
Chemically suppressing redox reaction at the NiOx/perovskite interface in narrow bandgap perovskite solar cells to exceed a power conversion efficiency of 20%†
Hongyu
Bian‡
ab,
Jiayu
You‡
a,
Cunyun
Xu
a,
Xiaofeng
He
ab,
Meng
Wang
c,
YanQing
Yao
 d,
Wenqi
Zeng
ab,
Pengju
Guo
a,
Hongyu
Zhou
ab,
Dengcheng
Lu
a,
Zhongjun
Dai
ab,
Sam
Zhang
*ab and
Qunliang
Song
*a
d,
Wenqi
Zeng
ab,
Pengju
Guo
a,
Hongyu
Zhou
ab,
Dengcheng
Lu
a,
Zhongjun
Dai
ab,
Sam
Zhang
*ab and
Qunliang
Song
*a
aInstitute for Clean Energy and Advanced Materials, School of Materials and Energy, Southwest University, Chongqing, 400715, China. E-mail: qlsong@swu.edu.cn; samzhang@swu.edu.cn
bCenter for Advanced Thin Films and Devices, School of Materials and Energy, Southwest University, Chongqing, 40075, China
cCollege of Materials Science and Engineering, Sichuan University, Chengdu, 610064, China
dSchool of Physics and Electronic Science, Zunyi Normal College, Zunyi 563002, China
First published on 19th November 2022
Abstract
NiOx as a type of inorganic hole-transporting layer (HTL) material in narrow bandgap perovskite solar cells (NBG PSCs) showed exceptional stability but suffered a considerably poorer performance compared with NBG PSCs with commonly used PEDOT:PSS as the HTL. Herein, we found that redox reactions would occur at the interface between Ni3+ on the NiOx surface and the easily oxidized Sn2+ in the perovskite, causing considerable non-radiative recombination centers. On this basis, we proposed a bifacial reduction strategy at the interface to boost the performance of NBG PSCs. By using a reductive reagent ascorbic acid to reduce the Ni3+/Ni2+ ratio on the surface of NiOx beforehand, the possibility of contact between Ni3+ on the surface of NiOx and perovskite is chemically reduced substantially, suppressing the redox reaction between them as well as the non-radiative recombination at the interface. By applying this strategy, the device's power conversion efficiency is elevated from 17.81% to 20.48%, with 91% remaining after 1128 hours of storage in a nitrogen-filled glovebox.
Introduction
With a reported power conversion efficiency (PCE) of 3.8%1 in 2009 and to a certified 25.7%2 in merely 12 years, the perovskite solar cell has attained much attention as one of the next-generation photovoltaic devices due to its advantages, such as high absorption coefficient, long carrier diffusion length, high carrier mobility, and adjustable bandgap.3–6 The active layer in PSCs has a structure of ABX3 (B = Pb, Sn). Partially substituting Pb with Sn can absorb long-wavelength photons, broadening the absorption spectral range and forming a narrow bandgap perovskite (NBG). Thus, a possibility to achieve a higher theoretical PCE is provided for a single junction solar cell7 and realize all-perovskite tandem solar cells after coupling with a wide bandgap perovskite solar cell to surpass the Shockley–Queisser limit of single junction solar cells.8–11The inferior performance of NBG PSCs can be ascribed to both perovskite intrinsic properties and matching of energy levels among functional layers. On the one hand, the existence of Sn2+ in perovskite impairs the device stability due to the inherent strong reducing property. To solve this problem, some reducing additives (such as ascorbic acid, tea polyphenol, caffeic acid, and 4-hydrazinobenzoic acid) have been reported to prolong the lifetime.12–15 On the other hand, the mismatch of the energy band between the perovskite film and carrier-transporting layers (CTLs) leads to nonradiative recombination and energy losses.16 Interface modification by inserting a thin layer of other materials between the perovskite layer and CTLs has also been proven to be an effective way for performance enhancement.17–19
