been demonstrated that electrodeposition of polyphenolic molecules on the surface of conductive materials induces the formation of a conformal coating without aggregates of reacted monomers in the bulk of the solution.18–22
The aim of this study was to fabricate a biosensing platform with antifouling ability using one-step electrodeposition of an antifouling polymer. A model binding system, avidin/biotin, was used to demonstrate the feasibility of the platform. One major distinguishing feature of the avidin/biotin system is its extraordinarily high affinity (the affinity constant, Ka, is 1013–15 M−1), which is extremely stable under harsh pH or temperature conditions.23 Avidin/biotin technology has been extensively applied to biotechnology, such as affinity chromatography, histochemistry, diagnostics, immunoassay, and drug delivery.24 Our group previously used the avidin/biotin system to immobilize cell adhesive peptides on polydopamine surfaces to elicit specific cell adhesion.25 In this study, biotin molecules were conjugated to polysulfobetaine as ligands, and then deposited on Indium tin oxide (ITO) glass via electrodeposition with pyrogallol. The ability of the modified ITO to resist nonspecific biointeractions and to detect avidin in serum solutions was demonstrated in this study.
Materials and methods
Materials
All chemicals were commercial products and were used without further purification. Sulfobetaine methacrylate (SBMA) was purchased from Taiwan Hopax Chemicals (Kaohsiung, Taiwan). Azobis(isobutyronitrile) (AIBN) and potassium hexacyanoferrate(II) trihydrate were purchased from Alfa Aesar (USA). Potassium hexacyanoferrate(III) was received from Acros Organics (USA). All other materials were obtained from Sigma-Aldrich (USA): 2-aminoethylmethacrylamide hydrochloride (AEMA), pyrogallol (PG), N-hydroxysuccinimide (NHS), N-(3-dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (EDC), dimethyl sulfoxide (DMSO), biotin, colorimetric biotin assay kit, avidin from egg white.The aqueous solutions were prepared from Milli Q water (Millipore, Molsheim, France) with a resistivity of 18.2 MΩ cm. Indium tin oxide (ITO) glass (0.7 mm in thickness and 7 Ω in resistivity) was purchased from Uni-Onward Corp., Taiwan.
Synthesis and characterization of poly(SBMA-co-AEMA) (pSBAE) and biotinylated pSBAE
A copolymer of SBMA and AEMA was synthesized by free radical polymerization, according to a previous protocol.17 The SBMA/AEMA ratio was optimized in our previous studies.17,26 Briefly, 850 mg (3 mmol) SBMA and 49 mg (0.3 mmol) AEMA were dissolved in 18 mL of deionized water and then mixed with 5 mg (0.03 mmol) AIBN that was dissolved in 2 mL of DMSO, followed by nitrogen purge. Polymerization was initiated at 70 °C and the reaction proceeded for 20 hours. The unwanted impurities were removed by dialysis with deionized water. The compositions of the synthesized copolymers were verified by 1H nuclear magnetic resonance (Bruker AVIII HD 400 NMR, Germany). The molecular weight of the copolymer was determined by gel permeation chromatography (Jasco, UV-2075, Japan). The Mn and Mw of pSBAE were 57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 787 Da and 130083 Da, respectively, with a polydisperse index of 2.25, while the molar ratio of SB/AE in the copolymer was 10.7.17
787 Da and 130083 Da, respectively, with a polydisperse index of 2.25, while the molar ratio of SB/AE in the copolymer was 10.7.17
For the synthesis of biotinylated pSBAE (b-pSBAE), 30 mg pSBAE was dissolved in 1 mL of 0.1 M MES buffer (pH 5.5) in the presence of biotin at 12, 36 or 100 μg mL−1. After the addition of 31 mg (0.2 M) EDC and 5.7 mg (0.05 M) NHS, the mixture was incubated in the dark for 20 hours with constant stirring. After the reaction, the solution was dialyzed against deionized water to remove impurities. The ratio of conjugated biotin to pSBAE was quantified using a biotin quantification kit (MK171, Sigma-Aldrich, USA). The grafting ratios for b1-pSBAE (12 μg mL−1), b2-pSBAE (36 μg mL−1) and b3-pSBAE (100 μg mL−1) were 8.89%, 16.30% and 45.93%, respectively.
Electrodeposition of PG/pSBAE
The ITO glass was cut into 1 cm × 2.5 cm slides, and cleaned under sonication with a series of solvents: methanol, 0.5 M K2CO3 in 3:1 methanol/deionized water, and finally deionized water.
The scheme of the electrodeposition setup is shown in Fig. 1. ITO was used as a working electrode, while a platinum coil and a silver chloride electrode (Ag/AgCl) were used as the counter and reference electrodes, respectively. Pyrogallol was electrodeposited on ITO slides. Electrodeposition was initiated by performing cyclic voltammetry between −0.8 V and 1.2 V versus the Ag/AgCl electrode with a scan rate of 0.02 V s−1 for 10 cycles. After electrodeposition, the modified ITO slides were washed copiously with deionized water and dried in air at room temperature. For codeposition of PG/pSBAE, PG (8 mg mL−1) and pSBAE (30 mg mL−1) dissolved in 0.1 M phosphate buffer, pH 5.0. All reactions were performed in an electrochemical cell at room temperature.
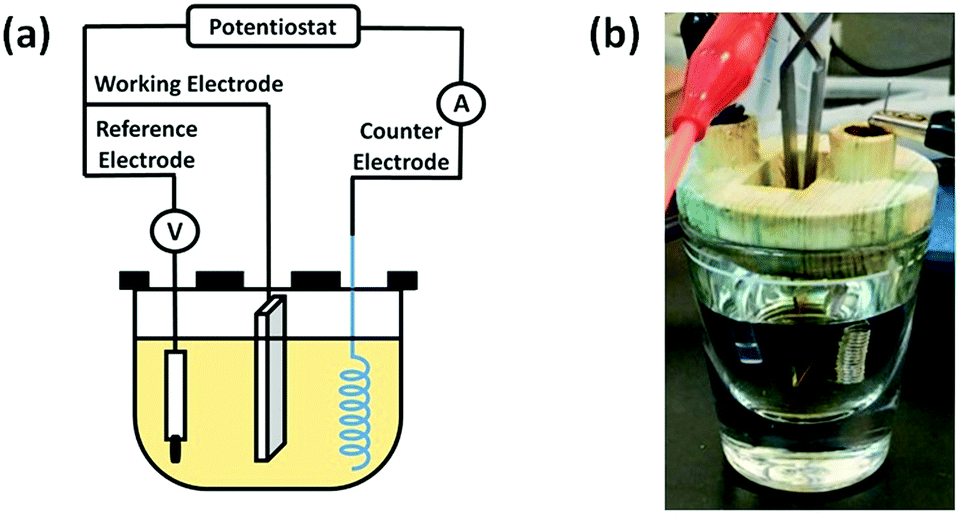 | ||
| Fig. 1 (a) Schematic diagram of an electrodeposition apparatus: ITO as the working electrode, platinum coil as the counter electrode, and an Ag/AgCl electrode as the reference electrode. (b) A photo of an electrodeposition setup. | ||
Characterization of electro-deposited ITO
The electrochemical characterization of the modified ITO was evaluated by cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) measurement using the Electrochemical Working Station Autolab System (ECO CHEMIE Metrohm, PGSTAT128N, Switzerland). CV was used to evaluate the coating characteristics by examining the changes in current values with coating cycles. The EIS measurement was used to determine the change in surface resistance with coating cycles. The electrolyte used was 10 mM K3Fe(CN)6/K4Fe(CN)6 and 0.1 M KCl.The static water contact angle (WCA) apparatus (FTA-125, First Ten Angstroms, USA) was used to determine the surface wettability. Water droplets (5 μL) were placed on the samples and then the digital images were collected. The water contact angles were measured from the images. For every sample, 10 water contact angle measurements were taken at different points. The reported WCA values were averaged from three samples of each type of surface.
The surface morphology and topography were examined using a scanning electron microscope (SEM, NovaTM NanoSEM 230, Scanservice, USA) and an atomic force microscope (AFM, NanoScope IIIa, Digital Instruments, USA). The samples sputter-coated with a ∼10 nm gold film prior to SEM examination.
Protein adsorption to modified ITO
The modified surfaces were incubated in the fetal bovine serum (FBS) solutions from 0.1%, 1%, 5% to 10% in phosphate buffered saline (PBS, pH 7.4) for 1 hour at 37 °C. After adsorption, the surface charge resistance was measured using the electrochemical setup with three electrode system.Cell adhesion to modified ITO
Before cell seeding, the samples were sterilized by UV irradiation for 2 hours and then soaked in 75% ethanol for 30 minutes. L929 cells were seeded on the samples with a cell density of 2 × 104 cells per cm2 and cultured in an incubator with a 5% CO2 atmosphere at 37 °C for 24 hours. Attached cells were fixed with 4% paraformaldehyde and then cell images were taken using a phase contrast microscope. The numbers of attached cells were counted from the images.Statistical analysis
Data were reported as mean ± standard deviation (SD). Statistical analyzes between different groups were determined using the Student's t test. Probabilities of p < 0.05 were considered a significant difference. All statistical analyses were performed using GraphPad Instat 3.0 program (GraphPad Software, La Jolla, CA).Results and discussion
CV electrodeposition of ITO with PG/pSBAE
Electrodeposition of PG or PG/pSBAE on ITO was performed via CV in 0.1 M phosphate buffer, pH 5.0. Acidic pH was chosen to avoid self-polymerization of pyrogallol in solution.27–29 Electric potential between −0.8 V–1.2 V vs. Ag/AgCl with a scan rate of 20 mV s−1 was used as fixed electrical parameters for several CV cycles (1, 2, 4, 6, 8 and 10). When a potential was applied, the transparent solution near the working electrode turned light brown to dark brown, indicating that an electrochemical event of PG oxidation took place around the working electrode (Fig. 1B).The first cycle in the PG cyclic voltammogram was examined in Fig. 2A. An oxidation peak appeared around 0.56 V, which is caused by the oxidation of hydroxyl groups in pyrogallol, while a small reduction peak occurs at ∼0.05 V on the reverse scan. Furthermore, the oxidation peak potential shifted toward more positive potentials, accompanied by a rapid decrease in the peak current with an increasing number of cycles. The result indicates that pyrogallol oxidation is irreversible, consistent with a previous report.30 The oxidation step leads to the formation of orthoquinones, which are transformed into ortho-semiquinones in the reduction step. Ortho-semiquinones form oligomers as the origin of the film deposition.31 A PG film gradually thickened on ITO during the oxidation process of pyrogallol, and resulted in a gradual decrease in the current. The cyclic voltammogram of PG/pSBAE was similar to that of PG (Fig. 2B).
