
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal–ligand complexation and clustering in mussel-inspired side-chain functionalized supramolecular hydrogels†
Amir
Jangizehi
a,
Mostafa
Ahmadi
 a,
Sarah
Pschierer
a,
Paola
Nicolella
a,
Hailong
Li
b,
Katrin
Amann-Winkel
b and
Sebastian
Seiffert
*a
a,
Sarah
Pschierer
a,
Paola
Nicolella
a,
Hailong
Li
b,
Katrin
Amann-Winkel
b and
Sebastian
Seiffert
*a
aJohannes Gutenberg University Mainz, Department of Chemistry, Duesberwerg 10–14, D-55128 Mainz, Germany. E-mail: Sebastian.seiffert@uni-mainz.de
bMax Planck Institute for Polymer Research, D-55128 Mainz, Germany
First published on 30th August 2022
Abstract
Byssus threads of mussels have high resistance against abrasion in wave-swept habitats because of their outer cuticle, which is rich in amino acid dopa complexes with Fe3+ ions. This stems from the transient nature of metal–ligand complexes that creates extra relaxation mechanisms. Inspired by this concept, in this work, supramolecular hydrogels based on poly(acrylic acid) functionalized with nitrocatechol groups are synthesized. Polymer chains are physically crosslinked via nitrocatechol–Fe3+ complexes. The hydrogels have different polymer volume fractions as well as different nitrocatechol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe3+ molar ratios. The strength of the supramolecular crosslinks strongly depends on the pH of the medium. The dynamics of these hydrogels are studied by stress relaxation experiments followed by calculation of the relaxation time spectrum. Generally, samples have three relaxation modes, including dissociation of distinct metal–ligand complexes, reptation of sticky polymer chains, and disengagement of network segments from supramolecular aggregates and clusters. Such clusters hinder the terminal relaxation and potentially increase the stability of supramolecular hydrogels.
:Fe3+ molar ratios. The strength of the supramolecular crosslinks strongly depends on the pH of the medium. The dynamics of these hydrogels are studied by stress relaxation experiments followed by calculation of the relaxation time spectrum. Generally, samples have three relaxation modes, including dissociation of distinct metal–ligand complexes, reptation of sticky polymer chains, and disengagement of network segments from supramolecular aggregates and clusters. Such clusters hinder the terminal relaxation and potentially increase the stability of supramolecular hydrogels.
Introduction
Supramolecular hydrogels are a class of hydrogels, in which the network strands are connected through transient interactions like hydrogen bonding, host–guest interactions, electrostatic interactions, or metal–ligand complexations.1–3 Considering the reversible/transient nature of such bonds, supramolecular hydrogels potentially have several interesting features like the ability to be reshaped/processed/injected, controlled sol–gel transition and self-healing, which makes this class of hydrogels promising for biological applications compared to chemically crosslinked hydrogels.2,4–7 Moreover, if supramolecular hydrogels are obtained through supramolecular crosslinking of precursor polymeric chains, beneficial properties of polymers with advantages of supramolecular chemistry can be combined. In addition, besides the crosslinking density and the polymer volume fraction, which are in common with chemically-crosslinked hydrogels, the dynamics and the mechanical properties of supramolecular hydrogels can be controlled by the strength or the equilibrium constant and the lifetime of the physical bonds.4,8,9 On top of that, if associative groups aggregate by the collective assembly into clusters, the time scale that polymer segments require to escape from these aggregates and clusters affects the overall dynamic and mechanical properties.10One of the key points that should be taken into account for the synthesis of supramolecular hydrogels is the stability of transient association in polar media like water. Generally, the transient interactions are weakening in water, for example, a single hydrogen bond can easily dissociate in water.1,7 To increase the stability of hydrogen bonds in water, several strategies like utilizing associative groups with multiple hydrogen bindings, hydrophobic shielding, and/or combining with other types of physical interactions like π–π conjugations have been studied.11–15 Besides such stabilization of supramolecular associations, the polymer volume fraction is a key factor for preparing a stable supramolecular hydrogel. This factor is specifically critical if the hydrogel is immersed in an excess amount of water after formation of a stable hydrogel at a defined concentration.13,16 In this case, the swelling capacity of the hydrogel and the stability of the physical crosslinks at the equilibrium swelling condition are key parameters. This might be one of the reasons that most of the studied supramolecular hydrogels contain less than 98% water (in equilibrium swelling or out of equilibrium swelling).1 However, in a few studies, supramolecular hydrogels with even more water content, like up to 99.7%, have been successfully prepared and investigated.17
One of the promising types of physical interactions that are utilized to prepare supramolecular hydrogels is metal–ligand complexation. This physical bonding covers a very broad range of binding energies (0–400 kJ mol−1), depending on the metal ion and the ligand, making them promising for the design of tough and stiff supramolecular hydrogels.18 In addition, via oxidation or reduction of the metal ion center, it is possible to trigger or avoid the self-assembly and the sol–gel transition.19,20 One of the well-known examples of metal–ligand-based supramolecular materials in nature is the byssus threads of mussels, in which histidine-rich domains coordinate to transition metal ions like zinc and copper, giving self-healing properties to this stiff material. In addition, the threads are protected by an outer cuticle, which is rich in amino acid dopa complexes with Fe3+ ions.21 Inspired by these concepts, Messersmith and coworkers prepared a series of hydrogels by metal–ligand complexation of histidine-functionalized tetra-arm poly(ethylene glycol), tetra-arm PEG, with transition metals like zinc, copper, cobalt, and nickel. The results revealed that the sol–gel transition (gel formation), and the relaxation time of the hydrogels can be controlled by the type of the metal ion and the pH of the gel formation medium.22 It is also shown that by rational use of the two metal ions in formation of a single network, the hierarchical self-assembly of the supramolecular motifs can be engineered, which has a significant impact on the mechanics and dynamics of the obtained hydrogels.23 The complex of catechol and Fe3+ shows an extremely large equilibrium constant (1040 M−1), making the required force for rapture of a single bond comparable with the one of covalent bonds. However, in contrast to covalent bonds, the dissociated catechol–Fe3+ bond can be reformed due to its achievable reformation rate.21 The stoichiometry of catechol–Fe3+ complexation strongly depends on the pH of the bond formation medium. By increasing pH, it changes from mono-, to bis-, and to tris-type assembly.24 In a case of tris-complexation, the elastic modulus of hydrogels with a bulk structure of catechol-functionalized tetra-arm PEG is comparable with a model covalent network prepared with the same precursor and at similar concentration.24 A similar polymer precursor has been also coordinated with vanadium and aluminum salts. The results revealed that variation of the bonding dynamics is a powerful tool to tune the viscoelastic properties over several orders of magnitude.25 In addition, by using derivatives of catechol like nitrocatechol (ND) or 3-hydroxy 4-pyridinone, it is possible to promote tris complexation to form stable and robust gels at lower pH, compared to that required for the catechol group, without significant reduction in binding strength. Investigation of the rheological properties of the corresponding hydrogels with an identical tetra-arm PEG scaffold demonstrated almost similar plateau moduli, but significantly different relaxation times ranging from 0.09 s (for plain catechol) to 125 s (for 3-hydroxy 4-pyridinone).26 Besides the effect of the ligands, Ma and Hou showed that by changing the microstructure of the hydrogels network to a side-chain supramolecule polymer, it is possible to modulate the dynamics and mechanics of the obtained hydrogels. To reach this target, the authors prepared catechol-functionalized (meth)acrylamide monomers and made precoordinated crosslinkers by mixing them with Fe3+ solution. Hydrogels were prepared by employing an appropriate photo-initiator. With this method, it was possible to prepare a hydrogel with high extensibility and fast self-healing properties.27 Nevertheless, the origin of this significant improvement in the hydrogel properties upon changing the microstructure of the polymeric network platform has not been discussed.
In this work, we investigate hydrogels with side-chain supramolecular polymeric structure formed by integrating nitrocatechol (ND)–Fe3+ complexes along sodium acrylate and hydroxyethyl acrylate random copolymers. The target of using sodium acrylate is to increase the theoretical potential of water adsorption. The metal–ligand complexation and the mechanical properties are probed by UV-vis spectroscopy and rheology. The analysis of the rheological data is done with specific attention to potential clustering of the associative groups. The results confirm aggregation and clustering of the metal–ligand complexes on top of entanglement of the sticky polymer chains. Such supramolecular aggregations influence the terminal relaxation of hydrogels and with that potentially their stability.