The hole-transporting layer (HTL) plays a pivotal role in PSCs, especially for the inverted device structure, in which some stringent criteria must be complied such as good physical and chemical stability, band alignment, and high transmittance. NBG inverted PSCs with PEDOT:PSS as HTL have received predominant PCE after years of efforts.20,21 However, PEDOT:PSS is characterized by acidity and hydrophilicity that would accelerate the degradation of the upper perovskite layer and thus damage the device stability.22,23 Additionally, the severe nonradiative recombination caused by the unaligned energy level at the interface of PEDOT:PSS and perovskite layer results in a large energy loss for carrier extraction and consequent VOC loss. Inorganic p-type materials, such as NiOx, CuSCN, and CuI, are considered to be more predominant HTLs than PEDOT:PSS because of the naturally robust chemical stability and relatively deeper valence band maximum (VBM).24,25 Particularly, NiOx is most notable for its various film preparation methods. These benefits might present a better performance and stability of PSCs with NiOx as HTL than PEDOT:PSS. However, few works have been reported for efficient NiOx-based NBG PSCs, resulting in the slow development of this field. Chi et al. compared PEDOT:PSS and NiOx as HTL in NBG PSCs in 2018, and found that NiOx would lead to a high-quality perovskite film with higher device performance.26 Han et al. applied NiOx prepared at low temperature into NBG PSCs, and received a PCE value of 17.6%. Moreover, 95% of the initial PCE still remained after storage in a glove box for 102 days.27 Although it is stable, the performance of the NiOx-based NBG PSCs is still relatively low compared with its counterpart. To comprehend the reasons why the NiOx-based NBG PSCs suffered such a poor performance, we have to turn our focus back on Pb-based PSCs since studies on the NBG PSCs based on NiOx are very limited. Boyd et al. believed that in inverted PSCs, Ni≥3+ on the surface of NiOx, acting as Brønsted proton acceptors and Lewis electron acceptors, would deprotonate and oxidize methylammonium iodide (MAI) and formamidinium iodide (FAI) (both provide protons and electrons) in the precursor solution during the fabrication process. Thus, I2, methylamine, and formamidine are produced and evaporated to form segregated PbI2 at the interface and in perovskite films due to the absence of A-site cations. To solve this issue, they proposed two strategies: one is to add excess MAI or FAI to compensate for the A-site ions deficiency; the other is to use A-site component post-treat NiOx films.28 Some inert polymers and small molecules were deposited on NiOx to separate both and to avoid their direct contact physically.29–34 Moreover, Wang et al. came up with an in situ bifacial passivation strategy using HI to simultaneously reduce the Ni3+/Ni2+ ratio and increase the crystallization of perovskite.35 In NBG PSCs, however, Sn2+ is easier to be oxidized and would similarly show deteriorated performance.
In this work, we demonstrated the reason why NBG PSCs with NiOx as HTL suffered from poor performance. It is attributed to the redox reaction that happens between Ni3+ on the NiOx surface and easily oxidizable Sn2+ in NBG. Accordingly, we propose a bifacial reduction strategy (BRS) to chemically suppress such redox reaction. The ratio of Ni3+/Ni2+ was decreased after a reductive reagent, ascorbic acid (AA, the structure is drawn in Fig. S1†) was modified on the NiOx film. In addition, excess AA can prevent the oxidation of Sn2+ in NBG perovskite. The device performance can be improved greatly with the aid of BRS. Finally, NBG PSCs with a champion PCE of 20.48% was achieved with a structure of indium tin oxide (ITO)/NiOx/AA/(FASnI3)0.6(MAPbI3)0.4/fullerene (C60)/bathocuproine (BCP)/Ag, which is a high PCE of inverted NiOx-based NBG PSCs. Furthermore, 91% of its initial PCE was retained after storage in a glove box for 1128 hours.
Results and discussion
Based on the Pb-based PSCs reported by McGehee et al.,28 we speculate that a similar redox reaction would happen in NBG PSCs at the interface of NiOx/perovskite between Sn2+ and Ni3+Ox, and all of the redox reactions can be described as follows:| HAPbI3 ↔ HA+ + PbI2 + I− | (1) |
| HASnI3 ↔ HA+ + SnI2 + I− | (2) |
| HA+ ↔ H+ + A(g) | (3) |
 | (4) |
| SnI2 + 2I− ↔ SnI4 + 2e− | (5) |
| Ni3+Ox + e− + H+ ↔ Ni2+OxH | (6) |
As is illustrated, Ni3+ can act both as Lewis electron acceptors and Brønsted proton acceptors, oxidizing the iodide species and deprotonating the cationic amines (HA+, represents MA+ or FA+). In inverted NBG PSCs with NiOx, obviously, eqn (5) is easier to proceed forward compared with eqn (4), as the standard redox potential of Sn4+/Sn2+ at 0.15 V is much lower than that of I2/I− at 0.54 V.36 In this case, the Sn4+ formed by the redox reaction at the interfaces will cause recombination centers.
In order to corroborate our hypothesis that Ni3+ would oxidize Sn2+ and rule out the reducing effect from I−, SnCl2 as the Sn2+ source and MACl as the H+ source were dissolved into DMF (Sn2+–H+ solution) and dropped onto a NiOx film for the reaction process. Afterward, DMF was used for rinsing, and then the substrate was annealed on a hotplate. The X-ray photoelectron spectrum (XPS) was recorded to quantitatively measure the Ni3+/Ni2+ ratio on the surface of the NiOx film. The result of Ni 2p3/2 presented in Fig. 1a displays three primary peaks: 853.89 eV corresponding to Ni2+, 855.69 eV for Ni3+, as well as the satellite peak at 860.87 eV.27,37 In comparison to bare NiOx, the result obtained from the NiOx film after the reaction shows a reduced Ni3+/Ni2+ ratio from 2.56 to 1.95 (Fig. 1a and b), demonstrating the redox reaction between Sn2+ and Ni3+. Such reaction would result in Sn2+ and HA+ vacancies at the interface, inducing p-doping, and thus leading to the formation of nonradiative recombination centers.38–40 We chose a widely used AA as a reducing reagent to provide both electrons and protons for the redox reaction needed on the NiOx surface prior to the deposition of the NBG perovskite layer. Dark black (characteristic color of Ni3+) NiOx NPs visibly turn into a green color (characteristic color of Ni2+) after AA is added (Fig. S2†), manifesting their redox reaction. We then prepared the AA modified NiOx film (rinsed) and measured them (the result is shown in Fig. 1c). A reduced Ni3+/Ni2+ ratio of 2.08 was also observed, further confirming the reducing effect from AA on NiOx. Furthermore, in order to testify whether the redox reaction between Ni3+ and Sn2+ happened after AA treatment, we used AA to modify NiOx (rinsed after), and then made the film react with the Sn2+–H+ solution. Fig. 1d displays a further decease of the Ni3+/Ni2+ ratio from 2.08 down to 1.96, which indicates that the redox reaction between bare NiOx and Sn2+–H+ solution is largely suppressed. In addition, the XPS results (Fig. S3a–d†) originating from the O 1s results confirm the decrease of the Ni3+/Ni2+ ratio. The reason of the decrease will be discussed in a later section.
 | ||
| Fig. 1 High resolution XPS of (a) bare NiOx film reacting with (b) Sn2+ and MA+, (c) AA, (d) AA, and then reacting with Sn2+ and MA+. | ||
We modified NiOx films with various concentrations of AA. To identify whether the modification would affect the optical properties of NiOx films, we used UV-vis for characterization and the structure of glass/ITO/NiOx/AA was made. No obvious difference is observed in the transmittance of NiOx (Fig. S4a†), which indicates that the NiOx film treated with AA does not influence its optical properties, and thus hardly affects the light incident into the upper perovskite layer. Furthermore, atomic force microscopy (AFM) was applied to test whether the surface properties were altered after AA treatment on the NiOx film. The morphologies of the NiOx film displays no significant change after AA treatment, as shown in Fig. S4b and c.† Furthermore, the root mean square (RMS) for NiOx and NiOx/AA are 9.38 and 9.10 nm, respectively. We also continued to test the absorbance of perovskites (Fig. S5a†) on NiOx substrates treated with different concentrations of AA, and found no distinct change. Moreover, the Tauc plot (Fig. S5b†) displays the same bandgap value, which further indicates that a minor mass of AA treatment would not affect the perovskite layer's optical properties.
Based on the unenhanced absorbance of the perovskite, we speculated that the crystallinity of the perovskite would not be changed after AA modification. To test our hypothesis, we examined the morphology of NBG grown on NiOx treated with the same AA concentrations as above via field emission scanning electron microscope (FESEM). As shown in Fig. S6a–e,† the top-view FESEM images of the NBG films show almost no difference regardless of the selected concentration. The cross-sectional FESEM images of the 0 and 0.8 mg per mL AA modification at the interface of the perovskite and NiOx with a structure of ITO/NiOx/AA/NBG are displayed in Fig. S6f and g.† Both exhibit vertical perovskite crystals without lateral grain boundaries. Moreover, all XRD patterns (Fig. S7a†) display similarly high diffraction peaks at 14.07° and 28.33°, corresponding to the preferred orientations at the (110) and (220) planes, respectively. These results are consistent with the negligible change on the grain sizes, and no peak shifts (Fig. S7b and c†) are observed for both (110) and (220) planes after AA treatments. These results are also the same as we presumed: the NiOx film surfaces treated with AA have a negligible impact on the growth of the perovskite crystals. We then considered that the low performance of the narrow-bandgap PSCs with NiOx as HTL could be attributed to the interface of NiOx/NBG, rather than the crystallinity of the bulk perovskite.
