
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Suppression of self-stratification in colloidal mixtures with high Péclet numbers†
M.
Schulz
a,
R.
Brinkhuis
b,
C.
Crean
 c,
R. P.
Sear
a and
J. L.
Keddie
*a
c,
R. P.
Sear
a and
J. L.
Keddie
*a
aDepartment of Physics, University of Surrey, Guildford, Surrey, GU2 7XH, UK. E-mail: j.keddie@surrey.ac.uk
bAllnex, Nieuwe Kanaal 7N, 6709 PA Wageningen, The Netherlands
cDepartment of Chemistry, University of Surrey, Guildford, Surrey, GU2 7XH, UK
First published on 16th March 2022
Abstract
The non-equilibrium assembly of bimodal colloids during evaporative processes is an attractive means to achieve gradient or stratified layers in thick films. Here, we show that the stratification of small colloids on top of large is prevented when the viscosity of the continuous aqueous phase is too high. We propose a model where a too narrow width of the gradient in concentration of small colloids suppresses the stratification.
Multi-layered films and coatings have numerous applications, ranging from pharmaceuticals1,2 to photovoltaic devices3 and automobiles.4 Stratified microstructures with designed surfaces5 are known to tailor coating properties, including the abrasion resistance,6,7 wetting,8,9 antifouling,10 corrosion resistance,11 and even flame retardancy.12 Self-stratifying coatings13 offer greater sustainability through efficient use of resources: materials, time and energy.14 In certain circumstances, binary mixtures of large and small colloids,15–17 or colloids with soluble polymers in a common solvent,18–20 stratify during drying such that a layer of the smaller species spontaneously assembles at the top of a film, called the “small-on-top” structure.21 One explanation (but not the only one) for the phenomenon is a diffusiophoresis mechanism22 in which the larger particles diffuse downward in a concentration gradient of the smaller species that develops during drying.
Experiments16,23–25 and simulations26,27 have both shown that the stratification of small particles above larger particles occurs when the volume fraction of small particles, ϕS, is sufficiently large, and the Péclet number, Pe, for the process is sufficiently high. Pe depends on the velocity, vev, at which an evaporating solvent surface moves downward, the collective diffusion coefficient of a particle, D, at the initial time, and the initial film thickness, H. It is given by Pe = Hvev/D, and is large when evaporation is faster than diffusion.
Most of the reports in the literature have investigated colloidal binary systems of large and small particles. However, there have been some experiments6 and simulations28 considering stratification in ternary systems consisting of colloids of three different sizes and also experiments on polydisperse systems.29 Elsewhere, mixtures of colloids with polymers in dilute solutions have been investigated in experiments,19 simulations,18,20 and theory30 with a Pe defined for the coiled chains.
Models developed by Zhou et al.,31 Sear and Warren,30 Sear,32 and Rees-Zimmerman and Routh33 make predictions for the parameter values for which stratification is observed. Rees-Zimmerman and Routh and Zhou et al. both used models in which the large and small colloids each obey diffusion-like equations. In contrast, Sear-Warren and Sear use models that assume a large size difference between the large and small colloids. They then compute the forces arising from the concentration gradient along the surface of the large colloid and hence find an induced motion of the large colloid due to the gradient in concentration of the small. Experiments have generally found good agreement with these models for colloid (and polymer/colloid) mixtures.21,34 However, in experiments on model systems of colloids dispersed in water or organic solvents, values of Pe > 100 are not typically obtained, and this regime has been largely unexplored.
Stratification with higher values of Pe were investigated using Langevin dynamic simulations, which suggested that there is an optimum value of Pe to obtain the maximum stratification.35 However, the optimum value was not found, and there were no experimental investigations. Tang et al.36 found in large scale molecular dynamics simulations that the amount of “small-on-top” stratification increased monotonically with Pe. The stratification was most enhanced at an intermediate value of Pe ≈ 300 for their system. They separately explored the effects of strong temperature gradients achieved by raising the temperature of the substrate and accounting for the effects of evaporative cooling.37 With very high evaporation rates (greater than are achievable in experiments), they found “large-on-top” stratification.
Here, we experimentally explore stratification in the high-Pe regime, seeing if there is an upper limit to the effect. We do not use excessively high evaporation rates or temperatures, but we slow the diffusivity of the colloids by raising the viscosity of the continuous aqueous phase. When used in waterborne coatings, colloidal dispersions contain additional components, such as thickeners, pigments and wetting agents.38 We studied a mixture of two colloids with a water-soluble polymer that is used as a non-associative thickener, as is illustrated in Fig. 1. As water evaporates and the polymer becomes more concentrated, Pe increases further. We propose a physical model to explain the experimental observations.
 | ||
| Fig. 1 Schematic diagram of the initial film system (thickness of H) consisting of three components in water. Large (acrylic) and small (polyurethane) colloids are represented by the purple and orange circles, respectively. A water-soluble polymer (thickener) is represented by the white coils. | ||
In our experiments, we mixed acrylic copolymer particles (mean diameter of 160 nm) with polyurethane particles (28 nm) in water (see Table S1 (ESI†) and a description of the materials in the ESI†). In all cases, the volume fraction of the small polyurethane particles, ϕS, in the initial mixture was 0.1. To achieve a range of Pe values for the smaller colloid, Pes, varying over four orders of magnitude (from 4 to 4 × 104), we raised the viscosity of the aqueous phase by adding a water-soluble, non-associative thickener at two volume fractions of ϕT = 0.01 and ϕT = 0.02 (based on the total volume of the dispersion). We varied the evaporation rate by increasing the temperature or by flowing air across the film surface, and adjusted the film thickness (Table S2, ESI†). For each sample, we used Raman depth profiling (described in ESI† and Fig. S1) to determine the extent to which the surface was enriched with the small polyurethane particles. We then classified the films as being stratified (having greater than a 15% excess concentration of the small particle than expected in a random mixture), intermediate (an excess of 12 to 15%), or non-stratified.
When no thickener is added, there is a value of Pes above which stratification is observed (see Fig. 2a). The transition point is very similar to what is found from a model by Sear32 (as is shown by the dotted line). This model assumes that stratification will occur when Pes is above a threshold value: PeS > ϕjam/ϕS, where ϕjam is the jamming volume fraction for spherical colloids. Because of the effects of jamming, ϕS must typically be <0.2 for stratification to occur, according to the model.
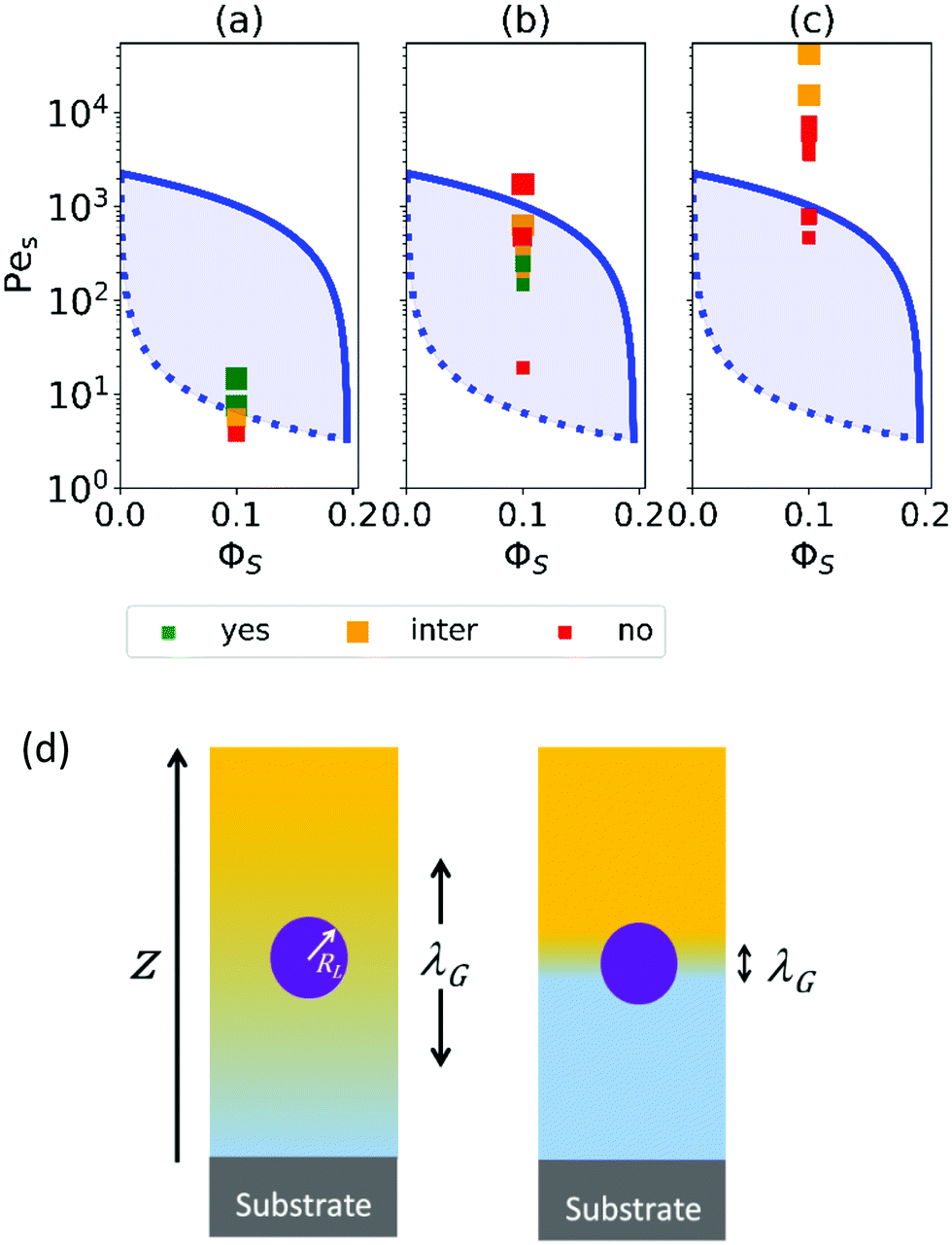 | ||
Fig. 2 Parameter maps (PeS and ϕs) for stratification for colloid mixtures (a) without thickener; (b) with thickener at ϕT = 0.01 and (c) ϕT = 0.02. Experimental data are shown with the symbols with their size being proportional to H. The colour of symbols indicates when stratification has occurred (green: yes) or has not occurred (red: no) or is intermediate between the two (yellow). The minimum PeS for stratification according to the Sear model32 is shown as a dotted line. The upper PeS limit, above which stratification does not occur according to our revised model, is dependent on the ratio of initial film thickness to the radius of the large colloid, 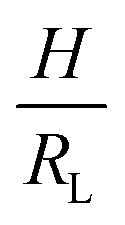 . The solid line at the upper boundary uses . The solid line at the upper boundary uses 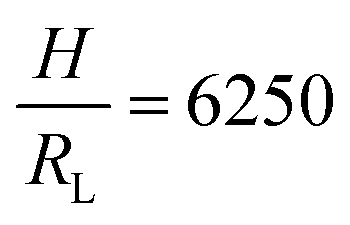 (corresponding to an average H of 500 μm). See the Jupyter notebook (Masterplot) in the ESI.† (d) Schematic diagrams showing a large colloid (purple) in a concentration gradient of small particles (yellow) in water (blue). At the left, width of the gradient layer, λG, is less than the diameter of the large colloid. (corresponding to an average H of 500 μm). See the Jupyter notebook (Masterplot) in the ESI.† (d) Schematic diagrams showing a large colloid (purple) in a concentration gradient of small particles (yellow) in water (blue). At the left, width of the gradient layer, λG, is less than the diameter of the large colloid. | ||
When thickener was added, PeS is always large enough so that Sear's model32 predicts stratification, and indeed with an initial thickener volume fraction of ϕT = 0.01 (Fig. 2b), stratification was observed for some values of PeS. However, at the highest values of Pes, no stratification was found, contrary to the expectations of Sear's and Zhou et al.'s models. With ϕT = 0.02 (Fig. 2c), only high values of Pes were obtained, and stratification was never observed. There was intermediate stratification only for very thick films.
The existence of an upper limit on stratification in our experiments at high PeS is a new discovery that has not been predicted by existing models in the literature. We therefore analyse our ternary system under high Pe and why the original model fails.
First, we reconsider our model for diffusiophoresis, which is the motion of one species, here the larger colloid, arising from a gradient in concentration in another species, here the smaller colloid. Hence, the diffusiophoretic speed U of the larger colloid is proportional to the spatial gradient in the volume fraction of the smaller colloid, ϕs, as given by
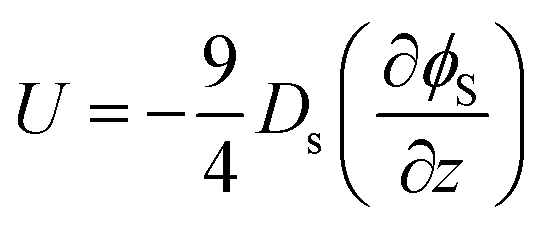 | (1) |
To achieve stratification, the diffusiophoretic speed U must be large enough to separate the large and small colloids during drying. This requires a large enough gradient of the concentration of the smaller colloid. At high PeS, the smaller colloid will rapidly accumulate below the descending water/air interface, and so its volume fraction will reach ϕjam. As Okozune et al.39 and Sear32 realised, when jamming occurs, a jammed layer of the smaller colloid descends at a speed 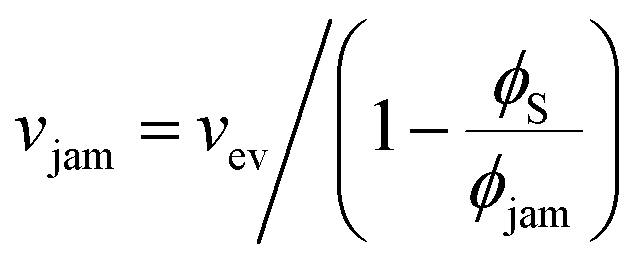 . Below this descending jammed layer is an accumulation zone of width λG = Ds/vjam wherein the volume fraction of the smaller colloid is
. Below this descending jammed layer is an accumulation zone of width λG = Ds/vjam wherein the volume fraction of the smaller colloid is
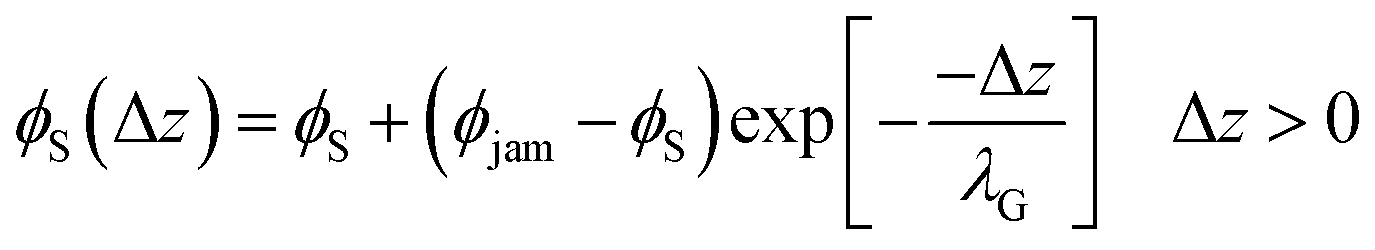 | (2) |
For stratification to happen, the large colloids must outrun the descending jammed layer of the smaller colloids, i.e., U(Δz = RL) > vjam. Here we consider a large colloid with top edge touching the descending jammed layer and so with a centre of mass at a distance of RL below this layer. In our model, the jamming front is descending at a constant speed vjam and with a concentration profile ahead of the front that moves at the same speed. Stratification then occurs if – at that front – the large colloids are being pushed down by this gradient at a speed that is at least as fast as vjam.
Using the derivative of the small colloid volume fraction to determine the diffusiophoretic speed, and requiring that it exceed vjam we have
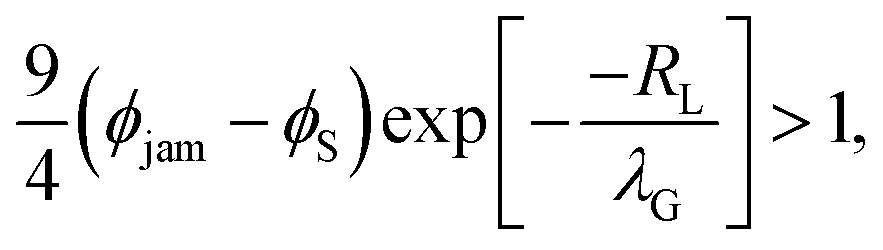 | (3) |
In earlier experimental work21 at moderate Pe values of order one to ten, the width of the accumulation zone λG was much larger than the size of the larger colloids, so that the exponential term,  , was close to 1. Sear and Warren30 approximated the ratio as 1. Thus, they and others31,33 neglected the effects that occur when the width of the accumulation zone for small particles becomes comparable to the size of the larger colloids. This neglect is typically reasonable for Pe values of order ten or less. However, in the current experiments, we are reaching very large values of PeS by greatly increasing the viscosity. The resulting strong decrease in Ds also dramatically decreases λG, meaning that λG can become comparable to the size of the larger colloid, thereby weakening diffusiophoresis.
, was close to 1. Sear and Warren30 approximated the ratio as 1. Thus, they and others31,33 neglected the effects that occur when the width of the accumulation zone for small particles becomes comparable to the size of the larger colloids. This neglect is typically reasonable for Pe values of order ten or less. However, in the current experiments, we are reaching very large values of PeS by greatly increasing the viscosity. The resulting strong decrease in Ds also dramatically decreases λG, meaning that λG can become comparable to the size of the larger colloid, thereby weakening diffusiophoresis.
To compare with experiment, we note that we can write PeS = vevH/(vjamλG), i.e. the Péclet number varies as 1/λG so that the left-hand side of eqn (3) decays exponentially with increasing Pe. If we replace the inequality by an equality, we obtain an upper limit on Pe for stratification. Here, we set H = 500 μm and RL = 80 nm, which is appropriate for our experiments. This upper limit is plotted in Fig. 2 as the solid curve.
From the experimental results we can see that only weak (yellow points) or no (red) stratification is observed at very high PeS in the systems containing thickener, therefore suggesting an upper PeS limit exists. Looking at the solid lines, which show the predictions of a stratification window from our revised model with an upper limit, a general agreement with the data is apparent.
When the thickener was introduced into the binary mixture, the initial viscosity of the continuous phase, η, increased, as presented in Fig. S2 (ESI†). This effect appears in our model as a smaller λG arising from the smaller DS. However, as the viscosity increases rapidly with thickener concentration during drying, the viscosity will rapidly increase. This effect is not in our very simple model. However, we can estimate the time dependence of the viscosity during drying, as follows.
Knowing the initial volumes of thickener and water, and having measured vev, we calculated the time evolution of the volume fraction of thickener in the aqueous continuous phase, ϕCP(t). With an experimental scaling relation of η ∼ ϕ4.6CP, found via rheology measurements at a low shear rate of 0.1 s−1 (Fig. S2 and Table S3, Viscosity_fitplot Jupyter notebook in ESI†), we transformed ϕCP(t) into η(t), which is the time-dependent, low-shear-rate viscosity of the continuous phase during water evaporation. See Fig. 3a. Note that due to the steep dependence of viscosity on concentration, the viscosity increases by orders of magnitude during drying.
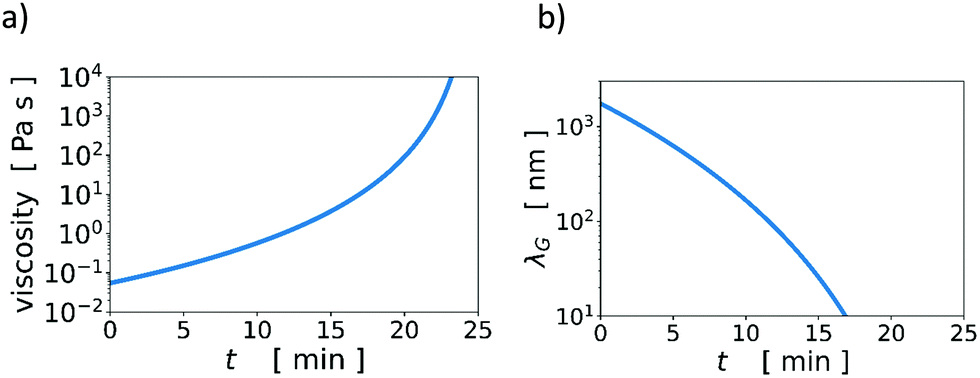 | ||
| Fig. 3 (a) Time evolution of the viscosity of a thickener solution, η(t), and (b) the width of the gradient layer, λG, during the evaporation of water at a rate E = 140 nm s−1 at T = 30 °C, in a film an initial thickness of H = 300 μm with ϕT = 0.01 initially. | ||
Using η(t) and the Stokes–Einstein expression to find the evolving Ds (Fig. S3, ESI†), we plot an estimate of λG as a function of time during evaporation in Fig. 3b. We estimated the change in λG for a film with ϕT = 0.01, H = 300 μm, and vev = 140 nm s−1. The evaporation velocity is approximately constant for drying times t < tjam, however, DS decreases strongly with t. We see in Fig. 3c that λG decreases by orders of magnitudes during drying.
The upper solid curve in Fig. 2 is the Péclet number above which even the initial width of the accumulation zone is too narrow to drive stratification. At lower initial viscosities, although λG may be initially wide enough for stratification, it may become too small during the evaporation process, thus switching off stratification during drying. Thus, for systems just below the solid curve in Fig. 2, there may be only very weak or partial stratification. Note that the time-dependent viscosity in Fig. 3a is only an estimate, which was obtained from measurements of low-shear viscosities of thickener solutions at a few different concentrations.
Our experiments clearly observe suppression of stratification at high Pe, and our model is consistent with that finding. However, within our simple model, the thickener solution is treated as a uniform continuous phase. The thickener's only effect in our model is to slow the diffusion of the large and small colloids strongly, via an increased viscosity that appears in the Stokes–Einstein expression for DS. The thickener molecules in the continuous phase could potentially stratify during film formation, as has been reported already for polymer/colloid mixtures.19 However, the swollen thickener particles (>300 nm) are large compared to the colloids and at high concentrations will raise the viscosity, which will act against their stratification. The Raman depth profiling method was not applied to the thickener molecules to explore this possibility. Our system is a complex four-component mixture of two colloids, highly-swollen thickener particles, and water. Hence, it is possible that other mechanisms are at play beyond those considered in our model. Further work is warranted.
Information on the evolving particle dynamics during the evaporation process, such as has been obtained elsewhere using laser speckle imaging,40 would provide essential information to understand the evolving effects of the thickener on stratification. Also, as the thickener is shear thinning, future work could consider both the colloidal diffusion and the diffusiophoresis in viscoelastic media.
In summary, we have shown that self-stratification does occur in drying bimodal colloid films with a thickener in the continuous phase, but only over a range of Péclet numbers. We discovered that when the value of PeS exceeds an upper limit, stratification is suppressed. Our simple model predicts that this upper limit depends on H/RL, and occurs when the width of the gradient layer becomes too narrow (less than RL). Our results provide a warning for the formulators of stratified systems to avoid the high-Pe limit.
Author contributions
Malin Schulz: conceptualization, formal analysis, investigation, methodology, visualization, writing original draft. Richard Brinkhuis: conceptualization, resources, supervision. Carol Crean: methodology, resources, writing – reviewing and editing. Richard P. Sear: conceptualization, formal analysis, writing – reviewing and editing. Joseph L. Keddie: conceptualization, methodology, supervision, writing – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge funding from EPSRC for the ‘High Spec Raman Spectrometer Regional Facility’ (EP/M022749/1) and from Allnex for the PhD studentship of MS. We thank Matthew Wearon, Violeta Doukova, Agata Gajewicz-Jaromin, and Rachida Bance-Soualhi (University of Surrey) for technical support in the laboratory. We benefited from useful discussions with Yujing Zhang and Javier Bohorquez (Allnex).References
- A. N. Zelikin, ACS Nano, 2010, 4, 2494 CrossRef CAS PubMed.
- K. C. Wood, H. F. Chuang, R. D. Batten, D. M. Lynn and P. T. Hammond, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10207 CrossRef CAS PubMed.
- Q. Xu, Y. Lv, C. Dong, T. Sreenivasan Sreeprased, A. Tian, H. Zhang, Y. Tang, Z. Yu and N. Li, Nanoscale, 2015, 7, 10883 RSC.
- M. Maric, W. van Bronswijk, S. W. Lewis, K. Pitts and D. E. Martin, Forensic Sci. Int., 2013, 228, 165 CrossRef CAS PubMed.
- G. E. Stein, T. S. Laws and R. Verduzco, Macromolecules, 2019, 52, 4787 CrossRef CAS.
- J. D. Tinkler, A. Scacchi, H. R. Kothari, H. Tulliver, M. Agraiz, A. J. Archer and I. Martin-Fabiani, J. Colloid Interface Sci., 2021, 581, 729 CrossRef CAS PubMed.
- J. S. Nunes, S. J. Bohórquez, M. Meeuwisse, D. Mestach and J. M. Asua, Prog. Org. Coat., 2014, 77, 1523 CrossRef CAS.
- A. B. López, J. C. de la Cal and J. M. Asua, Langmuir, 2016, 32, 7459 CrossRef PubMed.
- A. B. López, J. C. de la Cal and J. M. Asua, Polymer, 2017, 124, 12 CrossRef.
- R. B. Bodkhe, S. E. M. Thompson, C. Yehle, N. Cilz, J. Daniels, S. J. Stafslien, M. E. Callow, J. A. Callow and D. C. Webster, J. Coat. Technol. Res., 2012, 9, 235 CrossRef CAS.
- S. Zahedi and S. R. Ghaffarian, Trans. Inst. Met. Finish., 2021, 99, 133 CrossRef CAS.
- A. Beaugendre, S. Degoutin, S. Bellayer, C. Pierlot, S. Duquesne, M. Casetta and M. Jimenez, Coatings, 2018, 8, 448 CrossRef.
- A. Beaugendre, S. Degoutin, S. Bellayer, C. Pierlot, S. Duquesne, M. Casetta and M. Jimenez, Prog. Org. Coat., 2017, 110, 210 CrossRef CAS.
- J. Baghdachi, H. Perez, P. Talapatcharoenkit and B. Wang, Prog. Org. Coat., 2015, 78, 464 CrossRef CAS.
- R. E. Trueman, E. Lago Domingues, S. N. Emmett, M. W. Murray and A. F. Routh, J. Colloid Interface Sci., 2012, 377, 207 CrossRef CAS PubMed.
- A. J. Carr, W. Liu, K. G. Yager, A. F. Routh and S. R. Bhatia, ACS Appl. Nano Mater., 2018, 1, 4211 CrossRef CAS.
- A. Fortini, I. Martín-Fabiani, J. L. De La Haye, P.-Y. Dugas, M. Lansalot, F. D’Agosto, E. Bourgeat-Lami, J. L. Keddie and R. P. Sear, Phys. Rev. Lett., 2016, 116, 118301 CrossRef PubMed.
- M. P. Howard, A. Nikoubashman and A. Z. Panagiotopoulos, Langmuir, 2017, 33, 11390 CrossRef CAS PubMed.
- M. Schulz, R. W. Smith, R. P. Sear, R. Brinkhuis and J. L. Keddie, ACS Macro Lett., 2020, 9, 1286 CrossRef CAS.
- S. Cheng and G. S. Grest, ACS Macro Lett., 2016, 5, 694 CrossRef CAS.
- M. Schulz and J. L. Keddie, Soft Matter, 2018, 14, 6181 RSC.
- D. Velegol, A. Garg, R. Guha, A. Kar and M. Kumar, Soft Matter, 2016, 12, 4686 RSC.
- D. M. Makepeace, A. Fortini, A. Markov, P. Locatelli, C. Lindsay, S. Moorhouse, R. Lind, R. P. Sear and J. L. Keddie, Soft Matter, 2017, 13, 6969 RSC.
- M. Schulz, C. Crean, R. Brinkhuis, R. P. Sear and J. L. Keddie, Prog. Org. Coat., 2021, 157, 106272 CrossRef CAS.
- X. Liu, W. Liu, A. J. Carr, D. Santiago Vazquez, D. Nykypanchuk, P. W. Majewski, A. F. Routh and S. R. Bhatia, J. Colloid Interface Sci., 2018, 515, 70 CrossRef CAS PubMed.
- M. P. Howard, A. Nikoubashman and A. Z. Panagiotopoulos, Langmuir, 2017, 33, 3685 CrossRef CAS PubMed.
- Y. Tang, G. S. Grest and S. Cheng, Langmuir, 2018, 34, 7161 CrossRef CAS PubMed.
- A. Fortini and R. P. Sear, Langmuir, 2017, 33, 4796 CrossRef CAS PubMed.
- Y. Dong, N. Busatto, P. J. Roth and I. Martin-Fabiani, Soft Matter, 2020, 16, 8453 RSC.
- R. P. Sear and P. B. Warren, Phys. Rev. E, 2017, 96, 062602 CrossRef PubMed.
- J. Zhou, Y. Jiang and M. Doi, Phys. Rev. Lett., 2017, 118, 108002 CrossRef PubMed.
- R. P. Sear, J. Chem. Phys., 2018, 148, 134909 CrossRef PubMed.
- C. R. Rees-Zimmerman and A. F. Routh, J. Fluid Mech., 2021, 928, A15 CrossRef.
- J. Zhou, Z. Man, Y. Jian and M. Doi, Adv. Mater., 2017, 29, 1703769 CrossRef PubMed.
- R. Tatsumi, T. Iwao, O. Koike, Y. Yamaguchi and Y. Tsuji, Appl. Phys. Lett., 2018, 112, 053702 CrossRef.
- Y. Tang, G. S. Grest and S. Cheng, J. Chem. Phys., 2019, 150, 224901 CrossRef PubMed.
- Y. Tang, G. S. Grest and S. Cheng, Langmuir, 2019, 35, 4296 CrossRef CAS PubMed.
- T. F. Tadros, Colloids in Paints, Wiley-VCH, Weinheim, 2011, vol. 6 Search PubMed.
- T. Okuzono, K. Ozawa and M. Doi, Phys. Rev. Lett., 2006, 97, 136103 CrossRef PubMed.
- H. van der Kooij, R. Fokkink, J. van der Gucht and J. Sprakel, Sci. Rep., 2016, 6, 34383 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2sm00194b |
| This journal is © The Royal Society of Chemistry 2022 |