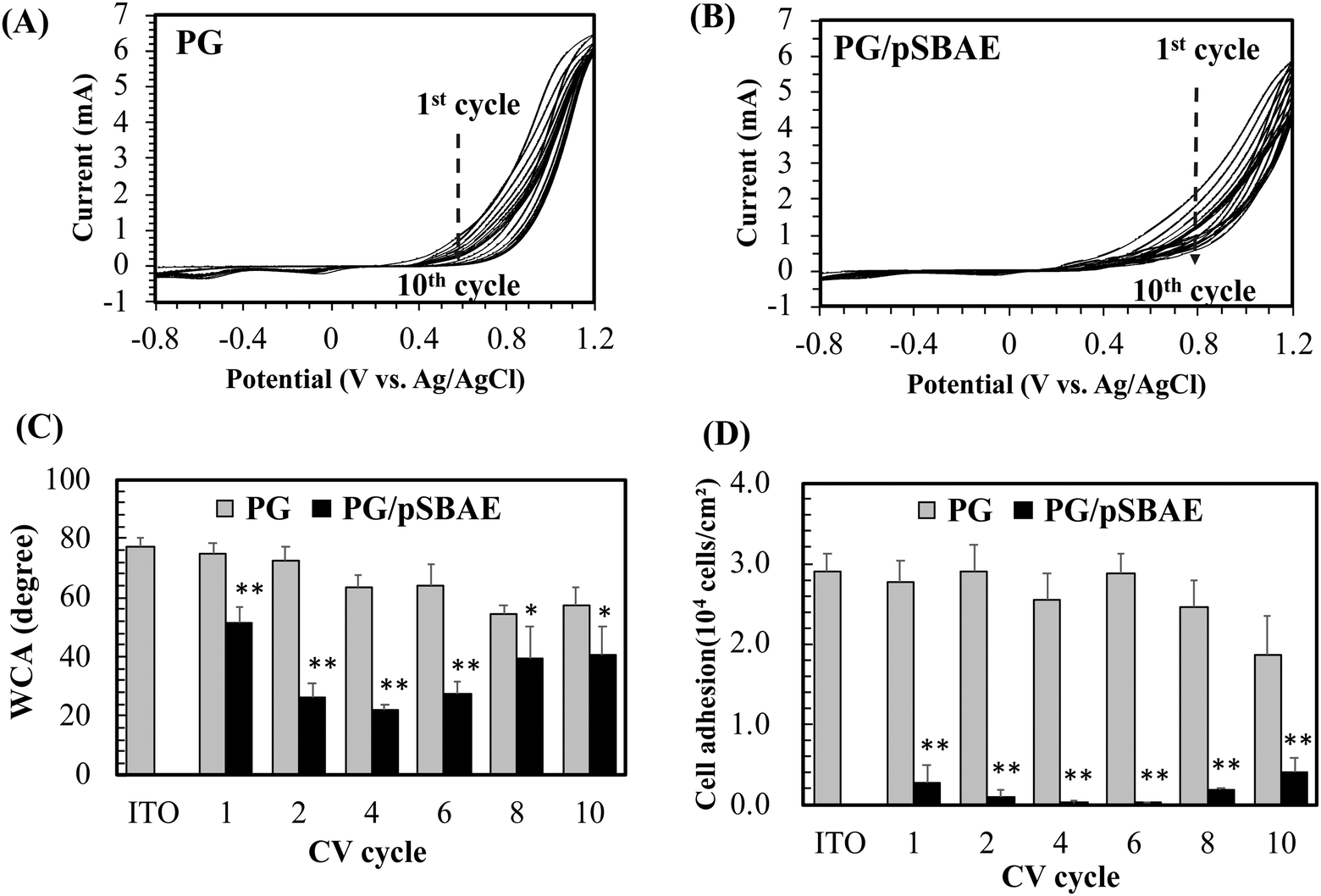 | ||
| Fig. 2 (A) The cyclic voltammograms of PG electrodeposition on ITO for 10 cycles; (B) the cyclic voltammograms of PG/pSBAE electrodeposition on ITO for 10 cycles; (C) static water contact angle (WCA) measurement on ITO electrodeposited with PG and PG/pSBAE. (D) Cell adhesion to ITO electrodeposited with PG and PG/pSBAE. * and ** indicate p < 0.05 and 0.001, respectively, vs. the corresponding PG. Value = mean ± standard deviation, n = 3. | ||
The surface hydrophilicity, determined by static water contact angle measurement, was monitored during CV cycles. Bare ITO was relatively hydrophobic with WCA of 77.3°. The PG deposition made ITO more hydrophilic. The WCA decreased to 75.1° after one cycle, and further decreased to 57.3° after ten cycles (Fig. 2C). The surface hydrophilicity increased further when the surface was modified with polyphenol-assisted pSBAE coatings. Deposition of PG/pSBAE further increased surface hydrophilicity. The WCA decreased to 51.6° after one cycle, reached a minimum of 21.8° after four cycles, and increased to 40.4° after 10 cycles (Fig. 2C). We guess that during the coating cycles, the concentration of pSBAE in the solution decreases faster than that of PG. Thus, at a higher cycle, e.g. 10, the ratio of PG to pSBAE in the solution was raised and thus more PG was deposited on the top layer, resulting in decreasing surface hydrophilicity. The trend of WCA measurement is similar to our study in the deposition of PG/pSBAE on PDMS through a one-step immersion deposition under alkaline conditions.17 WCA decreased to a minimum after 12 hours of soaking and increased slightly after 24 hours. Using electrodeposition, the time required for minimal WCA decreased greatly to less than 15 minutes (4 cycles) from spontaneous immersion deposition of 12 hours.
Zwitterionic molecules form a stable hydration shell via strong electrostatic induced hydration, which repels the nonspecific adsorption of biomolecules on the surface.32,33 A more hydrophilic surface tends to be more antifouling. Next, the resistance to cell adhesion on the modified ITO was studied. After one CV cycle, cell adhesion decreased greatly to 9.1% or 9.6% of that on ITO or PG (Fig. 2D). After 4 CV cycles, cell adhesion was almost inhibited, less than 0.5% of cell adhesion to ITO. Further CV cycles slightly increased cell adhesion. The decrease in anti-cell adhesion performance with increasing CV cycles is probably due to the local depletion of pSBAE in the PG/pSBAE solution around ITO. Therefore, an increased amount of PG was deposited on the topmost layers. The results indicated that PG/pSBAE could be deposited on ITO via CV. Compared to our previous immersion deposition study which showed that 12 hours is needed to reach a minimum cell adhesion,17 the fabrication time decreased to less than 15 minutes by electrodeposition. The results indicate that electrodeposition is an efficient method to deposit antifouling polymers on ITO. Because the best anti-cell adhesion effect appeared in the fourth cycle, the condition was used for subsequent biosensing experiments.
