
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTopotactic BI3-assisted borodization: synthesis and electrocatalysis applications of transition metal borides†
Katherine E.
Woo‡
ab,
Seongyoung
Kong‡
 ab,
Wei
Chen‡
c,
Tsz Hin
Chang
d,
Gayatri
Viswanathan
ab,
Aida M.
Díez
e,
Viviana
Sousa
eg,
Yury V.
Kolen'ko
e,
Oleg I.
Lebedev
f,
Marta
Costa Figueiredo
c and
Kirill
Kovnir
*ab
ab,
Wei
Chen‡
c,
Tsz Hin
Chang
d,
Gayatri
Viswanathan
ab,
Aida M.
Díez
e,
Viviana
Sousa
eg,
Yury V.
Kolen'ko
e,
Oleg I.
Lebedev
f,
Marta
Costa Figueiredo
c and
Kirill
Kovnir
*ab
aDepartment of Chemistry, Iowa State University, Ames, Iowa 50011, USA. E-mail: kovnir@iastate.edu; kewoo3@gmail.com; sykong@iastate.edu; gayatriv@iastate.edu
bAmes National Laboratory, U.S. Department of Energy, Ames, Iowa 50011, USA
cDepartment of Chemical Engineering and Chemistry, Eindhoven University of Technology, 5600 MB Eindhoven, Netherlands. E-mail: m.c.costa.figueiredo@tue.nl; w.chen2@tue.nl
dDepartment of Chemistry, University of California, Davis, California 95616, USA. E-mail: tchang9525@gmail.com
eNanochemistry Research Group, International Iberian Nanotechnology Laboratory, Braga 4715-330, Portugal. E-mail: aida.diez@inl.int; yury.kolenko@inl.int; viviana.sousa@inl.int
fLaboratoire CRISMAT, ENSICAEN, CNRS UMR 6508, 14050 Caen, France
gCenter of Physics of the Universities of Minho and Porto, University of Minho, Braga 4710-057, Portugal
First published on 15th September 2022
Abstract
A facile and universal route for synthesizing transition metal borides has been developed by reaction of boron triiodide (BI3) with elemental transition metals. This method employs relatively low synthesis temperatures to afford single-phase samples of various binary and ternary metal borides, such as Fe2B, Co2B, Ni3B, TiB2, VB2, CrB2, and Ni2CoB. This synthesis protocol can be utilized for the topotactic transformation of metal shapes into their respective borides, as exemplified by transformation of Ni foam to Ni3B foam. In situ powder X-ray diffraction studies of the Ni–BI3 system showed that the crystalline nickel borides, Ni4B3 and Ni2B, start to form at temperatures as low as 700 K and 877 K, respectively, which is significantly lower than the typical synthesis temperatures required to produce these borides. Ni3B synthesized by this method was tested as a supporting material for oxygen evolution reaction (OER) in acidic media. Composite electrocatalysts of IrO2/Ni3B with only 50% of IrO2 exhibit current densities and stability similar to pure IrO2 at mass loadings lower than 0.5 mg cm−2, indicating Ni3B could be a promising supporting material for acidic OER.
Introduction
Metal borides (MxBy) have found use in a wide variety of applications including Fe2B coatings,1 {V, Cr, Mn, Fe, Co, Ni, Mo}–B catalysts for electrocatalytic hydrogen evolution reaction (HER) and oxygen evolution reaction (OER),2–6 LaB6 as a hot cathode,7 and VB2 anodes for primary batteries8 to name a few. Robust and facile solid state synthesis methods for metal borides are, however, hindered by the inertness of boron and, oftentimes, the metal as well. Traditional methods to synthesize metal borides include arc-melting of the elements, borothermic reduction of metal oxides, or metal flux assisted synthesis.5 These methods require high temperatures over 1273 K and often require further purification due to the presence of oxygen or other contaminants from the flux or reaction vessel.The most common lower temperature synthesis of early transition metal borides was discussed by Rao and co-workers in 1995.9 This study was restricted to the synthesis of group 4–6 metal diborides, in which metal chloride precursors were reacted with MgB2 at 1123 K. This method was recently extended by Gillan et al. to late transition metal borides.10 The syntheses of hafnium diboride HfB2 and tungsten borides WBx were performed by the reaction of a resistive heated metal wire (Hf or W) with boron bromide instead of using a metal halide precursor.11,12 This method required a sophisticated apparatus to deal with the liquid boron bromide source and the Hf or W metal wire was only partially converted into boride, thus preserving a metallic core. Fokwa and co-workers developed a straightforward synthesis of nanoscale transition metal borides by reacting anhydrous metal chlorides and elemental boron in Sn flux, which served simultaneously as a high-temperature solvent and as a reducing agent.13 Another interesting approach to forming Li-containing borides was recently developed using LiH as a precursor.14 Despite the successful formation of metal borides, these methods are generally limited in that they only produce powders of metal borides (even when using a flux or metal hydrides) or they leave behind unreacted elemental metal from the metal source.
While lighter boron halides have already been employed to synthesize metal borides, boron triiodide (BI3) has the additional benefit of being a solid, greatly increasing the ease of handling it. BI3 thermally decomposes at a moderate temperature under reduced pressure and was shown to transport pure boron via heat gradient.17 In addition, unreacted BI3 hydrolyzes into H3BO3 and HI, which can be easily dissolved in water.18 In the present work, motivated by the lack of more efficient methods for making metal borides, we developed a novel borodization route by reaction of metals with BI3. By controlling the amount of BI3, several single-phase transition metal borides, namely Fe2B, Co2B, Ni3B, TiB2, VB2, and CrB2 were synthesized at relatively low temperature of 1073 K. In situ powder X-ray diffraction experiments were conducted to understand the formation of the metal borides starting from Ni and BI3 reagents. It was found that the crystalline metal borides starts to form at a relatively low temperatures (∼700 K), supporting the efficiency of this synthesis. Another advantage of this method is the ability to preserve the morphology of reactant metal after borodization, as we have demonstrated for Ni foam. Topotactic borodization reactions might be useful for converting not only powders, but films and nanocrystals of metals into borides, thus opening avenues to study applications of metal boride phases of various shapes and structures. In addition, topotactic conversion of metal foams into their respective metal phosphide foam electrocatalysts was shown to be of high importance for electrochemical HER and OER.15–17
One of the possible applications of metal borides is as a supporting material for electrocatalytic reactions. Acidic OER imposes substantial challenges regarding materials stability and most non-oxide 3d transition metals compounds are prone to severe degradation.20 However, metal borides show good OER stability in general and treating Ni3B powders with concentrated hydrochloric acid results in no detectable changes, making such borides good candidates for use in acidic conditions.21,22 In this work, we investigated the use of Ni3B as a support for oxygen evolution reaction (OER) electrocatalysis in acidic media. Composite electrocatalysts with different IrO2/Ni3B ratios were tested towards OER; this revealed that the Ni3B has low capacitance and can be used to effectively support IrO2 electrocatalysts. Importantly, IrO2/Ni3B composite electrocatalysts show remarkable specific activity, comparable to that of pure IrO2. Moreover, stability tests showed that the IrO2/Ni3B composite electrocatalyst has a similar stability to pure IrO2 during OER electrocatalysis in acidic media. These results suggest that the addition of Ni3B as a supporting material can be beneficial to reduce use of unsustainable IrO2 while retaining most of the original OER activity.
Experimental part
Caution : BI3 sublimes at low temperatures and can develop substantial autogenic vapor pressure, which may lead to rupturing and shattering of the reaction ampoule. Amounts of BI3 should be kept at a minimum. For the given volume of the sealed ampoule (16–19 cm3) at a reaction temperature of 1073 K, the amount of BI3 per reaction was limited to less than 650 mg to prevent any potential explosions. Additionally, it is recommended to wrap the ampoule in silica wool or to place it into a secondary container (alumina beaker) filled with sand. In either case, the furnace should be placed in a well-ventilated space, such as a fumehood.Synthesis of metal boride bulk powders
Boron triiodide BI3 (Sigma Aldrich, 99.8%) and metal powders, namely Ti (Alfa Aesar, 99.9%), V (Alfa Aesar, 99.5%), Cr (Alfa Aesar, 99.5%), Fe (Sigma Aldrich, 99.99%), Co (Alfa Aesar, 99.8%), Ni (Alfa Aesar, 99.996%), La (Alfa Aesar, 99.9%), and Ru (Alfa Aesar, 99.95%) were used as received and handled inside an argon glovebox. Reagents were sealed into 17 mm ID (19 mm OD) silica ampoules with a 14 mm × 16 mm silica plug for an enclosed length of roughly 9 cm. The amount of BI3 per reaction was limited to less than 650 mg. For VB2, CrB2, and Ni3B foam reactions, 9 mm ID (11 mm OD) silica tubes were used and the amount of BI3 was downscaled to less than 40 mg cm−3 of the sealed ampoule internal volume. The ampoules with reagents were cooled in an ice bath as they were evacuated to ≈10−2 mbar and then flame sealed under static vacuum. Starting ratios of reagents for selected borides are listed in Table 1.| Targeted boride | Metal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :BI3 ratio :BI3 ratio |
|---|---|
| TiB2 | 1 Ti:1 BI3 |
| VB2 | 3 V:2 BI3 |
| CrB2 | 1 Cr:1 BI3 |
| Fe2B | 7 Fe:2 BI3 |
| Co2B | 7 Co:2 BI3 |
| Ni3B | 7 Ni:2 BI3 |
The sealed ampoule was then laid on its side in a muffle furnace to achieve a slight temperature gradient with the reagents pointing to the back of the furnace where it would be slightly hotter. The default heating profile for the reactions was a 2 h ramp to 1073 K and annealing at that temperature for 24 h. After annealing, the sample was opened in the air (unless specified elsewhere) and washed in distilled water for 2 days. The product was then filtered using a nylon membrane filter and air dried. The Fe reaction was further washed in HCl solution (36–38%, Fisher Scientific) for a day to remove the FeO(OH) by-product and finally filtered using a polypropylene membrane filter.
Topotactic Ni foam transformation
A cut piece of Ni foam (110 pores per inch, 0.3 mm thick, Heze Jiaotong), approximately 7 × 17 mm2, was used instead of Ni powder to synthesize Ni3B. BI3 was loaded as powder with the ratio shown in Table 2 and the Ni foam was inserted vertically into the 9 mm ampoule. The above-described ratio and temperature profile was used.:BI3 starting reagents and corresponding products determined by powder XRD (see Fig. S1). Bold row indicates optimized ratio of starting materials
| Ni:BI3 molar ratio |
Crystalline reaction products detected by XRD |
|---|---|
| 1:3 |
Ni6BSi2 |
| 1:1 |
Ni6BSi2 + Ni3B + Ni2B |
| 4:2 |
Ni3B + Ni2B |
| 5:2 |
Ni3B + Ni2B |
7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :2 :2
|
Ni 3 B |
| 9:2 |
Ni + Ni3B |
| 11:2 |
Ni + Ni3B |
Powder X-ray diffraction (XRD)
Powder XRD experiments were performed using a 600 Miniflex diffractometer (Rigaku) equipped with Cu-Kα radiation and a Ni-Kβ filter. Samples were ground in an agate mortar and pestle, and then deposited on a thin layer of grease on a zero-background Si holder to minimize preferred orientation of particles.Synchrotron in situ powder XRD
Ni powder and BI3 crystals totaling 50 mg were ground together with an agate mortar and pestle inside the glovebox. The mixture was loaded into a silica capillary (0.5 mm ID/0.7 mm OD, Friedrich & Dimmock, Inc.). The capillary was then evacuated, and flame sealed. Beamline 17-BM-B at the Advanced Photon Source at Argonne National Laboratory was used for in situ experiments. The sample was loaded into a flow cell, equipped with electric heaters, and exposed to synchrotron radiation (λ = 0.24125 Å). Data were collected as the sample was heated at 30 K min−1 to 573 K, ramped at 10 K min−1 to 1073 K, cooled at 20 K min−1 to 573 K, and then cooled at 50 K min−1 to 348 K. Analysis of data was performed using GSAS-II software package.23Scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDX)
Elemental analyses were conducted using either a Quanta 250 field emission-SEM (FEI), equipped with a X-Max 80 EDX detector (Oxford), or a JSM-IT200 (JEOL) with built-in EDX detector. Powder samples were loaded into a graphite die, which was then filled with epoxy and polished smooth. A thin conductive carbon film was then deposited on the surface of the puck.Ni foam and Ni3B foam were loaded on carbon tape without further treatments. To process the data, the Aztec and SMILE VIEW software were used for FEI and JEOL, respectively.
Scanning transmission electron microscopy (STEM)
HAADF-STEM images and Electron Diffraction (ED) data were acquired using JEM ARM200F cold FEG double aberration corrected microscope operated at 200 kV, equipped with a large angle CENTURIO EDX detector, Orius CCD camera, and Quantum GIF. STEM samples were prepared by depositing ethanolic suspension of the target phase on the Cu holey carbon grid.Preparation of IrO2/Ni3B composite electrocatalysts
Synthesized phase-pure Ni3B and commercial IrO2 (Alfa Aesar, 99.99%) powders were mixed in 1:2, 1:1, and 2:1 molar ratios with a total amount of each mixture being 150 mg; the corresponding samples were labeled as 25%, 50%, and 75% of Ni3B. Once the samples were combined, they were ball-milled using a SPEX mixer/mill 8000M for an hour to ensure that the powders were completely blended and became fine enough for facile electrocatalytic ink formulation. For comparison purposes, samples of pure Ni3B and pure IrO2 were also ball-milled in a similar manner. The milled powder was then drop-cast on a home-made glassy carbon (GC) rotating disk electrode (RDE) with a geometric area of 0.2826 cm2, which served as the working electrode (WE) for electrochemical characterizations.
The ink containing the electrocatalysts was prepared by mixing water, isopropanol (volume ratio of water:isopropanol = 3:1), Nafion ionomer solution (5% in aliphatic alcohols and water, Sigma-Aldrich), and the composite electrocatalyst powders (50 mg mL−1). Before drop-casting, the GC RDE was polished with alumina powder for 5 min and then rinsed with water and ethanol. This procedure was repeated 3 times. After an ultrasonic treatment for 15 min, the as-formulated ink was drop-casted on the GC RDE and then left in a solvent-vapor-saturated (water + isopropanol) atmosphere overnight for drying. The mass loading of IrO2/Ni3B (0.25, 0.5, 1.0, 2.0 mg cm−2) was controlled by the volume of the drop-casted ink.
Characterization of OER activity and stability in acidic media
The electrochemical characterization of OER activity and stability was done in a conventional three-electrode glass cell. The counter electrode (CE) and reference electrode (RE) was a GC rod and a saturated Ag|AgCl electrode, respectively. All potentials are quoted in reference to the reversible hydrogen electrode (RHE). The electrolyte solution was 0.5 M H2SO4 prepared from H2SO4 (Merck, 99.999%) and ultrapure water (18.2 MΩ cm, Milli-Q). All the experiments were performed under N2 (5.0, LindeGas) saturated conditions. The applied potential was controlled by PGSTAT302N potentiostat (Metrohm) and the RDE rotating rate by an Autolab RRDE rotator (Metrohm). Before the electrochemical characterization, the WE (with electrocatalysts) was carefully rinsed with pure water, followed by electrochemical cleaning for 15 min by performing cyclic voltammetry from 0.05 to 1.1 VRHE at a scan rate of 200 mV s−1. All the data shown was corrected for a voltage drop (iR) of 80% unless specified elsewhere.Results and discussion
Synthesis
We have shown that the refractory boride, BP, can be synthesized using BI3 as the source of boron.24 Later, refractory metal borides such as HfB2 and WB2 have been prepared by reaction of the resistive heated metal wire with BBr3 vapor.11,12 In the current work, we extend BI3-assisted synthesis to first row transition metals. Similar to solid state metathesis reactions, the main driving force for this reaction is the formation of thermodynamically stable, water-soluble metal iodides: (nx + 3)M + nBI3 → nMxB + 3MIn (x = 1/6 to 3; n = 2, 3, and 4). For the selected metals (M = Ti, V, Cr, Fe, Co, and Ni) phase-pure polycrystalline metal boride powders or samples with minute amounts of admixtures (Fig. 1) can be successfully synthesized using the optimized ratios of the precursors, as described in Table 1. Co2B, Fe2B, and VB2 can be synthesized by a balanced reaction, i.e., no excess of metal or halide is expected in the products: 7Co + 2BI3 → 2Co2B + 3CoI2. In turn, Ni3B, CrB2, and TiB2 required an excess of BI3, indicating a formation of the gaseous iodine by-product: 7Ni + 2BI3 → 2Ni3B + NiI2 + 2I2↑, 4Cr + 4BI3 → 2CrB2 + 2CrI3 + 3I2↑, and 2Ti + 2BI3 → TiB2 + TiI4 + I2↑. Note that the melting points of the corresponding borides, 1429 K (Ni3B) and 3498 K (TiB2) are significantly higher than the utilized reaction temperature, 1073 K. | ||
| Fig. 1 Calculated (black) and experimental (red) powder XRD patterns of TiB2, VB2, CrB2, Fe2B, Co2B, and Ni3B synthesized through the reaction of metal powders and BI3. A peak at 2.79 Å−1Q present in all patterns, but most visible in the case of the VB2 sample, is attributed to contribution from the grease used for attaching powders to the XRD holder. | ||
For Ni, the reaction of 7 equivalents of Ni with 2 equivalents of BI3 provided single phase samples of Ni3B. These reaction ratios were utilized for the topotactic conversion of Ni foam, as well as for the preparation of the electrocatalysts discussed below. Additionally, a detailed study of the impact of metal to BI3 ratio was performed. Employing a large excess of BI3 resulted in the formation of Ni6BSi2 due to reaction with the silica ampoule. An unknown amorphous phase was detected in the reactions that produced Ni6BSi2, possibly also resulting from reaction with the silica ampoule. A stoichiometric reaction of 9Ni:2BI3 or reactions with higher Ni contents resulted in the presence of leftover metallic Ni in the products. As detailed in Table 2, Ni2B could be synthesized as well, in the presence of a slight excess of BI3, with Ni3B as a side product (Fig. S1†).
Other metals, namely La and Ru, were investigated using starting ratios for balanced reactions (i.e., La:BI3 = 7:6 and Ru:BI3 = 3:2), assuming formation of LaI3 and RuI2 co-products. Ru7B3 and LaB6 were successfully synthesized using the heating profile described in the experimental section, although an admixture of unreacted metal was present (Fig. S2†). CrB was also successfully synthesized using the same stoichiometric ratio with a single unidentified peak in the PXRD pattern at 2.44 Å−1Q (Fig. S3†). Further optimizations of reagents ratios and temperature profiles are required to produce phase-pure samples of these borides.
The herein developed method for synthesis of metal borides demonstrates universality by allowing for formation of materials with high diversity in B:M ratios, crystal structures, and degrees of B–B bonding (Scheme 1). As the B:M ratio decreases from 6 to 1/3, the dimensionality of the boron framework changes from 3D to 0D. For LaB6, the 3D boron framework is composed of covalently-linked octahedra. 2D graphene-like boron layers are present in the crystal structures of MB2 (TiB2, VB2, and CrB2), while 1D boron chains are found in MB (CrB) and M2B (Co2B and Fe2B). Further increasing the metal content stabilizes structures with isolated boron atoms surrounded by M6 trigonal prisms (as in Ru7B3) or M9 tricapped trigonal prism (as in Ni3B).
 | ||
| Scheme 1 Boron frameworks in the metal borides synthesized in this study with respect to the ratio of boron to metal. Boron: blue; metal: grey. | ||
In situ powder XRD
To understand the mechanism of borodization, in situ experiments were performed to study the formation of nickel boride (Fig. 2). The provided temperatures are approximate (±20 K) because the thermocouple is located outside the reaction capillary. Nevertheless, the series of phase transformations can still be determined with high reliability which further aids synthesis efforts and our knowledge of solid state reaction mechanisms.25–28In situ XRD studies were previously shown to be useful for synthesis of novel boron-containing compounds.29 In the studied synthesis of nickel boride, BI3 amorphized or went into the gaseous phase at 359 K, leaving only Ni to be detected by powder XRD. At 542 K, NiI2 is formed. New diffraction peaks appear in the powder patterns from 700 K to 877 K, which can be assigned to Ni4B3. It should be mentioned that the Ni4B3 intermediate was detected in other high-temperature in situ XRD studies involving the decomposition of ternary Li–Ni–B compounds.30 The detection of Ni4B3 demonstrates that the formation of NiI2 provides a strong enough driving force to make crystalline nickel boride at extremely low temperatures of ∼700 K. Ni4B3 may serve as an intermediate in the studied reaction. One can assume that an amorphous Ni–B phase is present in the capillary as well, at least in the 810–875 K range because nearly all the elemental Ni has been consumed by 812 K, and the formation of Ni2B is detected only at 877 K. Ni2B is the final thermodynamically stable phase and as soon as it forms, the peaks corresponding to Ni4B3 can no longer be detected. At 1053 K, NiI2 melts, in agreeance with its reported melting point, and Ni2B is the only crystalline phase present. Similarly, upon cooling, only Ni2B and an unknown phase were detected. Intense peaks around 2 Å−1 corresponding to NiI2 were no longer observed. Given the water solubility of the unidentified byproduct and the observation of NiI2 in the products of larger scale ex situ reactions, the unidentified phase is hypothesized to contain nickel and iodine. The persistent peaks in the powder patterns at ∼1 Å−1 are unaffected by temperature; thus these peaks are attributed to the experimental setup and not intrinsic to the studied system. The highly electrostatic nature of BI3 and the small scale of the reaction may have prevented accurate loading ratios of the starting materials. Additionally, increased vapor pressure or the lack of temperature gradient in the in situ set-up may have contributed to the reaction outcome as Ni2B rather than Ni3B as observed in larger scale ex situ reactions. Representative powder XRD patterns at different temperatures in the in situ study and calculated patterns of crystalline known phase are plotted in Fig. S5.†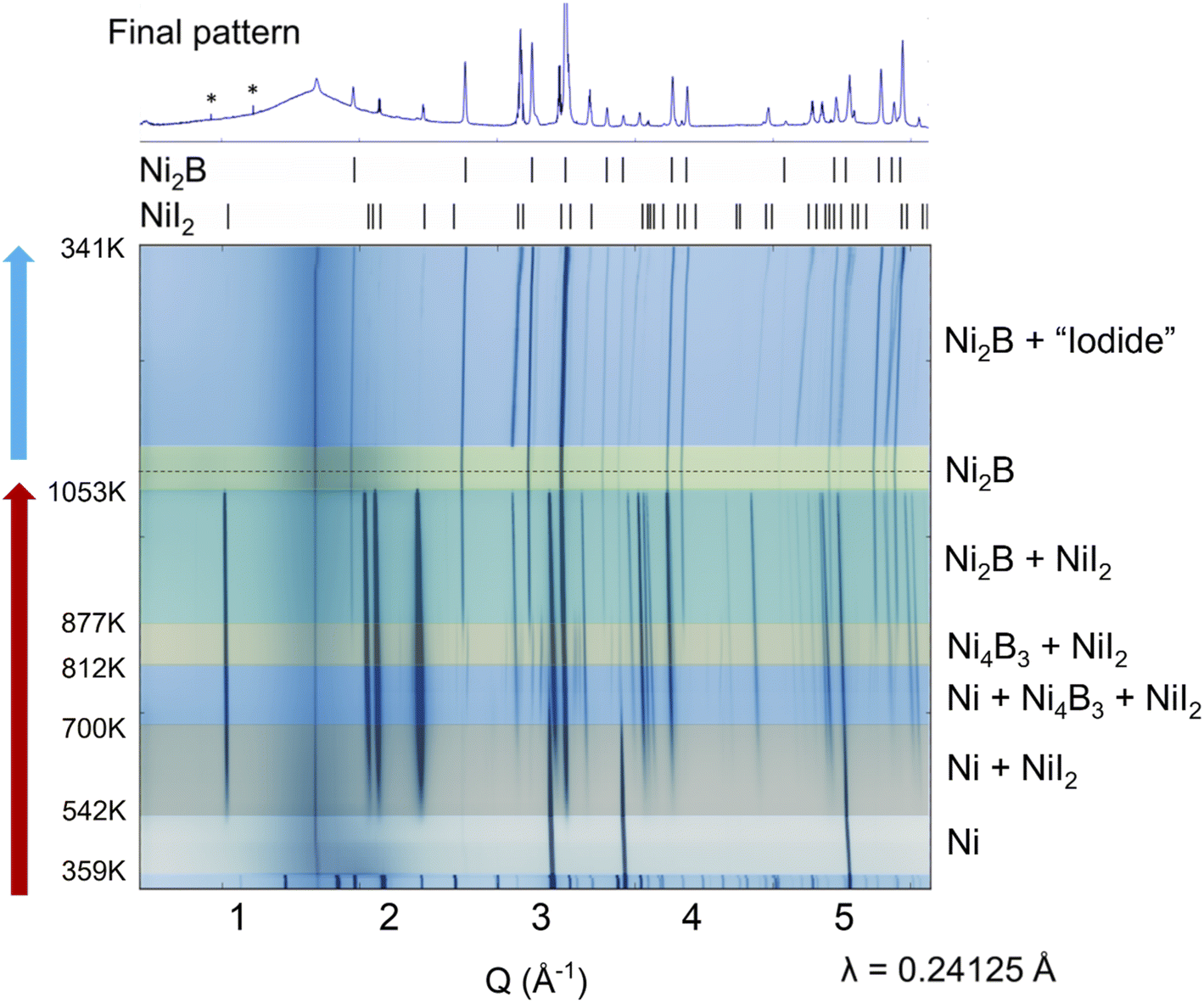 | ||
| Fig. 2 Contour plot of in situ powder XRD patterns upon reaction of Ni and BI3. The red arrow on the left indicates heating while the blue one corresponds to the cooling process. On top is the room temperature powder XRD pattern after completion of the in situ study, with tick marks corresponding to calculated positions for Ni2B and NiI2 diffraction peaks. Asterisks mark instrumental artifacts in the powder pattern. | ||
Topotactic reaction
The developed borodization method is suitable for generating metal borides of unique morphologies. A high surface area is known to be important for electrocatalysts,16 and Ni foam borodization was therefore attempted. The Ni3B reaction was repeated using Ni foam (Fig. 3). While a slight amount of Ni2B was detected by powder XRD, a predominantly Ni3B foam was successfully synthesized, albeit relatively brittle (Fig. 3d). Energy dispersive X-ray spectroscopy (EDX) showed no trace of iodine, demonstrating all iodide species were dissolved in water during the rinsing process (Fig. S4†). Fig. 3a shows the optical images of unreacted Ni foam (left) and the resultant Ni3B foam (right) synthesized using BI3. The original morphology of the Ni foam was preserved, and the color of the foam changed from shiny silver to dark brown. SEM images showed that the initial Ni foam was composed of Ni ligaments arranged in a tangled and porous 3D network (Fig. 3b). After borodization, this structure was retained, showing the topotactic ability of the reaction (Fig. 3c). Occasionally, the foam structure was altered during the course of the reaction such that the pores were eliminated (Fig. S6†). This suggests that location of the Ni foam in the ampoule (with or without direct contact with BI3) might impact the morphology. Increasing the Ni:BI3 ratio to 11:2 appeared to reduce the brittleness of the resulting Ni3B foam, although unreacted Ni was expectedly detected by powder XRD. Nevertheless, the as-produced Ni3B/Ni foam was still stiff and did not retain the flexibility of the original Ni foam. The aforementioned formation of Ni6BSi2 can also be detected by EDX as it crystallizes as well-shaped hexagonal plate-like crystals (Fig. S7†).
 | ||
| Fig. 3 (a) Photographs of pristine Ni foam (left) used as a reagent and the resultant product Ni3B foam (right) synthesized by a topotactic borodization. SEM backscattered electron images of (b) pristine Ni foam and (c) the resulting Ni3B foam. (d) Powder XRD patterns of the ground Ni3B foam (red) and calculated patterns for Ni3B (black). A peak at 2.52 Å−1Q marked with * corresponds to Ni2B admixture. | ||
Ternary mixed metal borides
Synthesis of ternary borides were approached via a proposed borodization of binary metal alloys. A Ni/Co alloy was produced by grinding a 1:1 molar ratio of Ni and Co powders together, which were then loaded in a silica ampoule and heated at 1173 K for 72 h. The resulting alloy ingot could not be crushed and was instead cut into smaller pieces using diagonal cutting pliers. These Ni/Co alloy pieces were then loaded in a 7:2 molar ratio of the alloy and BI3 in accordance with the optimized Ni3B synthesis (Table 2). The resulting product was a malleable foil and grey-brown powder. The powder XRD pattern of the latter resembles Ni3B or Co3B patterns with ∼0.01 Å−1Q peak shifts (Fig. S8†). Analysis by EDX found the ternary boride powder to contain a mixture of Ni and Co with 70/30 molar ratio, i.e. Ni2.1(1)Co0.9B (Fig. S9†). Different areas of the sample were analyzed, and the chemical composition was almost identical for all probed areas. Boron is too light to be accurately determined by EDX, and its signal overlaps with C and O signals, which are ubiquitous in all samples. The metal foil was most likely unreacted NiCo alloy and identified to be ∼1:1 Co:Ni (47.3(3)% Ni and 52.7(6)% Co) which is similar to the nominal composition of the alloy reactant. The synthesis of Ni2.1(1)Co0.9B suggests that ternary borides may be successfully synthesized by our methodology when using an alloyed precursor with BI3, rather than individual metal elements. In contrast, a controlled reaction starting from a mixture of Fe and Co metal powders and BI3 resulted in the formation of two individual borides rather than a ternary boride.
OER electrocatalysis
Metal borides have been widely explored as electrocatalysts. The Fokwa group showed applicability of various metal borides as HER electrocatalysts for H2 generation.5 For OER, different metal borides have been evaluated but these studies are limited to alkaline conditions.6,19–22,31 Alkaline OER currently has lower technological importance as all current industrial membrane-based electrolyzers operate in acidic media due to restrictions on ion-conductive membranes. IrO2 is state-of-the-art acidic OER catalysts, but is not sustainable due to the scarcity of Ir. Thus, the development of acidic OER electrocatalysts which can reduce the usage of noble and scarce Ir metal is of high relevance.32OER activity was evaluated for composites of IrO2/Ni3B with different ratios by measuring cyclic voltammetry (CV) at a scan rate of 50 mV s−1 in N2 saturated 0.5 M H2SO4 solutions. As shown in Fig. 4a, the onset potential for OER with pure IrO2 with mass loading of 1 mg cm−2 was ≈1.46 VRHE, which is consistent with previous reports.33–36 When compared with IrO2, the OER activities of pure Ni3B (mass loading of 2 mg cm−2) were negligible like the bare GC disk, and didn't show any additional reaction before OER (Fig. 4a). For these reasons, we assume that Ni3B works as a conductive supporting material with negligible activity and that IrO2 is the main contributor to the OER activities observed for the composite electrocatalysts.
 | ||
| Fig. 4 (a) Cyclic voltammogram of bare GC, Ni3B (loading = 1 mg cm−2), IrO2 (loading = 1 mg cm−2), IrO2 + Ni3B mixture (molar ratio of Ni3B = 25%, 50%, 75%, loading = 1 mg cm−2) in N2 saturated 0.5 M H2SO4. Scan rate = 50 mV s−1, electrode rotating rate = 1600 rpm. (b) OER current densities of Ni3B + IrO2 mixture with different molar percentage of Ni3B (0/25%/50%/75%) in N2 saturated 0.5 M H2SO4. The measured currents by CV at 1.6 V vs. RHE were normalized by ECSA. Electrode rotation rate = 1600 rpm. | ||
To allow for quantitative comparison of the acidic OER activity under different conditions, e.g., mass loading or molar ratio of Ni3B, the OER currents at a constant applied potential were normalized by electrochemical surface area (ECSA). The ECSA of IrO2 and IrO2/Ni3B were determined by integral charge (Q) from 0.05 VRHE to 1.0 VRHE:

where the subscripts total and GC refer to total measured current (or integral charge) for the electrocatalyst drop-casted WE and for the bare GC, respectively. The as-calculated current densities are shown in Fig. 4b. For pure IrO2 with 1 mg cm−2 mass loading, the current density at 1.6 VRHE is ≈0.4 mA cm−2, which is close to the reported values for polycrystalline IrO2,33 single crystalline IrO2,34–36 and nanosized IrO2 in proton exchange membrane electrolyzers.37 The uncertainties in IrO2 current densities were rather small, which indicates good control of the electrochemical experiments. With the decrease of mass loading per geometrical area from 1 mg cm−2 to 0.5 mg cm−2, the jECSA for IrO2 slightly decreases. However, the jECSA for IrO2 with 0.25 mg cm−2 mass loading is ∼25% of that for sample with 1 mg cm−2 loading.

The normalization by ECSA can omit the influence of the electrolyte accessibility to the electrocatalysts. However, normalization by mass loading, where the electrode accessibility would be taken into account, also presents some challenges,38 since mass loading can influence the Qref value (Qref for IrO2 changes from 320 μC cm−2 at mass loadings ≤1 mg cm−2 to 600 μC cm−2 at reasonably lower IrO2 loading, around 0.15 mg cm−2 (ref. 37 and 39)). Moreover, lower mass loadings can lead to higher dissolution or deactivation of IrO2 during the electrochemical cleaning process, even at potentials more negative than the onset potential of OER.33 Lastly, the effect of mass loading on particle distribution may originate from different derivations of the electron/ion transport. For these reasons, in the present manuscript, the currents were normalized by ECSA unless stated otherwise.
Fig. 4b shows that for the IrO2/Ni3B composite electrocatalysts, the OER current densities are close to pure IrO2, regardless of the Ni3B molar ratio and the total loading. This indicates that the addition of Ni3B helps to reduce the utilization of IrO2 while maintaining most of the OER activity. Thus, the synthesized Ni3B has the potential to become an alternative support to carbon materials, which are unstable at elevated temperatures and require high potentials in electrolyzers.40–43 The overpotential and Tafel slope of pure IrO2 and Ni3B/IrO2 composites are shown in Table S1 and Fig. S10.† IrO2/Ni3B samples showed higher uncertainties when compared to the pure IrO2 electrocatalyst. To investigate the origin of this uncertainty, contact angle studies for Ni and Ni3B foam with water were conducted (Fig. S11†). Results showed that Ni3B foam became hydrophilic compared to the hydrophobic precursor, Ni foam, indicating that the uncertainty of the composite may come from the deactivation of Ni3B, not from low interaction between the catalyst and electrolyte.
The stability of the composite electrocatalysts in acidic OER was evaluated using CV under RDE configuration from 0.8 VRHE to 1.8 VRHE at a scan rate of 50 mV s−1. The obtained results are plotted in Fig. 5. For the comparison, we plotted the OER current, ECSA, and OER current densities at different cycle numbers normalized to the first cycle (i.e., first cycle shows 100%). In this way, we can access the relative deactivation and/or changes caused by the increase in time (number of measurements). The OER current densities were normalized by the ECSA determined by the data obtain from the same cycle. In Fig. 5, the OER currents, ECSA, and OER current densities of all the tested electrocatalysts show a significant reduction with increasing the number of scanning CV cycles. We have carefully confirmed that no obvious electrocatalyst detachment from the GC or dropping into the electrolyte occurred during the whole measurement.
 | ||
| Fig. 5 Normalized (a) OER current at 1.8 VRHE, (b) ECSA and (c) current density at 1.8 VRHE for IrO2/Ni3B composite electrocatalyst under different scanning cycles from 0.8 VRHE to 1.8 VRHE (0/25%/50%/75% Ni3B). The data is normalized such that the value at the first cycle was set to 100%. Scan rate = 50 mV s−1. Electrolyte solution: N2 saturated 0.5 M H2SO4, electrode rotating rate = 1600 rpm. Mass loading = 1 mg cm−2. | ||
The observed OER deactivation on IrO2 has been previously attributed to an artifact caused by the insufficient removal of O2 bubbles in the porous electrode layer.44 The displacement of electrolyte within or near the electrocatalyst layer by O2 will lead to possible loss of ionic contact and a substantial reduction of the ECSA, as confirmed by Fig. 5b. In this regard, hereon, we will use the expression “apparent” stability. For IrO2/Ni3B composite electrocatalysts with different molar ratios, the observed behavior was different. In the case of 50% Ni3B, the “apparent” stability was found to be similar to that of pure IrO2, while in the cases of 25% and 75% Ni3B, the “apparent” stability decreased. Since the IrO2/Ni3B samples showed no difference in hydrophobicity or conductivity (results not shown), this behavior may originate from a combination of two hypothesized factors that operate in opposite directions:
(1) Ni3B shows some deactivation even in the absence of IrO2 (Fig. S12†). Therefore, the addition of Ni3B into IrO2 will introduce another component to the stability of the layer. Considering this factor alone, the higher the Ni3B molar ratio, the worse the “apparent” stability.
(2) Since IrO2 is more active toward acidic OER than pristine Ni3B (Fig. 4a), the addition of Ni3B to IrO2 will reduce the total OER current, and reduce O2 generation and accumulation within the electrocatalyst's layers. Considering this factor alone, the higher the Ni3B molar ratio, the better the “apparent” stability.
Therefore, the best “apparent” stability was achieved at 50% Ni3B molar ratio, which can be explained by a good balance between (1) the amount of support and (2) the reducing O2 production and consequent accumulation in the electrocatalyst layer. Further measurements are necessary to quantitatively assess these two competitive factors, which is out of the scope of this work.
To investigate potential degradation of the catalysts during the OER tests, a detailed scanning-transmission electron microscopy study was performed. A freshly prepared IrO2/Ni3B composite samples (Fig. 6) showed a good dispersion of Ir oxide and Ni boride crystallites without extreme agglomeration. Both IrO2 and Ni3B demonstrated high crystallinity.
 | ||
| Fig. 6 TEM results of Ni3B/IrO2 sample before the catalysis test. (a) Low magnification of high-angle annular dark-field (HAADF)-STEM image and simultaneously acquired EDX-STEM elemental mapping for N-K, Ir-M, O-K edges and overlaid color images. (b) Low magnification HAADF-STEM image and selected area electron diffraction (SAED) pattern is given as inset. (c) Bright field low magnification TEM image showing the agglomeration of IrO2 nanoparticles (dark contrast) and Ni3B support (light contrast); (d) HRTEM image of IrO2 nanoparticles on Ni3B support material; (e) Bright field HRTEM image of agglomeration of IrO2 nanoparticles. Inset: [110] HRTEM image of separated IrO2 nanoparticle faceted along the 〈111〉 plane. | ||
A sample taken from the same batch was used for aforementioned catalysis tests and afterwards subjected to a microscopy investigation (Fig. 7). EDX-STEM mapping showed a similar dispersion of Ir oxide and Ni boride before and after catalysis (compare Fig. 6a, b and 7a, b) indicating no significant sample degradation. An important difference is a higher degree of agglomeration of IrO2 nanocrystals was detected after OER. The observed aggregation might be another potential mechanism for a catalyst's deactivation. To probe this, methods other than ball-milling to prepare composites should be investigated, such as chemical precipitation of IrO2 nanocrystals on Ni3B, which is a subject for further study. The surface of nickel boride may be oxidized during OER reaction, nevertheless the crystalline Ni3B was still present in the sample after catalysis as shown by SAED (Fig. 7d inset).
 | ||
| Fig. 7 TEM results of Ni3B/IrO2 sample after the catalysis test. (a) Low magnification HAADF-STEM image and simultaneously acquired EDX-STEM elemental mapping for Ni-K, Ir-M, O-K edges and overlaid color images. (b) Low magnification HAADF-STEM image. (c) Low magnification TEM image of IrO2 nanoparticles agglomerated. (d) Bright field HRTEM image of agglomeration of IrO2 nanoparticles. Top right inset: cluster of IrO2 nanoparticles. Bottom left inset: selected area electron diffraction (SAED) pattern with highlighted diffraction spots of Ni3B. | ||
Overall, we have shown the potential of Ni3B as a potential supporting material for acidic OER. The addition of 50% Ni3B molar ratio can achieve a similar “apparent” stability as pure IrO2, while simultaneously maintaining most of the OER activity. Better anchoring of IrO2 into the support may improve the overall catalyst's stability. For further improvement, particle size control or nanostructuring of metal borides can be achieved by taking advantage of the topotactic nature of the proposed synthetic method, improving the surface interaction with IrO2.31,32,45,46 In addition, probing different metal borides and ternary mixed metal borides with better intrinsic stability might be valuable. For both purposes, the herein reported method is a viable synthetic avenue.
Conclusion
A facile and prompt low-temperature method of synthesizing various metal borides spanning across the transition metal group was developed. The borodization was achieved by reaction of a metal source with BI3, producing the metal boride and a washable metal iodide admixture. The method was shown to be able to produce mixed metal borides as well as to topotactically transform complex metal morphologies, such as Ni foam, into corresponding metal boride morphologies, i.e., Ni3B foam. In situ studies of the reaction mechanism revealed that the formation of the metal iodide byproduct is a sufficient driving force to initiate metal boride formation at temperatures as low as 700 K. From an applications point-of-view, we have shown the potential of Ni3B to become a viable stable supporting material for the state-of-the-art IrO2 electrocatalyst for OER in acidic conditions. The composite electrocatalyst with 50% Ni3B and 50% IrO2 exhibits similar activity and stability in acidic OER as pure IrO2 electrocatalyst. Further investigations on using metal borides alone as OER electrocatalysts under acidic conditions, or the addition of metal borides for promotion of OER activity while simultaneously reducing the usage of expensive platinum group metal electrocatalysts, will be of great interest.Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.Author contributions
The manuscript was written through contributions of all authors.Conflicts of interest
Authors declare no conflict of interests.Acknowledgements
We would like to thank Dr Wenqian Xu and Dr Andrey Yakovenko (APS ANL) for assistance with synchrotron data collection, Dr Julia V. Zaikina (ISU) for access to the ball-mill, and Dr Shannon Lee (ISU) for specimen preparation and analysis for SEM/EDX. This research is supported by the National Science Foundation under Grant No. 1955456. Use of the Advanced Photon Source at Argonne National Laboratory was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. A. M. D. is grateful to Xunta de Galicia for the postdoctoral fellowship (ED481B 2019/091). Yu. V. K. thanks the Portuguese Foundation for Science and Technology (FCT) for the support under the CritMag Project (PTDC/NAN-MAT/28745/2017). V. S. acknowledges support by the Portuguese Foundation for Science and Technology (FCT) PhD Fellowship (Grant No. SFRH/BD/143750/2019).References
- J. Jiang, Y. Wang, Q. Zhong, Q. Zhou and L. Zhang, Preparation of Fe2B Boride Coating on Low-Carbon Steel Surfaces and Its Evaluation of Hardness and Corrosion Resistance, Surf. Coat. Technol., 2011, 206(2–3), 473–478 CrossRef CAS.
- C. Wu, F. Wu, Y. Bai, B. Yi and H. Zhang, Cobalt Boride Catalysts for Hydrogen Generation from Alkaline NaBH4 Solution, Mater. Lett., 2005, 59(14–15), 1748–1751 CrossRef CAS.
- S. U. Jeong, E. A. Cho, S. W. Nam, I. H. Oh, U. H. Jung and S. H. Kim, Effect of Preparation Method on Co-B Catalytic Activity for Hydrogen Generation from Alkali NaBH4 Solution, Int. J. Hydrogen Energy, 2007, 32(12), 1749–1754 CrossRef CAS.
- F. Baydaroglu, E. Özdemir and A. Hasimoglu, An Effective Synthesis Route for Improving the Catalytic Activity of Carbon-Supported Co-B Catalyst for Hydrogen Generation through Hydrolysis of NaBH4, Int. J. Hydrogen Energy, 2014, 39(3), 1516–1522 CrossRef CAS.
- E. Lee and B. P. T. Fokwa, Nonprecious Metal Borides: Emerging Electrocatalysts for Hydrogen Production, Acc. Chem. Res., 2022, 55(1), 56–64 CrossRef CAS PubMed.
- D. K. Mann, J. Xu, N. E. Mordvinova, V. Yannello, Y. Ziouani, N. González-Ballesteros, J. P. S. Sousa, O. I. Lebedev, Y. V. Kolen'ko and M. Shatruk, Electrocatalytic Water Oxidation over AlFe2B2, Chem. Sci., 2019, 10(9), 2796–2804 RSC.
- P. H. Schmidt, L. D. Longinotti, D. C. Joy, S. D. Ferris, H. J. Leamy and Z. Fisk, Design and optimization of directly heated LaB6 cathode assemblies for electron-beam Instruments, J. Vac. Sci. Technol., 1978, 15(4), 1554–1560 CrossRef CAS.
- J. Stuart, M. Lefler, C. P. Rhodes and S. Licht, High Energy Capacity TiB2/VB2 Composite Metal Boride Air Battery, J. Electrochem. Soc., 2015, 162(3), A432–A436 CrossRef CAS.
- L. Rao, E. G. Gillan and R. B. Kaner, Rapid Synthesis of Transition-Metal Borides by Solid-State Metathesis, J. Mater. Res., 1995, 10(2), 353–361 CrossRef CAS.
- J. P. Abeysinghe, A. F. Kölln and E. G. Gillan, Rapid and Energetic Solid-State Metathesis Reactions for Iron, Cobalt, and Nickel Boride Formation and Their Investigation as Bifunctional Water Splitting Electrocatalysts, ACS Mater. Au, 2022, 2, 489–504 CrossRef CAS PubMed.
- A. Henschel, M. Binnewies, M. Schmidt, H. Borrmann and Yu. Grin, Crucible-Free Preparation of Transition-Metal Borides: HfB2, Chem.–Eur. J., 2017, 23, 15869–15873 CrossRef CAS PubMed.
- A. Henschel, M. Binnewies, M. Schmidt, R. Köppe, U. Burkhardt and Y. Grin, Tungsten Borides: On the Reaction of Tungsten with Boron(III) Bromide, Chem.–Eur. J., 2018, 24(40), 10109–10115 CrossRef CAS PubMed.
- P. R. Jothi, K. Yubuta and B. P. T. Fokwa, A Simple, General Synthetic Route toward Nanoscale Transition Metal Borides, Adv. Mater., 2018, 30(14), 1704181 CrossRef PubMed.
- V. Gvozdetskyi, G. Bhaskar, M. Batuk, X. Zhao, R. Wang, S. L. Carnahan, M. P. Hanrahan, R. A. Ribeiro, P. C. Canfield, A. J. Rossini, C. Wang, K. Ho, J. Hadermann and J. V. Zaikina, Computationally Driven Discovery of a Family of Layered LiNiB Polymorphs, Angew. Chem., Int. Ed., 2019, 58, 15855–15862 CrossRef CAS PubMed.
- J. Cueilleron and J. C. Viala, Purity of boron prepared by vacuum decomposition of purified boron triiodide, J. Less-Common Met., 1979, 65(2), 167–173 CrossRef CAS.
- J. Cueilleron and J. C. Viala, Direct synthesis of boron triiodide and the chemical transport of boron with iodine, J. Less-Common Met., 1978, 58(2), 123–131 CrossRef CAS.
- X. Wang, Y. V. Kolen'ko, X. Bao, K. Kovnir and L. Liu, One-Step Synthesis of Self-Supported Nickel Phosphide Nanosheet Array Cathodes for Efficient Electrocatalytic Hydrogen Generation, Angew. Chem., Int. Ed., 2015, 54, 8188–8192 CrossRef CAS PubMed.
- J. Xu, J. P. S. Sousa, N. Mordvinova, J. D. Costa, J. L. Lado, D. Y. Petrovykh, K. Kovnir, O. I. Lebedev and Yu. V. Kolen'ko, Al-induced In situ Formation of Highly Active Nanostructured Water-oxidation Electrocatalyst Based on Ni-phosphide, ACS Catal., 2018, 8, 2595–2600 CrossRef CAS.
- A. Serov, K. Kovnir, M. Shatruk and Y. V. Kolen'ko, Critical Review of Platinum Group Metal-Free Materials for Water Electrolysis: Transition from the Laboratory to the Market: Earth-Abundant Borides and Phosphides as Catalysts for Sustainable Hydrogen Production, Johnson Matthey Technol. Rev., 2021, 65(2), 207–226 CrossRef CAS.
- Z. P. Wu, X. F. Lu, S. Q. Zang and X. W. Lou, Non-Noble-Metal-Based Electrocatalysts toward the Oxygen Evolution Reaction, Adv. Funct. Mater., 2020, 30(15), 1910274 CrossRef CAS.
- J. Li, H. Chen, Y. Liu, R. Gao and X. Zou, In Situ Structural Evolution of a Nickel Boride Catalyst: Synergistic Geometric and Electronic Optimization for the Oxygen Evolution Reaction, J. Mater. Chem. A, 2019, 7(10), 5288–5294 RSC.
- N. Wang, A. Xu, P. Ou, S. F. Hung, A. Ozden, Y. R. Lu, J. Abed, Z. Wang, Y. Yan, M. J. Sun, Y. Xia, M. Han, J. Han, K. Yao, F. Y. Wu, P. H. Chen, A. Vomiero, A. Seifitokaldani, X. Sun, D. Sinton, Y. Liu, E. H. Sargent and H. Liang, Boride-Derived Oxygen-Evolution Catalysts, Nat. Commun., 2021, 12(1), 6089 CrossRef CAS.
- B. H. Toby and R. B. Von Dreele, GSAS-II: The Genesis of a Modern Open-Source All Purpose Crystallography Software Package, J. Appl. Crystallogr., 2013, 46(2), 544–549 CrossRef CAS.
- K. Woo, K. Lee and K. B. P. Kovnir, Synthesis and Properties of Boron Phosphide, Mater. Res. Express, 2016, 3(7), 074003 CrossRef.
- K. Kovnir, Predictive Synthesis, Chem. Mater., 2021, 33(13), 4835–4841 CrossRef CAS.
- G. Akopov, N. W. Hewage, P. Yox, G. Viswanathan, S. J. Lee, L. P. Hulsebosch, S. D. Cady, A. L. Paterson, F. A. Perras, W. Xu, K. Wu, Y. Mudryk and K. Kovnir, Synthesis-Enabled Exploration of Chiral and Polar Multivalent Quaternary Sulfides, Chem. Sci., 2021, 12(44), 14718–14730 RSC.
- P. Yox, S. J. Lee, L. L. Wang, D. Jing and K. Kovnir, Crystal Structure and Properties of Layered Pnictides BaCuSi2Pn3(Pn = P, As), Inorg. Chem., 2021, 60(8), 5627–5634 CrossRef CAS PubMed.
- B. Owens-Baird, J. Wang, S. G. Wang, Y. S. Chen, S. Lee, D. Donadio and K. Kovnir, III-V Clathrate Semiconductors with Outstanding Hole Mobility: Cs8In27Sb19 and A8Ga27Sb19 (A = Cs, Rb), J. Am. Chem. Soc., 2020, 142(4), 2031–2041 CrossRef CAS PubMed.
- K. E. Woo, J. Wang, J. Mark and K. Kovnir, Directing Boron-Phosphorus Bonds in Crystalline Solid: Oxidative Polymerization of P=B=P Monomers into 1D Chains, J. Am. Chem. Soc., 2019, 141(33), 13017–13021 CrossRef CAS PubMed.
- V. Gvozdetskyi, Y. Sun, X. Zhao, G. Bhaskar, S. L. Carnahan, C. P. Harmer, F. Zhang, R. A. Ribeiro, P. C. Canfield, A. J. Rossini, C. Z. Wang, K. M. Ho and J. V. Zaikina, Lithium Nickel Borides: Evolution of [NiB] Layers Driven by Li Pressure, Inorg. Chem. Front., 2021, 8(7), 1675–1685 RSC.
- L. M. Salonen, D. Y. Petrovykh and Y. V. Kolen'ko, Sustainable Catalysts for Water Electrolysis: Selected Strategies for Reduction and Replacement of Platinum-Group Metals, Mater. Today Sustain., 2021, 11–12, 100060 CrossRef.
- S. Cherevko, S. Geiger, O. Kasian, N. Kulyk, J. P. Grote, A. Savan, B. R. Shrestha, S. Merzlikin, B. Breitbach, A. Ludwig and K. J. J. Mayrhofer, Oxygen and Hydrogen Evolution Reactions on Ru, RuO2, Ir, and IrO2 Thin Film Electrodes in Acidic and Alkaline Electrolytes: A Comparative Study on Activity and Stability, Catal. Today, 2016, 262, 170–180 CrossRef CAS.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, Synthesis and Activities of Rutile IrO2 and RuO2 Nanoparticles for Oxygen Evolution in Acid and Alkaline Solutions, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- K. A. Stoerzinger, L. Qiao, M. D. Biegalski and Y. Shao-Horn, Orientation-Dependent Oxygen Evolution Activities of Rutile IrO2 and RuO2, J. Phys. Chem. Lett., 2014, 5(10), 1636–1641 CrossRef CAS.
- F. Zhao, B. Wen, W. Niu, Z. Chen, C. Yan, A. Selloni, C. G. Tully, X. Yang and B. E. Koel, Increasing Iridium Oxide Activity for the Oxygen Evolution Reaction with Hafnium Modification, J. Am. Chem. Soc., 2021, 143(38), 15616–15623 CrossRef CAS PubMed.
- S. Zhao, H. Yu, R. Maric, N. Danilovic, C. B. Capuano, K. E. Ayers and W. E. Mustain, Calculating the Electrochemically Active Surface Area of Iridium Oxide in Operating Proton Exchange Membrane Electrolyzers, J. Electrochem. Soc., 2015, 162(12), F1292–F1298 CrossRef CAS.
- L. Yu, S. Sun, H. Li and Z. J. Xu, Effects of Catalyst Mass Loading on Electrocatalytic Activity: An Example of Oxygen Evolution Reaction, Fundam. Res., 2021, 1(4), 448–452 CrossRef CAS.
- R. F. Savinell, R. L. Zeller III and J. A. Adams, Electrochemically Active Surface Area Voltammetric Charge Correlations for Ruthenium and Iridium Dioxide Electrodes, J. Electrochem. Soc., 1990, 137, 489–494 CrossRef CAS.
- S. Maass, F. Finsterwalder, G. Frank, R. Hartmann and C. Merten, Carbon Support Oxidation in PEM Fuel Cell Cathodes, J. Power Sources, 2008, 176(2), 444–451 CrossRef CAS.
- L. C. Colmenares, A. Wurth, Z. Jusys and R. J. Behm, Model Study on the Stability of Carbon Support Materials under Polymer Electrolyte Fuel Cell Cathode Operation Conditions, J. Power Sources, 2009, 190(1), 14–24 CrossRef CAS.
- I. S. Filimonenkov, C. Bouillet, G. Kéranguéven, P. A. Simonov, G. A. Tsirlina and E. R. Savinova, Carbon Materials as Additives to the OER Catalysts: RRDE Study of Carbon Corrosion at High Anodic Potentials, Electrochim. Acta, 2019, 321, 134657 CrossRef CAS.
- S. Möller, S. Barwe, J. Masa, D. Wintrich, S. Seisel, H. Baltruschat and W. Schuhmann, Online Monitoring of Electrochemical Carbon Corrosion in Alkaline Electrolytes by Differential Electrochemical Mass Spectrometry, Angew. Chem., Int. Ed., 2020, 59, 1585–1589 CrossRef.
- H. A. El-Sayed, A. Weiß, L. F. Olbrich, G. P. Putro and H. A. Gasteiger, OER Catalyst Stability Investigation Using RDE Technique: A Stability Measure or an Artifact?, J. Electrochem. Soc., 2019, 166(8), F458–F464 CrossRef CAS.
- Y. N. Regmi, E. Tzanetopoulos, G. Zeng, X. Peng, D. I. Kushner, T. A. Kistler, L. A. King and N. Danilovic, Supported Oxygen Evolution Catalysts by Design: Toward Lower Precious Metal Loading and Improved Conductivity in Proton Exchange Membrane Water Electrolyzers, ACS Catal., 2020, 10(21), 13125–13135 CrossRef CAS.
- E. Oakton, D. Lebedev, M. Povia, D. F. Abbott, E. Fabbri, A. Fedorov, M. Nachtegaal, C. Copéret and T. J. Schmidt, IrO2-TiO2: A High-Surface-Area, Active, and Stable Electrocatalyst for the Oxygen Evolution Reaction, ACS Catal., 2017, 7(4), 2346–2352 CrossRef CAS.
- J. M. V. Nsanzimana, L. Gong, R. Dangol, V. Reddu, V. Jose, B. Y. Xia, Q. Yan, J. M. Lee and X. Wang, Tailoring of Metal Boride Morphology via Anion for Efficient Water Oxidation, Adv. Energy Mater., 2019, 9(28), 1901503 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta04266e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |