
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Topotactic redox cycling in SrFeO2.5+δ explored by 3D electron diffraction in different gas atmospheres†
Maria
Batuk
 *a,
Daphne
Vandemeulebroucke
a,
Monica
Ceretti
b,
Werner
Paulus
b and
Joke
Hadermann
a
*a,
Daphne
Vandemeulebroucke
a,
Monica
Ceretti
b,
Werner
Paulus
b and
Joke
Hadermann
a
aUniversity of Antwerp, Groenenborgerlaan 171, Antwerp, 2020, Belgium. E-mail: maria.batuk@uantwerpen.be
bICGM, Univ. Montpellier, CNRS, ENSCM, Montpellier, France
First published on 23rd November 2022
Abstract
For oxygen conducting materials applied in solid oxide fuel cells and chemical-looping processes, the understanding of the oxygen diffusion mechanism and the materials' crystal structure at different stages of the redox reactions is a key parameter to control their performance. In this paper we report the first ever in situ 3D electron diffraction (ED) experiment in a gas environment and with it uncover the structure evolution of SrFeO2.5 as notably different from that reported from in situ X-ray and in situ neutron powder diffraction studies in gas environments. Using in situ 3D ED on submicron sized single crystals, we observe the transformation under O2 flow of brownmillerite SrFeO2.5 with an intra- and interlayer ordering of the left and right twisted (FeO4)∞ tetrahedral chains (space group Pcmb) into consecutively SrFeO2.75 with space group Cmmm (at 350 °C, 33% O2) and SrFeO3−δ with space group Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (at 400 °C, 100% O2). Upon reduction in H2 flow, the crystals return to the brownmillerite structure with intralayer order, but without regaining the interlayer order of the pristine crystals. Therefore, redox cycling of SrFeO2.5 crystals in O2 and H2 introduces stacking faults into the structure, resulting in an I2/m(0βγ)0s symmetry with variable β.
m (at 400 °C, 100% O2). Upon reduction in H2 flow, the crystals return to the brownmillerite structure with intralayer order, but without regaining the interlayer order of the pristine crystals. Therefore, redox cycling of SrFeO2.5 crystals in O2 and H2 introduces stacking faults into the structure, resulting in an I2/m(0βγ)0s symmetry with variable β.
Introduction
Perovskites with general formula La1−ySryFeOx show good mixed ionic and electronic conductivity1,2 and find their application in solid oxide fuel cells (SOFCs)3,4 and heterogeneous catalysis.5,6 Due to their high product selectivity for chemical-looping reactions and good cycling stability, they are also candidates for chemical-looping applications.7,8 In our research we focus on the y = 1 SrFeOx subsystem with x varying from 2.5 to 3 (an x = 2 phase9 is beyond the scope of the present study). In the SrFeOx system four distinct phases are known from ex situ redox experiments (Fig. 1): SrFeO2.5 with an orthorhombic brownmillerite structure; SrFeO2.75 (space group Cmmm); SrFeO2.875 (space group I4/mmm) and cubic Pmm perovskite SrFeO3−δ (δ indicates the presence of oxygen point vacancies in the structure).10–14 The SrFeO2.5 phase has a brownmillerite structure that consists of alternating layers of FeO4 tetrahedra and FeO6 octahedra.15–18 In brownmillerites, depending on the chemical composition the tetrahedra form (FeO4)∞ chains that can be twisted in two orientations, labelled left (L) and right (R).19,20 The SrFeO2.5 brownmillerite is subject to complex twinning,16 which made its exact structure a topic of disagreement,21–24 with contradicting results until electron diffraction (ED) and high resolution transmission electron microscopy (TEM) studies performed on SrFeO2.5 submicron single crystals15 clearly showed the alternation between L and R chains within the tetrahedral layers (so-called