Because the XRD results and FESEM images showed no discernible changes after AA modification, the optimal concentration must be figured out prior to further study. Fig. 2a displays the device fabrication process, and the device structure of ITO/NiOx/(AA)/NBG/C60/BCP/Ag is shown in Fig. 2b. As it can be seen clearly in Fig. S8,† the performance is enhanced firstly up to 0.8 mg per mL AA and then declines. To ensure the reproducibility, we fabricated 40 devices for each treatment condition, and made statistical graphs and performance chart (Fig. S9 and Table S1†). The statistical distribution on PCE with and without the optimal concentration of the AA-modified NiOx is drawn in Fig. 2c. Fig. 2d shows the J–V curves of the champion devices: the control champion device displays a rather low performance with a short-circuit current density (JSC) of 29.31 mA cm−2, open-circuit voltage (VOC) of 0.853 V, fill factor (FF) of 71.24% and a PCE of 17.81%, while the champion BRS device exhibits a JSC of 30.62 mA cm−2, VOC of 0.877 V, and FF of 76.27%, resulting in an elevated PCE of 20.48%. To the best of our knowledge, this is a relatively high PCE with NiOx as HTL in NBG PSCs, and we list the relevant information in Table S2.† Therefore, 0.8 mg mL−1 was defined as the optimal concentration for further characterization hereinafter. Fig. 2e displays the external quantum efficiency (EQE) spectra and integrated JSC of the champion devices based on bare NiOx and BRS-NiOx. The values of the integrated JSC are 28.97 and 30.01 mA cm−2, respectively, which is in good agreement with the JSC value from the J–V curves. To evaluate the stability of the device under operation, maximum power point tracking was measured with their bias voltages under one sun AM 1.5 G illumination (Fig. 2f). The steady-state PCE of the BRS device at a bias voltage is 20.18% after 200 s illumination and the control device displays a PCE of 17.44%. Both are consistent with the efficiencies obtained from the J–V curves. The J–V curves of the champion control and BRS devices measured under the reverse and forward scans are shown in Fig. S10.† The hysteresis index (HI) was calculated from the formula: HI = PCEreverse/PCEforward. The specific parameters are listed in Table S3.† Both control and BRS devices show the same HI of 1.10. The unchanged HI may be ascribed to the unmatched HTL with ETL.41
 | ||
| Fig. 2 (a) The fabrication process of PSC devices. (b) Structure of devices. (c) Statistical PCE distribution of the control and BRS PSCs. (d) The J–V curves measured under 100 mW cm−2 AM 1.5 G illumination, and (e) corresponding EQE spectrum and integrated JSC of the champion devices. (f) Maximum power point (MPP) tracking under 100 mW cm−2 AM 1.5 G illumination. (g) The J–V curves of the BRS devices with different rinsing times of DMF on the AA modified NiOx. | ||
As the AA is soluble in DMF and DMSO, a series of tiny amounts of AA was doped into the NBG perovskite precursor solutions to determine whether AA has a dominant effect in the bulk perovskite. Fig. S11a–c† show the XRD patterns of perovskite films doped with various concentrations of AA grown on NiOx film. As is clearly displayed, the crystallization peak strength remains unchanged. The peak centers of the (110) and (220) plane-preferred orientation do not shift, suggesting that AA did not enter the perovskite lattice. Then, Fourier transform infrared (FTIR) spectrometry was applied to find out whether PbI2 (representative for SnI2 here as it is extremely easy to oxidize with O2), MAI, FAI had interactions with AA. The results (Fig. S11d–f†) show that all of the main components in the NBG perovskite did not have interactions with AA. We then doped the same series of concentrations of AA into the NBG perovskite solution and fabricated devices. The performance is drawn in Fig. S12.† No changes are found between the samples with only minor amounts of doping and the control devices. This indicated that it would hardly affect the device performance although the trace amount of AA can be dissolved into the perovskite precursor solution. To further demonstrate that AA has a greater effect at the interface rather than in the perovskite layer, we then rinsed the AA–NiOx films for two, four and six times with DMF. The XRD patterns (Fig. S13a–c†) still show only a negligible variation for the peak position and the intensities. Furthermore, the device performance displayed in Fig. 2g suggests that the AA produces the main effect at the interface.
Based on the above results, we suppose that the decrease of the Ni3+/Ni2+ ratio is because AA provides both protons and electrons for the redox reaction needed, as mentioned in the chemical eqn (6) prior to the perovskite film deposition on NiOx. This prevents their reaction with each other, thus decreasing the HA+ and Sn2+ vacancies at the interface. In addition, AA as a reducing reagent with a tiny amount at the interface can prevent Sn2+ from oxidation.12 Thus, the scheme of the redox reaction at the interface and the BRS working mechanism presented in Fig. 3 can be deduced. FTIR measurements were conducted to detect the oxidation form of AA after it reacted with NiOx. In Fig. S14,† two peaks at 1797 and 1630 cm−1 corresponding to ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in dehydroascorbic acid (oxidized form of AA) appeared,42,43 which further confirm the redox reaction between AA and NiOx and provide evidence for our speculated mechanism.