SEM was used to observe the topography of the PG-modified surfaces. PG electrodeposition appeared as particulate aggregates, whose size increases with CV cycles (Fig. 3). After 10 cycles, a few fibrous structures appeared. On the other hand, electrodeposition of PG/pSBAE created a feature of marble stripes after 4 CV cycles, and the structure became denser after 10 CV cycles. The SEM images show that incorporation of pSBAE facilitates the formation of fibrous deposition on ITO.
 | ||
| Fig. 3 The SEM images of electrodeposited PG and PG/pSBAE coatings on ITO for 1, 4 and 10 cycles. | ||
A similar observation was also confirmed using AFM. No obvious change in surface topography was found after ITO was deposited with PG for 1 or 4 cycles (Fig. 4). After ten cycles of deposition, a feature of interwoven fibers was found on ITO, and surface roughness increased significantly from 5.7 nm for ITO to 50.9 nm. On the other hand, a fibrous feature was found on PG/pSBAE after one CV cycle, and became more prominent with increasing CV cycles (Fig. 4). The fiber length ranged from 50 to 250 nm. Surface roughness also increased rapidly from 45.1 nm after 1 CV cycle to 106 nm after 10 CV cycles.
 | ||
| Fig. 4 AFM images of surface topography of bare ITO, and ITO electrodeposited with PG and PGSB for 1, 4 and 10 cycles. Scale = 1.0 μm. | ||
It is interesting to note that when PG is electrodeposited, an interwoven fiber-like structure appears, as reported previously.30 On the other hand, the PG coating by immersion deposition does not exhibit interwoven fiber-like structures.34 This phenomenon suggests that PG polymerization/deposition on a substrate via electrodeposition and immersion deposition might occur differently. In immersion deposition, PG polymerization occurs in the bulk solution and then deposits onto a substrate as particulate forms. On at other hand, the electrodeposition the oxidation/reduction reaction occurs on the surface of the working electrode, so PG polymerization is initiated on the working electrode, and the PG polymerization develops into a fibrous structure.
Electrodeposition of PG/b-pSBAE
For biosensor application, the surface should be conjugated with ligands in order to detect the corresponding molecules. Biotin molecules were conjugated to pSBAE and then deposited on ITO. Three types of biotinylated pSBAE were synthesized: b1-pSBAE, b2-pSBAE and b3-pSBAE, of which the grafting ratios were 8.89, 16.30 and 45.93%, respectively. b-pSBAE was co-electrodeposited with PG to ITO. The measurement of WCA measurement indicated that the hydrophilicity of PG/b1-pSBAE was similar to that of PG/pSBAE, while PG/b2-pSBAE and PG/b3-pSBAE were more hydrophobic (Fig. 5A).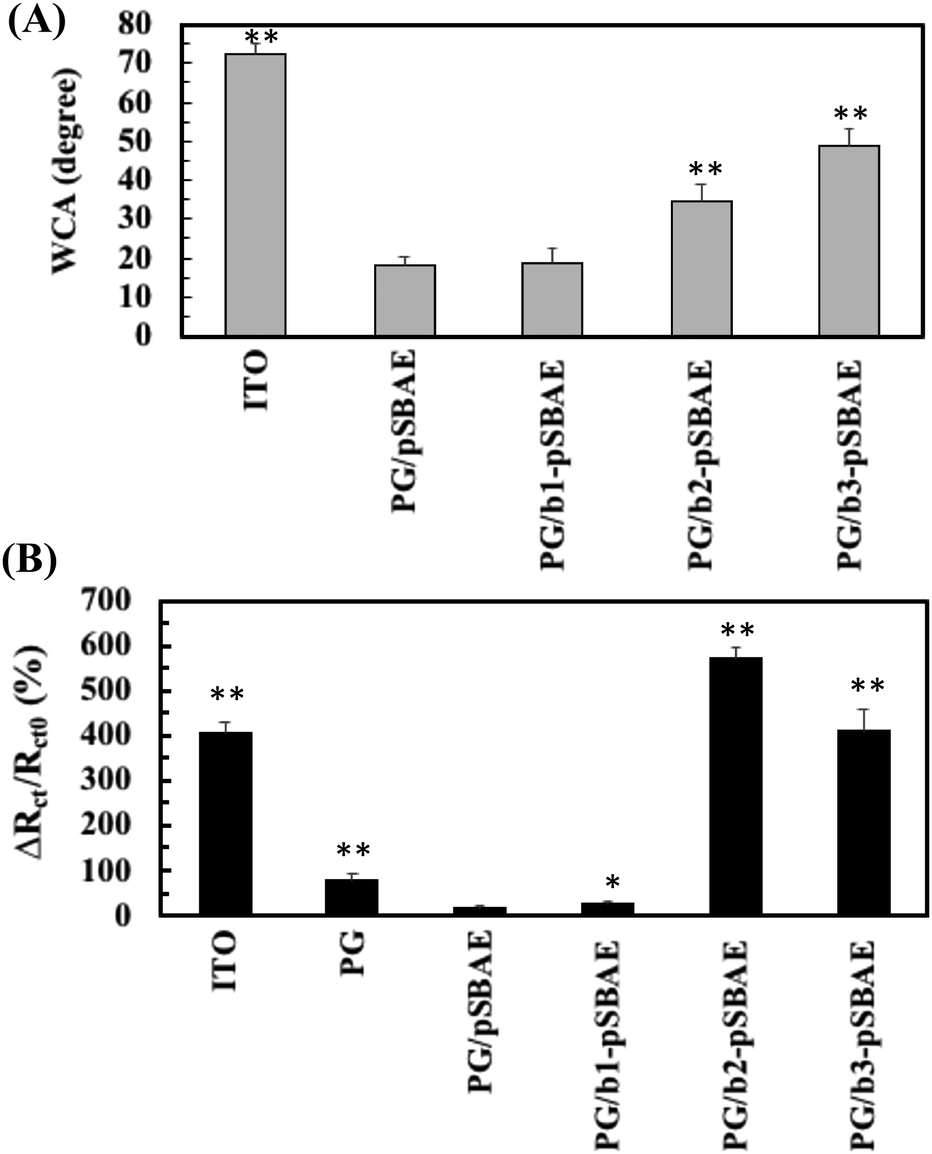 | ||
| Fig. 5 (A) Static water contact angles of PG/b-pSBAE electro-deposited ITO for 4 CV cycles. PG/b-pSBAE = 8/30 mg mL−1. (B) ITO and PG/pSBAE modified ITO was incubated in 0.1 mg mL−1 of avidin for 2 hours and the changes in charge transfer resistance were determined. * and ** represent p < 0.05 and < 0.001, compared to PG/pSBAE, respectively. Value = mean ± standard deviation, n = 3. | ||
Next, we determined the avidin capture ability of the PG/b-pSBAE modified ITO from 0.1 mg avidin per mL of PBS using EIS measurement. Pristine ITO adsorbed a large amount of avidin, while PG deposition decreased the change in Rct, that is, avidin adsorption, to ITO (Fig. 5B). The modification of PG/pSBAE modification further decreased the nonspecific adsorption of avidin to a very low level. The addition of b1-pSBAE did not show significant avidin capture, probably due to low biotin conjugation. The conjugation of b2-pSBAE greatly improved avidin attachment, higher than that of b3-pSBAE. The results proved that the PG/b-pSBAE coating could capture avidin. Since PG/b2-pSBAE gained maximal avidin attachment, it was used in subsequent experiments and indicated as b-pSBAE.
The conductivity of a biosensor is important for its sensing ability.35,36 A sensing element with a high conductivity could result in a high current and possess a high sensing ability without any lag. The conductivity of the PG/pSBAE modified surfaces was evaluated using the Rct by electrical impedance spectroscopy (EIS). The Nyquist plot shows that the deposition of PG greatly increased the impedance of ITO (Fig. 6A). Electrodes have been reported to lose their conducting properties due to the formation of a thin insulating layer by polymerization of polyphenolic molecules.38,39 However, the co-deposition of PG and pSBAE or b-pSBAE only slightly increased the impedance compared to the bare ITO (Fig. 6B). The result suggested that incorporation of pSBAE facilitates the transfer of Fe ions to the working electrode through a PG film. Thus, more Fe ions become oxidized and reduced at the PG/b-pSBAE-deposited working electrode. PG/b-pSBAE deposited ITO shows high potential for electrochemical biosensing.
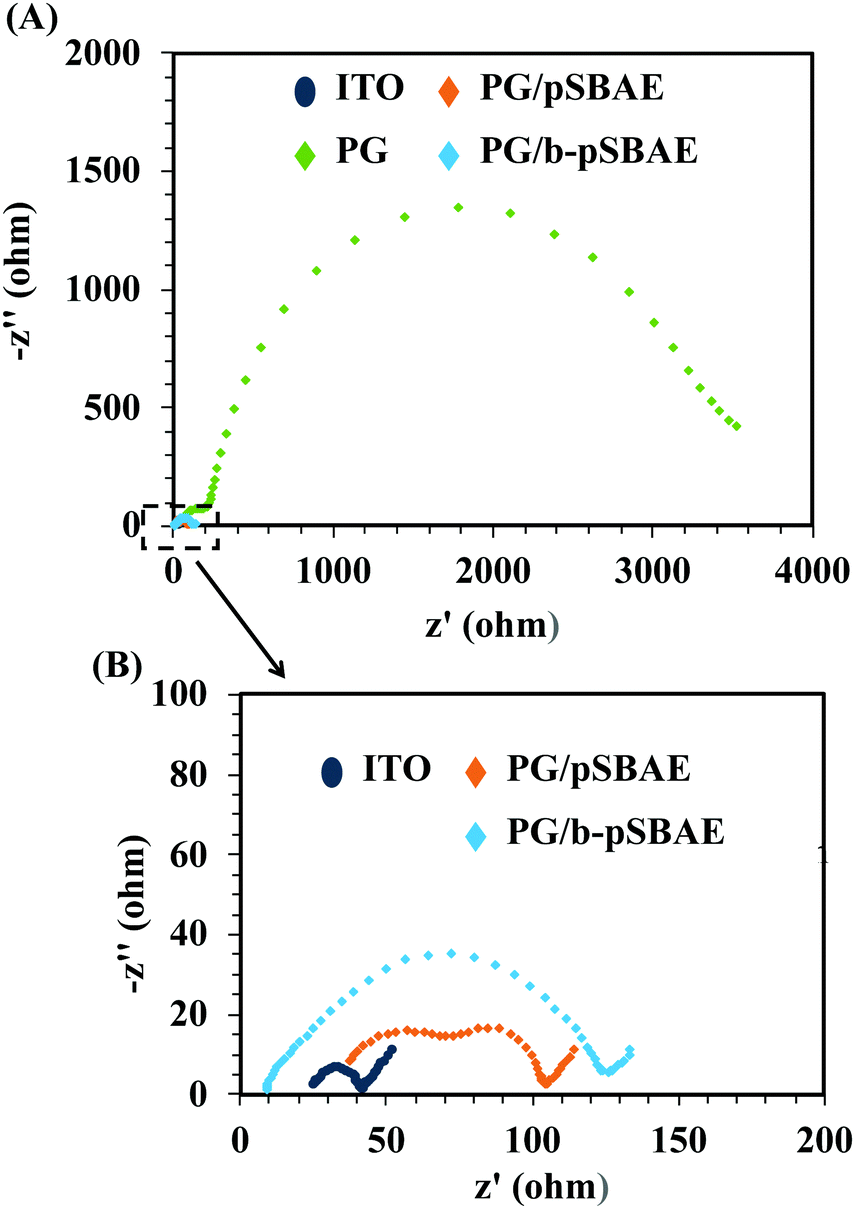 | ||
| Fig. 6 (A) Impedance Nyquist plots of the EIS obtained at different modified electrodes: ITO, PG (8 mg mL−1), PG/pSBAE and PG/b-pSBAE (8/30 mg mL−1). (B) The enlarged figure from the enclosed area in (A). Measurements were performed in PBS containing 10 mM [Fe(CN)6]3−/4− and 0.1 M KCl. | ||
The electrochemical sensing performance of the PG/b-pSBAE ITO for the determination of avidin was first investigated in PBS. Rct increased with increasing avidin concentrations (Fig. 7A). We found that the ΔRct/Rct0 (%) exhibited a good linear correlation with the logarithm of avidin concentrations with a regression equation ΔRct/Rct0 (%) = 48.66 log(C) (μg mL−1) − 4.81 (R2 = 0.9949) (Fig. 7B). The good sensing performance of the b-pSBAE–ITO indicates that the modified surface provides a good microenvironment to retain the binding affinity of the biotin–avidin pair.