Experiments
Materials
Copper(I) bromide, 4-dimethylaminopyridine, N,N′-disuccinimidyl carbonate, nitrodopamine hydrochloride, methyl 2-bromopropionate, and tris(2-dimethylaminoethyl)amine (Me6TREN) are purchased from Aldrich or Alfa-Aesar and utilized without further purification. tert-Butyl acrylate and hydroxyethyl acrylate are purchased from Aldrich and purified by a basic alumina column.Synthesis of nitrodopamine hydrogen sulfate
Nitrodopamine hydrochloride (3.02 g, 15.92 mmol) is dissolved in 90 mL water. Sodium nitrite (3.75 g, 54.36 mmol) is added to the solution and then cooled down to 3 °C by using an ice bath. Then, 30 mL of sulfuric acid (20% v/v) is added dropwise to the mixture. The ice batch is removed, and the mixture is stirred at room temperature overnight. The obtained yellow dispersion is filtered and washed with cold water (3 × 20 mL) and then dried under vacuum (2.23 g, yield 71%). 1H NMR (400 MHz, DMSO): δ = 3.06–3.20 (s, 4H, CCH2CH2), 6.89 (s, 1H, C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) COH), 7.46 (s, 1H, CCNO2) ppm.
COH), 7.46 (s, 1H, CCNO2) ppm.
Synthesis of poly(tert-butyl acrylate-ran-hydroxyethyl acrylate)
The copolymers are synthesized with 5, 10, or 15.5 mol% of hydroxyethyl acrylate by atom transfer radical polymerization (ATRP). For 5 mol%, for example, tert-butyl acrylate (28.90 g, 225.50 mmol), hydroxyethyl acrylate (1.09 g, 9.40 mmol), and Me6TREN (0.44 g, 1.92 mmol) are mixed in a 100 mL Schlenk flask. The mixture is degassed three times with a freeze–pump–thaw cycle. Copper bromide (0.14 g, 0.96 mmol) is added to the frozen mixture under nitrogen flow. After reaching room temperature, the mixture is stirred for 30 minutes. The flask is put in a preheated oil bath and then methyl 2-bromopropionate (0.008 g, 0.48 mmol) is added to the mixture. After stirring the mixture at 65 °C for 24 h, it is diluted by dichloromethane and passed through an alumina column to separate the catalyst. The solution is then dried to remove the solvent and the unreacted components (21.85 g, yield 73%). 1H NMR (400 MHz, CDCl3) δ = 1.43 (s, 9H, OCC3), 1.49 (d, 2H, C2CH, polymer backbone), 2.21 (s, 1H, CH2C, polymer backbone), 3.77 (s, 0.1 H, C2CH2OH), 4.17 (s, 0.1 H, CH2C2OH) ppm.
Synthesis of poly(tert-butyl acrylate-ran-NHS-ethyl acrylate)
To replace the hydroxy groups of synthesized poly(tert-butyl acrylate-ran-hydroxyethyl acrylate) copolymers with succinimidyl carbonate, each copolymer is reacted with 4-dimethylaminopyridine (DMAP) under argon atmosphere at room temperature. As an example, the copolymer with 5 mol% of the hydroxyethyl acrylate (21.80 g, 8.54 mmol) is dissolved in 100 mL dry dimethylformamide. This solution is added to a suspension of disuccinimidyl carbonate (4.38 g, 17.09 mmol) in 30 mL acetone under argon flow. To this mixture, a solution of 4-dimethylaminopyridine (2.02 g, 17.09 mmol) in 20 mL acetone is added dropwise. The mixture is then stirred at room temperature for two days. Afterwards, the functionalized copolymer is precipitated in 1.5 L mixture of methanol (70%) and water (30%) (21.14 g). 1H NMR (400 MHz, CDCl3): δ = 1.43 (s, 9H, OCC3), 1.52 (d, 2H, C2CH, polymer backbone), 2.23 (s, 1H, CH2C, polymer backbone). 2.84 (s, 0.10 H, NCOC2), 3.77 (s, 0.04H, C2CH2OH), 4.32 (s, 0.10H, CH2C2OH; C2CH2OCO), 4.52 (s, 0.07H, CH2C2OCO) ppm.
Synthesis of poly(acrylic acid-ran-NHS-ethyl acrylate)
To convert tert-butyl acrylate repeating units of poly(tert-butyl acrylate-ran-NHS-ethyl acrylate) copolymers obtained in the previous step into acrylic acid, the units should be hydrolyzed in an acidic medium. For this target, the polymer containing a total of 5 mol% comonomer, for example, (2.30 g, 17.13 mmol) is dissolved in 300 mL dichloromethane. Then, trifluoroacetic acid (14.80 g, 129.80 mmol) is added dropwise and the mixture is stirred overnight at room temperature. The precipitated polymer is filtered and dried under reduced pressure (1.93 g). 1H NMR (400 MHz, CDCl3): δ = 1.37 (s, 0.95H, OCC3), 1.61 (d, 2H, C2CH, polymer backbone), 2.20 (s, 1H, CH2C, polymer backbone), 2.81 (s, 0.10 H, NCOC2), 3.69 (s, 0.04H, C2CHOH), 4.26 (s, 0.10H, CH2C2OH; C2CH2OCO), 4.52 (s, 0.08H, CH2C2OCO) ppm.
Functionalization of copolymers with nitrodopamine groups
Reaction of the succinimidyl groups of the poly(acrylic acid-ran-NHS-ethyl acrylate) obtained in the previous step with nitrodopamine hydrogensulfate is done in a mixture of dimethyl sulfoxide (DMSO) and phosphate buffer. The sample (1.00 g, 0.34 mmol) is dissolved in dry 75 mL DMSO. Nitrodopamine hydrogen sulfate (0.18 g, 0.62 mmol), which is previously dissolved in 4 mL dry DMSO is added to this solution. Then, 50 mL buffer solution is added to the mixture. The solution is stirred overnight at room temperature. The solvent is removed under the vacuum, followed by the freeze drying. The product is purified for one week with dialysis in water, which is changed two times per day. 1H NMR (400 MHz, CDCl3): δ = 1.45 (s, 0.44H, OCC3), 1.82 (d, 2H, C2CH, polymer backbone), 2.40 (s, 1H, CH2C, polymer backbone), 4.39 (s, 0.10H, CH2C2OH; C2CH2OCO), 6.86 (s, 0.02H, CCCOH), 7.67 (s, 0.02H, CCNO2).
UV-vis spectroscopy
UV-vis spectroscopy is done on the solution of nitrodopamine hydrogensulfate or the solution of poly(acrylic acid-ran-hydroxyl ethyl acrylate) functionalized with nitro catechol groups, poly(AAc-ran-NC). In addition, the experiment is done on the mixture of nitrodopamine hydrogensulfate and Fe(III) sulfate or on the mixture of poly(AAc-ran-NC) and Fe(III) sulfate to investigate the complexation between Fe3+ ions and nitrodopamine in a low molar mass functional component and a functionalized polymeric template, respectively. In both mixtures, the molar ratio of the nitrodopamine and Fe3+ ions are 3:1. To prepare the probed solutions, a determined volume of Fe(III) sulfate solution in water with a concentration of 0.19 mol/L is added to the solutions of poly(AAc-ran-NC) or nitrodopamine hydrogensulfate in water with a concentration of 10−4 mol L−1. The probed solutions are stirred for 20 min. The pH is then adjusted by using hydrochloric acid solution (0.25 mol L−1) or sodium hydroxide solution (0.25 mol L−1).
SAXS
Small-angle X-ray scattering (SAXS) measurements are performed using CuKα radiation (RigakuMicroMax 007 X-ray generator, Osmic Confocal Max–Flux curved multilayer optics). Supramolecular hydrogels are sealed in capillaries with a thickness of 0.01 mm to avoid water evaporation under vacuum during the measurement. Two-dimensional (2D) diffraction patterns of these samples are recorded using an Mar345 image plate detector at a sample-detector distance of 2116 mm. Intensity profiles are recorded by radial averaging of the 2D datasets as a function of the scattering vector, q = (4π/λ) sin(2θ/2), where 2θ is the scattering angle.Rheology
The rheology measurements are done by stress-controlled Anton Paar 302 rheometer. To avoid solvent evaporation, a solvent trap is used during the measurements. To prepare the probed samples, poly(AAc-ran-NC) is dissolved in Milli-Q water with concentrations of 10 wt%, 15 wt%, or 20 wt%. Then, the required amount of Fe(III) sulfate such that the molar ratio of NC:Fe3+ ions set to 3:1, 2:1, or 4:1 is added to the polymer solution. After stirring for 30 min at room temperature, NaOH solution (10 wt%) is added to the mixture with a ratio of 1 μL solution per 10 mg of polymer solution. After 24 h of shaking at room temperature, the obtained gels are loaded onto the rheometer. For each sample, step strain measurement at T = 10 °C, 20 °C, 30 °C, and 40 °C followed by a frequency sweep measurement at 20 °C and 30 °C are done. In detail, each sample is equilibrated first at γ = 0% and T = 10 °C for 10 min. Then, the shear deformation is increased to γ = 10% for 34 min, during which the change of the stress is investigated. This protocol is repeated at 20 °C, 30 °C, and 40 °C. The sample is equilibrated at γ = 1%, ω = 0.1 rad s−1 for 10 min. At this shear rate and temperature, the frequency sweep measurement is done in the frequency range of 100–0.01 rad s−1. This protocol is repeated at 30 °C, 20 °C, and 10 °C.
Results and discussion
Synthesis of precursor polymers
Hydrogels studied in this work have a side-chain supramolecular polymeric structure, where the poly(acrylic acid-ran-hydroxyethyl acrylate) chains has pendant nitrocatechol functional groups, poly(AAc-ran-NC). The synthesis approach is summarized in Scheme 1 and the corresponding 1H NMR spectra are presented in Fig. S1–S5 in the ESI.† The copolymers are synthesized by atom transfer radical polymerization (ATRP) of tert-butyl acrylate and hydroxyethyl acrylate with a molecular mass average of Mn ≈ 39000 g mol−1 (see the GPC spectrum in Fig. S6 in the ESI†). The comonomer content is 5, 10, and 15.5 mol%, as determined by 1H NMR. The polydispersity of these copolymers increases upon increase of the comonomer content due to the general weakness of classical ATRP in the polymerization of polar monomers. The copolymers are reacted with disuccinimidyl carbonate to replace the hydroxy groups with N-hydroxy succinimidyl groups, thereby providing active sites for the subsequent click reaction with amine groups. The efficiency of this reaction is between 40–50%. In the next step, tert-butyl acrylate groups are hydrolyzed into acrylic acid groups by using trifluoroacetic acid dissolved in dichloromethane. Such a conversion depends on the comonomer content and gets lower with increasing the mol-percentage of hydroxyethyl acrylate. The non-hydrolyzed tert-butyl acrylate units limit the solubility of the copolymers in polar solvents like DMSO and water, which are the solvents in the following synthesis steps. Due to this limitation, only the copolymer with 5 mol% of comonomer could be used to prepare a hydrogel suitable for further investigations. This sample wherein 2.5 mol% of hydroxyl groups were replaced with succinimidyl groups is reacted with nitrodopamine hydrogensulfate in phosphate buffer medium to attached 2.5 mol% nitrocatechol groups to copolymer chains via urethane linkages. Considering the molar mass of the polymer determined by gel permeation chromatography and the integration peaks of 1H NMR, the average number of nitro catechol, NC, groups per chain is 6. This polymer is used to prepare supramolecular hydrogels at three different concentrations of 10 wt%, 15 wt%, and 20 wt% with a molar ratio of NC:Fe3+ = 3:1 as described in Experiments. In addition, at the highest concentration, two more hydrogels with different molar ratios of NC:Fe3+ = 2:1 or 4:1 are also prepared to investigate the effect of the ligands to the metal ions stoichiometry on hydrogel properties. Samples are designated as pAAX_NCY, where X denotes the polymer weight fraction and Y represents the molar ratio of NC to Fe3+ ions. For example, sample pAA10_NC3 is prepared by the polymer solution with a concentration of 10 wt% and a molar ratio of NC to Fe3+ of 3. This library of five as prepared supramolecular hydrogels, in which the concentration of precursor polymer and/or the molar ratio of the associative groups are varied are considered for further characterizations and analysis.
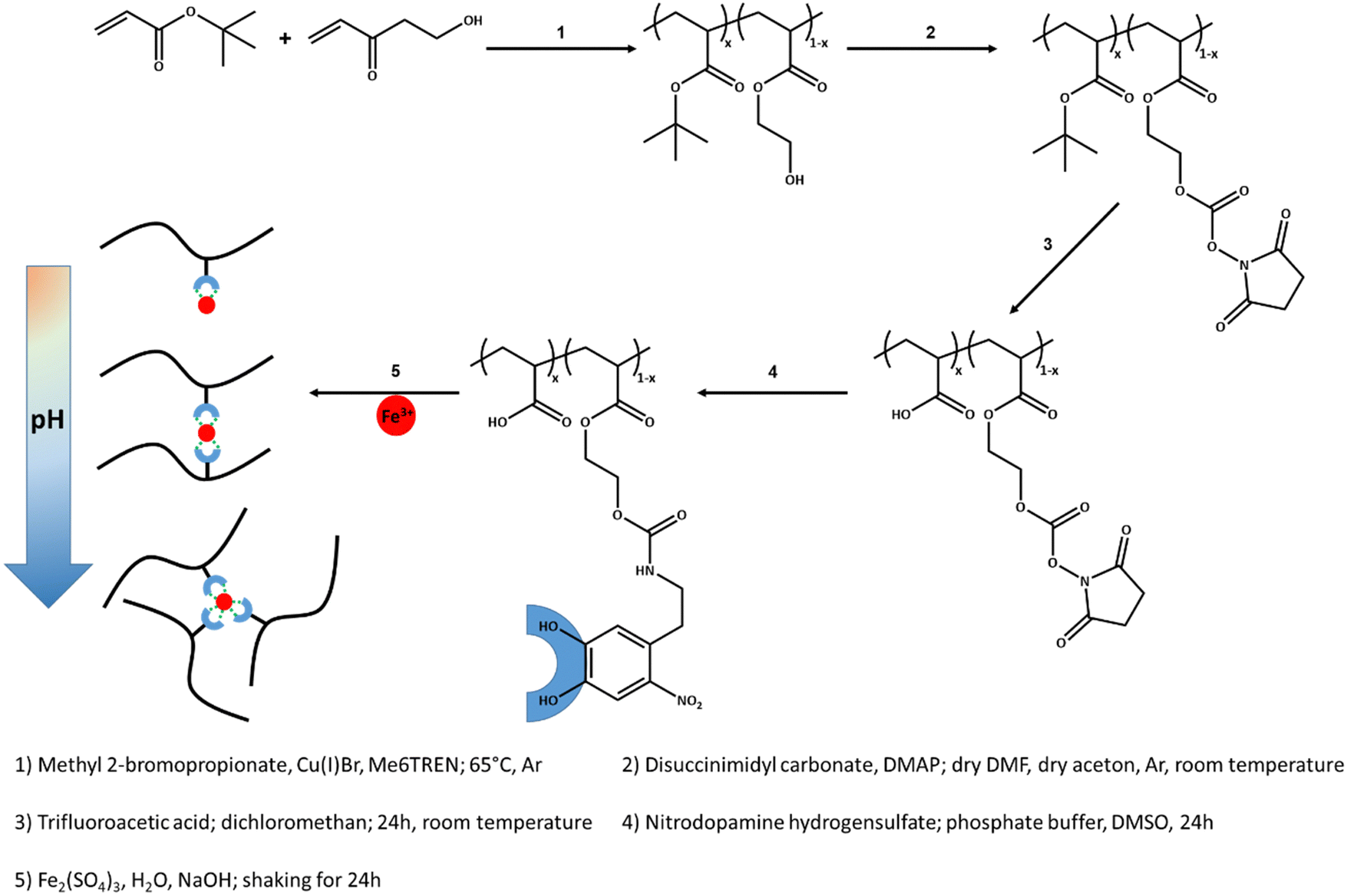 | ||
| Scheme 1 Preparation of supramolecular hydrogels. (1) Synthesis of random copolymers of tert-butyl acrylate and hydroxyl ethyl acrylate, (2) replacement hydroxyl groups of the random copolymers with succinimidyl groups. (3) Hydrolysis of tert-butyl acrylate units in the copolymers to acrylic acid groups, (4) functionalization of the copolymer with nitrocatechol groups, (5) preparation of the supramolecular hydrogels via complexation of nitrocatechol groups and Fe3+ ions. The type of the complexation (mono, bis, or tris) can be controlled by changing the pH. | ||
Deprotonation of nitrocatechol and its complexation with Fe3+ ions
The nitrocatechol group has two hydroxy substituents on its aromatic ring, which can be present in the form of OH (protonated state) or O− (deprotonated state), whereby the latter is more desired in a sense of complexation with Fe3+ ions. The protonation state of hydroxy substituents can be modulated by variation of the pH of the solution medium. To investigate this point in detail, solutions of nitrodopamine hydrogensulfate in Milli-Q water with different pH from 2.5 to 12.5 are investigated by UV-vis spectroscopy (Fig. 1A). The spectrum of each solution reveals one or two maxima between the wavelength of 350 and 500 nm. These peaks are related to the metal to ligand charge transfer (MLCT) and indicate the protonation state of the hydroxy groups on the aromatic ring of the nitrocatechol group. At pH = 2.5, one maximum appears at 350 nm, which indicates that both hydroxyl groups are at the protonated state (OH). With increasing pH, the maximum shifts to higher wavelengths, showing that one of the hydroxy substituents is deprotonated, while the other is still in the protonated state. By exceeding the pH above 11, two maxima can be detected. The first maximum, which appears at lower wavenumbers is related to nitrocatechol groups, with one deprotonated hydroxy substituent. The other maximum is related to nitrocatechol groups with two deprotonated hydroxy substituents. To see the effect of polymeric chains on the protonation state of hydroxy substituents and with that complexation of NC–Fe,3 a similar experiment is done for the solution of poly(AAc-ran-NC) at different pH values, as shown in inset of Fig. 1A. The overall trend of increasing the maximum wavelength upon increasing pH, which was found for the solution of nitrodopamine hydrogen sulfate is also recognized for the corresponding polymeric solution, however, with slight differences. First, the experiment cannot be done at pH lower than 4 due to the precipitation of the polymer. Second, deprotonation of hydroxy substituents on the aromatic ring of NC requires higher pH for polymeric solution compared to nitrodopamine hydrogen sulfate solution. For example, the pendant NC groups from polymeric chains have one deprotonated hydroxy substituent at pH = 10.8–11.5, while this state is observed for the solution of low-molar mass functional group at pH = 5.7–10.3.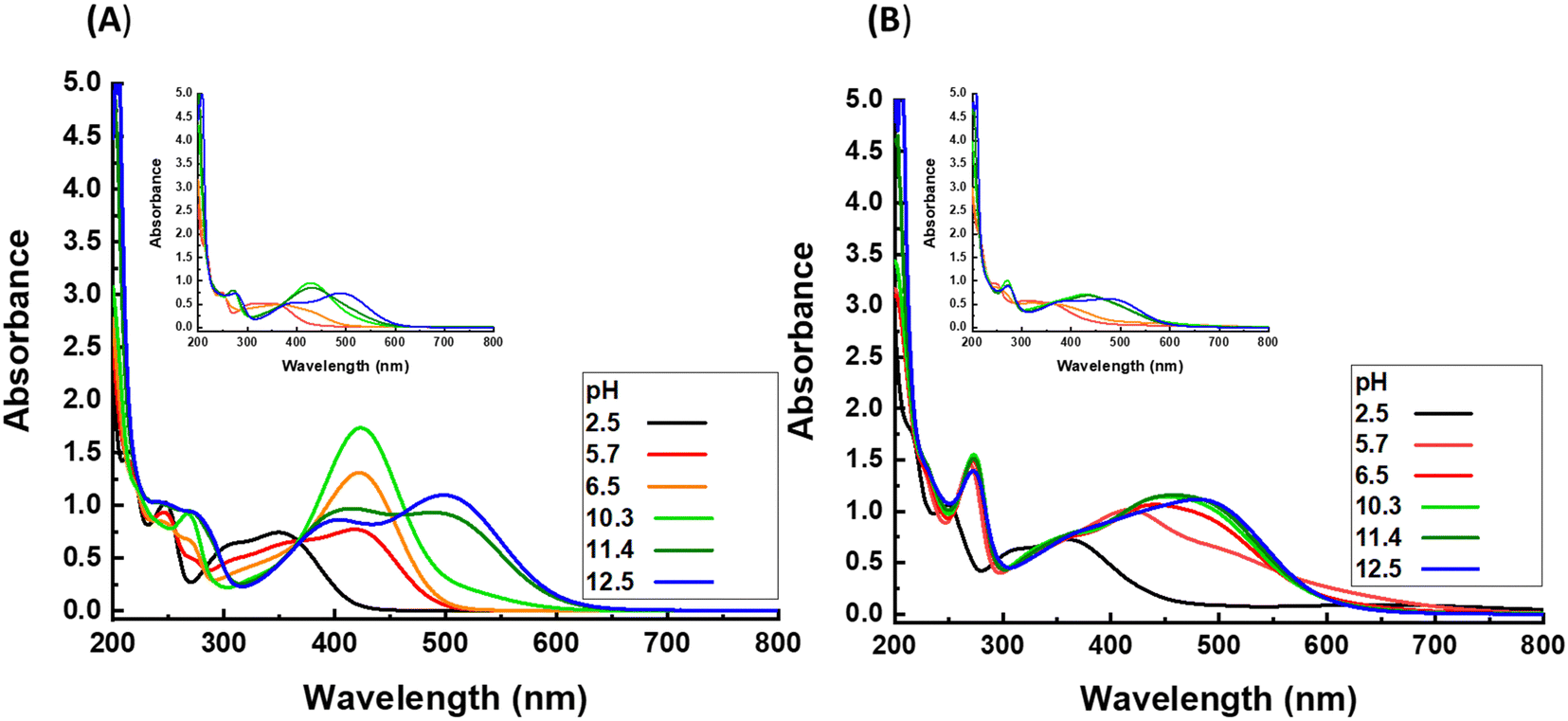 | ||
| Fig. 1 UV-vis spectra of nitrodopamine hydrogen sulfate and poly(AAc-ran-NC) (A) without and (B) with presence of Fe3+ ions. The insets represent the results of polymeric samples. | ||
Considering the nature of complexation between NC and Fe3+ ions, shifting from the fully-protonated over the mixture of fully- and mono-protonated to fully-deprotonated state in NC results in shifting from mono-complex over bis-complex to a mixture of bis- and tris-complexes, and eventually to almost pure tris-complex. Such a transformation can be recognized by tracking the variation of the maximum wavenumber in the UV-vis spectrum of the mixture of Fe2(SO4)3 with either nitrodopamine hydrogen sulfate or poly(AAc-ran-NC) solutions (Fig. 1B). Similar to that observed in the UV-vis spectra of plain solutions without iron ions, by increasing the pH, the maximum shifts to a higher wavelength, where such a shifting requires higher pH in the case of the polymeric solution compared to the small molecule solution. This correlation can be considered as an evidence that the protonation state of hydroxy substituents on the aromatic ring of NC group has a direct impact on the complexation type of NC and Fe3+ ions. According to the UV-vis data, nitrodopamine and Fe3+ ions form mono-, bis-, mixture of bis- and tris-, and tris-complexes in the pH of 2.5, 5.8, 6.5–11.4, and 12.5, respectively. In the case of poly(AAc-ran-NC), the pH range changes to 5.7–6.5, 10.3–11.4, and 12.5, respectively, without observing a mixture of bis- and tris-complexes.
Dynamics and mechanical properties of supramolecular hydrogels
The dynamics and mechanical properties of supramolecular hydrogels are studied by rheology with frequency sweep and stress relaxation experiments at 10 °C, 20 °C, 30 °C, and 40 °C. To check whether the construction of master curves is possible with these samples, the frequency sweep data at 20 °C are set as the reference and the other data are shifted accordingly. For all samples, the data for different temperatures are not completely overlapped by horizontal shifting, showing different levels of data scattering, as shown in Fig. 2A. This observation reveals that not all the relaxation mechanisms in these samples scale similarly with thermal energy, causing a thermo-rheological complex behavior and violation of the ability for time–temperature superposition. Therefore, analysis of the data for each temperature are done separately. For each sample, the relaxation modes are analyzed by conducting stress relaxation experiments at 20 °C and 30 °C; see Fig. 2B for sample pAA10_NC3. The stress relaxation experiment provides clear signatures of the relaxation processes, magnifies their temperature dependency, and makes it possible to extract different relaxation modes, which may overlap in frequency sweep experiments.28–30 To analyze relaxation modes, the shear stress curve is fitted by eqn (1):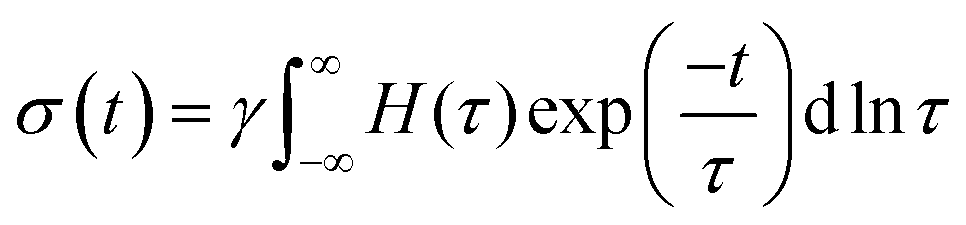 | (1) |
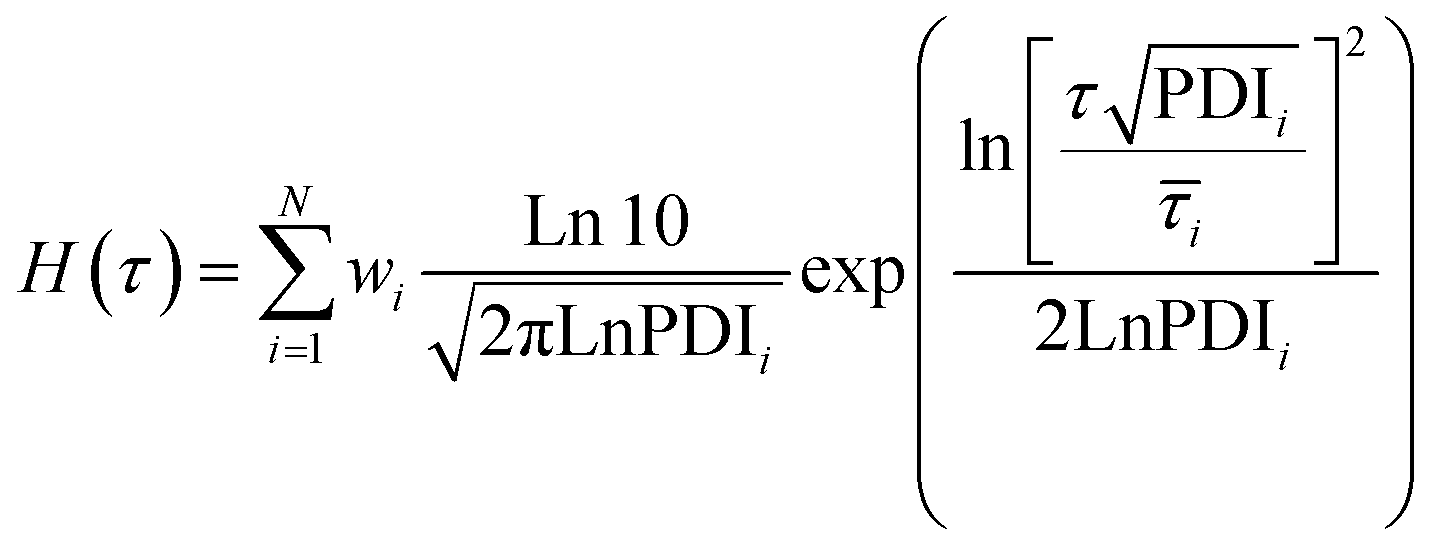 | (2) |
![[small tau, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d4.gif) i, wi, and PDIi represent the average relaxation time, its corresponding weight, and polydispersity index, respectively, for the relaxation mode i; all should be considered as fitting parameters. The number of the relaxation modes is estimated following the analysis of the dynamic moduli. The G′′ curve for all samples reveals a weak bump at high frequencies and a maximum at low frequencies. Directly after this maximum, samples show no terminal relaxation. Instead, dynamic moduli drop in parallel and as revealed by the stress relaxation data, there is even a tendency to form a plateau. Such rheological properties are commonly observed in supramolecular materials, in which on top of the distinct transient associations, aggregation of them into separated domains, known as clusters, occurs. The phase separation along with aggregation of transient associations hinder the terminal relaxation. Nevertheless, the terminal relaxation mechanism can still occur at very long time, which is not experimentally accessible. Therefore, we hypothesize that at least three relaxation modes are involved in the dynamics of such samples.31 By including the third relaxation mechanism, the results are generally better compared to those obtained with considering only two relaxation modes. Comparison of the calculated results with the experimental data and the obtained relaxation time spectrum for sample pAA10_NC3 are shown in Fig. 2B and C, respectively. The relaxation time spectrum at 30 °C involves three modes, but the fastest mode at ∼1.5 s is overlapped with the high-intensity mode at ∼105 s. To provide a quantitative view of the fitting procedure, all fit parameters are provided in Table S1 in the ESI.† Moreover, for other samples, the stress-relaxation graphs are shown in Fig. S7 in the ESI.†
i, wi, and PDIi represent the average relaxation time, its corresponding weight, and polydispersity index, respectively, for the relaxation mode i; all should be considered as fitting parameters. The number of the relaxation modes is estimated following the analysis of the dynamic moduli. The G′′ curve for all samples reveals a weak bump at high frequencies and a maximum at low frequencies. Directly after this maximum, samples show no terminal relaxation. Instead, dynamic moduli drop in parallel and as revealed by the stress relaxation data, there is even a tendency to form a plateau. Such rheological properties are commonly observed in supramolecular materials, in which on top of the distinct transient associations, aggregation of them into separated domains, known as clusters, occurs. The phase separation along with aggregation of transient associations hinder the terminal relaxation. Nevertheless, the terminal relaxation mechanism can still occur at very long time, which is not experimentally accessible. Therefore, we hypothesize that at least three relaxation modes are involved in the dynamics of such samples.31 By including the third relaxation mechanism, the results are generally better compared to those obtained with considering only two relaxation modes. Comparison of the calculated results with the experimental data and the obtained relaxation time spectrum for sample pAA10_NC3 are shown in Fig. 2B and C, respectively. The relaxation time spectrum at 30 °C involves three modes, but the fastest mode at ∼1.5 s is overlapped with the high-intensity mode at ∼105 s. To provide a quantitative view of the fitting procedure, all fit parameters are provided in Table S1 in the ESI.† Moreover, for other samples, the stress-relaxation graphs are shown in Fig. S7 in the ESI.†
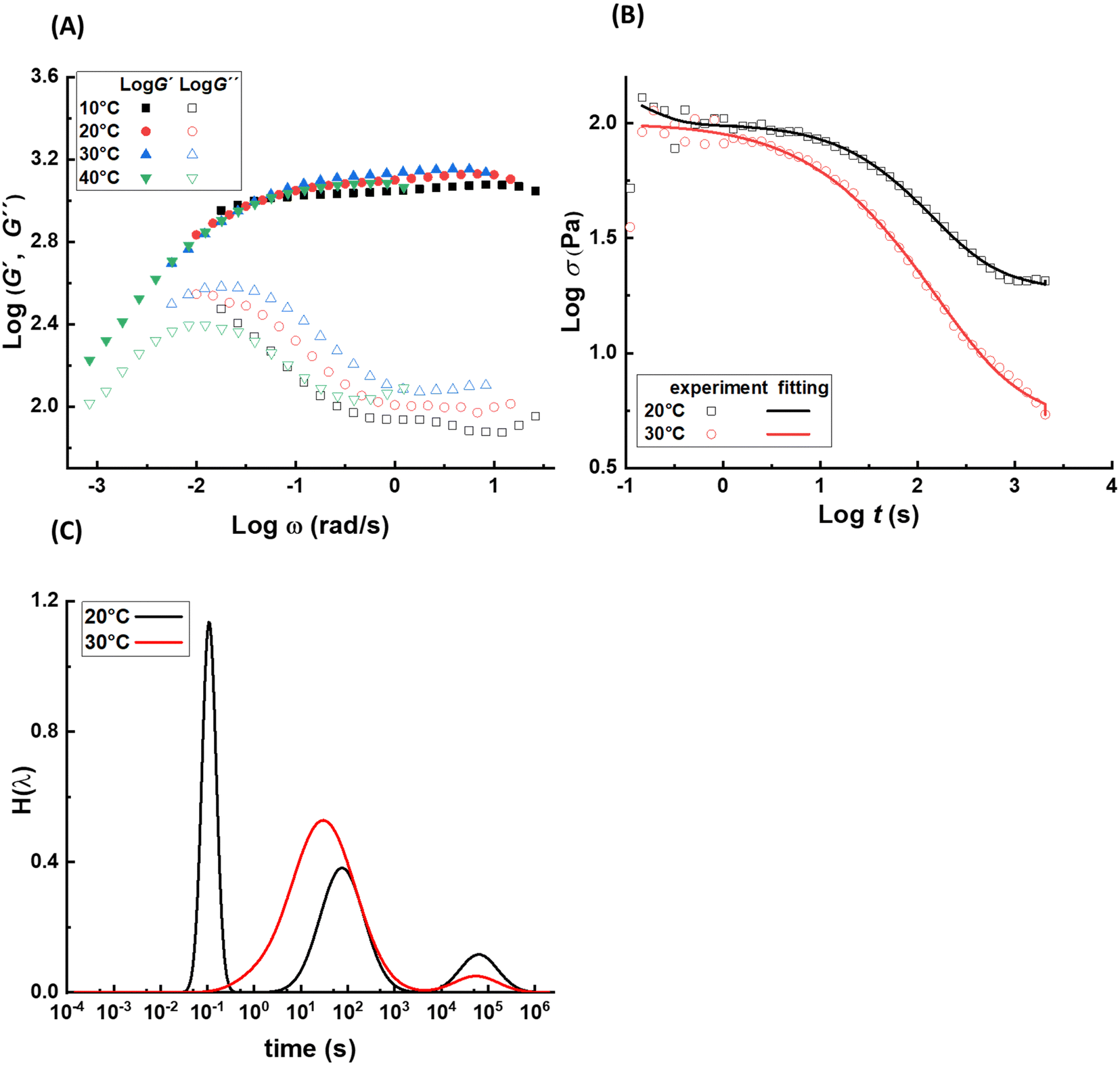 | ||
| Fig. 2 (A) Frequency sweep data of sample pAA10_NC3 at temperatures of 10, 20, 30, and 40 °C. The curve of 20 °C is set as the reference and other curves are shifted horizontally to form a master curve. (B) Comparison of the experimentally and calculated stress relaxation data for sample pAA10_NC3 at 20 °C and 30 °C. (C) Relaxation time spectrum for this sample at 20 °C and 30 °C, obtained from the fitting of stress relaxation data by considering three relaxation modes. | ||
The weight of each mode is a criterion showing how each mode affects the overall relaxation process. If wi is too small, for example less than 1%, relaxation mode i is neglected. For samples with NC:Fe3+ = 3:1, the times corresponding to the relaxation modes are 0.1–0.95 s for the fastest relaxation, 130–260 s for the intermediate one, and 9.8 × 104–1.0 × 105 s for the slowest relaxation mode, where the major relaxation mode is the intermediate one with the weight fraction of 85–90%.
In hydrogels with tetra-arm PEG bulk structure, crosslinked by the binary complexation of catechol/nitrocatechol with Fe3+ ions, the time scale of the relaxation that is caused by the dissociation of supramolecular crosslinks is between ∼0.07 s to ∼6 s.24–26 With this evidence, the fastest relaxation mode can be attributed to the dissociation of single, distinct NC–Fe3+ complexes.
To analyze the other two relaxation modes, it is necessary to estimate first if the precursor chains of the networks are entangled.9 For this target, the volume fraction threshold for the entanglement (ϕe) is estimated by32
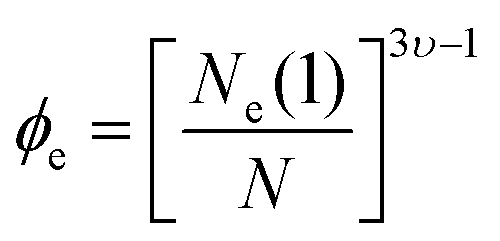 | (3) |
000 g mol−1) to the entanglement molar mass of poly(acrylic acid) (6900 g mol−1). Since hydrogels are prepared in basic pH, we consider υ = 1 leading to ϕe = 0.052. Therefore, the volume fraction of all hydrogels are above the threshold of entanglement, thereby, polymer chains could relax through disentanglement mechanism. For precursor polymer chains, the disentanglement time, calculated from the reptation theory, is in the scale of 10−6 s. Due to the presence of the transient associations in the side-chain position, however, the relaxation time corresponding to this mechanism is estimated by the sticky reptation theory34 as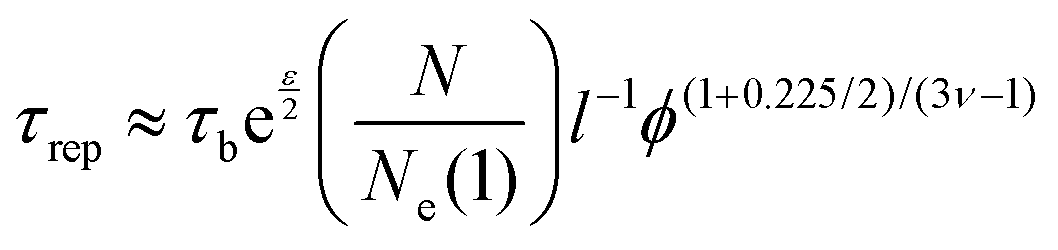 | (4) |
The third relaxation mode of the hydrogels is in the range of 105 s, which is out of the accessible time scale of our experiments. To investigate this relaxation mode in more detail, dynamic moduli of hydrogels at 20 °C and 30 °C are estimated from the relaxation spectrum by using eqn (5) and (6)
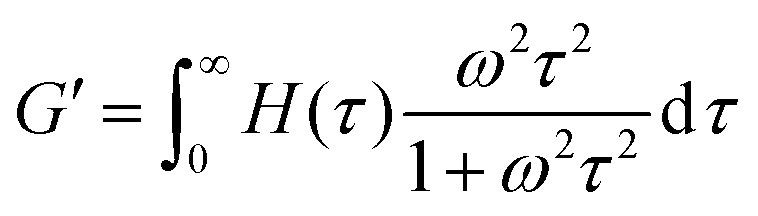 | (5) |
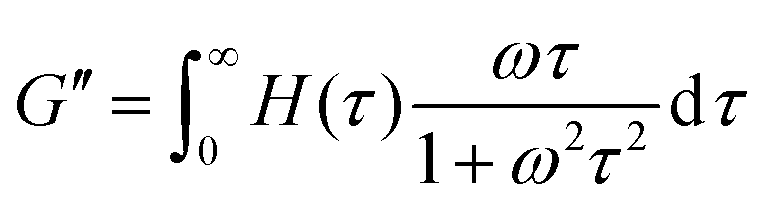 | (6) |
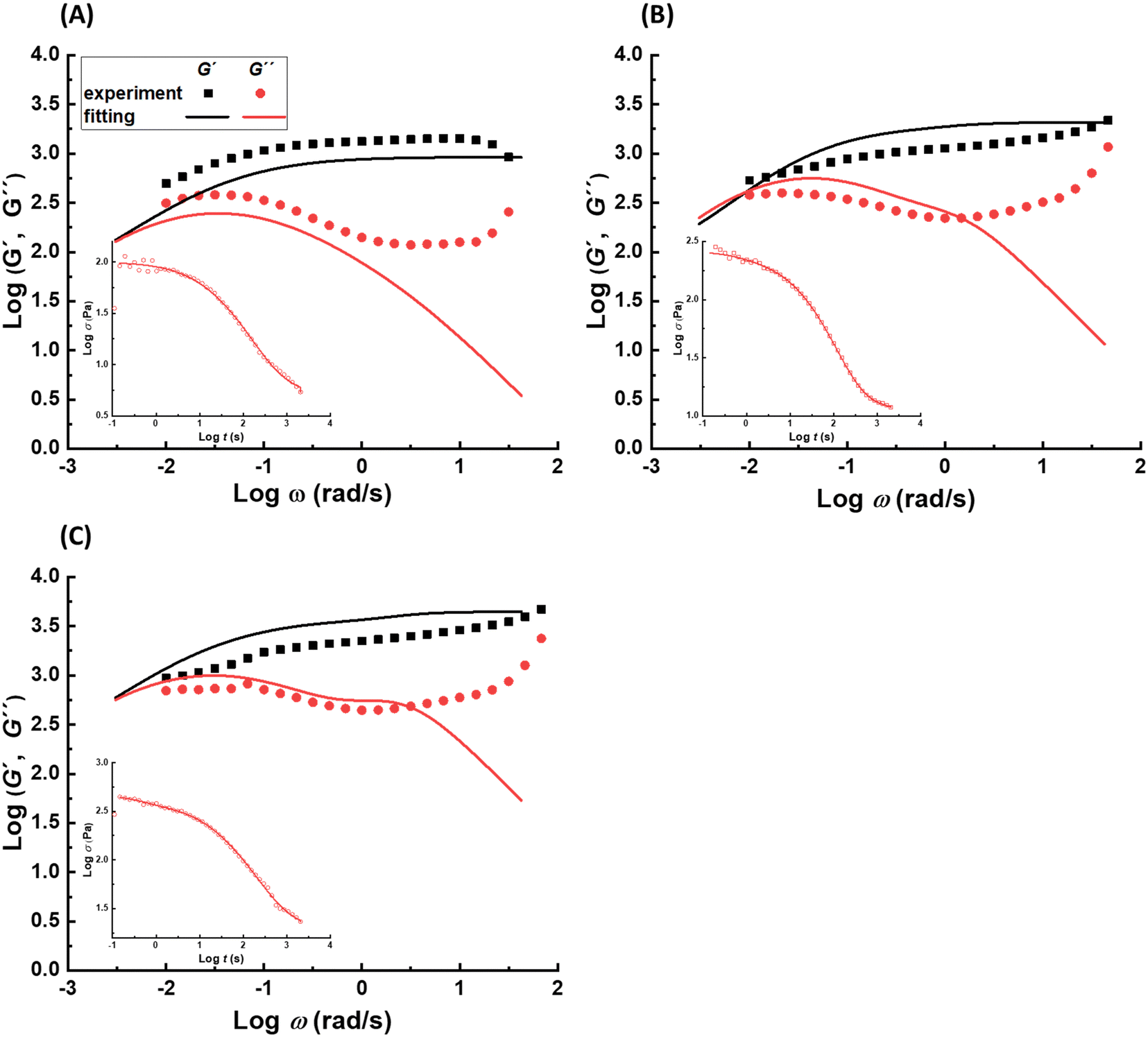 | ||
| Fig. 3 Experimental and calculated frequency sweep data for samples (A) pAA10_NC3, (B) pAA15_NC3, and (C) pAA20_NC3 at 30 °C. At frequencies lower than 1 rad s−1, a relatively good agreement between the experimental data and the calculated data based on fitting the stress–relaxation curves is obtained. Insets represent the experimental and calculated stress relaxation data for the corresponding samples. | ||
Considering the effect of the polymer concentration, no clear effect has been observed on the time scale of the fastest and longest relaxation modes. The former is expected as the lifetime of the distinct NC–Fe3+ complex does not depend on the polymer concentration. The latter might be related to the super large time scale, where the minor change in the values is not detectable by our fitting approach. In contrast, increasing the concentration increases the intermediate relaxation time as shown in Fig. 4A. This observation is related to the effect of polymer concentration on the sticky reptation time as suggested by eqn (4) (τrep ∝ ϕ0.55).
 | ||
| Fig. 4 (A) The effect of polymer concentration on rheological properties of studied supramolecular hydrogels. (B) Comparison of frequency sweep data for samples with 20 wt% concentration and different NC:Fe3+ molar ratios. The data of both panels are obtained from fitting the stress relaxation data. | ||
A similar analysis is done on samples with 20 wt% polymer concentration, in which the molar ratio of NC and Fe3+ ions is varied. The calculated frequency sweep graphs based on fitting stress relaxation data is demonstrated in Fig. 4B. The weight of the third relaxation is less than 1% for sample pAA20_NC2, which means that formation of stable clusters is negligible. The reason could be related to the presence of extra metal ions which push the complexation equilibrium towards formation of mono- and bis-complexes. This confirms that the stability of transient associations is one important factor that promote formation of clusters. This lower number of supramolecular crosslinking can explain the considerably lower plateau modulus of this sample compared to the sample pAA20_NC3. The lack of long-lasting clusters and the lower crosslinking density in this sample results in a more prominent crossover of G′ and G′′ at 30 °C compared to sample pAA20_NC3. The data at 30 °C also shows that this sample has a tendency for viscous flow at long time scales. The changing of stoichiometric ratio from 3:1 to 4:1 also influences the dynamics and mechanical properties of the hydrogel, however, this effect is considerably lower compared to what was observed for sample pAA20_NC2. The weight of the longest relaxation mode in sample pAA20_NC4 is comparable with that of sample pAA20_NC3. This is in agreement with no apparent viscous flow for sample pAA20_NC4 similar to sample pAA20_NC3. However, the plateau modulus of this sample is still lower than that of pAA20_NC3, which is related to the lower crosslinking density due to the lower amount of Fe3+ ions than what is needed to form tris-complexes.
In summary, we hypothesize that all studied hydrogels (except sample pAA20_NC2) have three relaxation mechanisms, with corresponding time scales that vary six orders of magnitudes from 0.1 s to 9.8 × 104 s. The fastest relaxation mode is attributed to the distinct, single NC–Fe3+ dissociation. The intermediate relaxation mode, with a time scale of 130–260 s and weight fraction of 85–90%, is related to the disentanglement of sticky polymer chains. The slowest relaxation mode is attributed to the disengagement of polymer segments from the aggregates of NC–Fe3+ complexations formed by the collective assembly of distinct transient bonds. The distinct association of NC–Fe3+, entanglement of stick chains, and the aggregation of supramolecular complexes are sketched in Scheme 2. The existence of long-lasting aggregates and clusters in our studied samples features in computational and experimental frequency sweep graphs by shifting the G′ and G′′ crossover to low frequency ranges and preventing the viscous flow of polymer chains in the terminal region.
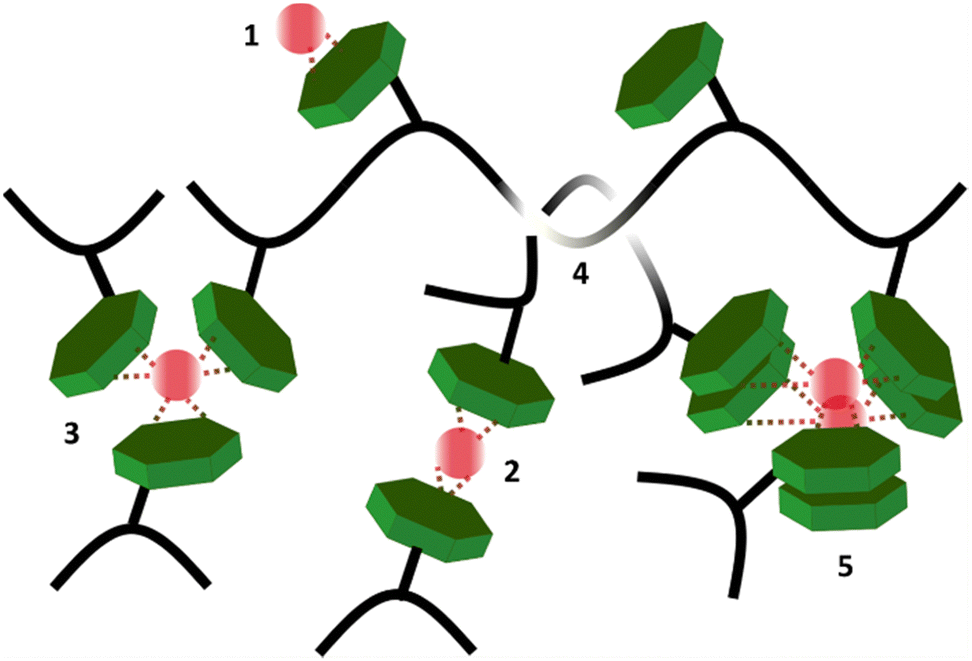 | ||
| Scheme 2 Different types of physical and topological interactions that are responsible for the relaxation modes, detected in this study. 1–3: Mono-complex, bis-complex, and tris-complex of NC–Fe3+, respectively. 4: Entanglement of sticky polymer chains. The onset concentration of entanglement significantly reduces by presence of transient associations. 5: Aggregation of distinct NC–Fe3+ complexes. | ||
In addition, this phenomenon can be confirmed by scattering techniques like small angle X-ray scattering (SAXS). The scattering intensity profile of sample pAA20_NC3 is represented in Fig. 5. At 0.0088 < q < 0.02 Å−1, the intensity profile demonstrates a power-law decay with a slope of −4, which could be related to the scattering objects that are too big to be resolved by this technique. In the following, at 0.02 < q < 0.1 Å−1, a shoulder emerges, which indicates the presence of scattering objects with the size of 1–5 nm. The appearance of this shoulder confirms our hypothesis about the presence of non-ordered aggregates of transient complexes in this sample. To have a better estimation of the size of these aggregates, the data is fitted with the modified Porod model as well as the modified Ornstein–Zernike model. The first model fits data well at q < 0.02 Å−1, where the intensity profile demonstrates a power-law decay. The second model fits well the middle q data, where the shoulder emerges. Therefore, the combination of both models can describe the scattering data in the whole covered range of q. The main outcome of the modified Ornstein–Zernike model is the correlation size of aggregates, which is predicted at 16 nm for sample pAA20_NC3. This value is larger than the covered size range, which might be related to the broadness of this shoulder and the absence of plateau at the very low q values.
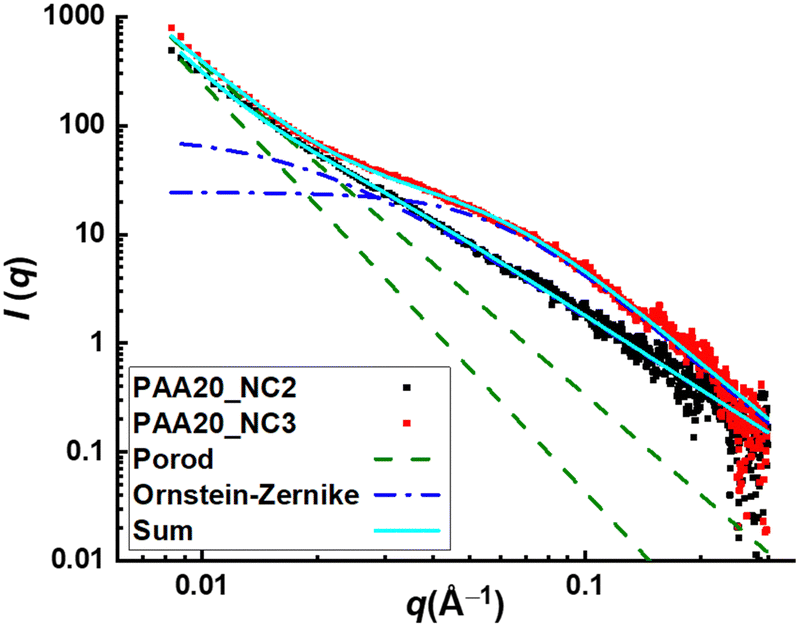 | ||
| Fig. 5 Scattering intensity profile of samples pAA20_NC2 and pAA20_NC3 obtained from SAXS measurement. The data are fitted with the modified Porod model as well as the Ornstein–Zernike model. The combination of both fittings (sum, cyan curve) can describe the data in the whole covered range of q. | ||
The aggregation of supramolecular associations into clusters and with that the stability and the relaxation time of the bulk network can be controlled by the pH of the hydrogel formation medium, the polymer concentration and the molar ratio of NC and Fe3+ ions. Regarding the latter parameter, the sample with a molar ratio of NC:Fe3+ = 2:1, reveals the tendency of viscous flow in the frequencies lower than that of the crossover of the dynamic moduli, which is hypothesized to be related to the lack of stable clusters. This result is confirmed by the absence of any shoulder in the middle q range of the scattering intensity profile of this sample as is represented in Fig. 5.
The influence of supramolecular clustering on the relaxation time of supramolecular hydrogels is an effective approach to form stable hydrogels. For example, the rational use of the collective assembly/stacking of the associative motifs for stabilizing single associated dimers has been extensively investigated in hydrogen-bonded hydrogels.11,12,14 This motivates us to investigate the stability of studied hydrogels in presence of an excess amount of water. While the studied hydrogels have no possibility for terminal relaxation within the studied time scales, the as prepared samples are not stable if they are immersed in an excess amount of water. This means that at some point during the water adsorption process, the gel–sol transition occurs before hydrogels could reach their swelling equilibrium. This instability limits the potential application of such materials for diverse applications, in which hydrogels should be stable in the desired environment (for example water), where the content of the surrounding medium is significantly greater than the amount of the water in as prepared hydrogels. One reason for this instability is the chemical nature of the bulk network of these hydrogels. The acrylic acid and its sodium salt are well known for the preparation of superabsorbers, which are able to retain a huge amount of water within their networks compared to the dry weight of the materials. One approach to prepare more stable hydrogels is to change their chemistry into a structure with less polarity and/or less osmotic pressure. In this sense, the method of hydrogel preparation should be modified, considering the limitation of solubility of less polar polymer chains in the aqueous medium, which is explained in Experiment. In addition, more stable hydrogels can be prepared by increasing the level of the aggregation of supramolecular motifs. This can be achieved, for example, by the increase of the molar mass of the polymer chains.37,38 Investigation of these parameters is the topic of our further studies.
Conclusion
The association of NC and Fe3+ with a molar ratio of 3:1 on a poly(AAc-ran-NC) network platform depends on the pH of the hydrogel preparation medium. By increasing the pH from 4 to 12, NC–Fe3+ ligand–metal association gets more stable by shifts from mono-complex over bis-complex to a mixture of bis- and tris-complexes, and eventually to almost pure tris-complex as visualized in Scheme 2. The best concentration range for the preparation of hydrogels is between 10 and 20 wt%. At lower polymer concentrations, the sol–gel transition does not occur at room temperature, whereas at higher concentrations, the formed hydrogels are very inhomogeneous because of the impossibility of mixing components during hydrogel formation due to the high viscosity of the polymer solutions. All samples with NC:Fe3+ = 3:1 have three relaxation modes. The fastest one is related to the dissociation of distinct NC–Fe3+ complexes. The intermediate relaxation mode, which is the major one, determined at time scales of 130–260 s, is related to the disentanglement of sticky polymer chains. The third relaxation mode, with extremely long relaxation time, is attributed to the long-lasting clusters. While the weight fraction of such clusters is less than 5%, the viscous flow with the fingerprint slopes of 2 and 1 for G′ and G′′, in the terminal region, is not observed in the log–log frequency sweep plots of these samples. The extent of clustering can be controlled by the molar ratio of NC and Fe3+ ions. For example, by changing the molar ratio of NC:Fe3+ to 2:1, the clustering of transient complexations is low enough such that the low-frequency G′–G′′ crossover and the terminal relaxation are experimentally accessible.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. J. and S. S. gratefully thank the Federal Ministry of Science and Education of Germany (BMBF) for the financial support under project HydroDeSal (Project Code 02WME1613). M. A. thanks the German Research Foundation (DFG) for financial support via research grant number 491930291. P. N. acknowledges the H2020 Program (Marie Skłodowska-Curie Actions) of the European Commission's Innovative Training Networks (H2020-MSCA-ITN-2017) under DoDyNet REA Grant Agreement No. 765811.References
- R. Dong, Y. Pang, Y. Su and X. Zhu, Biomater. Sci., 2015, 3, 937 RSC.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165 CrossRef CAS PubMed.
- L. Voorhaar and R. Hoogenboom, Chem. Soc. Rev., 2016, 45, 4013 RSC.
- J. L. Mann, C. Y. Anthony, G. Agmon and E. A. Appel, Biomater. Sci., 2018, 6, 10 RSC.
- J. Hoque, N. Sangaj and S. Varghese, Macromol. Biosci., 2019, 19, 1800259 CrossRef PubMed.
- M. J. Webber and P. Y. Dankers, Macromol. Biosci., 2019, 19, 1800452 CrossRef PubMed.
- E. Krieg, M. M. Bastings, P. Besenius and B. Rybtchinski, Chem. Rev., 2016, 116, 2414 CrossRef CAS PubMed.
- Y. Li, C. Zhu, Y. Dong and D. Liu, Polymer, 2020, 210, 122993 CrossRef CAS.
- S. C. Grindy, M. Lenz and N. Holten-Andersen, Macromolecules, 2016, 49, 8306 CrossRef CAS.
- A. Jangizehi, M. Ahmadi and S. Seiffert, Mater. Adv., 2021, 2, 1425 RSC.
- P. Y. Dankers, M. J. van Luyn, A. Huizinga-van der Vlag, G. M. van Gemert, A. H. Petersen, E. W. Meijer, H. M. Janssen, A. W. Bosman and E. R. Popa, Biomaterials, 2012, 33, 5144 CrossRef CAS PubMed.
- P. Y. Dankers, T. M. Hermans, T. W. Baughman, Y. Kamikawa, R. E. Kieltyka, M. M. Bastings, H. M. Janssen, N. A. Sommerdijk, A. Larsen and M. J. Van Luyn, Adv. Mater., 2012, 24, 2703 CrossRef CAS PubMed.
- C. L. Lewis and M. Anthamatten, Soft Matter, 2013, 9, 4058 RSC.
- M. Guo, L. M. Pitet, H. M. Wyss, M. Vos, P. Y. Dankers and E. Meijer, J. Am. Chem. Soc., 2014, 136, 6969 CrossRef CAS PubMed.
- S. I. Hendrikse, S. P. Wijnands, R. P. Lafleur, M. J. Pouderoijen, H. M. Janssen, P. Y. Dankers and E. Meijer, Chem. Commun., 2017, 53, 2279 RSC.
- S. Y. Zheng, H. Ding, J. Qian, J. Yin, Z. L. Wu, Y. Song and Q. Zheng, Macromolecules, 2016, 49, 9637 CrossRef CAS.
- E. A. Appel, X. J. Loh, S. T. Jones, F. Biedermann, C. A. Dreiss and O. A. Scherman, J. Am. Chem. Soc., 2012, 134, 11767 CrossRef CAS PubMed.
- T. Rossow and S. Seiffert, Supramolecular polymer networks: Preparation, properties, and potential. Supramolecular Polymer Networks and Gels, Springer: 2015; p. 1 Search PubMed.
- Y. Zhang, B. Zhang, Y. Kuang, Y. Gao, J. Shi, X. X. Zhang and B. Xu, J. Am. Chem. Soc., 2013, 135, 5008 CrossRef CAS PubMed.
- J. Brassinne, C.-A. Fustin and J.-F. Gohy, J. Inorg. Organomet. Polym. Mater., 2013, 23, 24 CrossRef CAS.
- M. J. Harrington, A. Masic, N. Holten-Andersen, J. H. Waite and P. Fratzl, Science, 2010, 328, 216 CrossRef CAS PubMed.
- D. E. Fullenkamp, L. He, D. G. Barrett, W. R. Burghardt and P. B. Messersmith, Macromolecules, 2013, 46, 1167 CrossRef CAS PubMed.
- S. C. Grindy, R. Learsch, D. Mozhdehi, J. Cheng, D. G. Barrett, Z. Guan, P. B. Messersmith and N. Holten-Andersen, Nat. Mater., 2015, 14, 1210 CrossRef CAS PubMed.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651 CrossRef CAS PubMed.
- N. Holten-Andersen, A. Jaishankar, M. J. Harrington, D. E. Fullenkamp, G. DiMarco, L. He, G. H. McKinley, P. B. Messersmith and K. Y. C. Lee, J. Mater. Chem. B, 2014, 2, 2467 RSC.
- M. S. Menyo, C. J. Hawker and J. H. Waite, Soft Matter, 2013, 9, 10314 RSC.
- S. Hou and P. X. Ma, Chem. Mater., 2015, 27, 7627 CrossRef CAS PubMed.
- M. Ahmadi and S. Seiffert, Soft Matter, 2020, 16, 2332 RSC.
- M. Ahmadi and S. Seiffert, Macromolecules, 2021, 54, 1388 CrossRef CAS.
- M. Ahmadi and S. Seiffert, Macromolecules, 2021, 54, 7113 CrossRef CAS.
- M. Ahmadi, A. Jangizehi and S. Seiffert, Macromolecules, 2022, 55, 5514 CrossRef CAS.
- M. Rubinstein and R. H. Colby, Polymer physics. Oxford university press, New York, 2003, vol. 23 Search PubMed.
- R. H. Colby, Rheol. Acta, 2010, 49, 425 CrossRef CAS.
- M. Rubinstein and A. N. Semenov, Macromolecules, 2001, 34, 1058 CrossRef CAS.
- M. A. Matin, M. M. Islam, T. Bredow and M. A. Aziz, Adv. Chem. Eng. Sci., 2017, 7, 137 CrossRef CAS.
- Y. Li, J. Wen, M. Qin, Y. Cao, H. Ma and W. Wang, ACS Biomater. Sci. Eng., 2017, 3, 979 CrossRef CAS PubMed.
- A. Jangizehi, M. Ahmadi and S. Seiffert, J. Polym. Sci., Part B: Polym. Phys., 2019, 57, 1209 CrossRef CAS.
- M. Ahmadi, A. Jangizehi, E. van Ruymbeke and S. Seiffert, Macromolecules, 2019, 52, 5255 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sm00666a |
| This journal is © The Royal Society of Chemistry 2022 |