O) in dehydroascorbic acid (oxidized form of AA) appeared,42,43 which further confirm the redox reaction between AA and NiOx and provide evidence for our speculated mechanism.
 | ||
| Fig. 3 Scheme of the redox reaction at the NiOx/NBG interface in the control and BRS device. Ni3+ on the NiOx surface would oxidize Sn2+ and deprotonate HA+ in the NBG perovskite precursor, resulting in both Sn2+ and HA+ vacancies at the interface after perovskite deposition. When AA with a good reducing effect is modified on the NiOx film prior to NBG deposition, both protons and electrons are provided for the redox reaction needed at the NiOx surface, preventing the redox reaction between Ni3+ among Sn2+, HA+. Thus, Sn2+ and HA+ vacancies at the interface are decreased. | ||
To quantitatively assess the defect densities of the perovskite films grown on bare NiOx and NiOx/AA, the hole-only devices were made (the structures are shown in the graph inseted in Fig. 4a and b). The space charge limited current (SCLC) measurement was carried out, and the trap density (nt) is calculated by the following equation:
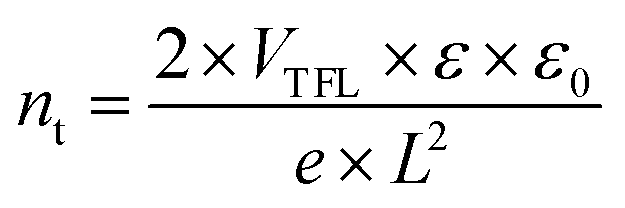 | (I) |
 | ||
| Fig. 4 The space-charge-limited-current (SCLC) module for (a) bare NiOx hole-only device and for (b) BRS hole-only device. (The inset is the device structure) (c) Mott–Schottky measurements of PSCs. (d) UPS measurements of the bare NiOx film and rinsed AA treated NiOx film. (e) Energy diagram of the layers in the PSC device. (f) VOC dependence on the light intensity of the control and BRS devices. | ||
To understand the origin of the upgraded VOC of the BRS devices, Mott–Schottky study was performed. By this manner, the built-in potential (Vbi) of the devices could be determined by fitting the linear part of the C−2–V curves according to eqn (II):
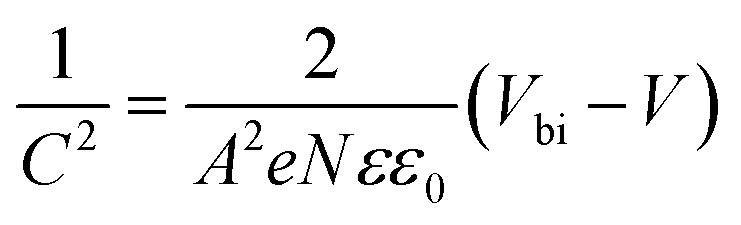 | (II) |
Fig. 5a displays the Nyquist plot of electrochemical impedance spectroscopy, and the inset graph is its equivalent circuit utilized for analyzing the data. The Rs and Rrec shown in Table S4† are the series resistance and recombination resistance, respectively. The Rrec of the BRS device with 1865 Ω is larger than that of the control device with 332 Ω, implying that the suppressed non-radiative recombination resulted from the pre-reduced Ni3+/Ni2+ ratio and less Sn2+ oxidation and HA+ deprotonation induced Sn2+ and HA+ vacancies at interface. Subsequently, steady-state photoluminescence (PL) and time-resolved PL (TRPL) spectra were performed to better understand the dynamics of the carrier at the interface. We deposited NBG perovskite films on bare NiOx and BRS NiOx. The results shown in Fig. 5b suggest that a larger PL quenching appeared after BRS, which could be caused by: (1) trap-assisted recombination states resulting from the intrinsic poor perovskite film quality and (2) the improvement of the hole extraction from the perovskite to NiOx. The first possibility can be excluded as the trap-states decrease greatly, as discussed above through the SCLC measurement. For further quantifying the carrier quenching lifetime and to better understand the extraction process of the carrier from the perovskite at the interface, the TRPL spectra of the NBG perovskite film grown on NiOx and AA–NiOx were carried out (Fig. 5c). The corresponding curves were fitted through a bi-exponent decay function. τ1 and τ2 correspond to the fast and slow decay lifetimes, respectively, and the average carrier decay lifetimes (τave) were calculated (Table S5†). A lower τave of BRS (26.259 ns) than that of the control (41.169 ns) shows a faster carrier decay, which is anastomotic with the PL results. The transient photocurrent (TPC) was measured. Fig. 5d displays a faster TPC attenuation (1.13 μs) after AA treatment. These results all suggest a better carrier extraction at the NiOx/NBG interface.
 | ||
| Fig. 5 (a) Nyquist plots of the corresponding PSCs under dark condition (embedded with the equivalent circuit). (b) Steady-state photoluminescence spectra and (c) time-resolved photoluminescence spectra of the perovskite film deposited on bare and BRS NiOx (the laser was incident from the glass side). (d) Transient photocurrent of control and BRS devices. | ||
Finally, the long-term stability of the unencapsulated NBG PSCs in a N2-filled glove box with an oxygen concentration of ≤10 ppm and with a H2O concentration of ≤0.1 ppm was measured. Fig. 6 shows that the PCE of the control device degrades to 83% of its initial value after 1128 h. Meanwhile, the BRS device retains 91% of its initial PCE value within the same storage time, suggesting that the BRS we proposed would not impair the stability of the NBG PSCs, while improving its performance.
 | ||
| Fig. 6 Stability of VOC, JSC, FF and PCE of the unencapsulated control and BRS PSCs in a nitrogen-filled glovebox with a H2O concentration of ≤0.1 and oxygen concentration of ≤10 ppm. | ||
Conclusions
We have demonstrated that the Ni3+ on the NiOx surface would react with Sn2+ in NBG perovskite. Such a reaction would introduce a mass of recombination centers at the interface of NiOx/NBG, leading to a poor performance of the NBG PSCs. After confirming this hypothesis, we proposed a bifacial reduction strategy to reduce Ni3+ on the NiOx surface to Ni2+ using AA to provide the protons and electrons needed for the redox reaction, thus chemically avoiding the contact between Ni3+ and Sn2+. Through this strategy, the redox reaction-induced recombination centers decreased, which was mainly attributed to the decreased Ni3+/Ni2+ ratio and Sn2+, HA+ vacancies at interface. This was better for the band alignment and carrier transportation. Thus, a lower energy loss in the extraction process of the interface and a high PCE with 20.48% were finally achieved. Our bifacial reduction strategy from the aspect of chemical separation provides a new train of thought to overcome the redox reaction at the interface of NiOx/NBG, contributing to the improvement of the NBG PSCs performance toward the development of future stable photoelectric devices.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors acknowledge financial support from the National Natural Foundation of China (grant no. 11774293 and 12074321), and the Chongqing Science and Technology Commission (cstc2020jcyj-msxm1120). This work was also supported by the Fundamental Research Funds for the Central Universities (SWU118105) and Academic Budding Project (grant number ZS-XM[2021]1-4). The authors would like to thank Prof. Xianju Zhou, from the College of Science, Chongqing University of Posts and Telecommunications, for help with the PL measurement. The authors also would like to thank Mr Chuanyao Luo for guidance in the synthesis of the NiOx nanoparticles.Notes and references
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS.
- https://www.nrel.gov/pv/assets/pdfs/cell-pv-eff-emergingpv-rev220630.pdf .
- P. P. Boix, K. Nonomura, N. Mathews and S. G. Mhaisalkar, Mater. Today, 2014, 17, 16–23 CrossRef CAS.
- M. A. Green, A. Ho-Baillie and H. J. Snaith, Nat. Photonics, 2014, 8, 506–514 CrossRef CAS.
- J. P. Correa-Baena, M. Saliba, T. Buonassisi, M. Gratzel, A. Abate, W. Tress and A. Hagfeldt, Science, 2017, 358, 739–744 CrossRef CAS PubMed.
- R. Prasanna, A. Gold-Parker, T. Leijtens, B. Conings, A. Babayigit, H. G. Boyen, M. F. Toney and M. D. McGehee, J. Am. Chem. Soc., 2017, 139, 11117–11124 CrossRef CAS PubMed.
- X. Y. Jiang, Z. H. Zang, Y. Y. Zhou, H. S. Li, Q. Wei and Z. J. Ning, Acc. Mater. Res., 2021, 2, 210–219 CrossRef CAS.
- P. Umari, E. Mosconi and F. De Angelis, Sci. Rep., 2014, 4, 4467 CrossRef PubMed.
- Y. Ogomi, A. Morita, S. Tsukamoto, T. Saitho, N. Fujikawa, Q. Shen, T. Toyoda, K. Yoshino, S. S. Pandey, T. L. Ma and S. Hayase, J. Phys. Chem. Lett., 2014, 5, 1004–1011 CrossRef CAS PubMed.
- G. E. Eperon, T. Leijtens, K. A. Bush, R. Prasanna, T. Green, J. T. W. Wang, D. P. McMeekin, G. Volonakis, R. L. Milot, R. May, A. Palmstrom, D. J. Slotcavage, R. A. Belisle, J. B. Patel, E. S. Parrott, R. J. Sutton, W. Ma, F. Moghadam, B. Conings, A. Babayigit, H. G. Boyen, S. Bent, F. Giustino, L. M. Herz, M. B. Johnston, M. D. McGehee and H. J. Snaith, Science, 2016, 354, 861–865 CrossRef CAS PubMed.
- Y. Q. Yao, F. Lv, L. Luo, L. P. Liao, G. Wang, D. B. Liu, C. Y. Xu, G. D. Zhou, X. S. Zhao and Q. L. Song, Sol. RRL, 2020, 4, 1900396 CrossRef CAS.
- X. B. Xu, C. C. Chueh, Z. B. Yang, A. Rajagopal, J. Q. Xu, S. B. Jo and A. K. Y. Jen, Nano Energy, 2017, 34, 392–398 CrossRef CAS.
- H. X. Ban, Q. Sun, T. Zhang, H. Li, Y. Shen and M. K. Wang, Sol. RRL, 2020, 4, 1900457 CrossRef CAS.
- H. Liu, L. Wang, R. Li, B. Shi, P. Wang, Y. Zhao and X. Zhang, ACS Energy Lett., 2021, 6, 2907–2916 CrossRef CAS.
- J. Cao, H. L. Loi, Y. Xu, X. Guo, N. Wang, C. k. Liu, T. Wang, H. Cheng, Y. Zhu, M. G. Li, W. Y. Wong and F. Yan, Adv. Mater., 2021, 34, 2107729 CrossRef.
- M. Wang, H. X. Wang, W. Li, X. F. Hu, K. Sun and Z. G. Zang, J. Mater. Chem. A, 2019, 7, 26421–26428 RSC.
- Y. Zhong, C. Li, G. Xu, C. Xu, J. Dong, D. Liu, D. Lu, J. You, C. Gao and Q. Song, Chem. Eng. J., 2022, 436, 135134 CrossRef CAS.
- X. Jiang, W. S. Subhani, K. Wang, H. Wang, L. J. Duan, M. Y. Du, S. P. Pang and S. Z. Liu, Adv. Mater. Interfaces, 2021, 8, 2001994 CrossRef CAS.
- B. B. Liu, H. Bi, D. M. He, L. Bai, W. Q. Wang, H. K. Yuan, Q. L. Song, P. Y. Su, Z. G. Zang, T. W. Zhou and J. Z. Chen, ACS Energy Lett., 2021, 6, 2526–2538 CrossRef CAS.
- R. X. Lin, J. Xu, M. Y. Wei, Y. R. Wang, Z. Y. Qin, Z. Liu, J. L. Wu, K. Xiao, B. Chen, S. M. Park, G. Chen, H. R. Atapattu, K. R. Graham, J. Xu, J. Zhu, L. D. Li, C. F. Zhang, E. H. Sargent and H. R. Tan, Nature, 2022, 603, 73–78 CrossRef CAS PubMed.
- G. Kapil, T. Bessho, Y. Sanehira, S. R. Sahamir, M. Chen, A. K. Baranwal, D. Liu, Y. Sono, D. Hirotani, D. Nomura, K. Nishimura, M. A. Kamarudin, Q. Shen, H. Segawa and S. Hayase, ACS Energy Lett., 2022, 7, 966–974 CrossRef CAS.
- M. Kim, M. Yi, W. Jang, J. K. Kim and D. H. Wang, Polymers, 2020, 12, 129 CrossRef CAS PubMed.
- S. Ma, W. Y. Qiao, T. Cheng, B. Zhang, J. X. Yao, A. Alsaedi, T. Hayat, Y. Ding, Z. A. Tan and S. Y. Dai, ACS Appl. Mater. Interfaces, 2018, 10, 3902–3911 CrossRef CAS PubMed.
- A. C. Nkele, A. C. Nwanya, N. M. Shinde, S. Ezugwu, M. Maaza, J. S. Shaikh and F. I. Ezema, Int. J. Energy Res., 2020, 44, 9839–9863 CrossRef CAS.
- X. C. Yang, H. X. Wang, B. Cai, Z. Yu and L. C. Sun, J. Energy Chem., 2018, 27, 650–672 CrossRef.
- D. Chi, S. H. Huang, M. Y. Zhang, S. Q. Mu, Y. Zhao, Y. Chen and J. B. You, Adv. Funct. Mater., 2018, 28, 1804603 CrossRef.
- Q. L. Han, Y. Wei, R. X. Lin, Z. M. Fang, K. Xiao, X. Luo, S. Gu, J. Zhu, L. M. Ding and H. R. Tan, Sci. Bull., 2019, 64, 1399–1401 CrossRef.
- C. C. Boyd, R. C. Shallcross, T. Moot, R. Kerner, L. Bertoluzzi, A. Onno, S. Kavadiya, C. Chosy, E. J. Wolf, J. Werner, J. A. Raiford, C. de Paula, A. F. Palmstrom, Z. S. J. Yu, J. J. Berry, S. F. Bent, Z. C. Holman, J. M. Luther, E. L. Ratcliff, N. R. Armstrong and M. D. McGehee, Joule, 2020, 4, 1759–1775 CrossRef CAS.
- X. M. Lian, J. H. Chen, S. Q. Shan, G. Wu and H. Z. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 46340–46347 CrossRef CAS PubMed.
- X. Y. Hou, F. J. Li, X. Zhang, Y. F. Shi, Y. X. Du, J. B. Gong, X. D. Xiao, S. Q. Ren, X. Z. Zhao and Q. D. Tai, Sol. RRL, 2021, 5, 2100287 CrossRef CAS.
- J. Zhang, J. Long, Z. Huang, J. Yang, X. Li, R. Dai, W. Sheng, L. Tan and Y. Chen, Chem. Eng. J., 2021, 426, 131357 CrossRef CAS.
- Y. X. Guo, J. Ma, H. B. Wang, F. H. Ye, L. B. Xiong, H. W. Lei and Z. J. Tan, Adv. Mater. Interfaces, 2021, 8, 2100920 CrossRef CAS.
- W. B. Han, G. H. Ren, Z. Q. Li, M. N. Dong, C. Y. Liu and W. B. Guo, J. Energy Chem., 2020, 46, 202–207 CrossRef.
- L. Zheng, Y. Xuan, J. Wang, S. Bao, X. Liu and K. Zhang, J. Mater. Chem. A, 2022, 10, 7251–7262 RSC.
- H. Y. Wang, Z. Q. Huang, S. Q. Xiao, X. C. Meng, Z. Xing, L. Rao, C. X. Gong, R. S. Wu, T. Hu, L. C. Tan, X. T. Hu, S. H. Zhang and Y. W. Chen, J. Mater. Chem. A, 2021, 9, 5759–5768 RSC.
- W. M. Latimer, Soil Sci., 1939, 48(4), 349 CrossRef.
- S. Wang, Y. Li, J. Yang, T. Wang, B. Yang, Q. Cao, X. Pu, L. Etgar, J. Han, J. Zhao, X. Li and A. Hagfeldt, Angew. Chem., Int. Ed., 2022, 61, e202116534 CAS.
- R. X. Lin, K. Xiao, Z. Y. Qin, Q. L. Han, C. F. Zhang, M. Y. Wei, M. I. Saidaminov, Y. Gao, J. Xu, M. Xiao, A. D. Li, J. Zhu, E. H. Sargent and H. R. Tan, Nat. Energy, 2019, 4, 864–873 CrossRef CAS.
- J. Xu, A. Maxwell, M. Y. Wei, Z. W. Wang, B. Chen, T. Zhu and E. H. Sargent, ACS Energy Lett., 2021, 6, 4220–4227 CrossRef CAS.
- Q. Y. Chen, J. C. Luo, R. He, H. G. Lai, S. Q. Ren, Y. T. Jiang, Z. X. Wan, W. W. Wang, X. Hao, Y. Wang, J. Q. Zhang, I. Constantinou, C. L. Wang, L. L. Wu, F. Fu and D. W. Zhao, Adv. Energy Mater., 2021, 11, 2101045 CrossRef CAS.
- D. H. Kong and N. G. Park, Adv. Mater., 2019, 31, 1805214 CrossRef PubMed.
- E. S. Wagner, W. White, M. Jennings and K. Bennett, Biochim. Biophys. Acta, 1987, 902, 133–136 CrossRef CAS PubMed.
- K. B. Kokoh, F. Hahn, A. Metayer and C. Lamy, Electrochim. Acta, 2002, 47, 3965–3969 CrossRef CAS.
- H. Chen, Z. J. Peng, K. M. Xu, Q. Wei, D. N. Yu, C. C. Han, H. S. Li and Z. J. Ning, Sci. China Mater., 2021, 64, 537–546 CrossRef CAS.
- B. Li, B. H. Chang, L. Pan, Z. H. Li, L. Fu, Z. B. He and L. W. Yin, ACS Energy Lett., 2020, 5, 3752–3772 CrossRef CAS.
- M. Du, S. Zhao, L. Duan, Y. Cao, H. Wang, Y. Sun, L. Wang, X. Zhu, J. Feng, L. Liu, X. Jiang, Q. Dong, Y. Shi, K. Wang and S. Liu, Joule, 2022, 6, 1931–1943 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta06211a |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |