
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEncapsulated droplet interface bilayers as a platform for high-throughput membrane studies†
D. K.
Baxani
 a,
W. D.
Jamieson
a,
D. A.
Barrow
b and
O. K.
Castell
*a
a,
W. D.
Jamieson
a,
D. A.
Barrow
b and
O. K.
Castell
*a
aCollege of Biomedical and Life Sciences, School of Pharmacy and Pharmaceutical Sciences, Cardiff University Redwood Building, King Edward VII Avenue, CF10 3NB Cardiff, UK. E-mail: CastellO@cardiff.ac.uk
bSchool of Engineering, Cardiff University, 14–17 The Parade, CF4 3AA Cardiff, UK
First published on 17th June 2022
Abstract
Whilst it is highly desirable to produce artificial lipid bilayer arrays allowing for systematic high-content screening of membrane conditions, it remains a challenge due to the combined requirements of scaled membrane production, simple measurement access, and independent control over individual bilayer experimental conditions. Here, droplet bilayers encapsulated within a hydrogel shell are output individually into multi-well plates for simple, arrayed quantitative measurements. The afforded experimental throughput is used to conduct a 2D concentration screen characterising the synergistic pore-forming peptides Magainin2 and PGLa. Maximal enhanced activity is revealed at equimolar peptide concentrations via a membrane dye leakage assay, a finding consistent with models proposed from NMR data. The versatility of the platform is demonstrated by performing in situ electrophysiology, revealing low conductance pore activity (∼15 to 20 pA with 4.5 pA sub-states). In conclusion, this array platform addresses the aforementioned challenges and provides new and flexible opportunities for high-throughput membrane studies. Furthermore, the ability to engineer droplet networks within each construct paves the way for “lab-in-a-capsule” approaches accommodating multiple assays per construct and allowing for communicative reaction pathways.
Introduction
Artificial lipid bilayers have provided unique insight into the vital functions of the cell membrane and the role of membrane proteins in maintaining cellular function, as well as their evaluation as drug targets in disease.1,2 Whilst there are many reports of lipid bilayer or liposome arrays, those that report the ability to make quantitative functional measurements across a range of experimental conditions are lacking by comparison. This is a consequence of the need to tackle the combined challenges of scaled membrane production, measurement access at scale, and to provide control over individual bilayer experimental conditions.The first membrane arrays3 made use of microfabrication technologies to create patterned surfaces to demarcate multiple individual supported lipid bilayer (SLB) patches. These approaches rely on bilayer formation upon a solid support, limiting mobility and insertion of trans-membrane components. Spacers of tethered polymers, or surface assembled monolayers,4,5 have been used to address this, but chemical access and solution volume remain limited, and measurement access can be challenging. Alternative means to mitigate interaction with an underlying substrate have been developed, including arrays of tethered vesicles creating hundreds or thousands of bilayers for individual6 or ensemble imaging.7–9 Whilst the measurement capacity has broken new ground in single molecule measurements,10,11 high content measurements to screen membranes under different conditions remains challenging due to a shared common external environment. Microfabricated architectures have been developed for the creation of arrays of suspended lipid bilayers,12–15 or the electro-formation of swollen vesicles over apertures,16 to address the twin challenges of mobility and access.
Droplet based approaches to lipid bilayer creation represent an attractive means to more readily segregate and tailor experimental conditions. In this regard Droplet Interface Bilayers (DIBs), formed by the contacting of aqueous droplets in oil in the presence of lipid (Fig. 1),17 represent a useful approach for artificial bilayer production. A number of microfluidic methods to produce large numbers of DIBs have been reported.18–24 The ease of formation, high stability, high electrical resistance25 together with potential ease of droplet manipulation make them attractive candidates for on-chip arrayed measurement.26,27 Automated approaches have been used for the detection and characterization of pore forming proteins.14,28 Devices for parallel formation of up to 16 DIBs for either alternating29,30 or parallel31 electrophysiology recordings have also been demonstrated.
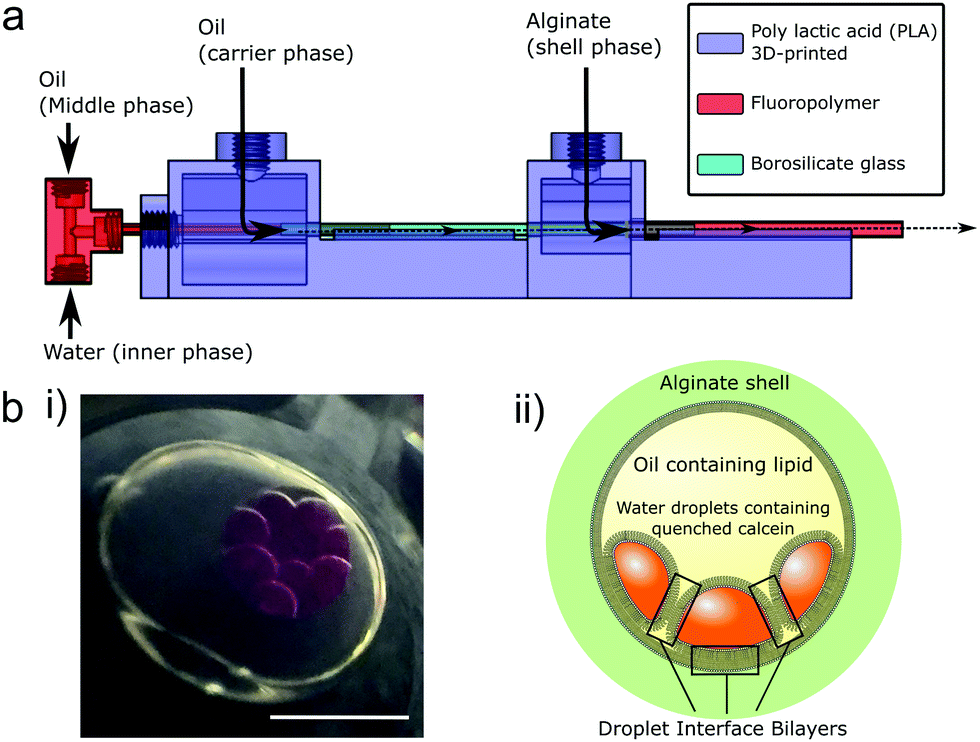 | ||
| Fig. 1 Encapsulated droplet interface bilayers (eDIBs) are nested droplet structures of water droplets in oil within a hydrogel shell. (a) Microfluidic device employed to produce eDIBs.20 (b) (i) Image of eDIB containing 10 aqueous cores. These cores are dyed with sulphorhodamine B for contrast. (ii) Diagram of an eDIB containing quenched calcein loaded aqueous cores, showing the presence of droplet interface bilayers between the internal cores as well as between the internal cores and the hydrogel shell. Scale bar = 1 mm. | ||
Optical measurement methods arguably provide greater opportunities to scale parallel measurements, as increasingly complex electronics are required to scale up electrophysiology set-ups.32 Fluorescent measurements in arrays of 18 DIBs have quantified ion flux and its inhibition through the membrane protein pore α-Hemolysin.33 This approach has been further scaled up, increasing temporal response34 and bilayer density.32 A DIB-like approach has also been used to create suspended bilayers over an array of micro-chambers35 for optical measurement of transport through α-Hemolysin.36,37 These approaches have all employed a common aqueous reservoir on one side of all membranes in the array, thus trading-off individual bilayer segregation and chemical control in favor of multiplexed optical access.
Whilst the benefit of microfluidics to scale DIB production has been demonstrated, the systems reported to date require the bilayers to be maintained on-chip,26,27,38 thus limiting their accessibility without the use of sophisticated liquid handling upstream and integration of measurement systems. We propose the use of microfluidically prepared hydrogel encapsulated DIBs (eDIBs),20 that can be output into standard multi-well plates for subsequent arrayed experimentation and analysis at the individual bilayer level. We previously demonstrated a microfluidic method for producing eDIBs which is employed in this study, comprising a hybrid glass capillary/3D-printed microfluidic device with successive coaxial droplet generation geometries (Fig. 1a). This device is able to produce DIB-forming aqueous droplets within an oil droplet, itself encapsulated within a hydrogel shell. Each encapsulated construct containing ≈10, ≈200 nL aqueous droplets.20 This approach affords the opportunity to contain, access and manipulate DIBs in a manner that is compatible with common liquid handling and analytical techniques, enabling the screening of large parameter spaces.
The afforded measurement throughput is demonstrated here via the performance of a 2D concentration screen of the antimicrobial peptides Magainin 2 (MAG2) and PGLa,39,40 from Xenopus Laevis, thought to act on phospholipid membranes and give rise to membrane pores or cause membrane rupture.39,41–43 We employ a calcein de-quenching assay contained within the aqueous membrane bound compartments of eDIBs to characterize membrane permeabilisation upon exposure to a 2D concentration screen of both peptides (6 × 64 conditions). In situ electrophysiology is also performed, demonstrating the amenability of the array to different methods of bilayer interrogation with differing, but complementary, strengths of throughput and sensitivity respectively. These experiments provide a dual route towards the characterisation of the synergistic mechanism of these peptides using artificial lipid membranes with single bilayer construct resolution, and sufficient throughput to study the peptide cooperativity.
Results
Droplet microfluidics was used to form nested droplet structures of water droplets within oil, encapsulated within a hydrogel shell.20 The presence of lipid dissolved in the oil phase facilities lipid monolayer self-assembly at the aqueous-oil interfaces. Where these interfaces contact, between internal droplets, and also with the hydrogel shell, lipid bilayers form (Fig. 1b). These encapsulated Droplet Interface Bilayers (eDIBs) are self-supporting and stable off-chip. The hydrogel shell provides structural support whilst maintaining diffusive contact between the lipid bilayers and the external environment (Fig. 1b). eDIBs were exited from the microfluidic device directly into individual wells of a 96-well plate (Fig. 2a). This format allowed pipette access to each well enabling all eDIBs to easily be exposed to different conditions (Fig. 2b), as well as allowing for parallel imaging or scanning using standard multi-well plate readers (Fig. 2c) or scanning microscopes.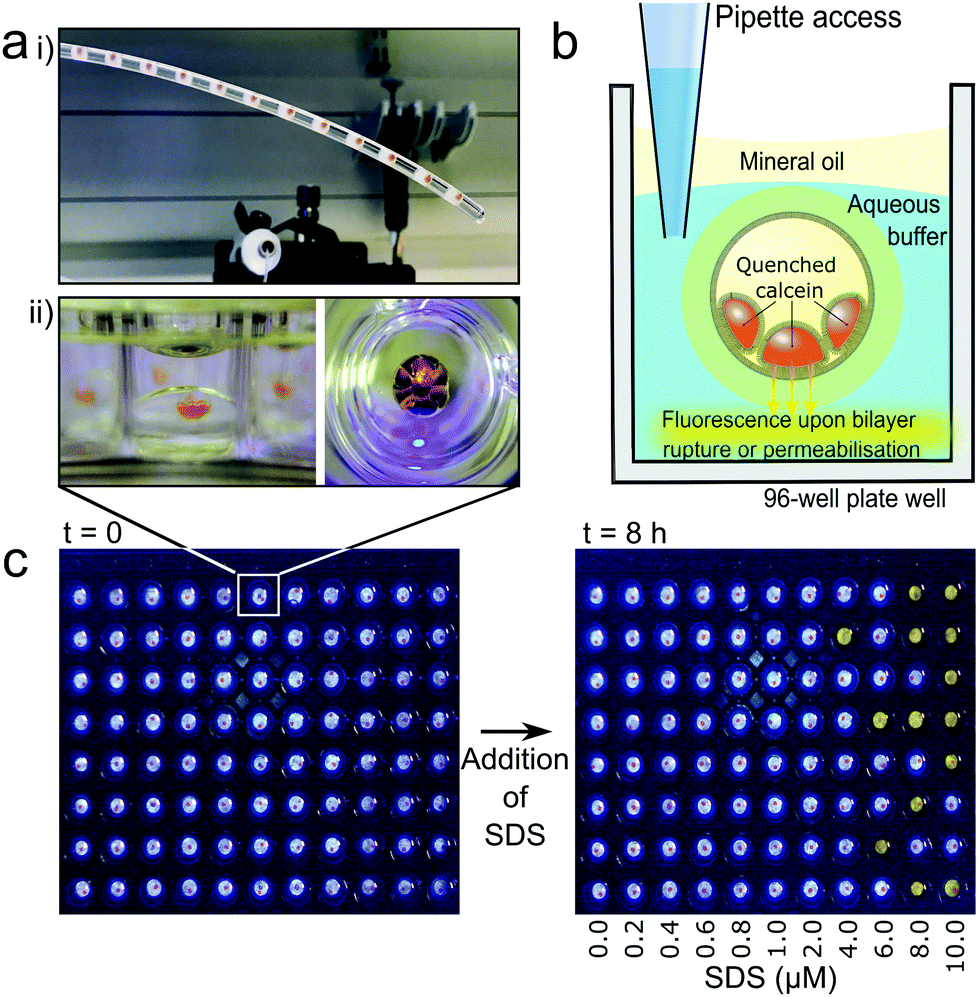 | ||
| Fig. 2 Individually addressable encapsulated droplet interface bilayers (eDIBs) in multi-well plate arrays. (a) (i) eDIBs in the outlet tube of their microfluidic production device are output into individual wells of a multi-well plate (ii) side view (left) and top-down view (right) of an individual eDIB within a plate well. (b) eDIBs reside in aqueous buffer in the well which is in diffusive contact with the encapsulated lipid bilayers enabling direct experimental additions to each well to interact with the lipid bilayer. A mineral oil layer prevents evaporation and allows storage. Self-quenched calcein (orange) within the internal aqueous droplets of eDIBs fluoresces upon leakage. (c) eDIB array demonstrated with fluorescent response in the well (yellow) to SDS induced bilayer permeabilisation upon exposure to increased SDS concentrations. | ||
Calcein encapsulated within the aqueous cores of the eDIBs at self-quenching concentrations (70 mM) was used to assess membrane leakage. Upon bilayer permeabilisation, or rupture, calcein can diffuse from the eDIB cores into the external aqueous media. At these lower concentrations the dye de-quenches and becomes fluorescent (Fig. 2b). Consequently, any increase in fluorescence within the well is indicative of bilayer permeabilisation or rupture. To demonstrate this, eDIBs were exposed to increasing concentrations of surfactant sodium dodecyl sulphate (SDS) (0–10 μM), a detergent known to cause bilayer solubilization and stochastic pore formation at sub-solubilizing concentrations.44 At concentrations above 4 μM a fluorescent response was detected due to bilayer leakage or rupture (Fig. 2c).
Fluorescent plate-reader time course measurements revealed two types of fluorescent leakage response of eDIB membranes in the presence of SDS (8 μM) (Fig. 3). Either slow leakage (Fig. 3a(ii)), indicative of bilayer permeabilisation and calcein eflux through otherwise intact membranes, giving rise to a slow steady increase in fluorescence, or sudden critical membrane rupture indicated by a sudden and saturating fluorescence increase (Fig. 3a(iii)). In contrast, eDIBs not exposed to SDS displayed little or no change in fluorescence (Fig. 3a(i)). In this way the stochastic nature of individual bilayer rupture or leakage behaviours could be observed through the measurement of single constructs in a way that is inaccessible with ensemble vesicle leakage assays. In such ensemble vesicle measurements it is challenging to disentangle the relative contributions of different behaviors across large numbers of vesicles that collectively give rise to the observed fluorescent response. Further characterisation of eDIBs displaying non-rupturing behaviour in response to SDS is shown in Fig. 3bvia quantitative fluorescence measurement (ESI† for callibration) of eDIBs exposed to 8 μM SDS and no SDS (n = 8 for both conditions). On exposure to SDS a steady increase in fluorescence is seen with time, indicative of the leakage of calcein from the internal eDIB cores, likely as a consequence of detergent bilayer permeabilisation (pore formation) without giving rise to critical membrane failure. The measured variance in leakage is likely due to the stochastic nature of bilayer pore formation by detergents, which can give rise to stable leakage or stochastic membrane failure (rupture) (Fig. 3a(iii)), which is measured here at the single eDIB level. Higher detergent concentrations increase the probability of membrane failure. Fig. 3c demonstrates the ability to use the same system as a low-cost, readily scalable, digital response assay. The fluorescence response of individual membrane rupture is easily detected via webcam or smart-phone camera under blue LED illumination. Photobleaching was deemed to be negligible due to low power illumination and high fluorophore concentrations. Here 432 individual eDIBs are imaged simultaneously (Fig. 3c).
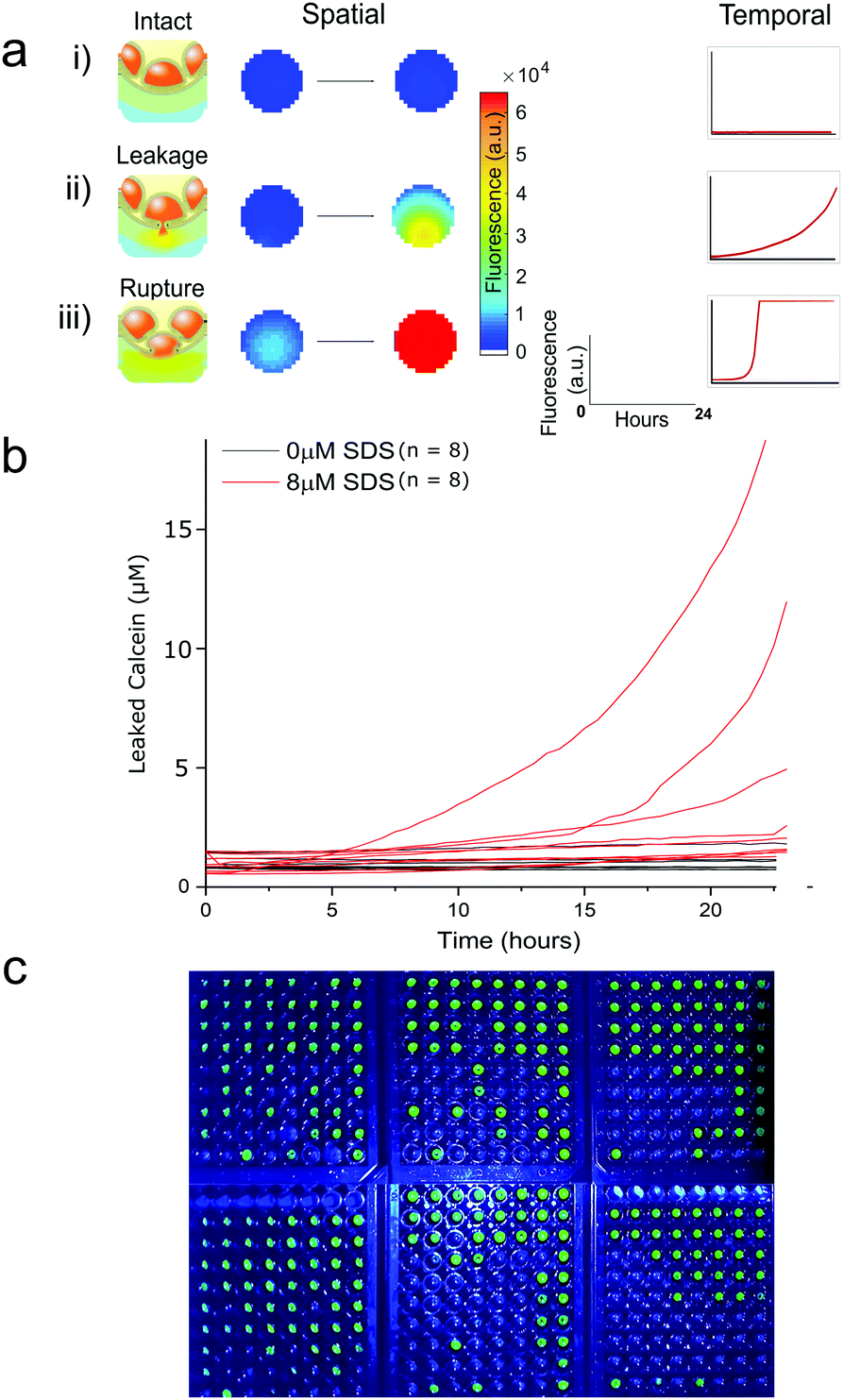 | ||
| Fig. 3 Fluorescence response from individual droplet interface bilayers (eDIBs) in arrayed multi-well plate measurements. (a) Arrayed eDIBs measured by quantitative fluorescent plate reader either by well scanning (imaging) or whole-well fluorescence providing spatial and temporal fluorescence data on calcein leakage and membrane integrity. (i) intact bilayers in eDIBs not exposed to SDS exhibit no appreciable change in fluorescence (n = 8). In eDIBs exposed to 8 μM SDS two types of fluorescent response are observed, either (ii) steady increase in fluorescence due to calcein leakage through membrane pores, or (iii) rapid increase in fluorescence due to calcein leakage on bilayer rupture, reflecting the stochastic nature of membrane pore formation and membrane failure (b) leakage of calcein from individual eDIBs into the aqueous volume within a well, when exposed to 8 μM SDS (red) and in the absence of SDS (black). The leaked calcein concentration is calculated from fluorescence intensity calibration (ESI†). (c) High content webcam simultaneous capture of individual eDIBs in wells of 6 × 96 well-plates imaged under blue LED illumination. | ||
Quantifying synergistic action of two membrane pore-forming peptides
Following validation of the approach with detergent mediated membrane activity, the platform was applied to quantify the synergistic action of the antimicrobial pore-forming peptides MAG2 and PGLa. These antimicrobial peptides form part of the innate immune system of Xenopus Laevis, offering antimicrobial action via membrane activity, in addition to reported antifungal, anti-viral and anti-tutor properties of the peptides.39 The afforded throughput of the eDIB array enabled a two-dimensional concentration screen to evaluate the synergistic activity of the two peptides at different concentrations and different ratios (0.22 to 28 μM for each peptide) (Fig. 4a and b).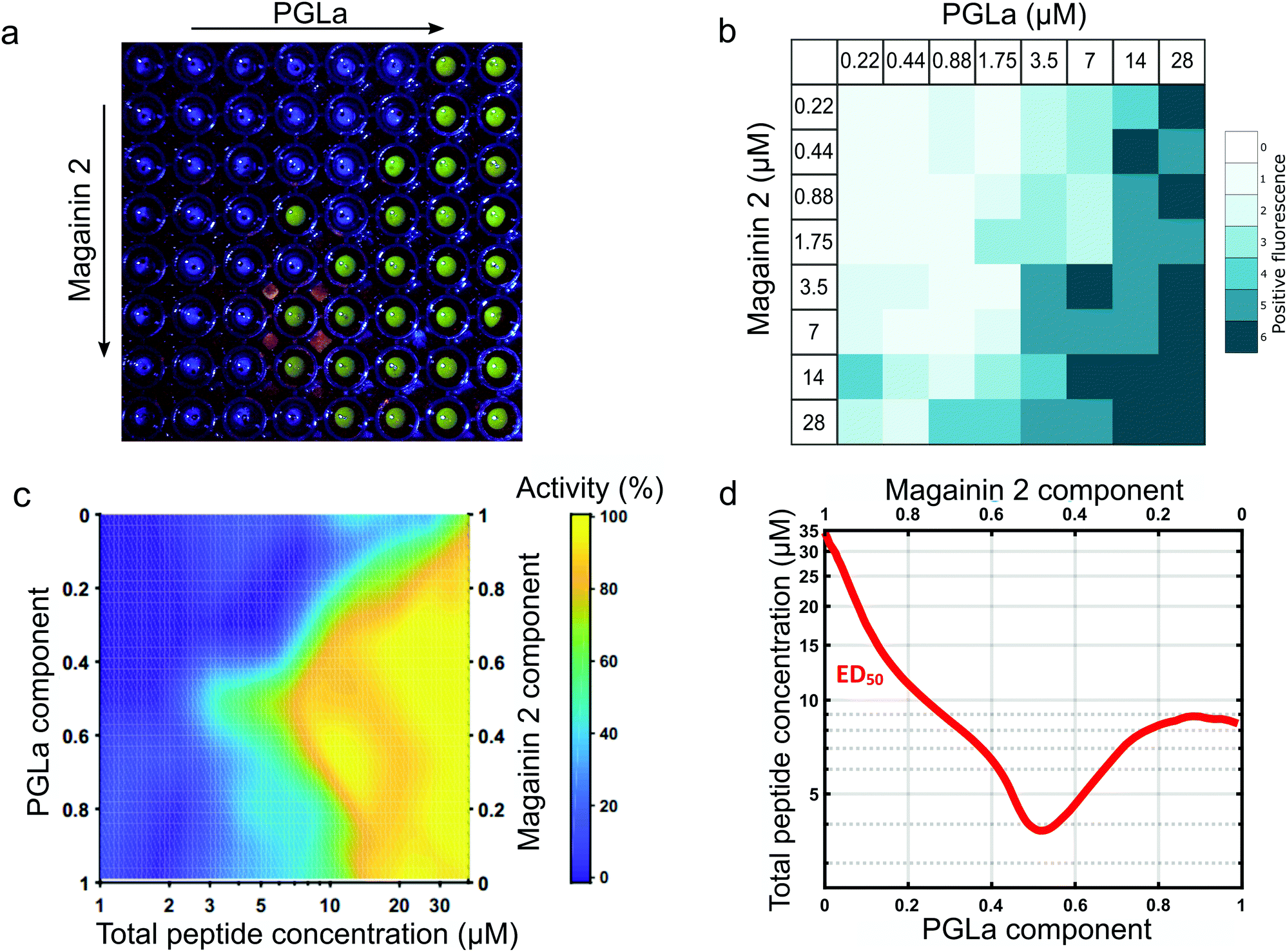 | ||
Fig. 4 Characterisation of the synergistic activity of the membrane pore forming peptides Magainin 2 and PGLa by a 2D-concentration screen array. (a) Example eDIB 2D array screen of increasing Magainin 2 (rows) and PGLa (columns) concentrations with increased visible fluorescence response observed due to rupture of the eDIB membranes containing quenched calcein and its subsequent dilution and dequenching in the external aqueous phase of the well. The peptide concentrations are as shown in (b). (b) Activity heatmap of peptide activity across the 2D combinatorial space. Heatmap reflects the proportion of eDIBs evidencing fluorescent increase associated with membrane rupture and calcein release (n = matrices of 64 conditions each, total 384 eDIBs, (a) constitutes one of such experiments). (c) Interpolated heatmap of relative activity with total peptide concentration (x-axis) and Magainin 2: PGLa ratio (y-axis). PGLa is found to be more active than Magainin 2 in the eDIB DPhPC membranes, with the two peptides displaying synergistic activity in combination, maximized at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. (d) ED50 plot depicting the effective total peptide concentration required to elicit 50% maximal response across peptide mixing ratios. Maximal synergy at equimolar concentrations is consistent with the proposed direct stoichiometric interaction between Magainin 2 and PGLa in their combined mechanism of membrane activity.39 :1 ratio. (d) ED50 plot depicting the effective total peptide concentration required to elicit 50% maximal response across peptide mixing ratios. Maximal synergy at equimolar concentrations is consistent with the proposed direct stoichiometric interaction between Magainin 2 and PGLa in their combined mechanism of membrane activity.39 | ||
Membrane rupture probability was observed to increase with increasing peptide concentration (Fig. 4a and b). A heat map of this activity was constructed from the replicate data (n = 6 for each condition) (Fig. 4b) which allowed construction of a 2D activity map across peptide mixing ratios and total combined peptide concentration (Fig. 4c). PGLa was found to be more active than MAG2 generating a more pronounced effect on eDIB membrane stability at concentrations above 3.5 μM.
The two peptides exhibited an enhancement in membrane activity in combination, with this maximized at a 1:1 stoichiometric ratio (Fig. 4c). The ability to explore the 2D combinatorial space enabled the determination of an effective active dose (ED50) plot (Fig. 4d) from the interpolated 2D map. In the ED50 plot (Fig. 4d) the red tie-line illustrates the total peptide concentration required to elicit a 50 per cent activity response across different peptide mixing proportions. The ED50 was found to display maximal activity (∼4 μM) at a 1:1 peptide ratio, with the ED50 concentration increasing to ∼8 μM for PGLa on its own, and 28 μM for MAG2 alone. The required peptide concentration to elicit 50% activity is observed to decrease as the peptide mixing proportions approach 1:1, indicating maximal synergistic activity at equimolar concentrations of the two peptide species. Such a finding is suggestive of a direct stoichiometric interaction between the two peptide species in a combined mode of action to bring about their synergy, rather than the result of two simultaneous, but independently acting mechanisms. These findings complement those of others, where stoichiometric PGLa-Mag2 interaction has been suggested following NMR measurements45 and molecular modelling.46 Previous combined activity studies have not presented exploration of both peptide ratio and concentration simultaneously in order to elicit ED50 response profiles, however the titration of the two species at a single fixed total peptide concentration has found maximal activity at around 1:1 ratio in vesicle leakage studies.42,43,47 corroborating our findings and demonstrating the utility of the platform.
To complement the optical characterisation of activity, the eDIB array platform was used to perform electrophysiology measurements in situ (Fig. 5). A custom Ag/AgCl electrode was used to pierce the eDIB alginate shell and access the internal droplets of the eDIB. A second electrode was placed in the external aqueous environment of the well (Fig. 5a and b). Single-channel electrophysiology ion-flux measurements of MAG2 and PGLa pore formation were made in eDIBs exposed to both peptides at a 1:1 stoichiometric ratio (3.5 μM total), performed within a select well of the array (Fig. 5c). This condition was selected for being just under the bilayer-rupturing concentration of peptides according to optical experiments. Under an applied potential (−30 mV), pore-formation was observed as step-like changes in conductance and fluctuation between multiple conductance states measured in an otherwise intact bilayer (marker represents no current ∼0 pA). Some of these current states appear to be long-lived (10 s of seconds), with measured current steps of 15 to 20 pA (Fig. 5c). Fluctuations of conductance states of short-duration (100 ms timescale) and of lower magnitude (∼4.5 pA) were observed within these periods (Fig. 5c exploded region and ESI†). Whilst the structure of MAG2-PGLa pores remains unknown, such behavior could be indicative of the presence of dynamic pores, with these representing the major conductance states, and their conductance modulated on the addition or loss of additional dimeric Mag2-PGLa subunits, a model consistent with recently postulated mechanisms based on NMR data.39
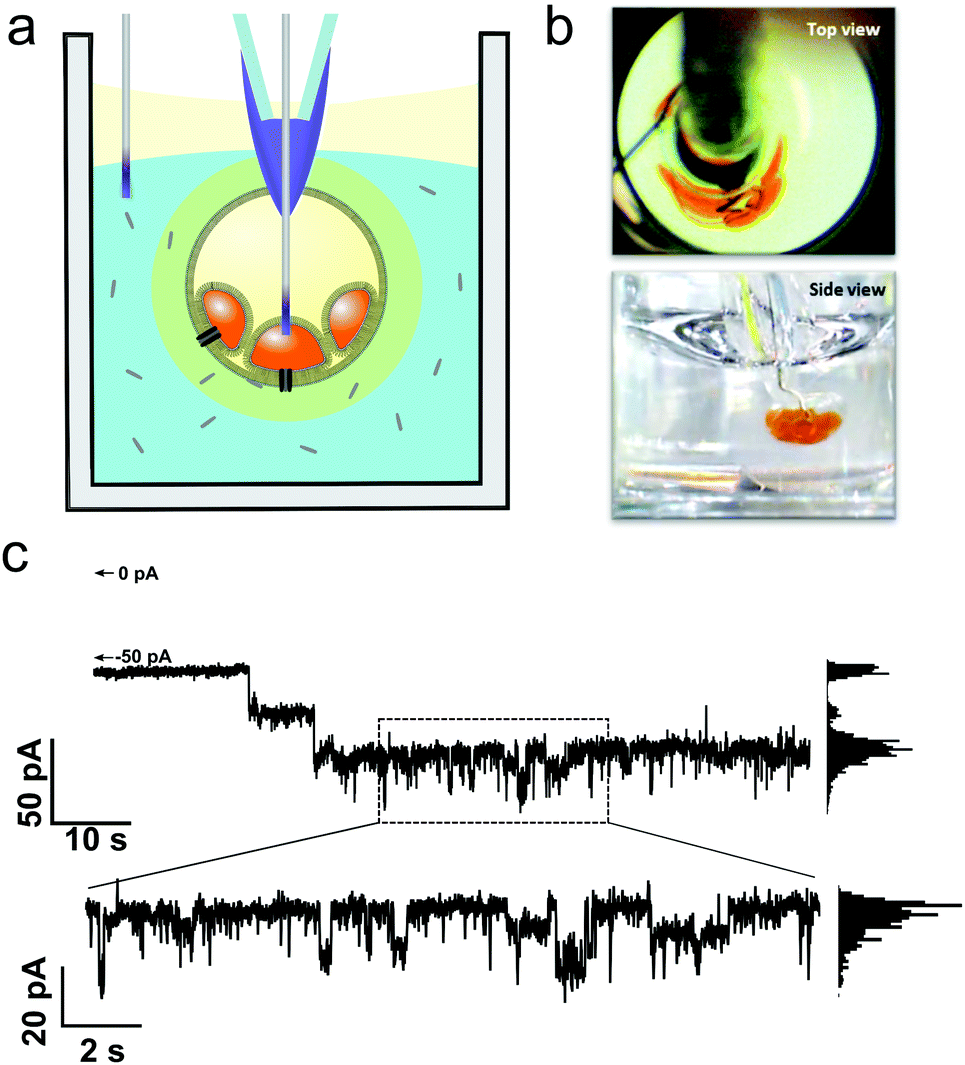 | ||
| Fig. 5 Electrophysiology of Magainin 2 (1.75 μM) and PGLa (1.75 μM) pores in eDIB membranes. (a) Electrophysiology can be performed in situ on eDIBs in selected wells of the array. Custom Ag/AgCl electrodes are inserted into internal aqueous droplets of eDIBs and placed in the external well environment. (b) Top and side view of electrode insertion viewed in a transparent 96-well plate. (c) Resultant single channel recording of membrane spanning pores in eDIBs on exposure to Magainin 2 (1.75 μM) and PGLa (1.75 μM) under an applied potential of −30 mV. Major, long-lived, conductance states of ∼15 to 20 pA are observed alongside shortlived step-like fluctuations of lower magnitude (∼4.5 pA). Histogram analysis of measured conductance states over short and long timescales is provided in the ESI.† Scale bars = 1 mm. | ||
Discussion and conclusions
The platform described here represents a unique approach for the formation of artificial lipid bilayer arrays by using hydrogel-encapsulated bilayer-bound droplet networks as individual, freestanding, membrane assay units. The ability to produce artificial bilayers at scale using microfluidics, with each membrane-unit being structurally rigid, yet able to maintain diffusive communication with the surrounding aqueous environment, means each can be output and individually manipulated. The experimental environment of each eDIB can be individually tailored and the eDIB interrogated with common laboratory equipment such as fluorescent plate-readers, automated microscopy platforms and liquid handling systems.This is demonstrated with the quantitative assessment of the synergy of two membrane active pore forming peptides, MAG2 and PGLa, in a 2D concentration screen with activity assessed by parallel optical measurement in conjunction with complementary ‘gold-standard’ electrophysiology on individually selected eDIBs within the array, affording opportunities for both high-throughput optical measurements and lower throughput but more sensitive electrophysiology measurements. eDIBs exposed to the peptides ruptured in a concentration and ratio dependent manner, displaying no discernable leakage by optical measurement prior to membrane rupture (ESI†). Whilst we measure pore formation by electrophysiology, this disparity could be due to the formation of pores that aren’t large enough for the transfer of calcein, or that the rate of calcein leakage was below the limit of detection of the optical methods employed here via plate-reader measurements. It is likely that higher resolution scanning optical microscopy approaches will afford greater measurement sensitivity, with single pore imaging demonstrated in droplet interface bilayers with sophisticated microscopy platforms.26,33
PGLa was found to be more active than MAG2, with the two peptides exhibiting an enhanced activity in combination, maximised at a 1:1 stoichiometry. Previous studies using NMR have suggested a synergistic mechanism via which surface-bound PGLa re-orients into a transmembrane position in the presence of MAG2.45 This has further been supported by peptide cross-linking studies,48 as well as vesicle leakage and bacterial death studies.39 The afforded experimental throughput here enabled elucidation of a quantitative 2D activity map of the two peptides, as well as the construction of a 50% effective dose (ED50) curve evaluating the influence of peptide concentration and ratio simultaneously. The positive charge of the two peptides (PGLa: +5, MAG2: +2) is thought to encourage electrostatic attraction to negatively charged lipids in bacterial cell membranes. A zwitterionic lipid, DPhPC, was used here as a benchmark lipid due to its ability to form highly stable DIBs, which was necessary due to the large number of DIBs employed in the study. Membrane surface coverage of MAG2 has been observed for both negatively charged (POPG) and zwitterionic (POPC) membranes,49 and SFG spectroscopy revealed symmetry breaking for both lipid systems, interpreted as potential membrane pore formation, in keeping with the electrophysiology reported here with DPhPC. The relative difference in peptide charge may explain the increased activity of PGLa measured here in comparison to MAG2.
The high-throughput nature of the eDIB platform proves the opportunity for a systematic screen of peptide ratios and concentrations simultaneously, elucidating maximal synergy at 1:1 peptide ratio. The electrophysiology experiments reported here demonstrate ion-flux across lipid bilayers when exposed to a fixed concentration of peptides, attributed to the formation of PGLa/MAG2 dynamic transmembrane pores. As such, analytical methods reporting on average peptide configuration at the membrane may not necessarily be representative of the peptide configuration in active pores. The array methodology reported here could help further elucidate the nature the peptide synergy by affording increased experimental throughput and individual membrane addressability. Functional transmembrane proteins have been widely reconstituted in DIBs50–55 and eDIBs,20 suggesting that similar studies may be performed on ion-channel or receptor membrane proteins.
This platform demonstrates the potential of membrane-based artificial cells for analytical applications. Whilst similar well plate DIB arrays have been published offering high-throughput electrophysiology recordings,28,30 the ability to carry out both high-throughput optical and in situ electrophysiology methods has not been demonstrated, and the use of freestanding eDIB constructs offers further experimental flexibility. The opportunity for formation of membrane segregated internal droplet networks may be used to facilitate more complex compartmentalised chemistry in such measurement systems. Furthermore, sophisticated liquid handling to tailor individual droplet contents or create continuous reagent or bilayer composition gradients will enable the prospect of additional experimental variable parameter space as well as multiplexed or multi-assay experiments. The ability to process large arrays simultaneously via optical methods is however offset somewhat by the relatively lengthy measurements performed here (up to 24 hours) in terms of sample throughput. However, miniaturisation of eDIB constructs is anticipated to increase assay speed, in a similar manner to that demonstrated by Tonooka et al.,32 where a 16-fold increase in assay speed was achieved compared to previous comparable approaches by volume reduction through miniaturisation.31 The free-standing nature of eDIBs should also allow for post measurement handling, where eDIBs could be extracted from wells for further processing, sample recovery or storage. Given eDIBs are stable over prolonged periods,20 it is feasible that pre-prepared assay plates could be stored or shipped for routine use. As such, eDIB arrays represent a promising approach at the intersection between high-throughput lipid membrane studies and programmable ‘lab-in-a-capsule’ technology platforms.
Materials and methods
All reagents were purchased from Sigma-Aldrich except where otherwise stated. Encapsulated Droplet Interface Bilayers (eDIBs) were produced by microfluidic methods using a hybrid 3D-printed and capillary device as described in our previous publication.20 The flow rates employed resulted in ca. 10 internal droplets per construct and were employed as follows: internal aqueous phase: 0.196 mLmin−1; internal oil phase: 0.196 mLmin−1; alginate phase: 2.5 mLmin−1; carrier oil phase: 6.67 mLmin−1. eDIB composition and assay: Aqueous inner cores comprised 0.5M NaCl, 70 mM calcein, 10 mM HEPES pH 7. The central oil phase comprised hexadecane:silicone oil AR20 (2:1) with dissolved 1,2-diphytanoyl-sn-glycero-3-phosphocholine (DPhPC) (4 mg mL−1). This solution was prepared by first dissolving DPhPC in chloroform, followed by solvent evaporation under a nitrogen gas stream and subsequent exposure to vacuum for 10 minutes. The resulting lipid cake was then dissolved in the hexadecane:silicone oil AR20 mixture. Liquid alginate (2% w/v) with suspended CaCO3 particles (75 mg mL−1) formed the outer shell phase. This was gelled during the microfluidic production process by the external droplet creating carrier mineral oil phase containing 0.5% dissolved glacial acetic acid. This liberates Ca2+ from the suspended CaCO3 particles in the contacting alginate phase, gelling the alginate.
Individual eDIBs were placed in wells of 96-well plates, with each well containing 150 μL of aqueous buffer (450 mM NaCl, 10 mM HEPES pH 7) and covered with a ∼150 μL of mineral oil. This helped prevent evaporation from the wells, enabled plate storage and encouraged uniformity of interfacial meniscus and eDIB positioning within wells. Stock solutions of Magainin 2 (Genscipt, USA) and PGLa (Generon, USA) (1 mM peptide, pH 7, 10 mM HEPES) were added directly into eDIB-containing wells to perform the 2D concentration screen assay. Fluorescence measurements were made by Fluostar Optima (BMG Labtech, Germany) plate reader (485 nm excitation and 520 nm emission filters) operating in either direct whole-well read mode or spatially resolved well-scanning mode (15 × 15 pixels per well). In the detergent mediated membrane leakage and rupture experiments a final concentration of 8 μM SDS was added directly into eDIB containing wells. Following range finding experiments 16 eDIBs were exposed to 8 μM SDS, the fluorescence data for the 8 non-ruptured eDIBs was plotted against 8 control eDIBs with no-SDS added (Fig. 3b). PGLa and Magainin II activity was assessed by an endpoint of membrane rupture of one or more of the 10 encapsulated droplet membranes, which would give rise to a positive fluorescent response. Unlike SDS, no passive leakage was observed by plate-reader detection as a preceding effect. Due to the binary nature of the fluorescent response, experiments were conducted by imaging eDIB-filled plates overnight and imaging using a commercial blue LED lamp. Each peptide concentration combination was evaluated on 6 replicate eDIBs and a 2D screen heatmap (Fig. 4b) constructed to reflect proportion of eDIBs eliciting a positive fluorescent response. Using Matlab 2018 (Mathsoft) this data was plotted to reflect total peptide concentration and PGLa:MagininII mixing ratio against observed activity (Fig. 4c). Interpolation of this 3D space to find the-tie line traversing 50% maximal activity determines the total peptide concentration eliciting a 50% maximal response across the range of peptide mixing ratios. This was plotted as an ED50 plot (Fig. 4d) showing the required total peptide concentration to elicit a 50% maximal response across peptide mixing ratios.
Electrophysiology
Custom Ag/AgCl electrodes consisted of a tapered 1 mm internal diameter glass capillary (CM Scientific, UK) with 0.75 mm length of exposed silver wire at the tapered end, sealed with PDMS (ESI†). Using a micromanipulator, the electrodes were inserted separately into the internal aqueous cores and the alginate shell to probe the bilayer segregating the internal droplet with the external well media. An Axopatch 200B (Molecular Devices, USA) was used for voltage clamped current measurements under an applied potential of −30 mV. Data was acquired with WinEDR (University of Strathclyde) and analogue filtered at 5 kHz. Electrophysiology traces were digitally filtered post-acquisition with either a 1 kHz or 100 Hz low-pass filter.Conflicts of interest
There are no conflicts to declare.Acknowledgements
O. K. C. is a Cardiff University SBP Research Fellow. We acknowledge funding from Wellcome Trust (ISSF 204824-Z-16-Z) in support of D. K. B. and W. D. J. during the course of this work and current funding from H2020-EU.1.2.2. - FET Proactive Grant agreement ID: 824060 – ‘ACDC’).References
- C. Peetla, A. Stine and V. Labhasetwar, Mol. Pharmaceutics, 2009, 6, 1264–1276 CrossRef CAS PubMed.
- C. G. Siontorou, G.-P. Nikoleli, D. P. Nikolelis and S. K. Karapetis, Membranes, 2017, 7(3), 38 CrossRef PubMed.
- J. T. Groves, N. Ulman and S. G. Boxer, Science, 1997, 275, 651–653 CrossRef CAS PubMed.
- J. Andersson and I. Köper, Membranes, 2016, 6(2), 30 CrossRef PubMed.
- X. Wang, Y. Zhang, H. Bi and X. Han, 2016.
- D. Stamou, C. Duschl, E. Delamarche and H. Vogel, Angew. Chem., 2003, 42, 5580–5583 CrossRef CAS PubMed.
- D. H. Kang, H.-S. Jung, N. Ahn, J. Lee, S. Seo, K.-Y. Suh, J. Kim and K. Kim, Chem. Commun., 2012, 48, 5313–5315 RSC.
- A.-E. Saliba, I. Vonkova, S. Ceschia, G. M. Findlay, K. Maeda, C. Tischer, S. Deghou, V. V. Noort, P. Bork, T. Pawson, J. Ellenberg and A.-C. Gavin, Nat. Methods, 2014, 11, 47–50 CrossRef CAS PubMed.
- A.-E. Saliba, I. Vonkova, S. Deghou, S. Ceschia, C. Tischer, K. G. Kugler, P. Bork, J. Ellenberg and A.-C. Gavin, Nat. Protoc., 2016, 11, 1021–1038 CrossRef CAS PubMed.
- S. Mathiasen, S. M. Christensen, J. J. Fung, S. G. F. Rasmussen, J. F. Fay, S. K. Jorgensen, S. Veshaguri, D. L. Farrens, M. Kiskowski, B. Kobilka and D. Stamou, Nat. Methods, 2014, 11, 931–934 CrossRef CAS PubMed.
- S. Veshaguri, S. M. Christensen, G. C. Kemmer, G. Ghale, M. P. Møller, C. Lohr, A. L. Christensen, B. H. Justesen, I. L. Jørgensen, J. Schiller, N. S. Hatzakis, M. Grabe, T. G. Pomorski and D. Stamou, Science, 2016, 351, 1469–1473 CrossRef CAS PubMed.
- S. C. Saha, F. Thei, M. R. R. de Planque and H. Morgan, Sens. Actuators, B, 2014, 199, 76–82 CrossRef CAS.
- B. Le Pioufle, H. Suzuki, K. V. Tabata, H. Noji and S. Takeuchi, Anal. Chem., 2008, 80, 328–332 CrossRef CAS PubMed.
- J. M. del Rio Martinez, E. Zaitseva, S. Petersen, G. Baaken and J. C. Behrends, Small, 2015, 11, 119–125 CrossRef CAS PubMed.
- S. Maher, H. Basit, R. J. Förster and T. E. Keyes, Bioelectrochemistry, 2016, 112, 16–23 CrossRef CAS PubMed.
- D.-H. Kang, W. B. Han, N. Choi, Y.-J. Kim and T. S. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 40401–40410 CrossRef CAS PubMed.
- H. Bayley, B. Cronin, A. Heron, M. A. Holden, W. L. Hwang, R. Syeda, J. Thompson and M. Wallace, Mol. BioSyst., 2008, 4, 1191–1208 RSC.
- C. E. Stanley, K. S. Elvira, X. Z. Niu, A. D. Gee, O. Ces, J. B. Edel and A. J. Demello, Chem. Commun., 2010, 46, 1620–1622 RSC.
- N. E. Barlow, G. Bolognesi, A. J. Flemming, N. J. Brooks, L. M. C. Barter and O. Ces, Lab Chip, 2016, 16, 4653–4657 RSC.
- D. K. Baxani, A. J. L. Morgan, W. D. Jamieson, C. J. Allender, D. A. Barrow and O. K. Castell, Angew. Chem., Int. Ed., 2016, 55, 14240–14245 CrossRef CAS.
- P. Carreras, Y. Elani, R. V. Law, N. J. Brooks, J. M. Seddon and O. Ces, Biomicrofluidics, 2015, 9, 064121 CrossRef CAS PubMed.
- T. Trantidou, M. Friddin, Y. Elani, N. J. Brooks, R. V. Law, J. M. Seddon and O. Ces, ACS Nano, 2017, 11(7), 6549–6565 CrossRef CAS PubMed.
- B. Schlicht and M. Zagnoni, 2013.
- V. C. Stimberg, J. G. Bomer, I. V. Uitert, A. V. D. Berg and S. L. Gac, Small, 2013, 9, 961 CrossRef.
- S. Leptihn, O. K. Castell, B. Cronin, E.-H. Lee, L. C. M. Gross, D. P. Marshall, J. R. Thompson, M. Holden and M. I. Wallace, Nat. Protoc., 2013, 8, 1048–1057 CrossRef CAS PubMed.
- M. A. Czekalska, T. S. Kaminski, S. Jakiela, K. T. Sapra, H. Bayley and P. Garstecki, Lab Chip, 2015, 15, 541–548 RSC.
- M. A. Czekalska, T. S. Kaminski, K. Makuch and P. Garstecki, Sens. Actuators, B, 2019, 286, 258–265 CrossRef CAS.
- J. L. Poulos, T.-J. Jeon, R. Damoiseaux, E. J. Gillespie, K. A. Bradley and J. J. Schmidt, Biosens. Bioelectron., 2009, 24, 1806–1810 CrossRef CAS PubMed.
- R. Syeda, M. A. Holden, W. L. Hwang and H. Bayley, J. Am. Chem. Soc., 2008, 130, 15543–15548 CrossRef CAS PubMed.
- J. L. Poulos, S. A. Portonovo, H. Bang and J. J. Schmidt, J. Phys.: Condens. Matter, 2010, 22, 454105 CrossRef CAS PubMed.
- R. Kawano, Y. Tsuji, K. Sato, T. Osaki, K. Kamiya, M. Hirano, T. Ide, N. Miki and S. Takeuchi, Sci. Rep., 2013, 3, 1995 CrossRef PubMed.
- S. Huang, M. Romero-Ruiz, O. K. Castell, H. Bayley and M. I. Wallace, Nat. Nanotechnol., 2015, 10, 986–991 CrossRef CAS.
- O. K. Castell, J. Berridge and M. I. Wallace, Angew. Chem., Int. Ed., 2012, 51, 3134–3138 CrossRef CAS PubMed.
- T. Tonooka, K. Sato, T. Osaki, R. Kawano and S. Takeuchi, Small, 2014, 10, 3275–3282 CrossRef CAS PubMed.
- R. Watanabe, N. Soga, T. Yamanaka and H. Noji, Sci. Rep., 2014, 4, srep07076 Search PubMed.
- R. Watanabe, N. Soga, D. Fujita, K. V. Tabata, L. Yamauchi, S. Hyeon Kim, D. Asanuma, M. Kamiya, Y. Urano, H. Suga and H. Noji, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- N. Soga, R. Watanabe and H. Noji, Sci. Rep., 2015, 5, srep11025 CrossRef PubMed.
- M.-A. Nguyen, B. Srijanto, C. P. Collier, S. T. Retterer and S. A. Sarles, Lab Chip, 2016, 16, 3576–3588 RSC.
- J. Zerweck, E. Strandberg, O. Kukharenko, J. Reichert, J. Bürck, P. Wadhwani and A. S. Ulrich, Sci. Rep., 2017, 7, 13153 CrossRef PubMed.
- M. Zasloff, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 5449–5453 CrossRef CAS PubMed.
- P. Tremouilhac, E. Strandberg, P. Wadhwani and A. S. Ulrich, J. Biol. Chem., 2006, 281, 32089–32094 CrossRef CAS PubMed.
- K. Matsuzaki, Y. Mitani, K. Y. Akada, O. Murase, S. Yoneyama, M. Zasloff and K. Miyajima, Biochemistry, 1998, 37, 15144–15153 CrossRef CAS PubMed.
- H. V. Westerhoff, M. Zasloff, J. L. Rosner, R. W. Hendler, A. De Waal, A. Vaz Gomes, P. M. Jongsma, A. Riethorst and D. Juretić, Eur. J. Biochem., 1995, 228, 257–264 CrossRef CAS PubMed.
- H. Ahyayauch, M. Bennouna, A. Alonso and F. M. Goñi, Langmuir, 2010, 26, 7307–7313 CrossRef CAS PubMed.
- E. Strandberg, P. Tremouilhac, P. Wadhwani and A. S. Ulrich, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 1667–1679 CrossRef CAS PubMed.
- E. Han and H. Lee, RSC Adv., 2015, 5, 2047–2055 RSC.
- J. Zerweck, E. Strandberg, J. Bürck, J. Reichert, P. Wadhwani, O. Kukharenko and A. S. Ulrich, Eur. Biophys. J., 2016, 45, 535–547 CrossRef CAS PubMed.
- T. Hara, Y. Mitani, K. Tanaka, N. Uematsu, A. Takakura, T. Tachi, H. Kodama, M. Kondo, H. Mori, A. Otaka, F. Nobutaka and K. Matsuzaki, Biochemistry, 2001, 40, 12395–12399 CrossRef CAS PubMed.
- K. T. Nguyen, S. V. Le Clair, S. Ye and Z. Chen, J. Phys. Chem. B, 2009, 113, 12358–12363 CrossRef CAS PubMed.
- P. M. Dijkman, O. K. Castell, A. D. Goddard, J. C. Munoz-Garcia, C. de Graaf, M. I. Wallace and A. Watts, Nat. Commun., 2018, 9, 1–14 CrossRef CAS PubMed.
- V. R. Schild, M. J. Booth, S. J. Box, S. N. Olof, K. R. Mahendran and H. Bayley, Sci. Rep., 2017, 7, 46585 CrossRef PubMed.
- M. J. Booth, V. R. Schild, A. D. Graham, S. N. Olof and H. Bayley, Sci. Adv., 2016, 2, e1600056 CrossRef PubMed.
- H. M. G. Barriga, P. Booth, S. Haylock, R. Bazin, R. H. Templer and O. Ces, J. R. Soc., Interface, 2014, 11, 20140404 CrossRef PubMed.
- S. Leptihn, J. R. Thompson, J. C. Ellory, S. J. Tucker and M. I. Wallace, J. Am. Chem. Soc., 2011, 133, 9370–9375 CrossRef CAS PubMed.
- A. Fischer, M. A. Holden, B. L. Pentelute and R. J. Collier, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16577–16581 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d1sm01111a |
| This journal is © The Royal Society of Chemistry 2022 |