intralayer ordering). Such ordered layers were stacked along the b axis in two different manners forming nanodomains with mainly Pcmb and C2/c symmetry (thus showing the short-range interlayer ordering). Also other stackings were observed, fitting in a general (3 + 1)D superspace model with I2/m(0βγ)0s with varying β for the different stacking order, and with γ = 1/2 indicating the L–R intralayer ordering.15 The Pcmb structure was afterwards confirmed as the main phase using neutron and synchrotron X-ray diffraction studies on single crystals of SrFeO2.5 (ref. 17, 18 and 25) and established to be a thermodynamic ground state of SrFeO2.5 by density functional theory (DFT) calculations.26,27 The structure could be refined from powder X-ray data, but with insignificant difference in R-values versus a disordered Imma structure.17
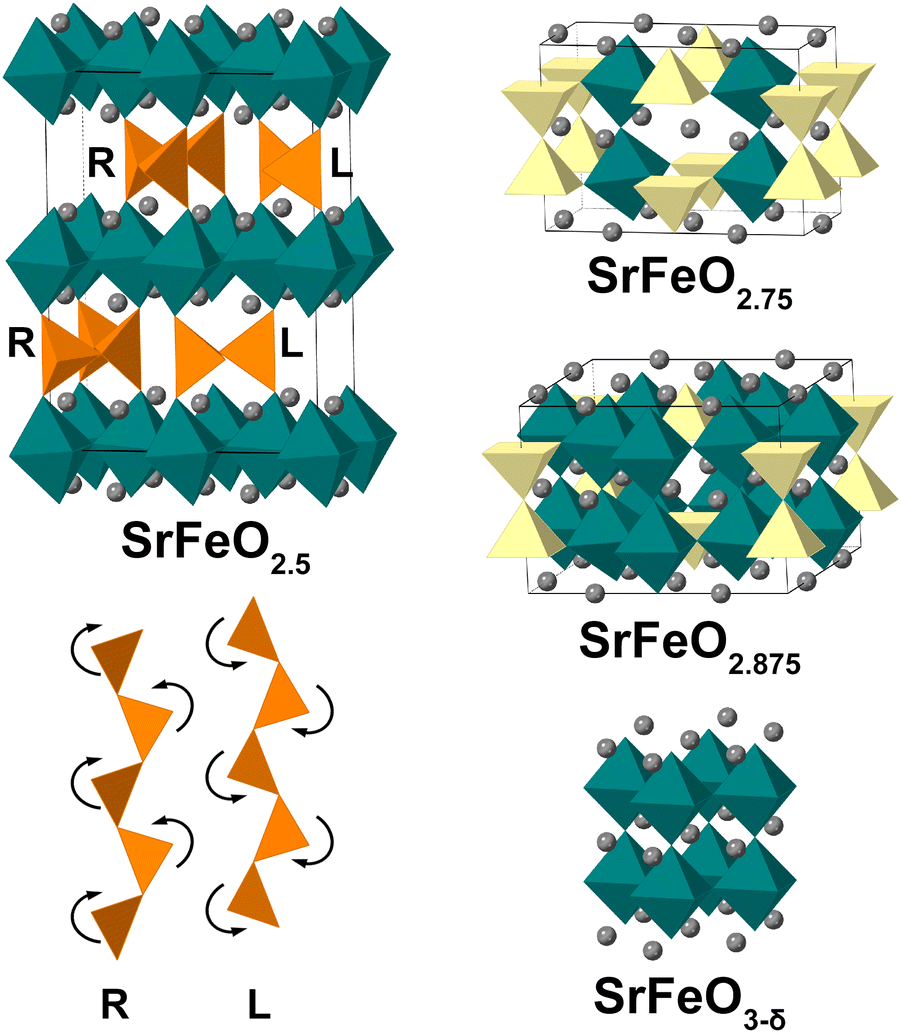 | ||
| Fig. 1 Crystal structures of the SrFeOx phases with x = 2.5, 2.75, 2.875 and 3−δ and two mirror-related configurations (right “R” and left “L”) of the tetrahedral chains in the brownmillerite structure. FeO4 tetrahedra are shown in orange; FeO5 square pyramids in yellow; FeO6 octahedra in green; Sr atoms as grey spheres, oxygen atoms are not shown for clarity. | ||
The four SrFeOx phases x = 2.5, 2.75, 2.875, 3.0 were synthesized and studied ex situ as individual compounds. However, for the applications in chemical-looping reactions or solid oxide fuel cells, an understanding of the oxygen diffusion mechanisms and the reversibility of oxygen order in the structure is crucial to optimize the performance of the materials and control their properties. To study the exact oxidation/reduction pathway, the reactions were monitored in situ on powder samples by X-ray and neutron diffraction (XRD and NPD) under the conditions of chemical-looping reactors, but no intermediate phases or different intermediate phases were detected.28,29 In ref. 28, it was demonstrated by in situ time-of-flight NPD that upon heating from 135 to 835 °C in methane, a I4/mmm SrFeO2.875 phasegradually transformed via an oxygen-vacancy disordered phase (Pmm) into the brownmillerite structure (Imma) and then into disordered Pmm again. In the same reference the crystal structure transformation was investigated during cycling at 700 °C by in situ synchrotron XRD , which showed that in methane the disordered Pmm structure gradually went to brownmillerite Imma, and when exposed to 20% O2/He, it immediately oxidized to the cubic perovskite (Pmm). In ref. 29, the SrFeO3−δ sample was heated in H2 at 900 °C and monitored by in situ XRD. In 60 minutes, it decomposed to SrO and Fe, but the intermediate states were not clear. The authors noted that only the cubic structure of SrFeO3−δ could be identified on the XRD patterns, though extra reflections were also present, which they could not ascribe to the intermediate orthorhombic or tetragonal space groups due to the lack of sensitivity of X-rays to the ordering of oxygen atoms. When exposed to CO2, SrO and Fe rapidly formed SrFeO3−δ with the cubic perovskite structure. The intermediate phases SrFeO2.75 and SrFeO2.875 did occur upon in situ electrochemical oxidation at room temperature of a SrFeO2.5 brownmillerite single crystal studied by synchrotron X-ray diffraction (SXRD).13,16 It is not clear whether the difference in phase transitions observed in in situ experiments in gas environments versus ex situ studies and in situ in electrochemical oxidation is due to the different temperature and environment. Another possibility is that the phases occurred but were not detected in the powder diffraction experiments due to peak broadening and peak overlap, for example. These discrepancies deserve to be cleared up.
Therefore, in this work, we studied the crystal structure evolution on SrFeO2.5 submicron sized single crystals during their in situ redox reaction in oxygen and hydrogen atmospheres at raised temperatures. To achieve this, we used three-dimensional electron diffraction (3D ED), which is based on collecting a series of ED patterns by tilting a crystal over small angle steps around a random rotation axis and combining the patterns into a 3D data set (see ref. 30–32 and references therein). The data were then used to analyze the reciprocal lattice of the crystal. For the simplicity of data analysis, we performed our experiments on the crushed monodomain SrFeO2.5 single crystals; however, the 3D ED experiment can be also performed on powder samples or thin films. Acquiring 3D ED data at different stages of the reaction, we could clarify the open question on the phase transitions during redox reactions in a gas environment: upon oxidation, we saw the formation of domains with Cmmm SrFeO2.75 intermediate state with further transformation to the perovskite Pmm structure. Thus, contrary to previous reports on in situ redox reactions in gas environments of SrFeOx, this oxygen-vacancy ordering does occur as an intermediate phase. Upon being reduced again, the brownmillerite phase lost the Pcmb interlayer ordering but retained the in-plane L–R order, which can alternatively be considered as the appearance of a large amount of stacking faults in the structure.
Experimental section
Single-crystal growth was carried out in an optical mirror furnace (NEC SC2, Japan) equipped with two 500 W halogen lamps as heat source and two ellipsoidal mirrors.33 Crystal growth was performed in high-purity Ar-atmosphere (99.999%) with a typical traveling rate of 1–2 mm h−1, while the upper and lower shafts were rotated in opposite directions at 35 rpm. The obtained as-grown crystals had a typical length of up to 10 cm and 8 mm in diameter with shiny metallic lustre. As-grown single crystals show the brownmillerite type structure and are usually highly twinned, related to the cubic to orthorhombic phase transition. Monodomain single crystals were obtained via post synthesis annealing at 1100 °C and 1 × 10−3 mbar (primary vacuum) for 1 h and subsequent slow furnace cooling to RT in a time window of 15 h. Single crystal X-ray diffraction studies were carried out on a STOE-STADIVARI diffractometer, equipped with a Mo μ-focus tube and 200 K Pilatus detector. Thereby different crystal parts, although treated under the same thermal conditions, showed a different twin behavior, and beside monodomain single crystals also crystals with up to four twin domains were obtained.Thermogravimetric (TG) analysis has been carried out on a 70 mg single crystal with up to 3 mm length on a PerkinElmer T8000 instrument, using a gas atmosphere consisting of 33% O2 together with 67% Ar. To assure to be close to equilibrium conditions for the oxygen uptake reaction, the heating rate was limited to 1 °C min−1.
Transmission electron microscopy (TEM) studies of the pristine SrFeO2.5 were performed using an FEI Tecnai G2 microscope operated at 200 kV and equipped with a Nanomegas DigiStar precession module. To prepare specimens for TEM analysis, a single crystal of SrFeO2.5 was crushed and ground in ethanol. A few drops of the suspension were deposited onto a holey carbon TEM grid. In-zone selected area electron diffraction (SAED) patterns were acquired using an FEI double tilt holder. The in situ heating in vacuum was done using a Gatan double tilt heating holder. The sample was heated from room temperature (RT) to 900 °C and in-zone SAED patterns were acquired every 100 °C.
To prepare the TEM specimen for the in situ study in the gas environment, the crushed sample was dispersed in ethanol or hexane by sonication during 4 intervals of 15 minutes. Between consecutive intervals, the water in the ultrasonic bath was replaced to counteract heating. After letting the solution sediment for 30 minutes, the suspension was drop-casted onto a MEMS-based bottom chip for the DENS solutions Climate holder. EDS-compatible top chips were used to ensure the highest possible tilting range (≈−30 to 30°) for the 3D ED experiments. The bottom chips were preliminary coated with an approximately 10 nm thick carbon layer at the outside of the chip to avoid charging as this can cause movement of the crystals, and cleaned with 75% Ar–25% O2 plasma for 2 minutes at 50% power. The bottom chip with the deposited sample was additionally cleaned with 75% Ar–25% O2 plasma for 15 seconds at 30% power to reduce contamination from the residuals of the organic solvent. The gas supply system (GSS) from DENS solutions was used to provide gasses (O2, H2, He), control the gas flow (0.4–0.5 ml min−1) and gas pressure (around 0.7 atm on the outlet). For the temperatures, we systematically tracked the whole temperature range from RT to 900 °C in O2/He flow with certain steps while focusing on the temperatures around 300–400 °C as determined by TG analysis (see below). The 3D ED series were acquired on an FEI Tecnai G2 microscope operated at 200 kV and FEI Titan 80-300 microscope operated at 300 kV. Electron diffraction patterns were obtained either each 0.5° or 1° by tilting manually, using a custom-made script for Digital Micrograph that allows automated tilt of the crystal and acquisition of the ED patterns, or using fast-ADT software.34 The PETS2.0 software was used to analyze the data and make the 3D reconstruction of the reciprocal space.35
Result and discussions
For this study, monodomain SrFeO2.5 single crystals were obtained via a slow furnace cooling under applied dynamic primary vacuum conditions. Note that this is the first time monodomain single crystals from this material are reported in literature. Fig. 2 shows the reconstructed sections of the [010]/[102] and [010]/[201] planes of SrFeO2.5, obtained by single crystal X-ray diffraction. These planes are chosen to evidence the monodomain nature of the crystal, and also clearly indicate the doubling of the c-axis related to intralayer (R, L)-(FeO4)∞ tetrahedral chain ordering by sharp and well-defined reflection profiles of half-integer (00l)-values. The absence of any diffuse scattering shows that no significant amount of stacking faults is present. The absence of additional and partially non-integer reflections related to the presence of twin domains in the left figure indicates that this is a pure monodomain SrFeO2.5 single crystal at room temperature. | ||
| Fig. 2 Reconstructed sections of the [010]/[102] (left) and [010]/[201] (right) single crystal X-ray diffraction planes (indexation given for the standard brownmillerite setting in the 5.54 × 15.62 × 5.70 Å3 unit cell) for SrFeO2.5, which are key planes for the attribution of different twin domains as well as (R, L)-(FeO4)∞ chain ordering. | ||
Selected area electron diffraction (SAED) patterns of SrFeO2.5 shown in Fig. 3 confirm the orthorhombic cell with the approximate cell parameters a = 5.53 Å, b = 15.59 Å, and c = 11.35 Å. The following reflection conditions can be derived: 0kl: l = 2n, hk0: k = 2n, 0k0: k = 2n, 00l: l = 2n, which agree with the Pcmb space group. The SAED patterns clearly show the reflections corresponding to the intralayer ordering (examples are outlined with orange circles) and interlayer ordering (examples are indicated by blue arrows) in the SrFeO2.5 structure. Extra extinctions are present that arise from symmetry properties of the tetrahedral chain superstructure as described in detail by D'Hondt et al.15 The Pcmb space group is also clear from the characteristic reflections at hkl: h, l = 2n + 1 in the [101] diffraction pattern, agreeing with the β = 0, γ = 1/2 case of the superspace I2/m(0βγ)0s model,15 and in conflict with any I-centered three-dimensional space group. All 20 crystals studied, obtained from the same parent crystal after crushing, yielded identical diffraction patterns, indicating a homogeneous interlayer order over the whole crystal. The crystals did not show any sign of twinning on the SAED patterns.
 | ||
Fig. 3 In-zone ED patterns of SrFeO2.5 indexed in the Pcmb space group. Corresponding perovskite zones are indicated as well with a subscript “p”. The basic perovskite unit cell ap is outlined in green for the 〈100〉 orientation and red for the 〈110〉 orientation. Blue arrows in the [101]Pcmb = [30![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ]p zone point to rows of superstructure reflections due to the interlayer ordering of (FeO4)∞ tetrahedral chains, while the reflections indicated by orange circles in the [010]Pcmb = [010]p and [211]Pcmb = [110]p zones are due to the intralayer ordering of (FeO4)∞ tetrahedral chains. ]p zone point to rows of superstructure reflections due to the interlayer ordering of (FeO4)∞ tetrahedral chains, while the reflections indicated by orange circles in the [010]Pcmb = [010]p and [211]Pcmb = [110]p zones are due to the intralayer ordering of (FeO4)∞ tetrahedral chains. | ||
To estimate the required oxygen uptake conditions for the in situ oxidation of SrFeO2.5, we performed a thermogravimetric (TG) study on a SrFeO2.5 single crystal (70 mg) in 33% O2–67% Ar atmosphere, using a heating and cooling rate of 1° min−1. As can be seen from Fig. 4, the oxygen uptake sets in already below 270 °C, showing a maximum at 325 °C and there is a continuous oxygen loss on further heating. Upon cooling, the continuous oxygen uptake remains constant below 250 °C, leading to a total weight increase of 3.42%. This weight uptake thus corresponds to a stoichiometry of SrFeO2.875, for which theoretically an increase of 3.27% is expected. Therefore, we expect to be able to see modifications of the brownmillerite phase with in situ 3D ED around 300 °C.
 | ||
| Fig. 4 Thermogravimetric study carried out on a SrFeO2.5 single crystal (70 mg) in 33% O2–67% Ar atmosphere. | ||
Prior to the experiments in the gas environment, we performed in situ heating of the pristine SrFeO2.5 in the vacuum of the TEM and confirmed that no phase transformations in the material are caused by heating in vacuum with and without electron beam exposure while heating up to 800 °C (see details in ESI Fig. S1†), i.e. the structure remains brownmillerite with both intra- and interlayer order of the twists of the tetrahedral chains. Then we performed several in situ experiments in gas with varying experimental conditions (temperature, heating rate, oxygen partial pressure, gases) and on crystals of different sizes and morphology. The obtained 3D ED sections were compared with the simulated ones for different SrFeOx phases given in Fig. S2–S6.† Note that the experimental patterns (Fig. S8–S21†) show a large missing wedge due to the limited tilt range of commercial in situ gas holders.
To illustrate our findings, we show in Fig. 5 close-ups of the informative fragments of the 3D ED sections for a representative crystal of about 200 nm size (Fig. S7–S17†). Each column corresponds to a different stage in the in situ experiment. For clarity we index the patterns in the perovskite “p” cell throughout the further text, and refer to any extra reflections due to the Pcmb or Cmmm ordering as superstructure reflections with respect to the perovskite reciprocal lattice. Upon slow (5° min−1) heating up to 300 °C in 33% O2, the crystal preserved its brownmillerite Pcmb structure. The superstructure reflections indicated by orange arrows in [010]p and [110]p prove the presence of the intralayer ordering of the (FeO4)∞ chains, while the sharp reflections indicated by the blue arrows in the [30]p zone confirm the interlayer ordering.
 | ||
| Fig. 5 Close-ups of the [010]p, [100]p, [110]p and [30]p reciprocal space sections reconstructed from the 3D ED series taken during the oxidation and reduction and the calculated [110]p zone. All patterns except “700 °C (10% H2)” are from the same crystal. Orange arrows point to the brownmillerite superstructure reflections due to the intralayer ordering of (FeO4)∞ tetrahedral chains. Blue arrows point to the rows of superstructure reflections due to the interlayer ordering of (FeO4)∞ tetrahedral chains. White circles outline the split reflections due to the formation of the second phase. | ||
At 350 °C some reflections become split (examples are circled in [010]p and [110]p) indicating the formation of domains with different interplanar distances along the direction of the split reflections. In the [110]p zone, the reflections corresponding to the brownmillerite phase faded (orange arrows) and new reflections emerged (pink arrows). These new reflections are typical for the SrFeO2.75 intermediate phase with Cmmm symmetry. When the corresponding patterns for the Pcmb and Cmmm phases are superimposed ([110]p calc.), the resulting pattern agrees well with the Experimental section [110]p.
Upon further heating to 400 °C in 33% O2, mainly the Cmmm reflections remained in the [110]p zone, while the Pcmb reflections faded. There was only partial oxidation of the crystal, since characteristic brownmillerite reflections in some other zones (e.g. [010]p and [30]p) were still preserved. When the oxygen concentration was increased up to 100% at 400 °C, all the superstructure brownmillerite Pcmb and orthorhombic Cmmm reflections disappeared, and the crystal obtained a cubic oxygen deficient perovskite-type structure within a few minutes. Note that going to 100% oxygen at 350 °C possibly might also give the phase transition to Pmm structure. However, it was not tested since in situ 3D ED does not allow for exhaustive testing of all different conditions as it is a manpower intensive technique where each crystal is checked manually at each chosen condition.
To check the stability of the crystals during the in situ reaction in TEM and their stability upon cycling, an additional cooling–heating cycle was performed. As can be seen in the sections in Fig. S13–S17 ,† the second cycle went similarly to the first one, and diffraction patterns showing pure Cmmm structure could be obtained prior to the transformation to the perovskite. The formation of the intermediate I4/mmm structure was not observed in any cycle.
After cooling down to RT in O2, the structure shows a lot of disorder, as can be seen by the presence of clear streaks in [100]p. The positions of the reflections and streaks show the best agreement with disordered brownmillerite (Imma) (compare to Fig. S2–S6†). The extra reflections centering the [010]p, [110]p and [10]p sections are the intersections of the streaks. No streaks or reflections at the characteristic places for (R, L)-ordering are present. The streaking occurs along the 〈100〉p directions of the perovskite subcell, and is possibly due to the presence of intergrowth layers without oxygen-vacancy order (perovskite type). Our hypothesis is as follows: when the crystal was oxidized at 400 °C, many oxygen vacancies were filled, and upon cooling the remaining vacancies tried to order and to return back to brownmillerite. However, due to the lack of vacancies left to restore the brownmillerite structure within the whole crystal, the layers of brownmillerite (vacancy-rich layers) alternate with layers of perovskite (vacancy-poor layers). This causes the streaks in the diffraction patterns along the direction that corresponds to the stacking direction.
If crystals in the perovskite phase (such as that obtained upon heating to 400 °C in 100% O2 in Fig. 5 and S18–S21†) are further heated up to 700 °C in 10% H2, then within a few seconds the structure returns to the brownmillerite structure (extra reflections in zones [100]p and [30]p) proving a good cycling capability of the material. The intralayer ordering of the tetrahedral chains is restored (weak reflections on the [010]p pattern marked by orange arrows). The interlayer order is not restored: the [30]p section of the reduced phase shows weak lines of diffuse intensity instead of sharp reflections. The patterns remained unchanged upon subsequent cooling in H2 atmosphere down to RT, meaning that the tetrahedral chain order across the layers is not restored.
While it was shown above that the pristine brownmillerite SrFeO2.5 crystal interlayer ordering persists up to 800 °C in vacuum, the structure after one cycle of in situ oxidation and reduction has a mix of layer stackings, as was the case for the SrFeO2.5 powder samples in the paper by D'Hondt et al.15 The absence of interlayer order could be a consequence of the faster cooling (15–20° min−1) than the 15 hours slow cooling that was used for obtaining the ordered pristine sample. The symmetry of the reduced structure corresponds to the I2/m(0βγ)0s space group with γ = 1/2 and varying β instead of Pcmb (which is I2/m(0βγ)0s with γ = 1/2 and β = 0 uniquely). The ability of the (FeO4)∞ chains in the brownmillerite structure to switch their orientation between the layers upon the oxidation–reduction cycle, can help to minimize the activation energy for oxygen diffusion processes.36
In contrast to what was reported from in situ powder XRD and NPD experiments, using 3D ED we observed a formation of an intermediate oxygen-vacancy ordered Cmmm phase during heating in O2. Our results also explain why this intermediate phase was not detected in the in situ powder diffraction experiments,28,29 since in the first cycle Cmmm appeared only in the form of small domains together with the brownmillerite structure. Thus, the coherence size of the domains would be too small to be detected by XRD or NPD. In our experiments we also saw the occurrence of nanoscale twinning when studying larger crystals (Fig. 6), which would impede obtaining clear crystal structures from the power diffraction patterns by broadening of the peaks. Electrochemical oxidation of the single crystal SrFeO2.5 performed at room temperature showed the formation of an extra I4/mmm phase,16 which we did not observe in our experiments. A possible explanation is the faster kinetics in the case of in situ 3D ED in a gas environment. A 3D ED series can be acquired within 5–10 minutes which is in the range of the cycling reaction time,28 while the electrochemical oxidation was performed over time intervals of more than 90 hours. In this case, the faster redox reaction implemented by in situ 3D ED would give the more relevant information for such applications as chemical looping, as the circumstances are closer to those of the applications. When the material was cooled down to RT in O2, all order in the twist of the tetrahedral chains disappeared and only disordered brownmillerite (Imma) remained.
 | ||
| Fig. 6 Fragments of the [101]p reciprocal space section reconstructed from the 3D ED series taken during the oxidation and reduction of a large crystal of 600 nm (top) and the calculated [101]p zone (bottom). The basic 〈110〉 perovskite unit cell ap is outlined in red. Reflections outlined by the red and orange circles correspond to 90° rotation twins around the 〈100〉p axes in brownmillerite Pcmb structure. | ||
We want to point out that the formation of all phases observed with ED are related to the specific kinetics for phase formation under the conditions applied. It is clear that the formation of monodomain single crystals, which has so far not been reported elsewhere, is the result of a sufficiently slow cooling process, under defined vacuum conditions. It becomes thus evident that the creation of inter- and intralayer ordering requires a longer time scale involving a more delicate structural evolution on a longer scale. This certainly also holds for the formation of the I4/mmm phase SrFeO2.875, which was clearly observed during electrochemical oxidation,16 while it is absent here during the ED experiments. Its formation is also depending on the interplay of oxygen partial pressure, together with the applied temperature. The easy switching between different order/disorder arrangements of the (FeO4)∞ tetrahedral chains suggest that they are energetically very close. This is probably also the reason why a faultless arrangement is rather difficult to obtain.
Such ordering processes are significant concerning the understanding of the oxygen diffusion mechanisms. That the R–L-tetrahedral (FeO4)∞-chain ordering persists up to 900 °C is an important structural detail, as it underlines the thermal stability of this configuration, which appears to be the ground state for stoichiometric SrFeO2.5. The underlying oxygen diffusion mechanisms is then supposed to be based on a two-step mechanism: due to large lattice dynamical displacements, the apical oxygen atoms first get sufficiently far from their equilibrium positions. Further on they jump into the empty vacancy channels, leaving behind a square pyramid and a modified tetrahedron. Then both relax towards a reoriented tetrahedral chain, evidencing the structural ability for a dynamical rearrangement of different FeOn polyhedral in agreement with the mechanism proposed by Paulus et al.37
Conclusions
Using 3D ED in situ studies of SrFeO2.5 in O2 and H2 environments, we obtained single crystal information on the structure transformation during redox reactions of individual particles with sizes of a few hundreds of nanometers. Our results clearly showed the formation of the SrFeO2.75Cmmm structure upon oxidation, followed by further oxidation to the perovskite SrFeO3−δPmm structure. This phase was not observed in previous in situ experiments in a gas environment with X-ray or neutron powder diffraction. We did not observe the formation of the I4/mmm SrFeO2.875 phase in the oxidation process contrary to what was reported from ex situ and in situ electrochemical oxidation, which can be explained by the faster kinetics of redox reactions of individual nanosized particles in a gas environment and differences in the experimental conditions (heating in O2vs. RT electrochemical oxidation), crystal sizes (200 nm vs. 70 μm) and duration of the reactions (few hours vs. 90 hours). Therefore, in situ 3D ED in gas (used in this paper for the first time in literature) proves to be a powerful tool to study gas–solid reactions of functional materials, as the fast reaction times in a gas environment are close to those used in applications such as chemical looping and solid oxide fuel cells. Furthermore, these studies can give single crystal diffraction data on individual submicron sized particles in powder samples, whereas X-ray and neutron diffraction in situ experiments on particles with relevant small sizes can only be done with powder diffraction.
Author contributions
MB and DV performed the TEM experiments, MB, DV and JH analyzed the TEM data and crystal structures, WP and MC synthesized the pristine sample, performed single crystal XRD and TGA measurements and analyzed the data thereof, initial version of the manuscript was written by MB, all authors completed, corrected and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support is acknowledged from the FWO-Hercules fund I003218N ‘Infrastructure for imaging nanoscale processes in gas/vapor or liquid environments’, from the University of Antwerp through grant BOF TOP 38689. This work was supported by the European Commission Horizon 2020 NanED grant number 956099. Financial support from the French National Research Agency (ANR) through the project “Structural induced Electronic Complexity controlled by low temperature Topotactic Reaction” (SECTOR No. ANR-14-CE36-0006-01) is gratefully acknowledged.References
- S. Stølen, E. Bakken and C. E. Mohn, Phys. Chem. Chem. Phys., 2006, 8, 429–447 RSC.
- M. V. Patrakeev, J. A. Bahteeva, E. B. Mitberg, I. A. Leonidov, V. L. Kozhevnikov and K. R. Poeppelmeier, J. Solid State Chem., 2003, 172, 219–231 CrossRef CAS.
- C. Sun, R. Hui and J. Roller, J. Solid State Electrochem., 2010, 14, 1125–1144 CrossRef CAS.
- J. M. Ralph, C. Rossignol and R. Kumar, J. Electrochem. Soc., 2003, 150, A1518 CrossRef CAS.
- J. Deng, H. Dai, H. Jiang, L. Zhang, G. Wang, H. He and C. T. Au, Environ. Sci. Technol., 2010, 44, 2618–2623 CrossRef CAS PubMed.
- A. Leontiou, A. Ladavos and P. Pomonis, Appl. Catal., A, 2003, 241, 133–141 CrossRef CAS.
- F. He, J. Chen, S. Liu, Z. Huang, G. Wei, G. Wang, Y. Cao and K. Zhao, Int. J. Hydrogen Energy, 2019, 44, 10265–10276 CrossRef CAS.
- H. Chang, E. Bjørgum, O. Mihai, J. Yang, H. L. Lein, T. Grande, S. Raaen, Y.-A. Zhu, A. Holmen and D. Chen, ACS Catal., 2020, 10, 3707–3719 CrossRef CAS.
- Y. Tsujimoto, C. Tassel, N. Hayashi, T. Watanabe, H. Kageyama, K. Yoshimura, M. Takano, M. Ceretti, C. Ritter and W. Paulus, Nature, 2007, 450, 1062–1065 CrossRef CAS PubMed.
- Y. Takeda, K. Kanno, T. Takada, O. Yamamoto, M. Takano, N. Nakayama and Y. Bando, J. Solid State Chem., 1986, 63, 237–249 CrossRef CAS.
- M. Takano, T. Okita, N. Nakayama, Y. Bando, Y. Takeda, O. Yamamoto and J. B. Goodenough, J. Solid State Chem., 1988, 73, 140–150 CrossRef CAS.
- J. P. P. Hodges, S. Short, J. D. D. Jorgensen, X. Xiong, B. Dabrowski, S. M. M. Mini and C. W. W. Kimball, J. Solid State Chem., 2000, 151, 190–209 CrossRef CAS.
- A. Nemudry, M. Weiss, I. Gainutdinov, V. Boldyrev and R. Schöllhorn, Chem. Mater., 2002, 10, 2403–2411 CrossRef.
- S. Schott, Z. Jia, A. K. Schaper, V. Thangadurai, W. Weppner and P. Schmid-Beurmann, Phys. Status Solidi, 2005, 202, 2330–2335 CrossRef CAS.
- H. D'Hondt, A. M. Abakumov, J. Hadermann, A. S. Kalyuzhnaya, M. G. Rozova, E. V. Antipov and G. Van Tendeloo, Chem. Mater., 2008, 20, 7188–7194 CrossRef.
- A. Maity, R. Dutta, B. Penkala, M. Ceretti, A. Letrouit-Lebranchu, D. Chernyshov, A. Perichon, A. Piovano, A. Bossak, M. Meven and W. Paulus, J. Phys. D: Appl. Phys., 2015, 48, 504004 CrossRef.
- J. E. Auckett, A. J. Studer, N. Sharma and C. D. Ling, Solid State Ionics, 2012, 225, 432–436 CrossRef CAS.
- J. E. Auckett, W. T. Lee, K. C. Rule, A. Bosak and C. D. Ling, Inorg. Chem., 2019, 58, 12317–12324 CrossRef CAS PubMed.
- A. M. Abakumov, A. S. Kalyuzhnaya, M. G. Rozova, E. V. Antipov, J. Hadermann and G. Van Tendeloo, Solid State Sci., 2005, 7, 801–811 CrossRef CAS.
- T. G. Parsons, H. D'Hondt, J. Hadermann and M. A. Hayward, Chem. Mater., 2009, 21, 5527–5538 CrossRef CAS.
- P. K. Gallagher, J. B. MacChesney and D. N. E. Buchanan, J. Chem. Phys., 1964, 41, 2429–2434 CrossRef CAS.
- C. Greaves, A. J. Jacobson, B. C. Tofield and B. E. F. Fender, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 641–646 CrossRef.
- J. P. Hodges, S. Short, J. D. Jorgensen, X. Xiong, B. Dabrowski, S. M. Mini and C. W. Kimball, J. Solid State Chem., 2000, 151, 190–209 CrossRef CAS.
- M. Schmidt and S. J. Campbell, J. Solid State Chem., 2001, 156, 292–304 CrossRef CAS.
- J. E. Auckett, A. J. Studer, E. Pellegrini, J. Ollivier, M. R. Johnson, H. Schober, W. Miiller and C. D. Ling, Chem. Mater., 2013, 25, 3080–3087 CrossRef CAS.
- J. Young and J. M. Rondinelli, Phys. Rev. B, 2015, 92, 174111 CrossRef.
- E. Heifets, E. A. Kotomin, A. A. Bagaturyants and J. Maier, Phys. Chem. Chem. Phys., 2019, 21, 3918–3931 RSC.
- D. D. Taylor, N. J. Schreiber, B. D. Levitas, W. Xu, P. S. Whitfield and E. E. Rodriguez, Chem. Mater., 2016, 28, 3951–3960 CrossRef CAS.
- E. Marek, W. Hu, M. Gaultois, C. P. Grey and S. A. Scott, Appl. Energy, 2018, 223, 369–382 CrossRef CAS.
- U. Kolb, T. Gorelik, C. Kübel, M. T. Otten and D. Hubert, Ultramicroscopy, 2007, 107, 507–513 CrossRef CAS PubMed.
- M. Gemmi and A. E. Lanza, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2019, 75, 495–504 CrossRef CAS PubMed.
- M. Gemmi, E. Mugnaioli, T. E. Gorelik, U. Kolb, L. Palatinus, P. Boullay, S. Hovmöller and J. P. Abrahams, ACS Cent. Sci., 2019, 5, 1315–1329 CrossRef CAS PubMed.
- A. Maljuk, A. Lebon, V. Damljanović, C. Ulrich, C. T. Lin, P. Adler and B. Keimer, J. Cryst. Growth, 2006, 291, 412–415 CrossRef CAS.
- S. Plana-Ruiz, Y. Krysiak, J. Portillo, E. Alig, S. Estradé, F. Peiró and U. Kolb, Ultramicroscopy, 2020, 211, 112951 CrossRef CAS PubMed.
- L. Palatinus, P. Brázda, M. Jelínek, J. Hrdá, G. Steciuk and M. Klementová, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2019, 75, 512–522 CrossRef CAS PubMed.
- R. Le Toquin, W. Paulus, A. Cousson, C. Prestipino and C. Lamberti, J. Am. Chem. Soc., 2006, 128, 13161–13174 CrossRef CAS PubMed.
- W. Paulus, H. Schober, S. Eibl, M. Johnson, T. Berthier, O. Hernandez, M. Ceretti, M. Plazanet, K. Conder and C. Lamberti, J. Am. Chem. Soc., 2008, 130, 16080–16085 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta03247c |
| This journal is © The Royal Society of Chemistry 2023 |