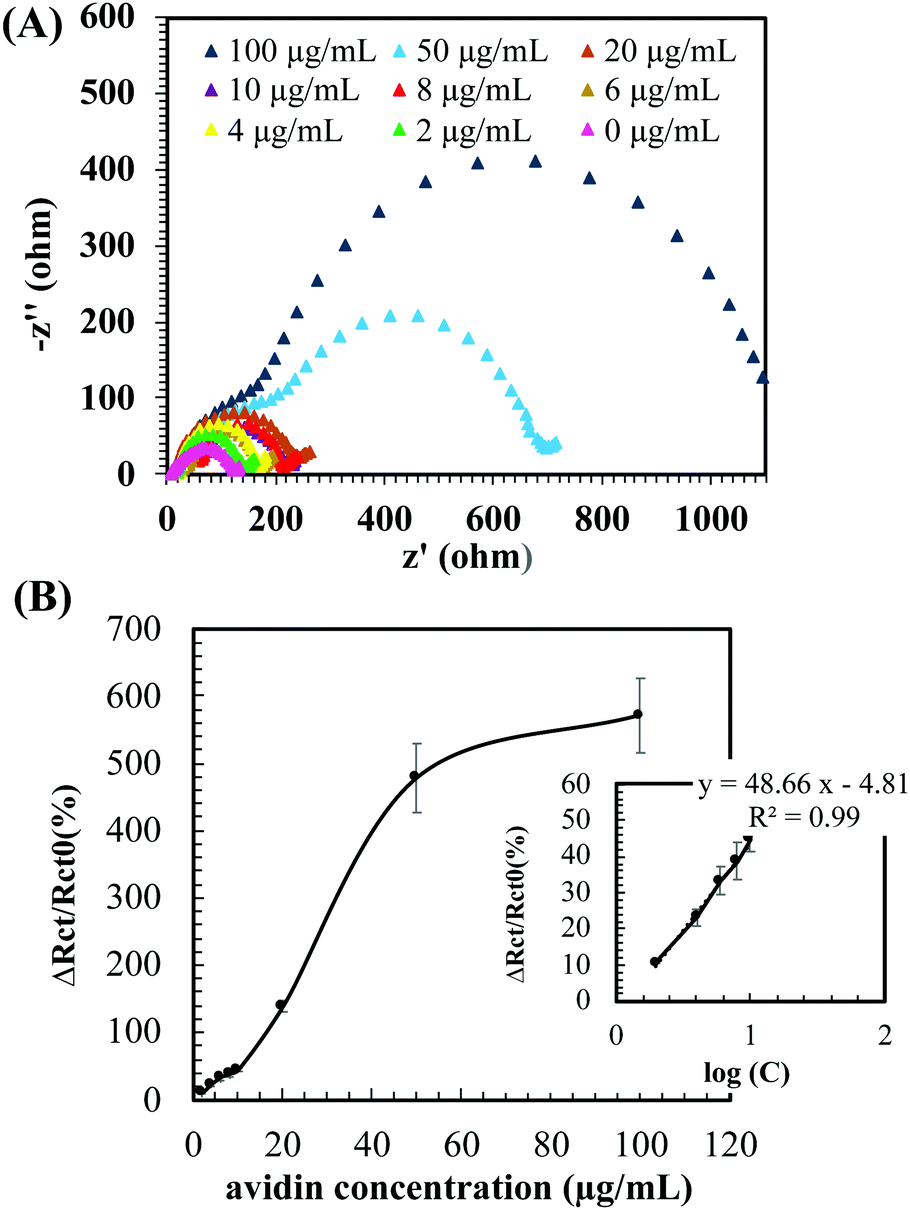 | ||
| Fig. 7 (A) Impedance Nyquist plots of the EIS from ITO modified with PG/b-pSBAE (8/30 mg mL−1) after immersion in avidin solution at various concentrations (0–100 μg mL−1). Measurements were performed in PBS containing 10 mM [Fe(CN)6]3−/4− and 0.1 M KCl. (B) Changes in the charge transfer resistance [ΔRct/Rct0 (%)] of the PG/b-pSBAE modified ITO after immersion in avidin solution at various concentrations. Insert: the corresponding regression curve of the PG/b-pSBAE modified ITO. C: Avidin concentration. Value = mean ± standard deviation, n = 3. | ||
Next, we evaluated avidin recognition of PG/b-pSBAE in complex media, such as serum. Complex media are prone to elicit significant nonspecific protein adsorption. We first tested the resistance of PG/b-pSBAE to nonspecific protein adsorption from fetal bovine serum (FBS) at various concentrations (Fig. 8A). When the PG/pSBAE and PG/b-pSBAE samples were immersed in 0.1% or 1% FBS, the increase in the change of Rct was very low, while significant amounts of nonspecific protein adsorption were found when the samples were immersed in 5% and 10% FBS. Furthermore, the PG/b-pSBAE coating elicits more nonspecific protein adsorption, indicating that biotinylation affects the antifouling efficacy of pSBAE.
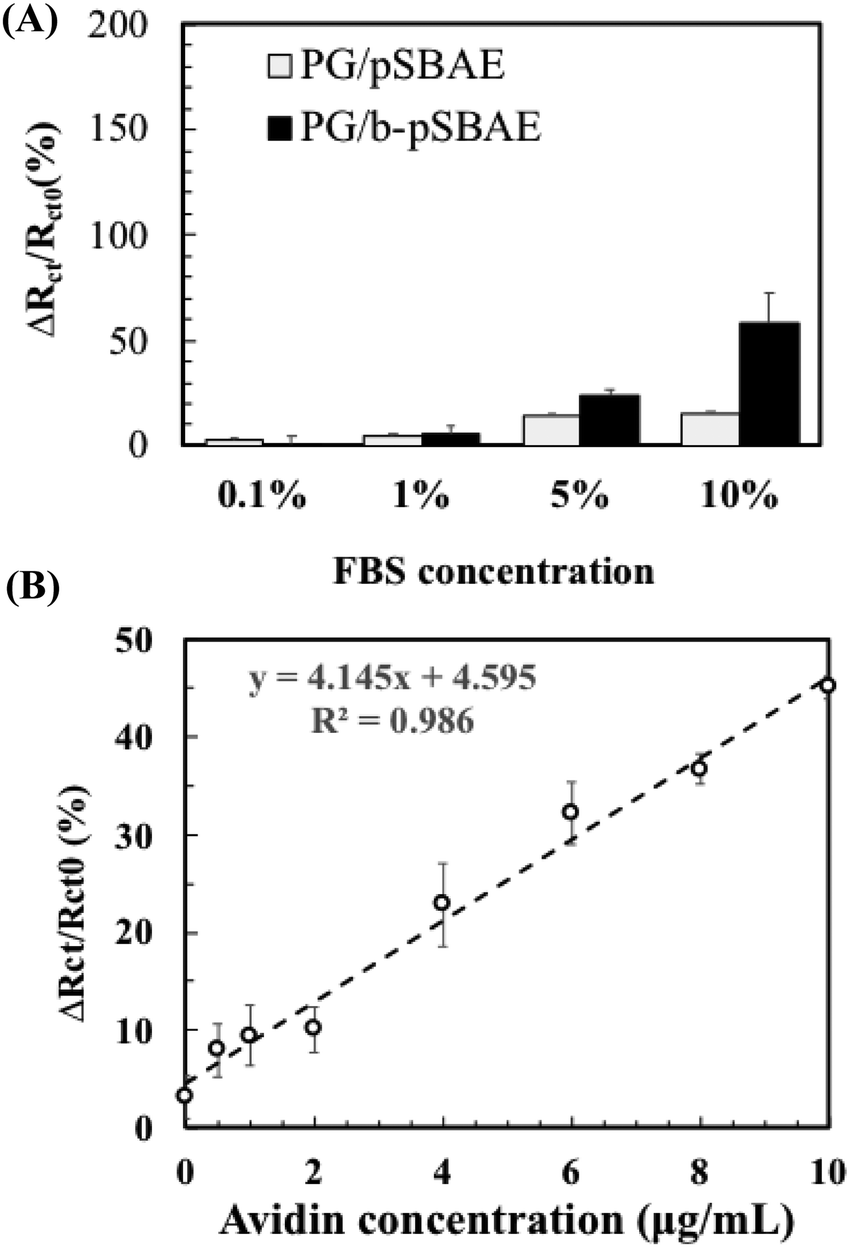 | ||
| Fig. 8 (A) Rct change [ΔRct/Rct0 (%)] on PG/pSBAE and PG/b-pSBAE (8/30 mg mL−1) modified ITO after incubation in FBS solutions at different concentrations for 2 hours. (B) Rct change [ΔRct/Rct0 (%)] of PG/b-pSBAE (8/30 mg mL−1) coated ITO after immersion in 1% FBS containing different amounts of avidin. | ||
Since the PG/b-pSBAE substrate resisted nonspecific protein adsorption from 1% FBS, we next tested the avidin recognition of PG/b-pSBAE from 1% FBS solution containing various amounts of avidin. Rct change increased with increasing avidin concentrations in a good linear relationship (Fig. 8B). However, the correlation of Rct changes and avidin concentrations is not as excellent as that in pure avidin concentration. The complex protein solution may affect the avidin/biotin binding.
Electrodeposition of polyphenolic molecules has been applied to the surface modification of biosensors.37–41 Many studies used a two-step process to conjugate antifouling molecules or biomolecules. For example, an impedimetric biosensor for the detection of breast cancer-related gene was first electrodeposited with polydopamine; tannic acid was deposited through Fe3+ mediation; finally PEG was grafted as an antifouling layer via a layer-by-layer technique.37 For immobilization of thiol-functionalized oligonucleotides, the platform was further electrodeposited with gold nanoparticles. Finally, the modified biosensor could detect the target gene. In another study, SBMA monomers were polymerized and deposited on a biosensor in the presence of dopamine and oxidizing agents, on which thiol-functionalized aptamers were conjugated to detect carcinoembryonic antigen even in the clinical sample.38 These preparation methods involve multiple time-consuming steps to create a non-fouling substrate and conjugate sensing probes. Here, we showed a facile one-step electrodeposition to immobilize antifouling polymers and detect molecules simultaneously over an electrode in 15 minutes. Biotin was used as the sensing element in this study to capture avidin molecules from a protein solution. Other sensing elements, such as antibodies, aptamers, and receptors, could be easily immobilized on a biosensor using our technology.
Conclusions
In this work, we developed a one-step electrodeposition method to immobilize biotinylated polysulfobetaine via pyrogallol on ITO via cyclic voltammetry. The PG/pSBAE coating was highly hydrophilic and possessed an antifouling property. Immobilization took less than 15 minutes to inhibit cell adhesion, compared to hours with conventional immersion deposition. Using biotinylated pSBAE, the modified surface resisted nonspecific protein adsorption from the fetal bovine serum solution and detected added avidin concentrations. Other sensing elements, such as antibodies, aptamers, and receptors, could be easily immobilized on a biosensor using our technology. Our results demonstrate an efficient platform for fabricating an electrochemical biosensor for the detection of biomarkers in a complex biological environment.Author contributions
Shang-Lin Yeh: investigation, formal analysis, design of methodology, writing – original draft. Piyush Deval: investigation, writing – original draft. Jhih-Guang Wu: investigation. Shyh-Chyang Luo: conceptualization, design of methodology. Wei-Bor Tsai: conceptualization, design of methodology, supervision, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by the Ministry of Science and Technology of Taiwan (MOST 107-3017-F-002-001 and 106-2221-E-002-166).References
- J. Halliwell, A. C. Savage, N. Buckley and C. Gwenin, Sens. Bio-Sens. Res., 2014, 2, 12–15 CrossRef.
- B. Y. Chang and S. M. Park, Annu. Rev. Anal. Chem., 2010, 3, 207–229 CrossRef CAS PubMed.
- F. Lisdat and D. Schafer, Anal. Bioanal. Chem., 2008, 391, 1555–1567 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- Y. L. Liu, K. L. Ai and L. H. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed.
- H. W. Chien, W. H. Kuo, M. J. Wang, S. W. Tsai and W. B. Tsai, Langmuir, 2012, 28, 5775–5782 CrossRef CAS PubMed.
- C. Y. Chien and W. B. Tsai, ACS Appl. Mater. Interfaces, 2013, 5, 6975–6983 CrossRef CAS PubMed.
- B. Stöckle, D. Y. W. Ng, C. Meier, T. Paust, F. Bischoff, T. Diemant, R. J. Behm, K. E. Gottschalk, U. Ziener and T. Weil, 2014.
- S. Kim, Y. Jang, L. K. Jang, S. H. Sunwoo, T.-i. Kim, S.-W. Cho and J. Y. Lee, J. Mater. Chem. B, 2017, 5, 4507–4513 RSC.
- G. Wang, Q. Xu, L. Liu, X. Su, J. Lin, G. Xu and X. Luo, ACS Appl. Mater. Interfaces, 2017, 9, 31153–31160 CrossRef CAS PubMed.
- S. Kim, L. K. Jang, H. S. Park and J. Y. Lee, Sci. Rep., 2016, 6, 1–8 CrossRef CAS PubMed.
- M. Martin, P. Salazar, R. Alvarez, A. Palmero, C. Lopez-Santos, J. L. Gonzalez-Mora and A. R. Gonzalez-Elipe, Sens. Actuators, B, 2017, 240, 37–45 CrossRef CAS.
- Z. Zhang, T. Chao, S. F. Chen and S. Y. Jiang, Langmuir, 2006, 22, 10072–10077 CrossRef CAS PubMed.
- Z. Y. Xu, R. Han, N. Z. Liu, F. X. Gao and X. L. Luo, Sens. Actuators, B, 2020, 319 Search PubMed.
- D. G. Barrett, T. S. Sileika and P. B. Messersmith, Chem. Commun., 2014, 50, 7265–7268 RSC.
- T. S. Sileika, D. G. Barrett, R. Zhang, K. H. A. Lau and P. B. Messersmith, Angew. Chem., Int. Ed., 2013, 52, 10766–10770 CrossRef CAS PubMed.
- S. L. Yeh, T. C. Wang, S. Yusa, H. Thissen and W. B. Tsai, ACS Omega, 2021, 6, 3517–3524 CrossRef CAS PubMed.
- V. Ball, Colloids Surf., A, 2019, 578, 123530 CrossRef CAS.
- A. Rohanifar, A. M. Devasurendra, J. A. Young and J. R. Kirchhoff, Anal. Methods, 2016, 8, 7891–7897 RSC.
- J. Wei, J. He, C. Chen and X. Wang, Anal. Methods, 2015, 7, 5641–5648 RSC.
- Q. Shi and S. Mu, J. Power Sources, 2012, 203, 48–56 CrossRef CAS.
- C. H. Hung, W. T. Chang, W. Y. Su and S. H. Cheng, Electroanalysis, 2014, 26, 2237–2243 CrossRef CAS.
- A. Chilkoti, P. H. Tan and P. S. Stayton, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 1754–1758 CrossRef CAS PubMed.
- W. B. Tsai and M. C. Wang, Biomaterials, 2005, 26, 3141–3151 CrossRef CAS PubMed.
- W.-B. Tsai, C.-Y. Chien, H. Thissen and J.-Y. Lai, Acta Biomater., 2011, 7, 2518–2525 CrossRef CAS PubMed.
- W.-H. Chen, T.-Y. Liao, H. Thissen and W.-B. Tsai, ACS Biomater. Sci. Eng., 2019, 5, 6454–6462 CrossRef CAS PubMed.
- M. Mochizuki, S.-i. Yamazaki, K. Kano and T. Ikeda, Biochim. Biophys. Acta, Gen. Subj., 2002, 1569, 35–44 CrossRef CAS.
- G. Loget, J. B. Wood, K. Cho, A. R. Halpern and R. M. Corn, Anal. Chem., 2013, 85, 9991–9995 CrossRef CAS PubMed.
- M. Akagawa, T. Shigemitsu and K. Suyama, Biosci., Biotechnol., Biochem., 2003, 67, 2632–2640 CrossRef CAS PubMed.
- S. Mu and C. Chen, J. Phys. Chem. C, 2012, 116, 3065–3070 CrossRef CAS.
- V. Ball, Colloids Surf., A, 2017, 518, 109–115 CrossRef CAS.
- B. Li, P. Jain, J. Ma, J. K. Smith, Z. Yuan, H.-C. Hung, Y. He, X. Lin, K. Wu and J. Pfaendtner, Sci. Adv., 2019, 5, eaaw9562 CrossRef CAS PubMed.
- Y. Hu, B. Liang, L. Fang, G. Ma, G. Yang, Q. Zhu, S. Chen and X. Ye, Langmuir, 2016, 32, 11763–11770 CrossRef CAS PubMed.
- S. Hong, J. Yeom, I. T. Song, S. M. Kang, H. Lee and H. Lee, Adv. Mater. Interfaces, 2014, 1, 1400113 CrossRef.
- J. Yoon, S. N. Lee, M. K. Shin, H.-W. Kim, H. K. Choi, T. Lee and J.-W. Choi, Biosens. Bioelectron., 2019, 140, 111343 CrossRef CAS PubMed.
- L. Cao, G.-C. Han, H. Xiao, Z. Chen and C. Fang, Anal. Chim. Acta, 2020, 1096, 34–43 CrossRef CAS PubMed.
- L. Chen, X. Liu and C. Chen, J. Electroanal. Chem., 2017, 791, 204–210 CrossRef CAS.
- Z. Xu, R. Han, N. Liu, F. Gao and X. Luo, Sens. Actuators, B, 2020, 319, 128253 CrossRef CAS.
- G. Wang, R. Han, X. Su, Y. Li, G. Xu and X. Luo, Biosens. Bioelectron., 2017, 92, 396–401 CrossRef CAS PubMed.
- G. Wang, X. Su, Q. Xu, G. Xu, J. Lin and X. Luo, Biosens. Bioelectron., 2018, 101, 129–134 CrossRef CAS PubMed.
- S. Wang, Y. Ma, Y. Wang, M. Jiao, X. Luo and M. Cui, Colloids Surf., B, 2020, 186, 110706 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |