
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Weak polyelectrolyte brushes
Mark
Geoghegan

School of Engineering, Newcastle University, Merz Court, Newcastle-upon-Tyne NE1 7RU, UK. E-mail: mark.geoghegan@newcastle.ac.uk
First published on 17th March 2022
Abstract
I review experimental developments in the growth and application of surface-grafted weak polyelectrolytes (brushes), concentrating on their surface, tribological, and adhesive and bioadhesive properties, and their role as actuators.
 Mark Geoghegan | Professor Mark Geoghegan has held the Roland Cookson Chair in Engineering Materials at Newcastle University since 2019. He obtained his BA in Physics at the University of Oxford in 1988 and an MSc on cold gaseous plasmas at the University of Birmingham in 1990. He undertook his PhD at the University of Cambridge under the supervision of (now) Professor Richard Jones. He submitted his doctoral thesis on polymer blend films in 1994. He spent two years at CEA-Saclay working as a postdoc with Dr François Boué before being awarded a Humboldt fellowship at the Universität Freiburg. Two years later, he moved to Bayreuth to undertake another postdoctoral fellowship in Bayreuth under the supervision of Professor Georg Krausch. In 2000 he moved to the University of Sheffield where he remained for nineteen years, being awarded a personal chair in 2011. |
1 Introduction
A polymer brush is a layer of surface-grafted polymers. These are generally end-grafted, but that does not need to be the case. By chemically attaching the chains to a surface, the layer can remain stable even in a good solvent. This gives them an important advantage over films cast from solvents in coatings technologies. Further benefits arise, for example, in colloidal stabilization due to the limited swelling available for the polymers, which are already extended in forming a brush.1 Hydrophilic polymer brushes are particularly useful in aqueous media to combat fouling.2,3Polyelectrolytes are a class of polymers which contain ionized or ionizable groups. Examples of weak polyelectrolytes include poly(methacrylic acid) [PMAA] and poly(2-vinylpyridine) (P2VP) whereas polystyrene sulfonic acid and poly(allylamine hydrochloride) are examples of strong polyelectrolytes. The strong acid and base retain their charge in aqueous solution over a wide range of pH, whilst the weak acid (here, PMAA) is neutral at low pH and the weak base (P2VP) is neutral at high pH. The much more pH-sensitive behaviour of weak polyelectrolytes has the potential for significant utility in nanoscience because the charged state of a polyelectrolyte brush is intimately related to its conformation. Change the charge and, under many conditions, the conformation changes too. This conformational change brings with it a concomitant capacity to do mechanical work, which offered great promise to the nascent field of soft nanotechnology at the beginning of the millennium.
Weak polyelectrolytes have been known as “smart” polymers for some time due to their pH responsive behaviour. For example, PMAA may be used as a coating in some oral medications because it can protect the drug from the harsh acidic environment of the stomach.4 In the gut, the pH is significantly higher, and the acid can dissolve, releasing the cargo. Similarly, dried hydrogels of sodium polyacrylate are used in nappies (diapers), and their capacity for absorbance allows for a five-hundred-fold increase in mass before the hydrogels become saturated and leak.
This review will cover the different synthesis routes of weak polyelectrolyte brushes and their characterization. Different topologies and their importance will also be discussed. Since polyelectrolytes have a rich conformational space covered by pH and added salt,5 some effort will be spent covering how these parameters control brush conformation. The physical properties of the surfaces thus created through contact mechanics will be covered. The review will conclude with a discussion of the various applications of polyelectrolyte brushes, where actuation is required. Applications in areas of bioadhesion are of growing importance and these will also be included. Related areas of research involving strong polyelectrolytes and polyzwitterions are not covered.
2 Synthetic routes
Brushes may either be made in situ (grafting from) or the polymer may be synthesized first and then a functional end group used to attach the polymer to the surface (grafting to). Both have advantages and disadvantages. The grafting-to method allows for much better characterization of the polymer, because it lends itself to techniques such as gel permeation chromatography and light scattering to find the size of the polymer prior to grafting. However, it is difficult to obtain dense and thick brushes using grafting-to techniques because the first chains to arrive at the surface form a mushroom conformation (Fig. 1).6 At a grafting certain density (the mushroom-to-brush transition), polymers face an entropic barrier preventing them from reaching the substrate, because they need to elongate to navigate through the chains already at the surface. Possible routes to increase the density of brushes grafted to the substrate are discussed below, as are attempts to characterize brushes grown from the substrate.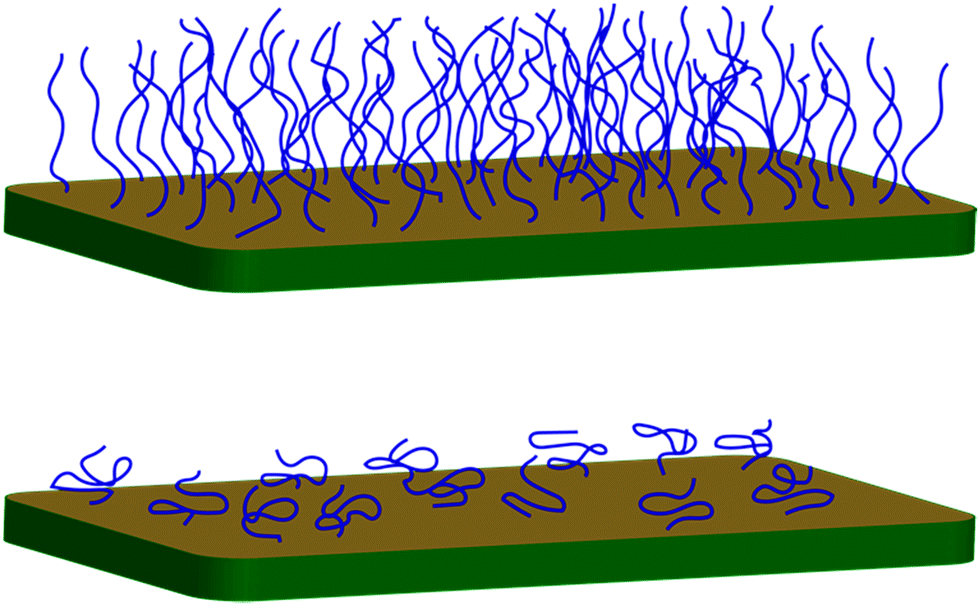 | ||
| Fig. 1 If the polymer grafting points are further away from each other than the size of the polymer chain in a self-avoiding work configuration, the polymers exhibit a mushroom conformation (bottom). If they are closer together, then a brush conformation is observed (top). | ||
Grafting-from techniques
The use of controlled polymerizations dominates the synthesis of polymer brushes grafted from surfaces. These are used because of their anticipated low dispersities. Free radical polymerization has been used with success to produce weak polyelectrolyte brush layers,7 but controlled polymerization methods are not significantly less convenient given that much of the effort in growing brushes involves depositing a suitable initiating monolayer.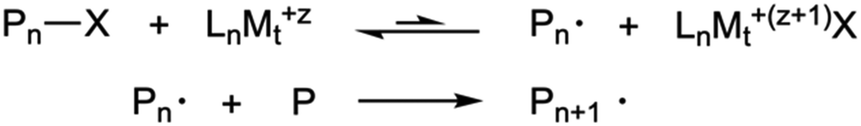 | ||
| Scheme 1 ATRP process. The polymer is activated by the metal (Mt) complexed ligand (Ln), which allows the chain to grow. | ||
The direct synthesis of polyelectrolyte brushes using ATRP is routinely performed with polycations, such as poly[2-(dimethyl-amino)ethyl methacrylate] (PDMAEMA)9 or poly[2-(diethyl-amino)ethyl methacrylate] (PDEAEMA).10 On the other hand, the growth of carboxylic polyanions in this manner is not easily achieved because they easily associate with the catalyst.11 Standard polyanions such as poly(acrylic acid) (PAA) and PMAA can be synthesized by polymerizing tert-butyl acrylate or tert-butyl methacrylate followed by a hydrolysis step.8,12 However, it has been shown that it is possible to grow PMAA brushes directly by using methacrylic acid sodium salt.13,14
The main development of ATRP is that of Activators ReGenerated by Electron Transfer (ARGET) ATRP, which reduces the quantity of heavy metal catalyst required by the addition of a reducing agent, often ascorbic acid. This agent reduces Cu(II) to Cu(I), which is the active catalyst. Since oxygen is generally responsible for the oxidation to Cu(II), the sensitivity of ATRP to oxygen is reduced, which is another advantage of the ARGET technique. ARGET ATRP is not routinely used in the production of polyelectrolyte brushes and it has been demonstrated that the tertiary amine groups present in PDMAEMA and, one supposes, PDEAEMA act effectively as reducing agents.15
New developments in ATRP have largely passed by those interested in the synthesis of polyelectrolyte brushes. For example, Initiators for Continuous Activator Regeneration (ICAR) ATRP is another method of synthesizing polymers in an aqueous environment16 and (like ARGET ATRP) reducing the amount of metal catalyst. This has not been embraced by those creating polyelectrolyte brushes. Indeed the metal catalyst can be eliminated using more specialized forms of ATRP,17 but this is currently rather niche. Ultimately, however, metal catalysts have an environmental footprint and create problems regarding the cleaning of the brushes. If ATRP is to be routinely used in biological systems, then perhaps more effort will need to be directed towards reducing or eliminating the metal catalysts that are used.
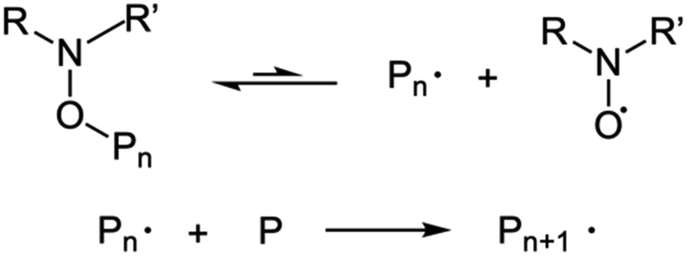 | ||
| Scheme 2 NMP process. | ||
 | ||
| Scheme 3 RAFT process. The chain transfer agent reacts with an activated radical or growing polymer to extend the chain. The notation here follows that of Perrier.25 | ||
Although many researchers prefer to use their favoured techniques, the development of chemical routes to grafting polymers from surfaces is ongoing. Clean chemistries that limit the use of metals are particularly under development and whichever dominates in the future will depend on such benefits as well as reliability and ease of synthesis. Of course, new developments are by no means restricted to the routes discussed here.37
Alternatives to grafting-from techniques
Although individual chains grafted to surfaces do not usually present dense brush layers due to the entropic barrier inherent in grafting-to approaches, it is possible to graft multiple chains to a surface. This has been demonstrated by the creation of combs of PDMAEMA pre-synthesized from a poly(methyl methacrylate) (PMMA) backbone. After deposition in a Langmuir trough, the hydrophobic PMMA sits on the water surface, while the PDMAEMA chains extend into the water.38 This layer can be compressed to achieve a desired grafting density and can be later deposited on a substrate of choice to create a brush layer.An alternative approach involves the self-assembly of polymers to form micelles with a polyelectrolyte corona. This has been achieved with diblock copolymers of the temperature-responsive polymer poly(N-isopropylacrylamide) (PNIPAm) and PDMAEMA. Here the polymers can self-assemble above 40 °C to form micelles with the hydrophobic PNIPAm block forming the core of the micelle and the PDMAEMA a rather dense corona.39 P2VP coronas have been grown from polyferrocenyldimethylsilane cores40 using crystallization-driven self-assembly.41 These micellar brushes form by hydrogen bonding or electrostatic interaction to a variety of surfaces and have been functionalized to demonstrate, for example, the separation of an oil from an aqueous emulsion.
The biopolymer hyaluronic acid is grown in vivo using a hyaluronan synthase enzyme. This process has been replicated to form hyaluronic acid brushes, which have been shown to reach layers of up to 15 μm in thickness,42 which is perhaps fifty times greater than the thickest brush layers readily grown by standard synthetic routes.
Characterization
The characterization of polymer brushes requires primarily confirmation that the intended polymer is grown on the surface; the molar mass of the polymer; and the grafting density. A measurement of the thickness of the brush in the dry state links the molar mass, Mw, and grafting density, σ, bywhere NA is the Avogadro constant and ρ the density of the polymer. If h is known, then only one of Mw or σ needs to be measured to determine the other. Direct measurement of grafting density is extremely difficult,43 so grafting density is usually calculated from knowledge of the molar mass, which is not in itself easy to determine. A common route to obtaining the molar mass is to grow the polymer ex situ (from an unconstrained initiator in solution) under the same conditions as the brush and assume that these constitute the same polymer.8 This has been questioned from computer simulations which reveal that brushes grow faster in solution than from surfaces.44 Experimental work on a variety of polymers show that PMMA grown in solution can be up to ∼30% longer than those densely grafted from the surface.45 For less dense polymers there is little difference in molar mass between solution-grown and surface-grafted polymers. The most accurate means of measuring molar mass is to de-graft the polymer and characterize it using standard techniques,45,46 but this method requires large substrates to obtain enough polymer to use for chromatography measurements.
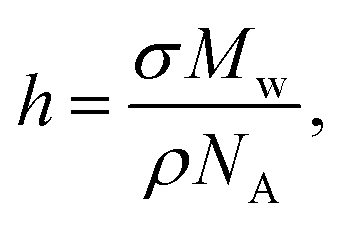
It is possible to measure the polymer molar mass in situ using an atomic force microscope (AFM). Here, the AFM tip is retracted from the surface of the polymer brush in solution. The length of tip retraction before it detaches from the brush can be correlated with the molar mass of the polymer. This technique is only suitable for solvent-swollen brushes and has had some success with neutral brushes.47 Although this AFM-based method (force spectroscopy) can be readily applied to weak polyelectrolyte brushes, it is more challenging because of the effect of pH23,48 and ionic strength48,49 on polymer conformation. Experiments on weak polyacids found good agreement between gel permeation chromatography data on polymers grown from free initiators and force spectroscopy on brushes at neutral pH. However, at low pH the collapsed brushes presented complex spectra and at high pH the highly charged brushes adhered poorly to the AFM tip.23 Poor adhesion of polycationic brushes to AFM tips was proposed as a reason for difficulty in determining the conformation in high ionic strength solutions.48 These polycationic brushes (PDMAEMA) were grafted from a poly(methyl methacrylate) macroinitiator using ATRP,38 and could be characterized by size-exclusion chromatography and so lend themselves well to a comparison of this nature. The pH dependence of PDMAEMA chain length determined by force spectroscopy,48 gave a similar result to that for PAA.23 This means that force spectroscopy can be used to provide a determination of polyelectrolyte molar mass (and dispersity) providing that the polymers are soluble but not too charged, which typically means near neutral pH. This unsatisfactory recommendation is compounded by the best PAA measurements being obtained in 100 mM NaCl in order to control polymer–AFM tip interactions.23
Ultimately, force spectroscopy works as a means of chain length (molar mass) characterization, but so much preparation is needed that calibration is critical for such a route to be pursued. Given the difficulties involved, if chromatographic characterization of the product of a synthesis from free initiator is not undertaken, then it is common for grafted-from polymers to be assumed to take a surface density of 0.5 chains per nm2, following from earlier work.20,50 This assumption is not absurd and can reasonably be applied to polyelectrolyte monomers. If the monomer is not overly bulky, then assuming a 0.5 chains per nm2 surface density is reasonable, given that the grafting density is set early in the synthesis and can only be reduced if the initiator layer is compromised, which may be by a protecting species,51–54 by mixing the initiator layer with inactive species,55 by using a Langmuir–Blodgett trough,56–58 or by otherwise reducing surface initiator concentration.50
Conformation
Brushes are defined by their density and molar mass. A polymer generally undertakes a self-avoiding random walk in a good solvent. This means that its size scales as N0.6,59,60 where N is the number of steps (often taken to mean monomers). This self-avoiding walk creates a greater power law dependence than a standard random walk (N0.5) due to excluded volume interactions between the monomers. A surface perturbs this (self-avoiding) random walk only a little, by ensuring that the grafted chain end lies at the edge of the polymer. This is the ‘mushroom’ conformation. When the grafting points are separated by less than the width of the mushroom (Fig. 1), the chains become compressed and are forced away from the substrate to form brushes.61,62 The height of the brush is then expected to increase monotonically with grafting density.Polyelectrolyte brushes also increase in thickness with increased grafting density, but, given that solvent quality is dramatically affected by pH and salt concentration, there are other parameters to consider when discussing their conformation.63 Changes in pH generally cause swelling or collapse in weak polyelectrolyte brushes,7,64–66 but the addition of salt complicates an analysis of their conformation.
Salt contributes more ions to the brush in addition to those already there. Given the accepted definition of counterion in colloid chemistry,67 salt ions with charges opposite to those of the polymer chain can also act as counterions. However, when considering chemical equilibrium, there is an important difference, because, by adding salt, some “native” counterions may be displaced from the brush, which may in turn cause the polyelectrolyte to become more charged. At low salt concentrations, chains do become more ionized and this results in an increase in brush swelling.68 Here, each chain can be considered as a series of blobs of a Debye length in size. Within each blob the chain exhibits salt-free behaviour, but on larger length scales the chains are extended to counter the addition of salt, causing an osmotic swelling. This behaviour has been given a fuller theoretical foundation based on self-consistent field theory.69 These brushes are referred to as osmotic brushes, but for brushes with reduced grafting density (or less charge) osmotic effects are reduced and the brush is often referred to as a Pincus brush (Fig. 2).
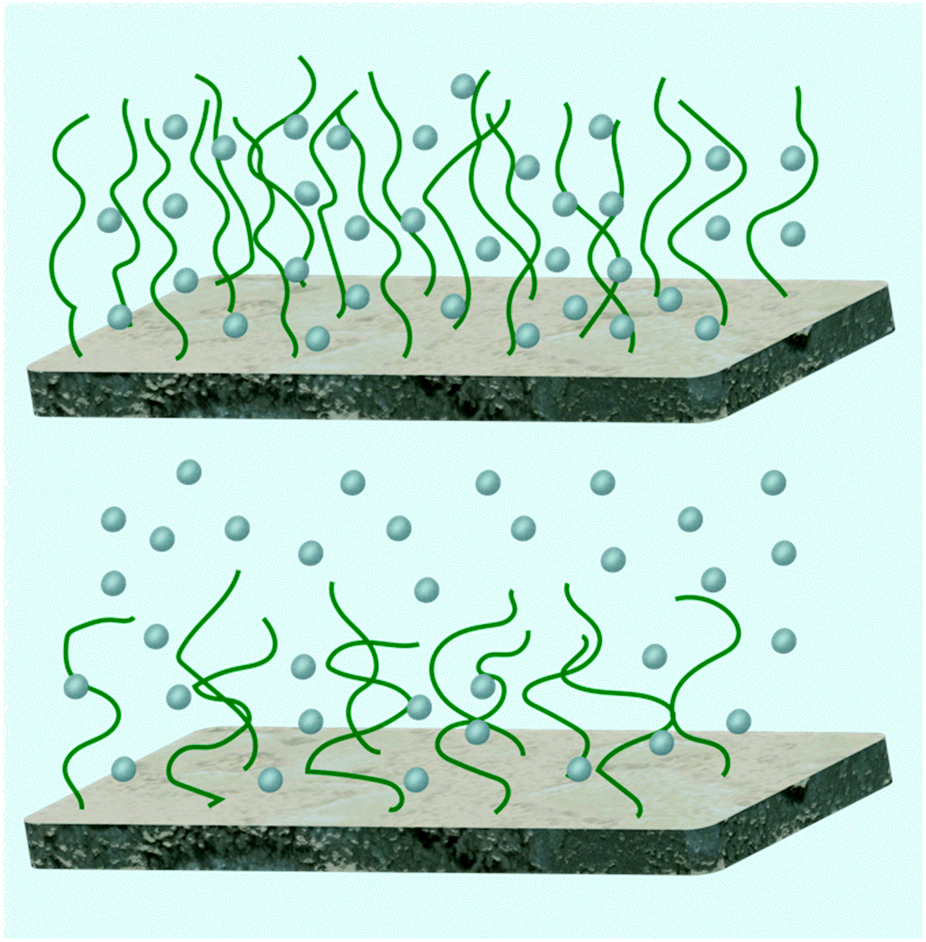 | ||
| Fig. 2 Osmotic brushes (top) retain counterions within the brush whereas Pincus brushes (bottom) do not, but have a layer of counterions above the brush. | ||
For Pincus brushes, the shielding increases with increasing ion concentration, which acts to reduce the osmotic pressure in the brush and cause a concomitant reduction in thickness.70,71
At large ionic strength, the shielding of charges is such that excluded volume interactions dominate electrostatic behaviour.5 This is known as the quasi-neutral brush regime because the presence of charges is irrelevant to the polymer conformation. Experimentally, there are examples of abrupt brush collapse at salt concentrations approaching 1 M.72–76 Other experiments present data that show a more gradual decline in brush thickness over two orders of magnitude of increasing salt concentration,7,77–80 again up to 1 M salt. With these conflicting results, it is only possible to assert that weak polyelectrolyte brushes tend to have a collapsed structure at high salt concentrations.
Osmotic brushes have been proposed to grow with ionic strength as I1/3,68 but experimental support for this prediction is limited7 with other studies offering a power law exponent of 0.1 or less.72,73,75–77,79,80 The origin of this small exponent is not clear and is likely to depend on several factors. For example, the behaviour may be restricted to a limited range of salt concentration, which makes an accurate assessment of power law or other behaviour difficult. Furthermore, brush swelling and collapse are known to depend on the nature of the salt used. While experiments on PMAA showed qualitatively the same behaviour for different salt cations,80 different polycations exhibited different behaviours when the anions were varied.78,81,82 In the polycation studies brush collapse was more pronounced for chaotropic ions.
Although brush conformation as a function of pH is expected to exhibit a monotonic increase in thickness as the polymers become more solvated due to increased charging, experiments show some subtlety in composition-depth profiles obtained by neutron reflectometry. As an example (Fig. 3), neutron reflectometry experiments on PDEAEMA83 and PDMAEMA38 brushes showed unusual composition depth profiles, whereby the brush concentration increased with distance from the substrate. Different explanations have been provided for this, noting that, in the middle of the film, there is increased polymer swelling, i.e., a reduced polymer volume fraction. Consequently, there is a greater polymer density both close to the substrate and towards the solution interface. These regions of less swelling (greater polymer density) neighbour either the substrate or the bulk medium, which have fewer charges and therefore limit chain stretching.84 Alternatively, it could be considered that the brush contained intrinsic entanglements that caused knotting as the brush swells,85,86 rather like trying to comb greasy hair. This would leave a high density of brush near the solution interface if swelling were initiated at the substrate. This explanation was provided for the earlier measurements of brushes created by ATRP from initiator-covered wafers,83 but it is not clear that it can explain the conformation of polymers created from a hydrophobic macroinitiator.38 It has also been proposed that hydrophobic interactions caused kinetic trapping of the brush near the solvent interface.87 Recent experiments on DMAEMA copolymerized with a non-polyelectrolyte, 2-(2-methoxyethoxy)ethyl methacrylate, also show similar concentration profiles,88,89 and the behaviour has even been observed using ellipsometry90 and a quartz crystal microbalance (QCM)91 for PDMAEMA and PAA respectively. There have been other attempts to explain the volume fraction profiles of weak polyelectrolytes, with good comparison being made between self-consistent field theory92 and neutron reflectometry experiments.93 These neutron experiments did not reveal pronounced swelling in the middle of the brush, however. Theoretical work and simulations suggest that solvent entropy is a factor that needs greater consideration.63,94
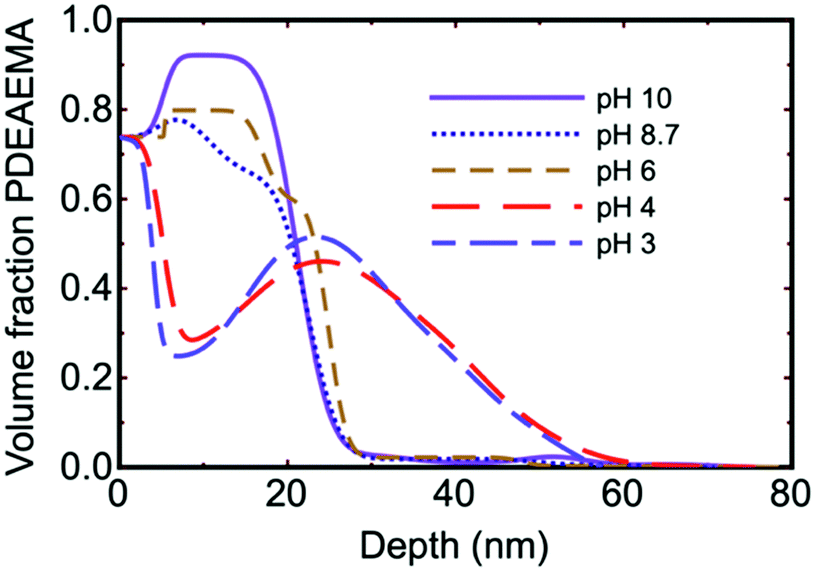 | ||
| Fig. 3 Brush conformation as a function of pH for a PDEAEMA brush as obtained using neutron reflectometry.83 The dry brush is 13 nm thick. The silicon substrate is equivalent to a volume fraction of 0.74. | ||
Other experiments of PDMAEMA concentration profiles have also shown non-monotonic behaviour that may be related to the measurements described above. Certainly, a strong adsorption of the brush at the substrate is noted,95 which is common to other measurements.83,89 (This strong adsorption may be electrostatic as it is present on sapphire surfaces at pH 9, but not at pH 3,96 which straddle the substrate isoelectric point.97) Given that, the conformation of weak polyelectrolyte brushes as a function of pH is not fully understood, a full understanding of the effects of salt is still some way off.
Electric fields
First experiments on PDMAEMA brushes showed that an electric field applied perpendicular to the brush could control its thickness.99 When a positive voltage was applied to the substrate (relative to the aqueous medium above the brush), brushes were swollen. This potential at the substrate repelled the polycations, causing swelling. Subsequent experiments using PDEAEMA, but a different experimental geometry produced similar findings (Fig. 4).98 In this case the data were interpreted as showing that the electric field effectively changed the pH, which is reasonable, although it is to be anticipated that the removal of the electroneutrality requirement in an electric field may cause additional effects such as a conformational phase separation of the brushes,100 a phenomenon that is expected to increase with grafting density.101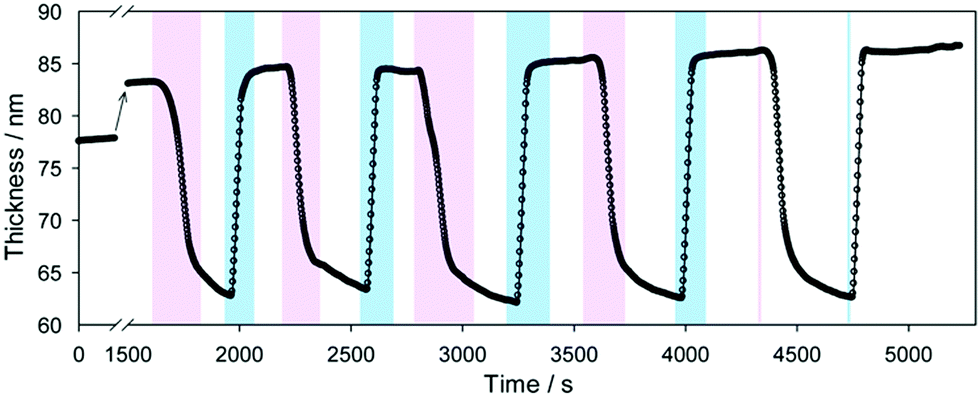 | ||
| Fig. 4 The swelling and collapse of a PDEAEMA brush as an applied voltage across the brush is changed from +5 V (pink) to –5 V (blue). Figure reproduced with permission from the original article.98 Copyright 2013 American Chemical Society. | ||
The behaviour of weak polyelectrolyte brushes in electric fields is sensitive to different parameters, and so comparing different experiments is rather tricky. It is noted, for example, that small changes in solution pH have resulted in a significant difference in the applied potential to bring about conformational change in PAA brushes.102,103 This was interpreted as being due to the distance from the pKa,103 but full studies of the effect of grafting density or chain molar mass are necessary to be confident of any interpretation.
Biological brushes
Biomacromolecules often have complex architectures, but sometimes this complexity hides simple phenomena. For example, the pH-dependent behaviour of Rhodococcus sp. RC291 attached to a colloidal probe on an AFM cantilever has been successfully modelled by considering that the extracellular substances act like a weak polyelectrolyte brush.104Given the insights into biomacromolecules that may arise from synthetic weak polyelectrolytes, it is worth noting that hyaluronic acid brushes, for example, have been created that can be analysed using the expected behaviour for polyelectrolytes,42 but other work has shown that hyaluronic acid on the surface of chondrocytes is much less tractable to simple modelling.105 Nevertheless, hyaluronic acid brushes have been the subject of numerous studies.106–110 It is perhaps worth noting that the salt dependence of initially swollen hyaluronic acid brushes was more like that of strong polyelectrolytes,106 which may simply reflect the low pKa (≈ 3.0) of hyaluronic acid.111 Biomacromolecular weak polyelectrolyte brushes are not restricted to polysaccharides, with one study of a protein brush demonstrating a pH dependence of height commensurate with a weak polyacid.112 Generally, however, proteins exhibit more complicated behaviour due to their polyampholytic nature.113
Contact mechanics
Friction is an energy loss process when two surfaces move relative to each other. Its description is largely attributed to da Vinci, but it was formalized at the end of the seventeenth century, some 200 years later, by Amontons.118 This is the well-known law that the force due to friction is linearly proportional to the load applied. Limitations are shown on the nanoscale where Amontons’ law needs to be supplemented by terms that are dependent upon the contact area.119 Such area-dependent behaviour is readily tested for brushes in contact with a nanoscale asperity such as an AFM tip. Here, the frictional force is the sum of a load dependent term and a shear term. This area dependence is not visible in the load dependent term because the contact area scales with load. The shear term includes, for example, lateral forces and so effectively describes deviations from Amontons’ law.
For weak polyelectrolytes, there is a variation across pH, which provides information about the mechanical properties of the brush. The brush may be analysed in terms of Amontons’ law, or alternatively a formalized contact mechanics approach may be used. If the interaction is soft and elastically deformable, the JKR (Johnson–Kendall–Roberts) approach121 is used, whereas for more rigid or stiff interactions, an approach based on DMT (Derjaguin–Muller–Toporov) theory122 is required. Both models have their origin in Hertzian mechanics123 corrected for adhesion and can be seen as limiting behaviours, which may be unified.124
In intermediate pH solutions, a PDMAEMA brush has a soft elastic contact with AFM tips coated with hydrophobic and hydrophilic monolayers (Fig. 5).120 These experiments were performed using a form of scanning force microscopy known as friction force microscopy.125–127 The extremes of pH are harder to predict, with mercaptoundecanoic acid-coated tips exhibiting elastic behaviour in contact with the brush over 3 ≤ pH ≤ 11, but the PDMAEMA brush presents a stiffer interaction (DMT behaviour) in contact with a dodecanethiol-coated tip at low pH. Furthermore, a gold-coated tip increasingly exhibits DMT behaviour with added salt, the concentration dependence of which is different for different anions.81 At the extremes of pH (1, 2, and 12), these PDMAEMA brushes obey Amontons’ law.
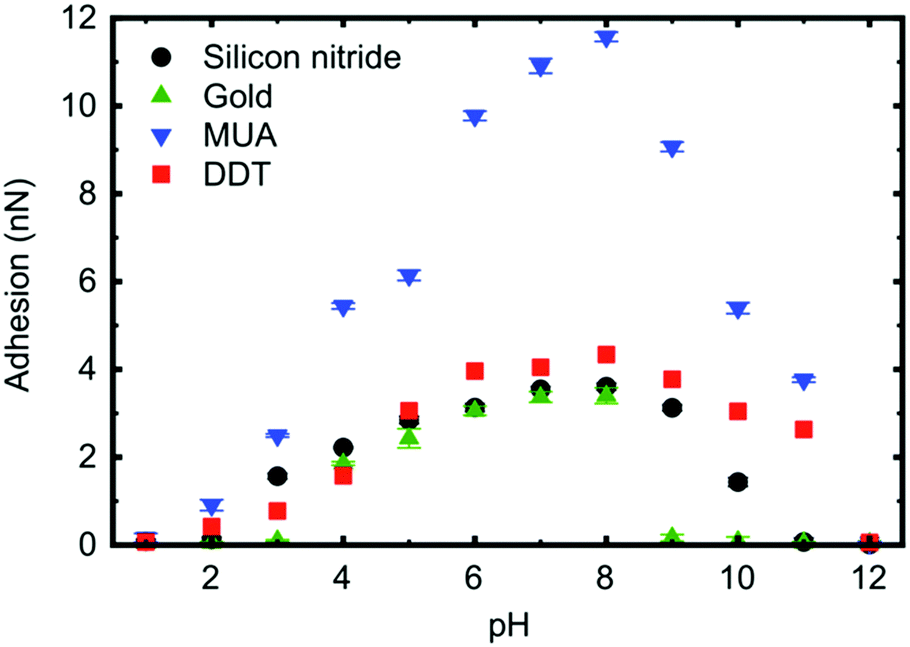 | ||
| Fig. 5 The adhesion of a PDMAEMA brush to a silicon nitride AFM tip as received or coated with mercaptoundecanoic acid (MUA), dodecanethiol (DDT), or gold. This is the maximum pull-off force as the AFM tip retracts from the brush. The figure is taken from the original article,120 and is reproduced under a CC BY 3.0 licence. | ||
Other experiments have shown a linear relationship between force and applied load for gold coated colloidal probes of 10 μm nominal diameter.128 Such discrepancies show the importance of geometry and length scale in measuring friction.
The FFM approach to friction is consistent with an adhesion-based approach where Hertzian (non-adhesive) contact mechanics is equivalent to Amontons’ behaviour once a correction129,130 is made for the multiple asperities that are important for the linear relationship between force and load predicted by Amontons. The shear term of the friction behaviour is mirrored by adhesive terms, which are dependent upon the elasticity of the layer.124
Mechanical properties
Little work has been done to understand the mechanical properties of weak polyelectrolyte brushes partly because of their perceived inaccessibility to experimental investigation and partly because there is little work on the mechanical properties of polymer brushes in general. The mechanical properties of a polymer film are largely dominated by its substrate, and it is difficult to find any application of these brushes whereby the properties of the film are separate from those of the substrate. However, it can often be necessary for the mechanical properties of the film to match the substrate, especially when the substrate itself is soft, e.g., for bioengineering applications.131,132The most important mechanical property that may be measured is an elastic modulus, which is obtained using indentation tests. Measurements using an AFM are possible, but determining the area of contact is challenging, although for non-adhesive contacts and relatively thick films, a methodology is available.133 Measurements on thin films (∼50 nm) of poly[(aminopropyl)siloxane], a weak polyelectrolyte,134 have been performed by interfacial force microscopy (a form of nanoindentation), assuming a non-adhesive contact.135 These measurements have all been performed in dry conditions, and so do not test the responsive (pH-dependent) properties of the brush.
Assessing the mechanical properties of brushes is intrinsically difficult because they are determined by perturbing the film, which is complicated by the film often being a similar thickness to the depth of any indentation. In such cases the substrate contributes to the measured modulus, causing an overestimation of the modulus for substrates harder than the film and underestimating it for relatively soft substrates. This problem was exemplified by work considering the mechanical properties of PAA brushes using a colloidal probe technique.136 Here, without substrate correction, a PAA brush in mildly acidic conditions (pH 4.0) had a greater (Young's) modulus than at neutral pH. With correction, the behaviour was inverted, with the PAA brush modulus increasing from ∼200 Pa at pH 4.0 to ∼1.1 kPa at pH 7.4. Consequently, this highly hydrated brush has much greater incompressibility than that at pH 4, which is supported by earlier QCM measurements.137
Experiments in which charged brushes have a smaller modulus than neutral brushes have been reported for both a polycation and polyanion, where a surface forces apparatus was used.138 Here, similar brushes were brought into contact, and the compression was measured. In this case very large compressibility moduli of up to 40 MPa were obtained, which is a surprising result. Also, that the charged brush had the lower modulus, while again surprising, is not inconsistent with the recognition that elasticity can vary at the extremes of pH, depending on how the system is measured.120,139 It is therefore unwise to declare that the modulus of such nanoscale systems is solely dependent upon pH, because it is also dependent on the experimental geometry of the measurement being performed.
Actuation
The pH-dependent conformational transition of weak polyelectrolytes lends them well to applications as nanoactuators. After all, a chain that can change from being collapsed to swollen can do mechanical work. This is not a new subject and it is not restricted to polyelectrolytes,140 but weak polyelectrolytes are attractive candidate materials for exploiting such behaviour. The response of polyelectrolyte brushes to changes in pH is usually not rapid, but it can be made to occur on the time scale of a few minutes.141An appealing and simple demonstration of the capacity of weak polyelectrolyte brushes to actuate is by placing them in an oscillatory chemical reaction.142 Here a chemical reaction occurs away from equilibrium, where the products of one reaction produce reactants for the reverse process. When this occurs in a redox system, protons are produced and consumed, realizing pH-oscillatory behaviour. This has been demonstrated for PAA brushes by QCM.137 Oscillatory chemical reactions can be put into reverse by a process of mechanotransduction. Here, work is done on the brushes to cause a chemical effect. Some work has been done on strong polyelectrolytes,143–145 but this subject has remained largely dormant in recent years.
By charging or neutralizing a brush-coated surface, it is possible to control electrochemical behaviour at that surface.146 A charged PMAA brush has been shown to attract redox probes,147 which is a route for electron transfer. This feature can be removed by lowering pH.
The actuating possibilities of weak polyelectrolyte brushes were postulated as giving them a significant role as components in a “molecular toolbox for soft nanotechnology”.12 The realization of this vision has (at least thus far) failed and it is worth speculating why. Polyelectrolyte brushes are nanoscale objects and therefore need to be addressed at the nanoscale. They need to be carefully grown using rather routine synthetic techniques, for which experiments at scale have not been performed. The work that a single polymer chain has been shown to produce during a collapse and expansion cycle is of the order of 2 eV,148 which limits potential applications. Nevertheless, it is still possible that early promise of polyelectrolyte brushes for adhesion could still be fulfilled and furthermore, responsive polymers have significant applications in biosensing.149,150
Adhesion
Different categories of adhesives include structural adhesives,151 including, for example, epoxies or cyanoacrylates, for high-load applications and pressure-sensitive adhesives,152,153 where a high density of van der Waals interactions gives sufficient strength for temporary and low-load bonds. Coulombic interactions are not commercially important in adhesives, but charged polymers have two important advantages. They are predominantly water-based and furthermore, they are environmentally controllable.Water-based adhesion of soft materials had historically been rather difficult to quantify,154 but these problems have been addressed in recent years and experiments are now quite tractable.155–157 The first experiments showed that a polycationic brush would adhere to a polyanion (PMAA) hydrogel in water and that, when the pH was reduced, the adhesion would fail.156 Deductive reasoning using contact mechanics experiments with oppositely charged polyelectrolyte brushes suggests that this adhesion was dominated by electrostatic forces.139 These forces are so strong that their failure in water is cohesive, i.e. one of the components fractures rather than the adhesive interface itself.158 However, replacing the PMAA gel with a double-network hydrogel created a robust system that could continuously be attached and detached in water without switching pH.159
Other attempts to use brushes for adhesive applications have used different bonding. The pH-dependent adhesion of PAA with poly(N,N-dimethylacrylamide) (PDMAc) hydrogels was attributed to hydrogen bonding. Electrostatic interactions are readily eliminated since PDMAc is not a polybase even if it often exhibits pH dependent behaviour.160 Reversible adhesion has been shown to also work with strong polyelectrolyte155 and polyzwitterionic161 brushes, but in these cases debonding was achieved by the addition of salt, a method that works equally well for weak polyelectrolyte brushes.162
Adhesion between the PAA brush and the PDMAc hydrogel fails with increasing pH because as the polyanion becomes charged it loses its H-bond donor. This system therefore showed significant adhesion only at low pH (∼2).157 PMAA adheres to PDMAEMA only in the neutral pH region (∼7),139 and its failure at the extremes of pH mean that hydrogen bonding cannot explain the adhesion and so it can be concluded that this system adheres electrostatically. The electrostatic adhesion between PMAA and PDMAEMA156 (and PMAA and PDEAEMA158) is a little greater than the hydrogen bonding between PAA and PDMAc,157 which probably reflects water competing for hydrogen bonding sites in the latter case.
The challenges of making progress with polyelectrolyte adhesion using brushes are significant. The adhesion is likely to be strong and robust, at least where electrostatic bonding dominates but the synthesis of brushes from surfaces is not appealing to the end-user. Pre-synthesized polymers that will self-assemble to form a brush layer is one approach that could be useful,58 but the efficient self-organization of a polyelectrolyte brush is not an easy challenge. The ability to make these adhesive layers detach is appealing because of the opportunities it opens in recycling technologies.163 However, the equilibration time of solutions required to change the pH has been prohibitively large thus far,156,157 although technologies can be improved.159 Replacing the hydrogel with another brush layer could improve equilibration speed, as might patterning the surface to create channels to expedite solvent flow.
Bioadhesion
Polyelectrolytes have possibilities for interacting with biological cells, and their responsive nature allows adhesion and repulsion to be tuned depending on the local environment. As a rule, polyanions such as PAA and PMAA have reasonably good biocompatibility whereas polycations are more likely to be cytotoxic. Nevertheless, both have advantages in biological applications. PMAA is hydrophobic at low pH and can under such conditions attach to a target, with an increase in pH allowing, for example, the release of a cargo.164 Conversely, because bacterial surfaces are typically negatively charged, polycations can readily attach to them. PDMAEMA does have the capacity to damage organisms,165 which may help in biofouling applications, but an important use may well be for the delivery of molecules to a target. PDMAEMA-grafted nanoparticles have been shown to deliver a cargo of indocyanine green (ICG, a molecule used for phototherapy applications) to kill a pathogen, Porphyromonas gingivalis (Fig. 6).166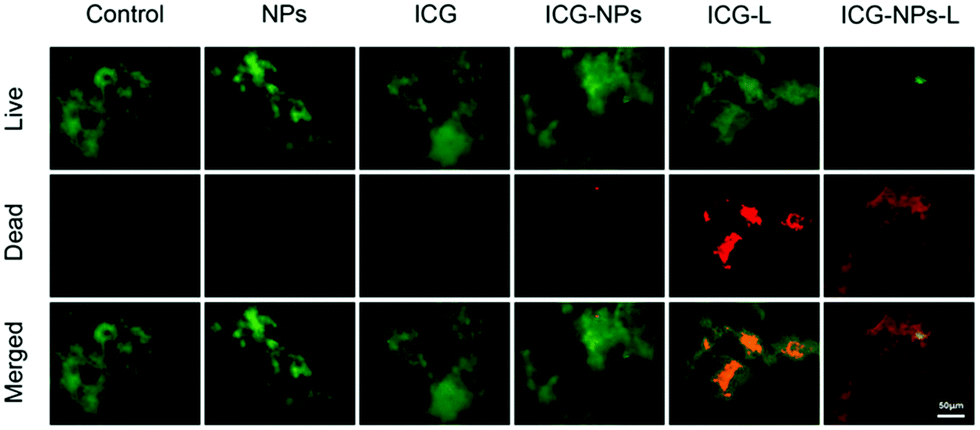 | ||
| Fig. 6 Live (green)–dead (red) staining of P. gingivalis. When PDMAEMA-grafted nanoparticles (NPs) are present there is no significant change from the control experiment. This is also the case in the presence of ICG or ICG-loaded nanoparticles. Cells are killed if ICG is present and laser (L) irradiated. However, the effectiveness of this is at its greatest when the ICG is loaded in the nanoparticles, which attach to the bacteria. The figure is taken from the original article,166 and is reproduced under a CC BY 4.0 licence. | ||
Although charge is a useful means of interacting with organisms, pH can be used with weak polyelectrolytes to turn on and off adhesion. This phenomenon has been used with Staphylococcus epidermis deposited on uncharged PAA brushes.167 When the pH is increased, the charged PAA rejects the bacteria from the surface. The challenge in these experiments is to ensure that sufficient conformational change is possible in environments with high salt concentrations. Currently the use of temperature-responsive polymer brushes (not without their own problems) seems more appropriate for such purposes.168
Bacteria are straightforward organisms for adhesion to polyelectrolyte brushes because they can be treated as negatively charged colloids. Eukaryotic cells are much more complex and the range of interactions that dictate their behaviour in contact with cationic and anionic weak polyelectrolyte brushes has been shown to be difficult to unravel.169 Nevertheless here PAA brushes were shown to be adherent to keratinocyte cells which shows that weak polyelectrolytes may have some utility in their interactions with mammalian cells.
Bottlebrushes
The bottlebrush polymer geometry is a new area in which significant interest is being shown. These are essentially molecular brushes, because chains are grafted from a backbone rather like the comb geometry, although significantly more densely.170,171 They have structural similarities to articular biopolymers such as lubricin172 or aggrecan,173,174 which have impressive lubrication and load-bearing properties. Bottlebrushes are not easy to graft from a surface and experiments are currently dominated by polymers grafted to the surface. Work on charged polymers is largely focussed on polyzwitterions, because of their highly lubricious nature.175,176 Surface-grafted weak polyelectrolyte bottlebrushes are less studied but are likely to have interesting properties that are expected to be developed in the coming years.3 Conclusions and outlook
Over the past decade there has been a significant amount of work developing polyelectrolyte brushes in areas of adhesion and lubrication. It is likely that much of this will continue, but the lack of new developments in actuation has been noticeable, despite early enthusiasm at the turn of the millennium. It is likely that future focus will lie in the interaction of polyelectrolyte brushes with biological materials and the further development of new geometries, such as bottlebrushes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
I am grateful to the reviewers for numerous useful comments and suggestions in the preparation of the manuscript.Notes and references
- S. T. Milner, Science, 1991, 251, 905–914 CrossRef CAS PubMed.
- Y. Higaki, M. Kobayashi, D. Murakami and A. Takahara, Polym. J., 2018, 48, 325–331 CrossRef.
- W. Yang and F. Zhou, Biosurf. Biotribol., 2017, 3, 97–114 CrossRef.
- L. Liu, W. Yao, Y. Rao, X. Lu and J. Gao, Drug Delivery, 2017, 24, 569–581 CrossRef CAS PubMed.
- O. V. Borisov, E. B. Zhulina and T. M. Birshtein, Macromolecules, 1994, 27, 4795–4803 CrossRef CAS.
- R. A. L. Jones and R. W. Richards, Polymers at Surfaces and Interfaces, Cambridge, Cambridge, UK, 1999 Search PubMed.
- M. Biesalski, D. Johannsmann and J. Rühe, J. Chem. Phys., 2002, 117, 4988–4994 CrossRef CAS.
- K. Matyjaszewski, P. J. Miller, N. Shukla, B. Immaraporn, A. Gelman, B. B. Luokala, T. M. Siclovan, G. Kickelbick, T. Vallant, H. Hoffmann and T. Pakula, Macromolecules, 1999, 32, 8716–8724 CrossRef CAS.
- R. R. Bhat, M. R. Tomlinson and J. Genzer, Macromol. Rapid Commun., 2004, 25, 270–274 CrossRef CAS.
- P. D. Topham, J. R. Howse, C. J. Crook, A. J. Parnell, M. Geoghegan, R. A. L. Jones and A. J. Ryan, Polym. Int., 2006, 55, 808–815 CrossRef CAS.
- J. Qiu and K. Matyjaszewski, Acta Polym., 1997, 48, 169–180 CrossRef CAS.
- A. J. Ryan, C. J. Crook, J. R. Howse, P. Topham, R. A. L. Jones, M. Geoghegan, A. J. Parnell, L. Ruiz-Pérez, S. J. Martin, A. Cadby, A. Menelle, J. R. P. Webster, A. J. Gleeson and W. Bras, Faraday Discuss., 2005, 128, 55–74 RSC.
- P. Chapman, R. E. Ducker, C. R. Hurley, J. K. Hobbs and G. J. Leggett, Langmuir, 2015, 31, 5935–5944 CrossRef CAS PubMed.
- A. Y. Sankhe, S. M. Husson and S. M. Kilbey II, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 566–575 CrossRef CAS.
- H. Dong and K. Matyjaszewski, Macromolecules, 2008, 41, 6868–6870 CrossRef CAS.
- D. Konkolewicz, A. J. D. Magenau, S. E. Averick, A. Simakova, H. He and K. Matyjaszewski, Macromolecules, 2012, 45, 4461–4468 CrossRef CAS.
- E. H. Discekici, C. W. Pester, N. J. Treat, J. Lawrence, K. M. Mattson, B. Narupai, E. P. Toumayan, Y. Luo, A. J. McGrath, P. G. Clark, J. Read de Alaniz and C. J. Hawker, ACS Macro Lett., 2016, 5, 258–262 CrossRef CAS.
- A. D. Jenkins, R. G. Jones and G. Moad, Pure Appl. Chem., 2010, 82, 483–491 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed.
- M. Husseman, E. E. Malmström, M. McNamara, M. Mate, D. Mecerreyes, D. G. Benoit, J. L. Hedrick, P. Mansky, E. Huang, T. P. Russell and C. J. Hawker, Macromolecules, 1999, 32, 1424–1431 CrossRef CAS.
- O. Garcia-Valdez, T. Brescacin, J. Arredondo, J. Bouchard, P. G. Jessop, P. Champagne and M. F. Cunningham, Polym. Chem., 2017, 8, 4124–4131 RSC.
- J. Glasing, J. Bouchard, P. G. Jessop, P. Champagne and M. F. Cunningham, Polym. Chem., 2017, 8, 6000–6012 RSC.
- L. Sonnenberg, J. Parvole, O. Borisov, L. Billon, H. E. Gaub and M. Seitz, Macromolecules, 2006, 39, 281–288 CrossRef CAS.
- G. Moad and E. Rizzardo, in Nitroxide Mediated Polymerization: From Fundamentals to Applications in Materials Science, ed. D. Gigmes, Royal Society of Chemistry, Cambridge, 2016, vol. 19, pp. 1–44 Search PubMed.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- F. Chen, Y. Zhu, W. Li, J. Yang, P. Fan, Z. Fei, M. Zhong, L. Chang and T. Kuang, Appl. Surf. Sci., 2019, 464, 273–279 CrossRef CAS.
- B. Wang, Z. Ye, Y. Tang, Y. Han, Q. Lin, H. Liu, H. Chen and K. Nan, Int. J. Nanomed., 2017, 12, 111–125 CrossRef CAS PubMed.
- M. K. Cho, H. J. Seo, J. H. Lee, W. K. Cho and K. Son, Polym. Chem., 2021, 12, 7023–7030 RSC.
- J. H. Lee, H. J. Seo, S. Y. Lee, W. K. Cho and K. Son, Macromol. Chem. Phys., 2021, 222, 2100270 CrossRef CAS.
- D. Li, L. Xu, J. Wang and J. E. Gautrot, Adv. Healthcare Mater., 2021, 10, 2000953 CrossRef CAS PubMed.
- M. Li and C. W. Pester, Polymers, 2020, 12, 1553 CrossRef CAS PubMed.
- A. Bratek-Skicki, P. Eloy, M. Morga and C. Dupont-Gillain, Langmuir, 2018, 34, 3037–3048 CrossRef CAS PubMed.
- N. Houbenov, S. Minko and M. Stamm, Macromolecules, 2003, 36, 5897–5901 CrossRef CAS.
- T. L. Fox, S. Tang, J. M. Horton, H. A. Holdaway, B. Zhao, L. Zhu and P. L. Stewart, Langmuir, 2015, 31, 8680–8688 CrossRef CAS PubMed.
- D. Li, X. Sheng and B. Zhao, J. Am. Chem. Soc., 2005, 127, 6248–6256 CrossRef CAS PubMed.
- A. Johnson, J. Madsen, P. Chapman, A. Alswieleh, O. Al-Jaf, P. Bao, C. R. Hurley, M. L. Cartron, S. D. Evans, J. K. Hobbs, C. N. Hunter, S. P. Armes and G. J. Leggett, Chem. Sci., 2017, 8, 4517–4526 RSC.
- C. Liu, F. Cheng, B. Liu, D. Gao, G. Cheng, C. Li, H. Wang and W. He, Langmuir, 2022, 38, 1001–1010 CrossRef PubMed.
- M. R. Tomlinson, F. Cousin and M. Geoghegan, Polymer, 2009, 50, 4829–4836 CrossRef CAS.
- L. Xu, Z. Zhu, O. V. Borisov, E. B. Zhulina and S. A. Sukhishvili, Phys. Rev. Lett., 2009, 103, 118301 CrossRef PubMed.
- J. Cai, C. Li, N. Kong, Y. Lu, G. Lin, X. Wang, Y. Yao, I. Manners and H. Qiu, Science, 2019, 366, 1095–1098 CrossRef CAS PubMed.
- W. Xu, Y. Zheng and P. Pan, J. Polym. Sci., 2022 DOI:10.1002/pol.20210789.
- J. L. Faubel, R. P. Patel, W. Wei, J. E. Curtis and B. K. Brettmann, ACS Macro Lett., 2019, 8, 1323–1327 CrossRef CAS.
- L. Michalek, L. Barner and C. Barner-Kowollik, Adv. Mater., 2018, 30, 1706321 CrossRef.
- S. Turgman-Cohen and J. Genzer, J. Am. Chem. Soc., 2011, 133, 17567–17569 CrossRef CAS PubMed.
- C. Kang, R. Crockett and N. D. Spencer, Polym. Chem., 2016, 7, 302–309 RSC.
- R. R. Patil, S. Turgman-Cohen, J. Šrogl, D. Kiserow and J. Genzer, ACS Macro Lett., 2015, 4, 251–254 CrossRef CAS.
- D. Goodman, J. N. Kizhakkedathu and D. E. Brooks, Langmuir, 2004, 20, 6238–6245 CrossRef CAS PubMed.
- A. AL-Baradi, M. R. Tomlinson, Z. J. Zhang and M. Geoghegan, Polymer, 2015, 67, 111–117 CrossRef CAS.
- J. D. Willott, T. J. Murdoch, G. B. Webber and E. J. Wanless, Macromolecules, 2016, 49, 2327–2338 CrossRef CAS.
- B. Lego, M. François, W. G. Skene and S. Giasson, Langmuir, 2009, 25, 5313–5321 CrossRef CAS PubMed.
- S. Alang Ahmad, G. J. Leggett, A. Hucknall and A. Chilkoti, Biointerphases, 2011, 6, 8–15 CrossRef PubMed.
- C. Schuh, S. Santer, O. Prucker and J. Rühe, Adv. Mater., 2009, 21, 4706–4710 CrossRef CAS.
- M. Steenackers, R. Jordan, A. Küller and M. Grunze, Adv. Mater., 2009, 21, 2921–2925 CrossRef CAS.
- Z. J. Zhang, M. Moxey, A. Alswieleh, S. P. Armes, A. L. Lewis, M. Geoghegan and G. J. Leggett, Langmuir, 2017, 33, 706–713 CrossRef CAS PubMed.
- S. Christau, T. Möller, Z. Yenice, J. Genzer and R. von Klitzing, Langmuir, 2014, 30, 13033–13041 CrossRef CAS PubMed.
- C. Devaux, J. P. Chapel, E. Beyou and P. Chaumont, Eur. Phys. J. E: Soft Matter Biol. Phys., 2002, 7, 345–352 CrossRef CAS PubMed.
- M. Ejaz, S. Yamamoto, K. Ohno, Y. Tsujii and T. Fukuda, Macromolecules, 1998, 31, 5934–5936 CrossRef CAS.
- W. D. Seddon, L. Alfhaid, A. D. F. Dunbar, M. Geoghegan and N. H. Williams, Langmuir, 2020, 36, 13843–13852 CrossRef CAS PubMed.
- P.-G. de Gennes, Scaling Concepts in Polymer Physics, Cornell University Press, Ithaca, 1979 Search PubMed.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, Ithaca, 1953 Search PubMed.
- S. Alexander, J. Phys., 1977, 38, 983–987 CrossRef CAS.
- P. G. de Gennes, Macromolecules, 1980, 13, 1069–1075 CrossRef CAS.
- J. D. Willott, T. J. Murdoch, G. B. Webber and E. J. Wanless, Prog. Polym. Sci., 2017, 64, 52–75 CrossRef CAS.
- S. W. An, P. N. Thirtle, R. K. Thomas, F. L. Baines, N. C. Billingham, S. P. Armes and J. Penfold, Macromolecules, 1999, 32, 2731–2738 CrossRef CAS.
- J. Zhang, H. Cai, L. Tang and G. Liu, Langmuir, 2018, 34, 12419–12427 CrossRef CAS PubMed.
- E. B. Zhulina and O. V. Borisov, J. Chem. Phys., 1997, 107, 5952–5967 CrossRef CAS.
- D. H. Everett, Pure Appl. Chem., 1972, 31, 577–638 CrossRef.
- E. B. Zhulina, T. M. Birshtein and O. V. Borisov, Macromolecules, 1995, 28, 1491–1499 CrossRef CAS.
- E. B. Zhulina and O. V. Borisov, Langmuir, 2011, 27, 10615–10633 CrossRef CAS PubMed.
- N. A. Kumar and C. Seidel, Macromolecules, 2005, 38, 9341–9350 CrossRef CAS.
- P. Pincus, Macromolecules, 1991, 24, 2912–2919 CrossRef CAS.
- D. Bendejacq, V. Ponsinet and M. Joanicot, Eur. Phys. J. E: Soft Matter Biol. Phys., 2004, 13, 3–13 CrossRef CAS PubMed.
- E. P. K. Currie, A. B. Sieval, G. J. Fleer and M. A. Cohen Stuart, Langmuir, 2000, 16, 8324–8333 CrossRef CAS.
- G. Ferrand-Drake del Castillo, R. L. N. Hailes and A. B. Dahlin, J. Phys. Chem. Lett., 2020, 11, 5212–5218 CrossRef CAS PubMed.
- A. J. Parnell, S. J. Martin, C. C. Dang, M. Geoghegan, R. A. L. Jones, C. J. Crook, J. R. Howse and A. J. Ryan, Polymer, 2009, 50, 1005–1014 CrossRef CAS.
- T. Wu, P. Gong, I. Szleifer, P. Vlček, V. Šubr and J. Genzer, Macromolecules, 2007, 40, 8756–8764 CrossRef CAS.
- X. Guo and M. Ballauff, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2001, 64, 051406 CrossRef CAS PubMed.
- J. D. Willott, B. A. Humphreys, G. B. Webber, E. J. Wanless and W. M. de Vos, Langmuir, 2019, 35, 2709–2718 CrossRef CAS PubMed.
- J. D. Willott, T. J. Murdoch, B. A. Humphreys, S. Edmondson, G. B. Webber and E. J. Wanless, Langmuir, 2014, 30, 1827–1836 CrossRef CAS PubMed.
- H. Zhang and J. Rühe, Macromolecules, 2005, 38, 4855–4860 CrossRef CAS.
- M. Raftari, Z. J. Zhang, S. R. Carter, G. J. Leggett and M. Geoghegan, Tribol. Lett., 2018, 66, 11 CrossRef PubMed.
- J. D. Willott, T. J. Murdoch, B. A. Humphreys, S. Edmondson, E. J. Wanless and G. B. Webber, Langmuir, 2015, 31, 3707–3717 CrossRef CAS PubMed.
- M. Geoghegan, L. Ruiz-Pérez, C. C. Dang, A. J. Parnell, S. J. Martin, J. R. Howse, R. A. L. Jones, R. Golestanian, P. D. Topham, C. J. Crook, A. J. Ryan, D. S. Sivia, J. R. P. Webster and A. Menelle, Soft Matter, 2006, 2, 1076–1080 RSC.
- F. von Goeler and M. Muthukumar, Macromolecules, 1995, 28, 6608–6617 CrossRef CAS.
- L. Ruiz-Pérez, PhD thesis, University of Sheffield, 2006.
- M. P. Weir and A. J. Parnell, Polymers, 2011, 3, 2107–2132 CrossRef CAS.
- R. Toomey and M. Tirrell, Annu. Rev. Phys. Chem., 2008, 59, 493–517 CrossRef CAS PubMed.
- E. C. Johnson, I. J. Gresham, S. W. Prescott, A. Nelson, E. J. Wanless and G. B. Webber, Polymer, 2021, 214, 123287 CrossRef CAS.
- E. C. Johnson, J. D. Willott, I. J. Gresham, T. J. Murdoch, B. A. Humphreys, S. W. Prescott, A. Nelson, W. M. de Vos, G. B. Webber and E. J. Wanless, Langmuir, 2020, 36, 12460–12472 CrossRef CAS PubMed.
- S. Rauch, P. Uhlmann and K.-J. Eichhorn, Anal. Bioanal. Chem., 2013, 405, 9061–9069 CrossRef CAS PubMed.
- K. Ehtiati, S. Z. Moghaddam, A. E. Daugaard and E. Thormann, Langmuir, 2020, 36, 2339–2348 CrossRef CAS PubMed.
- J. P. Mahalik, Y. Yang, C. Deodhar, J. F. Ankner, B. S. Lokitz, S. M. Kilbey, B. G. Sumpter and R. Kumar, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 956–964 CrossRef CAS.
- M. Moglianetti, J. R. P. Webster, S. Edmondson, S. P. Armes and S. Titmuss, Langmuir, 2010, 26, 12684–12689 CrossRef CAS PubMed.
- R. J. Nap, M. Tagliazucchi and I. Szleifer, J. Chem. Phys., 2014, 140, 024910 CrossRef CAS PubMed.
- H. Jia, A. Wildes and S. Titmuss, Macromolecules, 2012, 45, 305–312 CrossRef CAS.
- M. Moglianetti, J. R. P. Webster, S. Edmondson, S. P. Armes and S. Titmuss, Langmuir, 2011, 27, 4489–4496 CrossRef CAS PubMed.
- N. Li, R. K. Thomas and A. R. Rennie, J. Colloid Interface Sci., 2012, 378, 152–158 CrossRef CAS PubMed.
- G. J. Dunderdale and J. P. A. Fairclough, Langmuir, 2013, 29, 3628–3635 CrossRef CAS PubMed.
- M. P. Weir, S. Y. Heriot, S. J. Martin, A. J. Parnell, S. A. Holt, J. R. P. Webster and R. A. L. Jones, Langmuir, 2011, 27, 11000–11007 CrossRef CAS PubMed.
- B. M. Okrugin, R. P. Richter, F. A. M. Leermakers, I. M. Neelov, E. B. Zhulina and O. V. Borisov, Polymers, 2020, 12, 898 CrossRef CAS PubMed.
- Y.-F. Ho, T. N. Shendruk, G. W. Slater and P.-Y. Hsiao, Langmuir, 2013, 29, 2359–2370 CrossRef CAS PubMed.
- O. V. Borisova, L. Billon, R. P. Richter, E. Reimhult and O. V. Borisov, Langmuir, 2015, 31, 7684–7694 CrossRef CAS PubMed.
- V. Sénéchal, H. Saadaoui, J. Rodriguez-Hernandez and C. Drummond, Langmuir, 2017, 33, 4996–5005 CrossRef PubMed.
- Y. Pen, Z. J. Zhang, A. L. Morales-García, M. Mears, D. S. Tarmey, R. G. Edyvean, S. A. Banwart and M. Geoghegan, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 518–526 CrossRef CAS PubMed.
- H. Boehm, T. A. Mundinger, C. H. J. Boehm, V. Hagel, U. Rauch, J. P. Spatz and J. E. Curtis, Soft Matter, 2009, 5, 4331–4337 RSC.
- S. Attili, O. V. Borisov and R. P. Richter, Biomacromolecules, 2012, 13, 1466–1477 CrossRef CAS PubMed.
- S. Attili and R. P. Richter, Langmuir, 2012, 28, 3206–3216 CrossRef CAS PubMed.
- J. L. Faubel, W. Wei and J. E. Curtis, ACS Nano, 2021, 15, 4268–4276 CrossRef CAS PubMed.
- R. P. Richter, K. K. Hock, J. Burkhartsmeyer, H. Boehm, P. Bingen, G. Wang, N. F. Steinmetz, D. J. Evans and J. P. Spatz, J. Am. Chem. Soc., 2007, 129, 5306–5307 CrossRef CAS PubMed.
- J. Seror, L. Zhu, R. Goldberg, A. J. Day and J. Klein, Nat. Commun., 2015, 6, 6497 CrossRef CAS PubMed.
- R. L. Cleland, J. L. Wang and D. M. Detweiler, Macromolecules, 1982, 15, 386–395 CrossRef CAS.
- N. Srinivasan, M. Bhagawati, B. Ananthanarayanan and S. Kumar, Nat. Commun., 2014, 5, 5145 CrossRef CAS PubMed.
- A.-S. Yang and B. Honig, J. Mol. Biol., 1993, 231, 459–474 CrossRef CAS PubMed.
- U. Raviv, S. Glasson, N. Kampf, J.-F. Gohy, R. Jérôme and J. Klein, Nature, 2003, 425, 163–165 CrossRef CAS PubMed.
- M. Chen, W. H. Briscoe, S. P. Armes, H. Cohen and J. Klein, Eur. Polym. J., 2011, 47, 511–523 CrossRef CAS.
- M. Chen, W. H. Briscoe, S. P. Armes and J. Klein, Science, 2009, 323, 1698–1701 CrossRef CAS PubMed.
- J. Yu, N. E. Jackson, X. Xu, Y. Morgenstern, Y. Kaufman, M. Ruths, J. J. de Pablo and M. Tirrell, Science, 2018, 360, 1434–1438 CrossRef CAS PubMed.
- G. Amontons, Hist. Acad. R. Sci., 1699, 206–227 Search PubMed.
- B. Weber, T. Suhina, T. Junge, L. Pastewka, A. M. Brouwer and D. Bonn, Nat. Commun., 2018, 9, 888 CrossRef CAS PubMed.
- M. Raftari, Z. Zhang, S. R. Carter, G. J. Leggett and M. Geoghegan, Soft Matter, 2014, 10, 2759–2766 RSC.
- K. L. Johnson, K. Kendall and A. D. Roberts, Proc. R. Soc. London, Ser. A, 1971, 324, 301–313 CAS.
- B. V. Derjaguin, V. M. Muller and Y. P. Toporov, J. Colloid Interface Sci., 1975, 53, 314–326 CrossRef CAS.
- H. Hertz, J. Reine Angew. Math., 1881, 92, 156–171 Search PubMed.
- R. W. Carpick, D. F. Ogletree and M. Salmeron, J. Colloid Interface Sci., 1999, 211, 395–400 CrossRef CAS PubMed.
- R. W. Carpick and M. Salmeron, Chem. Rev., 1997, 97, 1163–1194 CrossRef CAS PubMed.
- G. J. Leggett, N. J. Brewer and K. S. L. Chong, Phys. Chem. Chem. Phys., 2005, 7, 1107–1120 RSC.
- E. Liamas, S. D. Connell, S. N. Ramakrishna and A. Sarkar, Nanoscale, 2020, 12, 2292–2308 RSC.
- N. Nordgren and M. W. Rutland, Nano Lett., 2009, 9, 2984–2990 CrossRef CAS PubMed.
- J. F. Archard, Proc. R. Soc. London, Ser. A, 1957, 243, 190–205 Search PubMed.
- M. Ciavarella, J. Joe, A. Papangelo and J. R. Barber, J. R. Soc., Interface, 2019, 16, 20180738 CrossRef CAS PubMed.
- F. J. O'Brien, Mater. Today, 2011, 14, 88–95 CrossRef.
- L. A. Osório, E. Silva and R. E. Mackay, Bioengineering, 2021, 8, 113 CrossRef PubMed.
- J. Domke and M. Radmacher, Langmuir, 1998, 14, 3320–3325 CrossRef CAS.
- M. Etienne and A. Walcarius, Talanta, 2003, 59, 1173–1188 CrossRef CAS PubMed.
- H. Cabibil, H. Celio, J. Lozano, J. M. White and R. Winter, Langmuir, 2001, 2160–2166 CrossRef CAS.
- P. C. Nalam, H.-S. Lee, N. Bhatt, R. W. Carpick, D. M. Eckmann and R. J. Composto, ACS Appl. Mater. Interfaces, 2017, 9, 12936–12948 CrossRef CAS PubMed.
- G. Liu and G. Zhang, J. Phys. Chem. B, 2008, 112, 10137–10141 CrossRef CAS PubMed.
- K. Kurihara, Adv. Colloid Interface Sci., 2010, 158, 130–138 CrossRef CAS PubMed.
- M. Raftari, Z. J. Zhang, S. R. Carter, G. J. Leggett and M. Geoghegan, Macromolecules, 2015, 48, 6272–6279 CrossRef CAS.
- S. A. Prokhorova, A. Kopyshev, A. Ramakrishnan, H. Zhang and J. Rühe, Nanotechnology, 2003, 14, 1098–1108 CrossRef CAS.
- G. Ferrand-Drake del Castillo, G. Emilsson and A. Dahlin, J. Phys. Chem. C, 2018, 122, 27516–27527 CrossRef CAS.
- A. Isakova and K. Novakovic, Eur. Polym. J., 2017, 95, 430–439 CrossRef CAS.
- O. Azzaroni, B. Trappmann, P. van Rijn, F. Zhou, B. Kong and W. T. S. Huck, Angew. Chem., Int. Ed., 2006, 45, 7440–7443 CrossRef CAS PubMed.
- J. Bünsow, J. Erath, P. M. Biesheuvel, A. Fery and W. T. S. Huck, Angew. Chem., Int. Ed., 2011, 50, 9629–9632 CrossRef PubMed.
- J. Bünsow, T. S. Kelby and W. T. S. Huck, Acc. Chem. Res., 2010, 43, 466–474 CrossRef PubMed.
- J. Anthi, V. Kolivoška, B. Holubová and H. Vaisocherová-Lísalová, Biomater. Sci., 2021, 9, 7379–7391 RSC.
- G. Panzarasa, M. Dübner, V. Pifferi, G. Soliveri and C. Padeste, J. Mater. Chem. C, 2016, 4, 6287–6294 RSC.
- S. Zou, M. A. Hempenius, H. Schönherr and G. J. Vancso, Macromol. Rapid Commun., 2005, 27, 103–108 CrossRef.
- L. Hu, Q. Zhang, X. Li and M. J. Serpe, Mater. Horiz., 2020, 6, 1774–1793 RSC.
- G. Kocak, C. Tuncer and V. Bütün, Polym. Chem., 2017, 8, 144–176 RSC.
- R. A. Pethrick, Proc. Inst. Mech. Eng., Part L, 2015, 229, 349–379 CAS.
- C. Creton, MRS Bull., 2003, 28, 434–439 CrossRef CAS.
- S. Sun, M. Li and A. Liu, Int. J. Adhes. Adhes., 2013, 41, 98–106 CrossRef CAS.
- C. Loskofsky, F. Song and B. Zhang Newby, J. Adhes., 2006, 82, 713–730 CrossRef CAS.
- M. Kobayashi, M. Terada and A. Takahara, Soft Matter, 2011, 7, 5717–5722 RSC.
- R. La Spina, M. R. Tomlinson, L. Ruiz-Pérez, A. Chiche, S. Langridge and M. Geoghegan, Angew. Chem., Int. Ed., 2007, 46, 6460–6463 CrossRef CAS PubMed.
- G. Sudre, L. Olanier, Y. Tran, D. Hourdet and C. Creton, Soft Matter, 2012, 8, 8184–8193 RSC.
- L. Alfhaid, R. La Spina, M. R. Tomlinson, A. R. Hall, W. D. Seddon, N. H. Williams, F. Cousin, S. Gorb and M. Geoghegan, J. Adhes., 2018, 94, 58–76 CrossRef CAS.
- L. Alfhaid, W. D. Seddon, N. H. Williams and M. Geoghegan, Soft Matter, 2016, 12, 5022–5028 RSC.
- Z. Zhang, M. R. Tomlinson, R. Golestanian and M. Geoghegan, Nanotechnology, 2008, 19, 035505 CrossRef CAS PubMed.
- M. Kobayashi and A. Takahara, Polym. Chem., 2013, 4, 4987–4992 RSC.
- L. Alfhaid, N. H. Williams and M. Geoghegan, J. Appl. Polym. Sci., 2020, 137, 49130 CrossRef CAS.
- J. M. Garcia and M. L. Robertson, Science, 2017, 358, 870–872 CrossRef CAS PubMed.
- E. Koh and Y. Taek Lee, Appl. Surf. Sci., 2021, 562, 150133 CrossRef CAS.
- W. Yandi, S. Mieszkin, A. di Fino, P. Martin-Tanchereau, M. E. Callow, J. A. Callow, L. Tyson, A. S. Clare and T. Ederth, Biofouling, 2016, 32, 609–625 CrossRef CAS PubMed.
- E. Shi, L. Bai, L. Mao, H. Wang, X. Yang, Y. Wang, M. Zhang, C. Li and Y. Wang, J. Nanobiotechnol., 2021, 19, 413 CrossRef PubMed.
- V. Yadav, Y. A. Jaimes-Lizcano, N. K. Dewangan, N. Park, T.-H. Li, M. L. Robertson and J. C. Conrad, ACS Appl. Mater. Interfaces, 2017, 9, 44900–44910 CrossRef CAS PubMed.
- J. Collett, A. Crawford, P. V. Hatton, M. Geoghegan and S. Rimmer, J. R. Soc., Interface, 2007, 4, 117–126 CrossRef PubMed.
- E. J. Cozens, D. Kong, N. Roohpour and J. E. Gautrot, Soft Matter, 2020, 16, 505–522 RSC.
- R. Verduzco, X. Li, S. L. Pesek and G. E. Stein, Chem. Soc. Rev., 2015, 44, 2405–2420 RSC.
- Y. Xu, F. Plamper, M. Ballauff and A. H. E. Müller, Adv. Polym. Sci., 2010, 228, 1–38 CAS.
- G. D. Jay and K. A. Waller, Matrix Biol., 2014, 39, 17–24 CrossRef CAS PubMed.
- C. Kiani, L. Chen, Y. J. Wu, A. J. Yee and B. B. Yang, Cell Res., 2002, 12, 19–32 CrossRef PubMed.
- L. Ng, A. J. Grodzinsky, P. Patwari, J. Sandy, A. Plaas and C. Ortiz, J. Struct. Biol., 2003, 143, 242–257 CrossRef CAS PubMed.
- X. Banquy, J. Burdyńska, D. W. Lee, K. Matyjaszewski and J. Israelachvili, J. Am. Chem. Soc., 2014, 136, 6199–8202 CrossRef CAS PubMed.
- J. Faivre, B. R. Shrestha, G. Xie, M. Olszewski, V. Adibnia, F. Moldovan, A. Montembault, G. Sudre, T. Delair, L. David, K. Matyjaszewski and X. Banquy, Chem. Mater., 2018, 30, 4140–4149 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |