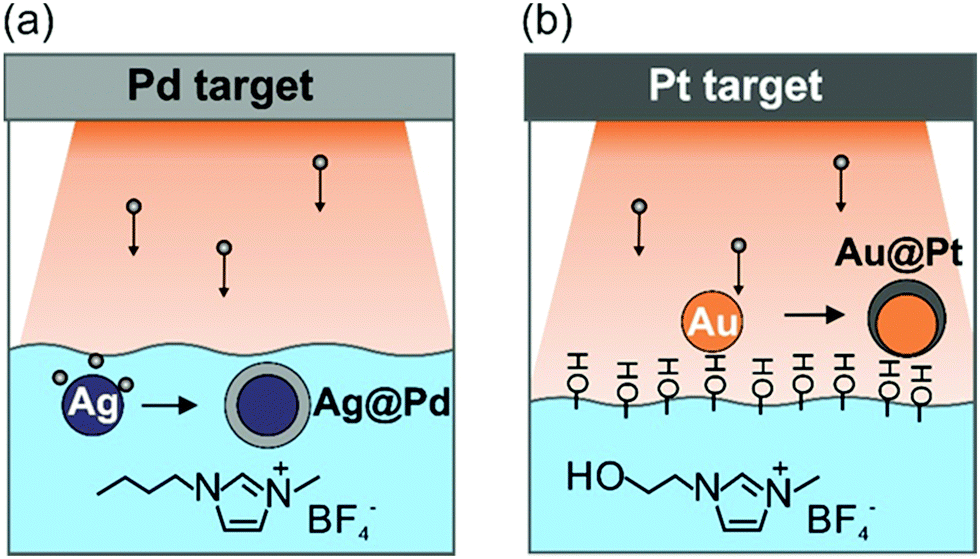
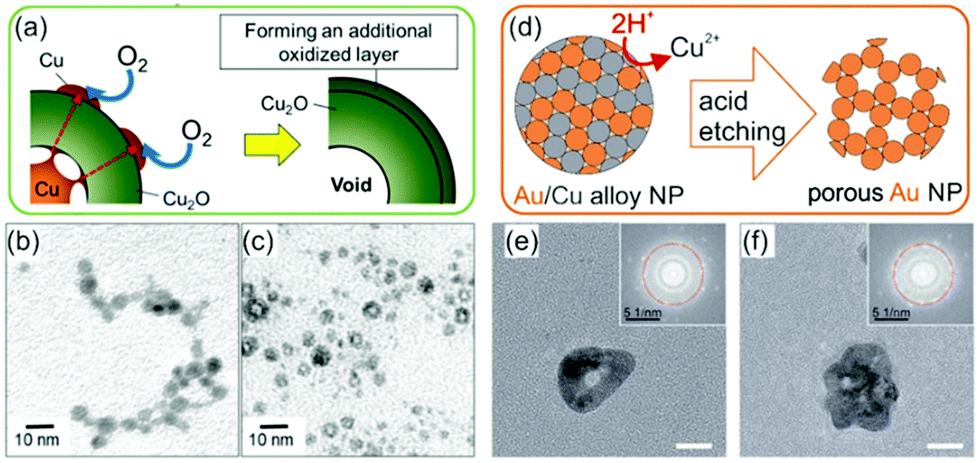
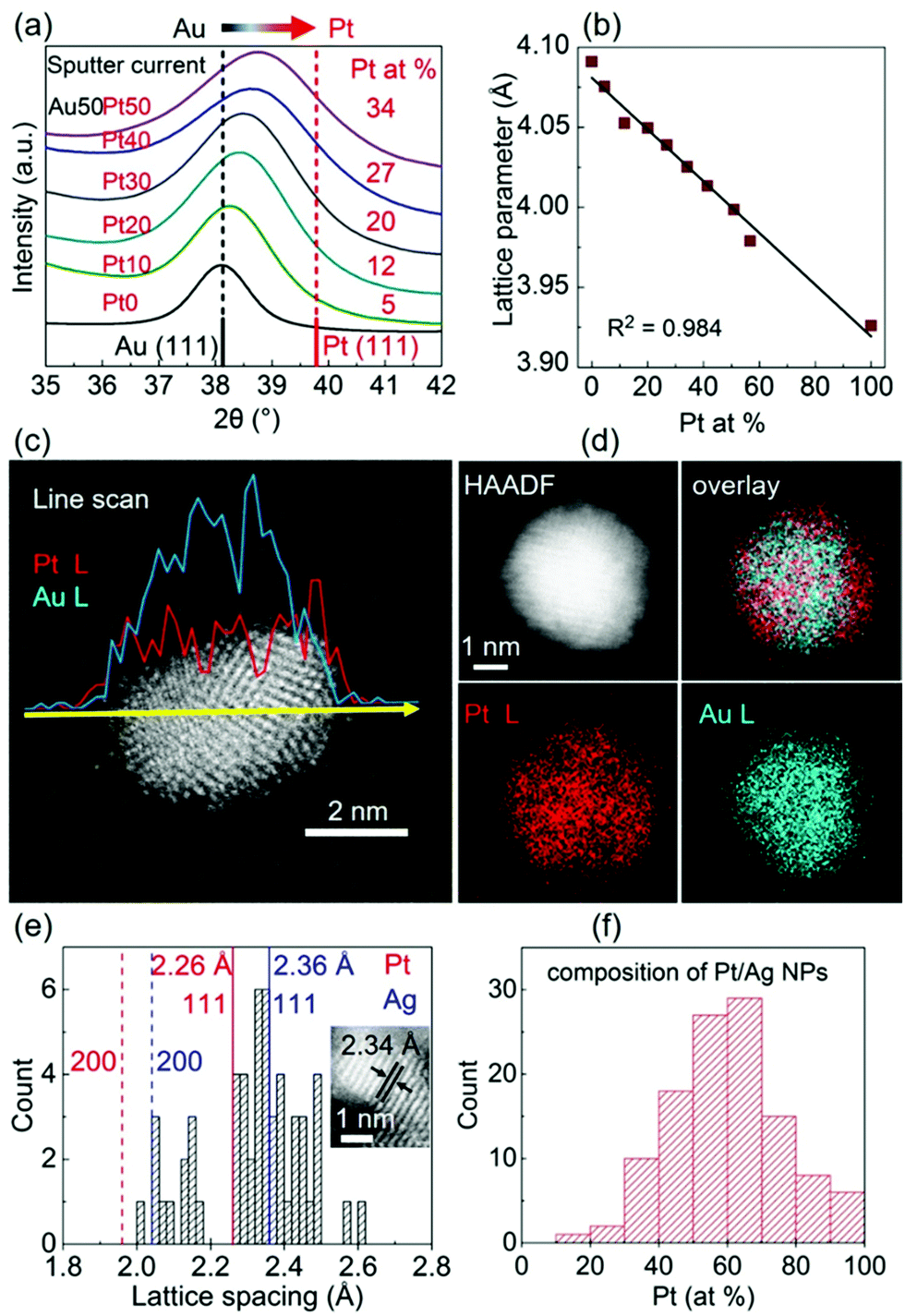
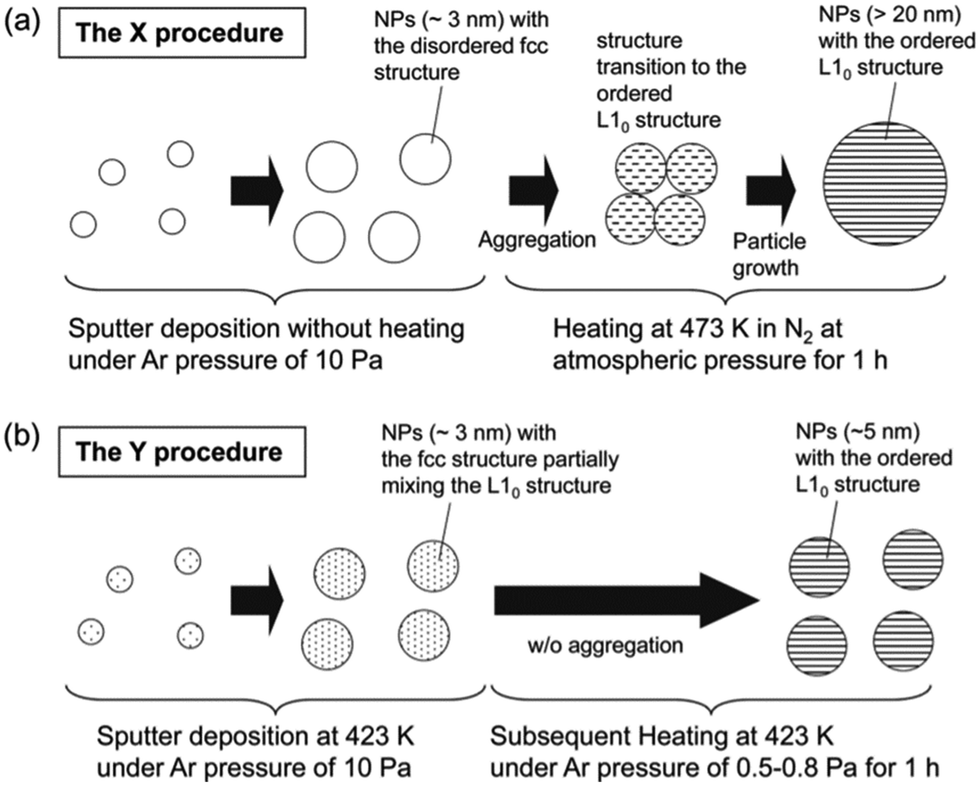
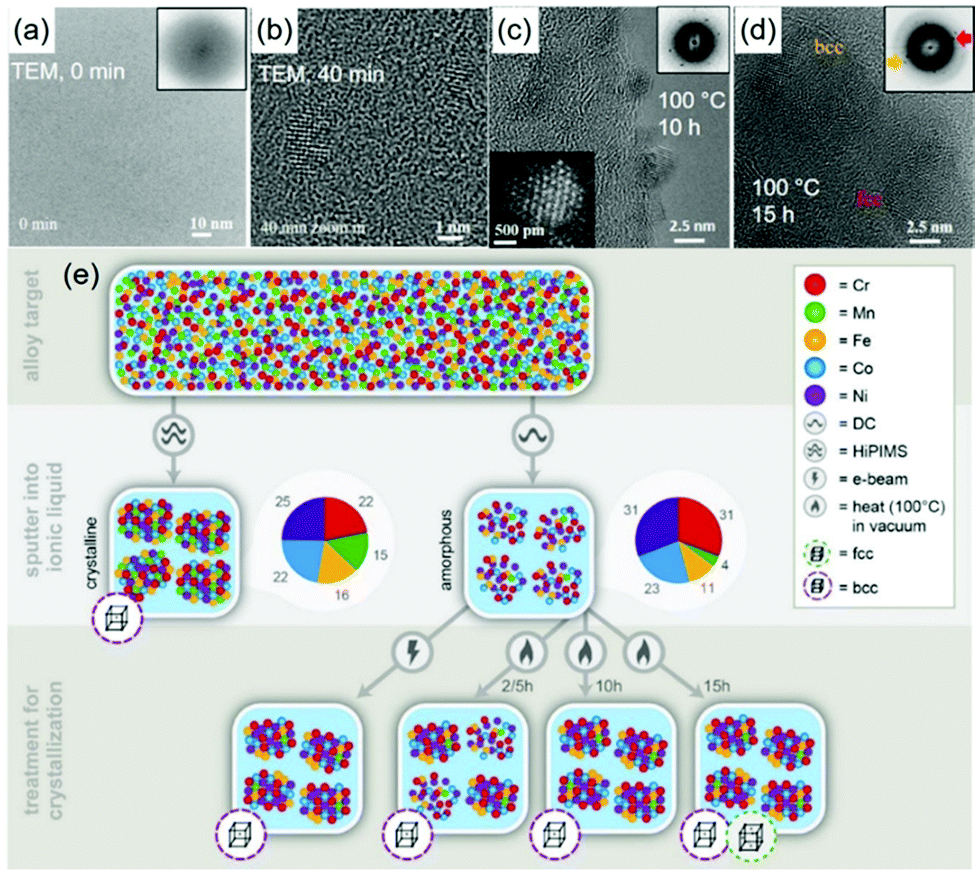
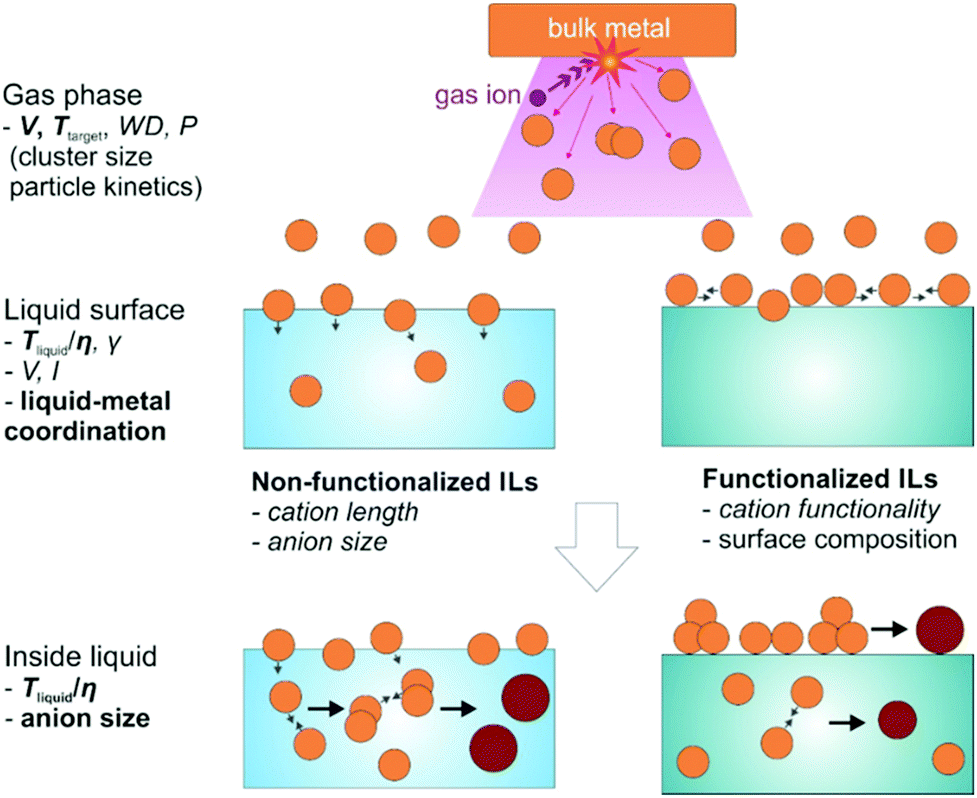
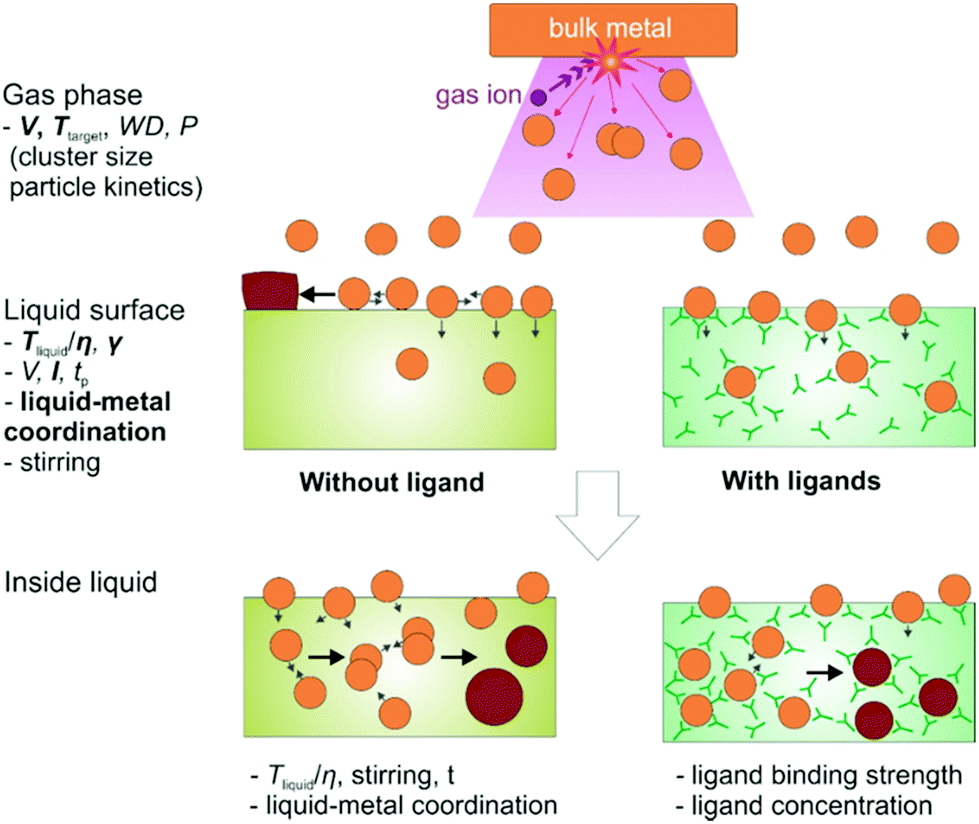
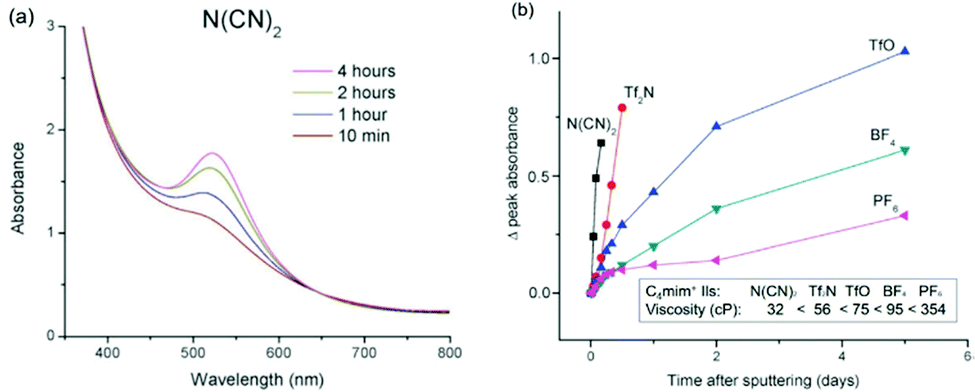
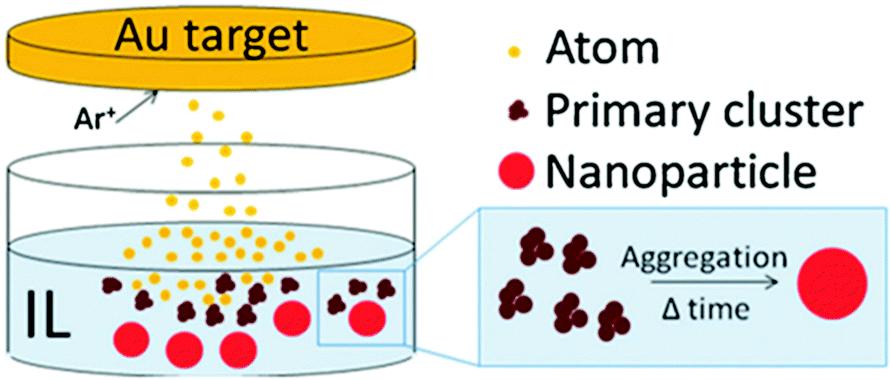
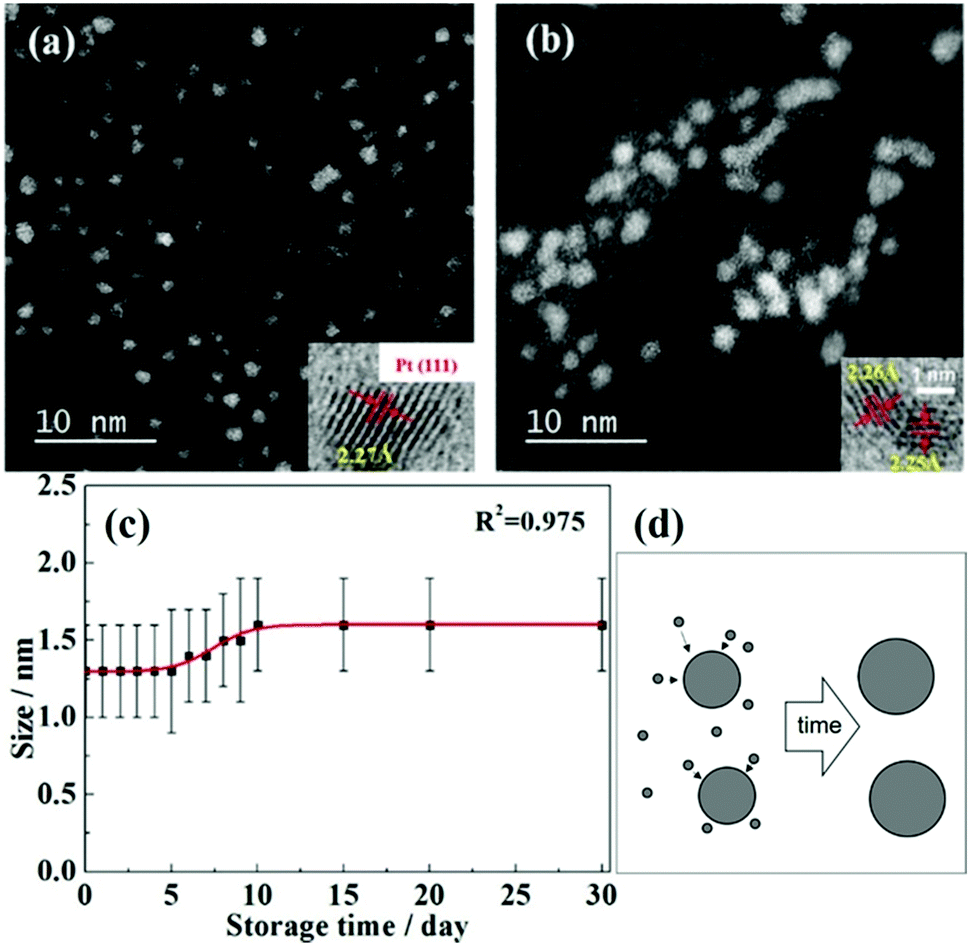
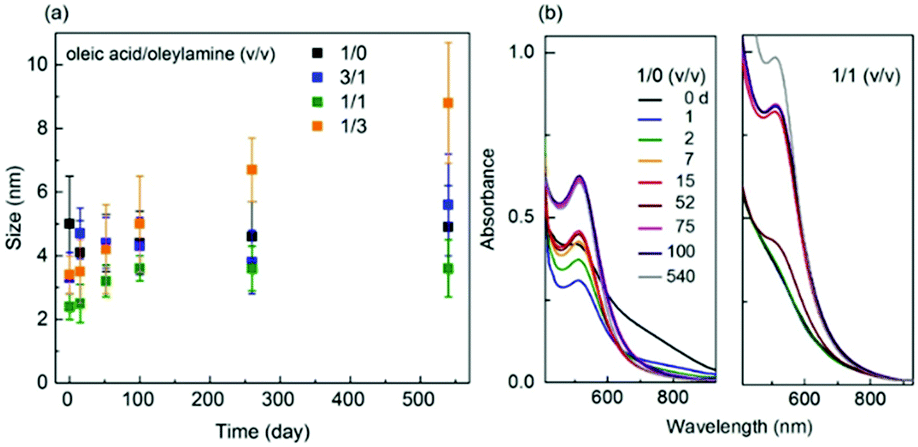
DOI:
10.1039/D1SM01002F
(Review Article)
Soft Matter, 2022,
18, 19-47
Control of nanoparticles synthesized via vacuum sputter deposition onto liquids: a review
Received
7th July 2021
, Accepted 22nd November 2021
First published on 23rd November 2021
Abstract
Sputter deposition onto a low volatile liquid matrix is a recently developed green synthesis method for metal/metal oxide nanoparticles (NPs). In this review, we introduce the synthesis method and highlight its unique features emerging from the combination of the sputter deposition and the ability of the liquid matrix to regulate particle growth. Then, manipulating the synthesis parameters to control the particle size, composition, morphology, and crystal structure of NPs is presented. Subsequently, we evaluate the key experimental factors governing the particle characteristics and the formation of monometallic and alloy NPs to provide overall directions and insights into the preparation of NPs with desired properties. Following that, the current understanding of the growth and formation mechanism of sputtered particles in liquid media, in particular, ionic liquids and liquid polymers, during and after sputtering is emphasized. Finally, we discuss the challenges that remain and share our perspectives on the future prospects of the synthesis method and the obtained NPs.
1. Introduction
1.1. Nanoparticles
Nanoparticles (NPs), objects with at least one dimension at the nanoscale, 1–100 nm, can enable new phenomena and novel properties.1–4 In 1987, Haruta et al. reported Au NPs of sizes below 5 nm as an effective catalyst for CO oxidation below 0 °C despite the long-term well-known “inert” characteristics of bulk Au.5 This is due to the large number of surface atoms with unsaturated bonds compared with the bulk counterpart.6,7 This finding marks renewed research interest and boosts various applications of NPs.8,9 The assembly formation or crystallization of metal nanoparticles has been intensively studied with various organic templates.10–13 Besides, adding or varying the metal components of NPs also helps create and tailor their properties.14,15 Bi-, tri-, and multi-metal NPs can form solid solution alloys, intermetallic compounds, and heterogeneous structures such as core@shell, island, and Janus structures, and precipitates of one phase in the others.16–30 In 1989, Toshima and Yonezawa pioneered core–shell bimetallic structured metal nanoparticles for effective hydrogenation catalysts.14,31,32 Their non-linear composition and structure-dependent properties and novel electronic structures have been observed due to the presence of two or more metals in a certain configuration.18,20,26–32 Hence, the size, structure, and composition can jointly regulate the particle properties.16–32 A large number of combinations of these parameters provide high versatility and capability for designing properties to such an extent that one does not have with monometallic NPs.16–32 On the other hand, the optical properties of metal nanoparticles are also interesting. Gold nanoparticles covered with fluorescent organic compounds show strong fluorescence.33 Surface enhanced Raman scattering (SERS) of metal nanoparticles can also be achieved by controlled aggregation of metal nanoparticles.34 Following this, precise control of NP characteristics for desired properties becomes crucial, highly necessary, and challenging in NP synthesis.
1.2. Synthetic approaches
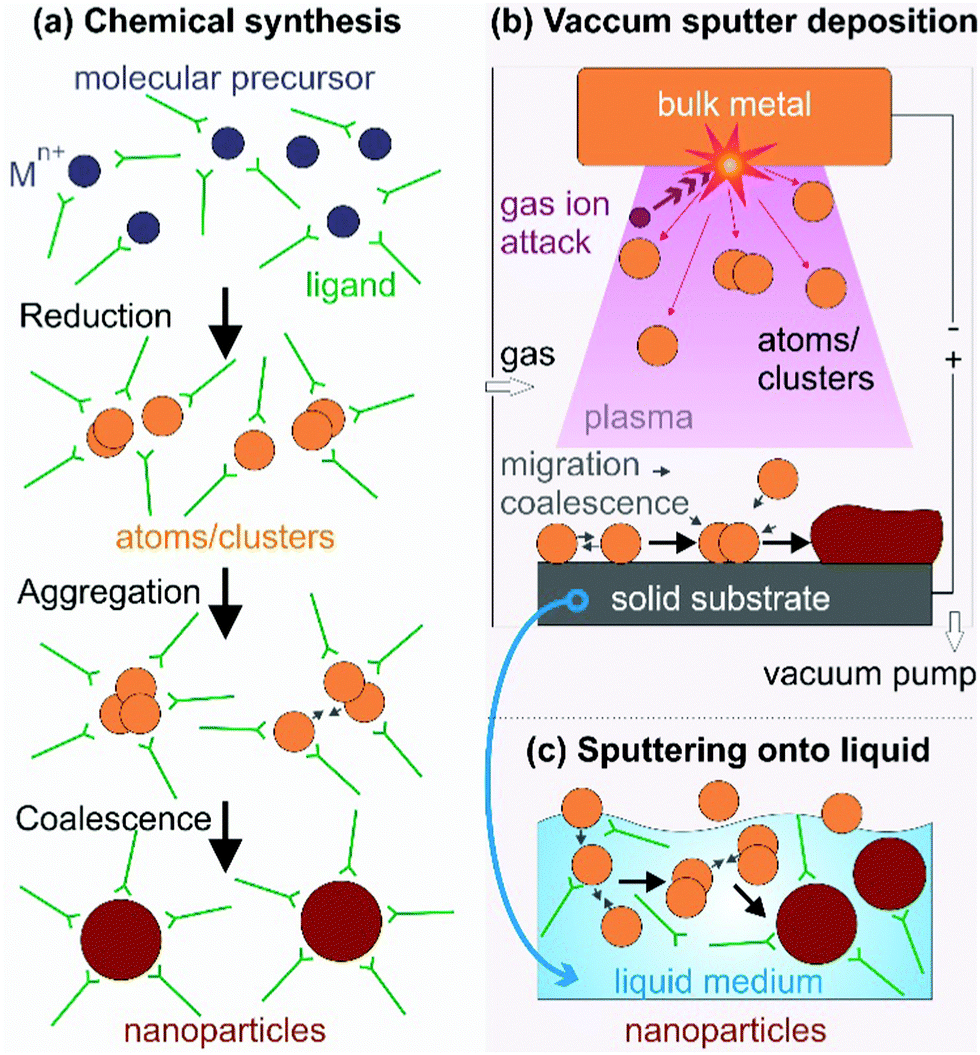
Synthesis of NPs generally includes chemical and physical methods (Fig. 1). Below the key features of the synthesis methods are discussed in terms of how atoms/clusters form, particle growth, and the capability of control of NP characteristics as the basics for understanding the fundamental and novel aspects of our method mainly focused on in this review, sputter deposition onto liquids, discussed in Section 1.3.
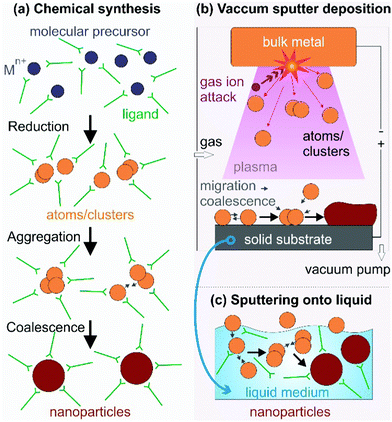 |
| | Fig. 1 NP synthesis via (a) reduction of the molecular metal precursor and growth regulated with capping ligands, (b) a top-down technique based on vacuum sputter deposition of a bulk metal to form metal atoms/clusters and their growth on a solid substrate, and (c) sputter deposition onto a liquid when the solid substrate in (b) is replaced with a low/non-volatile liquid. The capping ligand can be added to the liquid matrix in (c) for further control of particle growth. | |
1.2.1. Chemical syntheses.
Zero-valent metal atoms/clusters (aggregation of several atoms) are generated from molecular sources by thermal decomposition and/or photodecomposition of metal complexes or reduction of metal precursors (Fig. 1a).4,16,17,21,25–37 In chemical reductions, reductants, e.g., hydrazine, sodium borohydride, elemental hydrogen, and alcohols, donate electrons to the metal element in the oxidized states to form metal atoms/clusters.16,17,21,25–37 The subsequent nucleation and growth of the metal atoms/clusters result in metallic NPs. In this process, capping agents are applied to direct the particle growth, to bind to particle surfaces for compensating for the high surface energy of the NPs, and to support precise control of particle morphologies, dispersibility, and chemical/colloidal stability. They can be surfactants (hexadecyltrimethylammonium bromide (CTAB), sodium citrate),16 organic molecules (oleic acid, carboxylic acids,38 oleylamine, trioctylphosphine (TOP), 3-(N,N-dimethyldodecylammonio)propanesulfonate (SB12)),17 and polymers (poly(vinyl pyrrolidone) (PVP), polyvinyl alcohol (PVA), polyethylenimine (PEI), gelatin, peptides).4,16,25,31,39–41 The synthesis of bi-, tri-, and multi-metal NPs is often designed based on the desired structures with high versatility. For instance, solid solution NPs were obtained by co-reduction,24,26–32 whereas step-wise reduction and growth was applied for metal core@shell structures.16,19,25
1.2.2. Physical techniques.
Physical techniques are also called top-down methods for creating metal NPs. They are related to dividing bulk metals into smaller units (atoms, clusters, particles) in physical processes.6,7,18,20,23,42–48 Typical physical techniques include physical vapor deposition (PVD),6,7,23,42 laser ablation (PLA),19 pulsed laser deposition (PLD),43,44 microwave irradiation,45,46 ultra-sonication,47 and ball milling,48 to name a few. Taking metal atoms/clusters from the bulk (condensed phase) requires an enormous amount of energy. The energy can be supplied by a heat source (high temperature) and/or highly kinetic gas ions (PVD, Fig. 1b), a laser source (PLA, PLD), microwave power, and high temperature local heating (ultra-sonication), among others. Fig. 1b illustrates a PVD process, i.e., sputter deposition in a vacuum chamber. The gas molecules are ionized (positive charge) and accelerated by the applied negative electric potential to the bulk metal target. They hit the target and exchange their momentum to eject metal atoms and/or clusters (neutral charge), which then migrate, coalesce, nucleate, and grow to form NPs or a film on a solid substrate. Physical methods are advantageous in terms of high reproducibility and the absence of toxic solvents, reductants, and by-products. Some methods, e.g., molecular beam epitaxy (MBE),49 allow for high purity and fine growth of nanostructures at the molecular level. However, compared with chemical synthesis, physical methods are often less versatile in control of the particle morphology and higher cost.
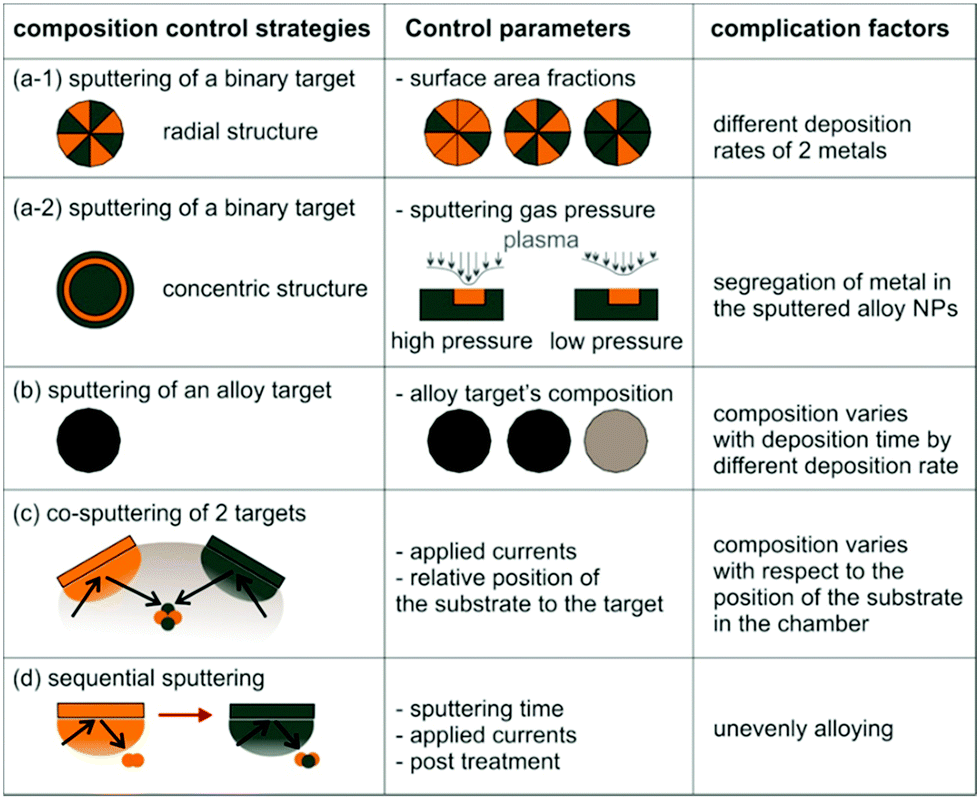
1.3. Sputter deposition onto liquids to produce metal and alloy NPs
Sputter deposition onto liquids (Fig. 1c) has important features of both the conventional physical technique (PVD) and wet chemical synthesis. The metal atoms/clusters are created via vacuum sputtering (Fig. 1b). Direct current (DC), radiofrequency (RF), and magnetron sputtering are applicable for ionizing gases and for metal targets.50–60 The DC sputtering conducted at typical tens of mPa to tens of Pa is cost-effective. However, positive charge accumulation can occur on the surfaces of dielectric targets (semiconductors, insulators such as oxides), causing arc-discharge or termination of sputtering.52 The charge-up effect is prevented in RF sputtering by applying an alternating electric potential of current in a vacuum at a radiofrequency to periodically supply electrons to the positively charged-up target surface after ionized gas bombardment.52–54 Magnetron sputtering supplies a magnetic field to trap electrons by directing them to travel around the magnetic field lines and to collide with neutral gas near the target surface.55,56 This saves energy loss via gas collisions, helps generate plasma at low pressures, and, hence, gives higher sputtering rates.56 Metal atoms/clusters of various metal elements can be produced simultaneously (co-sputtering) or subsequently (step-wise sputtering) for making desired alloy or core/shell nanostructures.57–60 Atomization can apply to almost all metals despite the difference in their redox potentials. This is crucial and beneficial for synthesis of alloy and core@shell NPs.57–60 By introducing non- or low volatile liquids with or without additional capping agents into the vacuum sputtering chamber, the nucleation and growth of the metal atoms/clusters to form NPs have been regulated in a liquid matrix (Fig. 1c).61–80Fig. 2 shows the TEM images of the sputtered NPs in various liquids. The particle growth on the liquid surface and in the liquid61–82 can be different from the typical model of thin film growth processes on solid substrates.83–85 Varying the chemical and physical properties of liquids can mediate particle growth, and the size, structure, and dispersibility of NPs can be tuned by varying the liquid–particle and particle–particle interaction in the liquid.61–80 Thus, sputtering onto liquids provides new control of particle characteristics (size, structure, composition, dispersibility, stability) compared with conventional chemical and physical methods.
 |
| | Fig. 2 TEM images of (a) 15 nm Fe NPs sputtered in silicone oil,62 (b) 5.5 nm Au NPs in C2mim+/BF4−,63 (c) 1–10 nm Au NPs in PEG (MW = 600),72 (d) 2.4 nm Au NPs in oleic acid and oleylamine,80 and (e) 1.3 nm Au NPs in molten salt by sputtering.70 After sputtering onto liquids, the NPs were diluted and collected onto TEM grids then dried; their TEM images were taken with a normal TEM. (Reproduced with permission from ref. 62, 63, 70, 72 and 80. Copyright 1999 Elsevier, 2006 AIP Publishing, 2011 The American Chemical Society, 2020 The American Chemical Society, and 2010 The Royal Society of Chemistry, respectively.) | |
Liquid medium for vacuum sputtering can be any liquid with a sufficiently low vapor pressure to be introduced inside a vacuum chamber (Table 1). Sputtering onto silicone oil with and without surfactant was initially used (Fig. 2a).61,62 However, without surfactant aggregation of NPs in silicone oil was observed owing to the weak binding of the oil to the NPs.62 Ionic liquids (ILs) are clean media for vacuum sputtering of NPs and thin films.63–69 This is advantageous for obtaining stable dispersions of uniform metal and alloy NPs without using stabilizing agents (Fig. 2b).63,65–69 Thus, an intensive study of sputtering onto ILs was conducted. Biocompatible liquids such as polymers (polyethylene glycol, PEG), vegetable oils, and carboxylic acids,71,72,79,80 which are cheaper than ILs, have also been studied as liquid matrices in sputtering (Fig. 2c and d). The above-mentioned liquids often resulted in NPs of about 2 nm or more. Strong capping agents and functionalized liquids, e.g., pentaerythritol tetrakis(3-mercaptopropionate) (PEMP), thiocholine chloride (TC), and mercaptoundecanoic acid (MUA), on the other hand, enabled obtaining nanoclusters of sizes below 2 nm with fluorescent emission (Fig. 2e).70,73,75–77 For catalyst synthesis, support materials (C, TiO2, SiO2) were added to liquid media for decorating metal NPs on the support in a single step.86–95
Table 1 Liquid media used in vacuum sputtering
| Year |
Liquid |
Metal |
Structure |
Particle size, d (nm) |
Ref. |
| 1996 |
Silicone oil |
Ag |
Film |
|
61
|
| 1999 |
Silicone oil |
Ag |
NPs |
5–15 (with sarcosyl oleic acid) |
62
|
| Fe |
|
15 |
| 9 (with sarcosyl oleic acid or a polyalkylene amine derivative) |
| 2006 |
Ionic liquids |
Au |
NPs |
1.9 ± 0.46, 5.5 ± 0.86 |
63
|
| 2007 |
Ag |
Film, NPs |
|
64
|
| 2008–2010 |
Au/Ag, In, Pt, Pd |
NPs |
2–10 (Au, Ag), 8 (In, InOx), 2.2–3 (Pt, Pd) |
65–69
|
| 2010 |
Molten salt |
Au |
Nanoclusters |
1.3 ± 0.3 |
70
|
| 2010 |
Vegetable oil |
Au |
NPs |
3.6 ± 1.0 (castor oil) |
71
|
| 2011 |
PEG |
Au |
NPs |
<10 nm (Fig. 2c) |
72
|
| 2011 |
PEMP |
Au |
Nanoclusters |
<1 |
73
|
| 2011 |
PEEL |
Au |
NPs |
2.1 ± 0.7 |
73
|
| 2014 |
TC–urea |
Au |
NPs |
5.0 ± 0.5 |
74
|
| 2015 |
TC/diglycerol (DG) |
Au |
Nanoclusters |
<2 |
75
|
| 2015 |
MUA/PEG |
Ag |
Nanoclusters |
2.2–2.4 |
76
|
| 2016 |
Cu |
1.6 |
77
|
| 2018 |
Glycerol, squalene |
Au, Ag |
Film, particles |
|
79
|
| 2020 |
Oleic acid + oleylamine |
Au |
NPs |
2.4 ± 0.4 |
80
|
2. Control of particle size and uniformity
Sputter deposition onto liquids to produce NPs has been investigated in terms of sputtering parameters, liquid media, their interaction with the sputtered particles and their impact on particle growth. The results obtained over about 3 decades help in gaining basic knowledge of control of particle size, structure, alloy formation, and the growth of metal and metal alloy NPs. Size control could be realized by varying experimental parameters. Two main groups of parameters include: (i) parameters related to the sputtering process which affect the generation of atoms/clusters, their size, and collision in the gas phase and penetration into the liquid and (ii) parameters related to the growth of NPs in the liquid (Fig. 3). The first group of parameters includes the kind of gas and gas pressure (P), target–substrate distance (working distance, WD), temperature of the target (Ttarget), discharge voltage (V), sputtering current (I), and kind of metal used as the target. The second group includes the temperature of the liquid (Tliquid), surface tension (γ), viscosity (η), type of liquid and functional group, stirring speed (vstir), sputtering time (t), and deposition period (tp). Parameters such as Tliquid, the functional group, the type of liquid, and vstir can be varied to adjust the physical and chemical properties of the liquid, and thus the interaction of the liquid with the NPs. The sputtering time and period are not directly related to the liquid physicochemical properties, but they take part in control of the concentration of the deposited particles on the surface and inside the liquid. Therefore, together with the surface tension, viscosity, temperature, and functionality of the liquid, they also affect the particles’ growth, size, and size distribution. Because the particles grow on the liquid–vacuum interface and inside the liquid, discussion of the effect of the first group of parameters cannot be done independently from that of the second group.
 |
| | Fig. 3 Experimental parameters regulate particle size in sputtering onto liquids. | |
2.1. Impact of the vacuum sputtering parameters
Perhaps the studies of the impact of vacuum sputtering parameters on the size of deposited NPs in liquids contribute to the foundation of the field. However, it is also challenging to draw a general conclusion of the effect of each sputtering parameter on particle size. This is because different experimental set-ups, liquid matrices, and sets of deposition parameters have been used by research groups, and hence, a direct comparison of various studies is not always possible and care must be taken. Hatakeyama et al. (2011) reported the effect of various sputtering parameters on the particle size of Au NPs in an ionic liquid.96 Wender et al. (2013) reviewed the impact of vacuum deposition parameters on the size of NPs prepared using ionic liquids and vegetable oils.97 Cha et al. (2016) added in a short review of NPs sputtered onto polymers and supporters in polymers.98 Here we discuss the impact of each synthesis parameter with up-to-date knowledge and for a wider range of liquid matrices with a focus on size control and the formation mechanism.
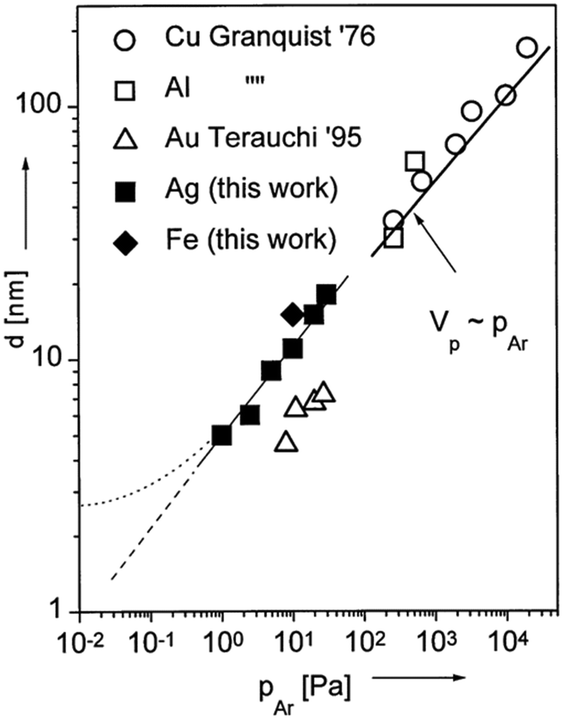
2.1.1. Gas pressure and the kind of gas.
The gas pressure was varied among publications (several Pa to tens of Pa) and there are not always enough comparative data for making a solid judgment of pressure-dependent particle size.62,63,65–80,86–96,99,100 Under identical synthesis conditions except for the gas pressure, Wagener et al. reported that the particle size, d, increased from 5 to 15 nm for an increase in the Ar gas pressure, PAr, from 1 to 30 Pa (Fig. 4).62 They also observed a linear relation between d and PAr for Ag and Fe sputtered onto silicone oil. The trend resembles the tendency observed in gas-phase synthesis of other NPs (Fig. 4).62 Thus, they attributed the increase in particle size under a higher pressure to more condensation and growth of particles in the gas phase via collision. The kind of gas used in sputtering can have some influence on the particle size; for example, smaller Pt NPs were obtained when using Ar (2.24 ± 0.36 nm) compared with those obtained using N2 (3.28 ± 0.60 nm).99 Air was sometimes used for sputtering of Au NPs63,100 and so far Ar has been used in sputtering onto liquids of metal NPs in most reports.
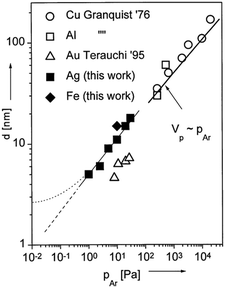 |
| | Fig. 4 Relation between the particle diameters of different metals and working pressures. The references given in the figure are mentioned in ref. 62 and “this work” refers to the results of ref. 62. (Reproduced with permission from ref. 62. Copyright 1999 Elsevier.) | |
2.1.2. Distance between the metal target and the liquid surface.
The Nishikawa group studied the relation between the WD and the size of Au NPs sputtered onto C4mim+/BF4− (I = 20 mA, V = 1000 V, t = 50 min, Tliquid = 20 °C, and without cooling of the target).96 The size of the Au NPs was measured with small angle X-ray scattering (SAXS). There is negligible change in the particle size and full-width at maximum (FWHM) of the size distribution when varying the WD from 25 to 75 mm. This indicates little effect of the WD on the particle size. In other words, the collision and growth in the gas phase are not significant under their sputtering conditions. In another experiment conducted by Asanithi et al., the WD was varied from 100 to 200 mm when Ag was sputtered for 1 s at 50 mA and 350 V onto a Si substrate in a fixed sputtering chamber.101 This resulted in a decrease in particle size from 5.9 ± 1.8 (WD = 100 mm) to 3.8 ± 0.7 nm (WD = 200 mm). The authors hypothesized that the larger WD helped reduce the deposition rate of Ag onto the substrate, which would result in smaller NPs on the substrate. The WD, V, and substrates used by Hatakeyama et al. and Asanithi et al. are considerably different to draw a general conclusion of the impact of the WD on particle size.96,101 In other publications of Nishikawa, Torimoto, Ludwig, Yonezawa, and Wang, a WD of <110 mm was often used. Hence, the effect of the WD can be considered to be insignificant among other factors.
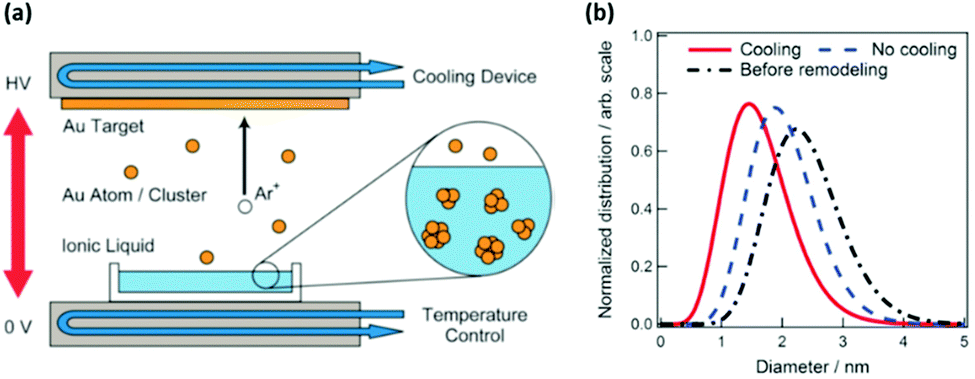
2.1.3. Target temperature.
Hatakeyama et al. were the first and have so far been the only one to evaluate the effect of Ttarget on particle size, in particular, of Au NPs deposited onto an IL, C4mim+/BF4−, when they cooled the target to 20 °C (Fig. 5a) and when no cooling was applied (Ar, P = 13 Pa, WD = 25 mm, V = 1000 V, I = 20 mA, t = 50 min, Tliquid = 50 °C).96 They found that the target without cooling produced larger Au NPs compared with the cooled target (Fig. 5b). Further, without cooling, the target of a smaller surface area (labelled “Before remodelling” in Fig. 5b), which suffered a higher increase in temperature, resulted in even larger Au NPs (Fig. 5b). The authors explained that the higher Ttarget could increase the kinetic energy of the sputtered Au particles, and thus, their mobility and collision in the IL, and the clustering of the particles; both of which lead to the formation of bigger Au NPs.
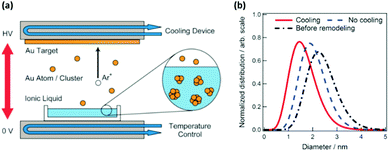 |
| | Fig. 5 (a) Illustration of the sputter deposition device with temperature control of the target and liquid matrix. (b) The size distributions obtained from SAXS measurements of Au NPs synthesized at different Ttarget values by applying cooling (red), no cooling (blue) to the target, and no cooling to a smaller surface-area target (black). (Reproduced with permission from ref. 96. Copyright 2011 The Royal Society of Chemistry.) | |
2.1.4. Applied voltage.
The impact of the applied voltage and current on particle size will be discussed based on the key results obtained by Torimoto, Nishikawa, Dupont, Wang, and Yonezawa (Tables 2 and 3). Particularly, the first four groups studied the voltage/current-dependent particle size with single target sputtering (monometallic, alloy) mainly onto ILs and the last one reported for both single- and double-target head sputtering of mono- and bi-metal alloy NPs onto liquid PEG.
Table 2 Summary of particle sizes varying with deposition parameters
| Metal/year |
Liquid matrix |
P
|
WD |
T
target
|
T
liquid
|
t
|
V
|
I
|
d
|
Ref. |
| Pa |
mm |
°C |
°C |
min |
V |
mA |
nm |
| IL cations: C4mim+: 1-butyl-3-methylimidazolium; TMPA+: N,N,N-trimethyl-N-propylammonium; IL anions: BF4−: tetrafluoroborate; NTf2−: bis(trifluoromethylsulfonyl)amide; TFSA−: bis(trifluoromethanesulfonyl)amide; RT: room temperature. |
| Au, 2011 |
C4mim+/BF4− |
— |
25 |
— |
20 |
50 |
1000 |
20 |
0.75–0.77 (SAXS) |
96
|
| 2 cm2/15.9 cm2 |
75 |
| Ag, 2012 |
Si |
— |
100 |
— |
— |
1 s |
350 |
50 |
5.9 ± 1.8 |
101
|
| 200 |
3.8 ± 0.7 |
| Au, 2010 |
Castor oil |
2 |
50 |
— |
RT |
5 |
260–405 |
8–90 |
2.4–3.7 |
71
|
| 1.15 mL/7 cm2 |
| Au, 2010 |
C4mim+/NTf2− |
2 |
50 |
— |
— |
2.5 |
299–410 |
30–110 |
3.2 ± 0.5 |
102
|
| 4.6 ± 0.7 |
| Au, 2011 |
C4mim+/BF4− |
13–30 |
25 |
20 |
50 |
50 |
700 |
20–40 |
1.7–1.8 |
96
|
| 2 cm2/15.9 cm2 |
1000 |
20–40 |
1.4–1.5 (SAXS) |
| Pd, 2015 |
MWCNTs in C4mim+/BF4− (3 mg/2 mL) |
3 |
— |
— |
RT |
5 |
— |
20–40 |
1.92 ± 0.37–2.03 ± 0.37 |
90
|
| Ag, 2009 |
C4mim+/PF6− |
5 |
85 |
— |
RT |
5 |
— |
20–40 |
5.6–11 |
103
|
| 0.6 mL/10 cm2 |
| Ag, 2012 |
Si |
— |
20 |
— |
— |
2 s |
— |
100 |
More coalescence |
101
|
| 150 |
| Au, 2015 |
PEG |
2–30 |
25 |
— |
RT |
10 (∼40) |
200 |
10 |
4.8 ± 0.8 |
100
|
| 20 |
5.4 ± 1.1 |
| 30 |
5.1 ± 2.8 |
| Pt, 2018 |
PEG, 80 rpm |
2 |
50 |
0 |
30 |
30 |
— |
5–50 |
0.9 ± 0.3–1.4 ± 0.3 |
105
|
| 10 mL/31 cm2 |
| TEM grid |
1s |
5–50 |
0.9 ± 0.3–1.6 ± 0.4 |
|
|
| PtCu, 2019 |
PEG, 80 rpm |
2 |
50 |
0 |
30 |
30 |
— |
5–50 |
0.9 ± 0.3–2.2± 0.6 |
106
|
| 10 mL/31 cm2 |
1s |
|
5–50 |
0.9 ± 0.4–1.6 ± 0.5 |
| TEM grid |
|
|
|
|
| Au/Cu, 2016 cosputtering |
PEG, 80 rpm |
2 |
110 |
0 |
|
|
— |
Au10–20 |
No clear tendency |
109
|
| 10 mL/31 cm2 |
Cu30–50 |
| Pd/Cu, 2020, cosputtering |
PEG, 80 rpm |
2 |
110 |
0 |
30 |
30 |
— |
10–50 |
2.4 ± 0.7–2.5 ± 0.6 |
110
|
| 10 mL/31 cm2 |
| Au/Pt, 2020 cosputtering |
PEG, 80 rpm |
2 |
110 |
0 |
30 |
30 |
— |
Au50 |
Au > Au/Pt |
112
|
| 10 mL/31 cm2 |
Pt 0–50 |
| AuPd, 2015 |
MWCNTs in C4mim+/BF4− (3 mg/2 mL) |
3 |
— |
— |
RT |
5 |
— |
20–40 |
∼2 nm (alloy target) |
90
|
Table 3 Particle size depends on the kind of metal and the effect of alloying
| Metal |
d/nm |
Liquid matrix (target system) |
P/Pa |
WD/mm |
T
target/°C |
T
liquid/°C |
t/min |
I/mA |
Ref. |
| Au/Ag |
3.1 ± 0.6 |
C4mim+/PF6− (single head) |
— |
35 |
— |
— |
5 |
40 |
65
|
| Au |
2.6 ± 0.3 |
| Ag |
6.0 ± 1.5 |
|
|
| Au |
Aggregate of ≥5 nm |
PEG 600 |
2 |
110 |
0 |
30 |
30 |
20–40 |
112
|
| Ag |
2.8 ± 0.7 |
10 mL/31 cm2 (double head) |
50 |
111
|
|
|
| Au/Pt |
1.5–3.5112 |
PEG 600 |
2 |
110 |
0 |
30 |
30 |
10–50 |
112
|
| Au |
≥5 nm112 |
10 mL/31 cm2 (double head) |
112
|
| Pt |
1.3–1.6111 |
|
111
|
|
|
| Au/Pt Au |
1.5 ± 0.4 |
TMPA+/TFSA− (single head) |
10 |
20 |
— |
RT |
5 |
40 (500 V) |
113
|
| 2.9 ± 0.7 |
| Pt |
1.0 ± 0.3 |
|
|
| Au/Pd |
1.8 |
C4mim+/TFSA− 0.6 cm3/10 cm2 (single head) |
20 |
35 |
— |
RT |
5 |
40 |
115
|
| Au |
2.2 |
| Pd |
1.7–2.5 |
|
|
| Au |
6.68116 |
GO/C4mim+/BF4− (single head) |
— |
— |
— |
RT |
5116 |
— |
116
|
| 5.7 ± 1.2117 |
117
|
| Pd |
1.8–2.5116,117 |
|
|
|
| Au/Cu |
3.1 ± 1.0 |
C2mim+/BF4− (single head) |
10 |
38 |
— |
RT |
5 |
40 (500 V) |
119
|
| Au |
3.4 ± 0.5 |
| Cu |
2.4± 0.5 |
|
|
| Cu/Pt |
2.2 ± 0.6106 |
PEG 600 |
2 |
50 |
0 |
30 |
30 |
50 |
106
|
| Cu |
3.1 ± 0.8120 |
10 mL/31 cm2 (single head) |
120
|
| Pt |
1.4 ± 0.3105 |
|
105
|
Dupont and Nishikawa reported reversed tendencies of the influence of the voltage on the particle size of deposited Au NPs. Dupont and coworkers observed smaller Au NPs when applying a lower sputtering voltage and current for both liquid substrates, that is, an IL, 1-n-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)amide, C4mim+/NTf2− (Ar, P = 2 Pa, WD = 50 mm, V = 299–410 V, I = 20–110 mA, t = 150 s, 1.23 g IL/7 cm2), and castor oil (Ar, P = 2 Pa, WD = 50 mm, V = 260–405 V, I = 8–90 mA, t = 300 s, Tliquid = RT, 1.15 cm3 liquid/∼7 cm2).71,102 The Ttarget or control of Ttarget was not stated and the voltage (current) increased accompanied by an increase in applied current (voltage). The results were interpreted based on the kinetic energy of the sputtered Au. A higher voltage produced particles with higher kinetic energies, and thus, particles penetrated into the bulk liquid for nucleation and growth. In contrast, particles with lower kinetic energies could stay and grow at the vacuum–liquid interface to become bigger in size. The authors also suggested that the higher current/voltage allowed for a higher concentration of the deposited metal atoms/clusters, which facilitated the formation of bigger NPs.
In contrast, Nishikawa found that using V = 1000 V resulted in smaller Au NPs (1.4–1.5 nm, Fig. 6a) in C4mim+/BF4− compared with those using V = 700 V (1.7–1.8 nm) for I = 20–40 mA (Ar, P = 13–30 Pa that increases with V, WD = 25 mm, t = 50 min, Ttarget = 20 °C, Tliquid = 50 °C, 2 mL IL/15.9 cm2).96 However, the cause of the voltage impact on the particle size is an open question. It should be noted that the voltage range used by the Nishikawa group is significantly higher than others, which can make the particle growth on liquid surfaces and inside the liquid different from that occurring in low voltage sputtering for a large difference in the kinetic energy of the ejected particles. In addition, there is lack of control or no published data of Tliquid and Ttarget, which are important in particle growth, during sputtering in other research studies. These factors can contribute to the different trends of the voltage-dependent particle-size published in the literature.
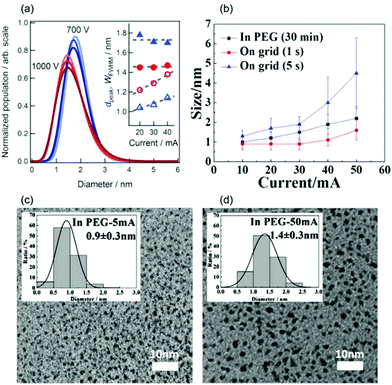 |
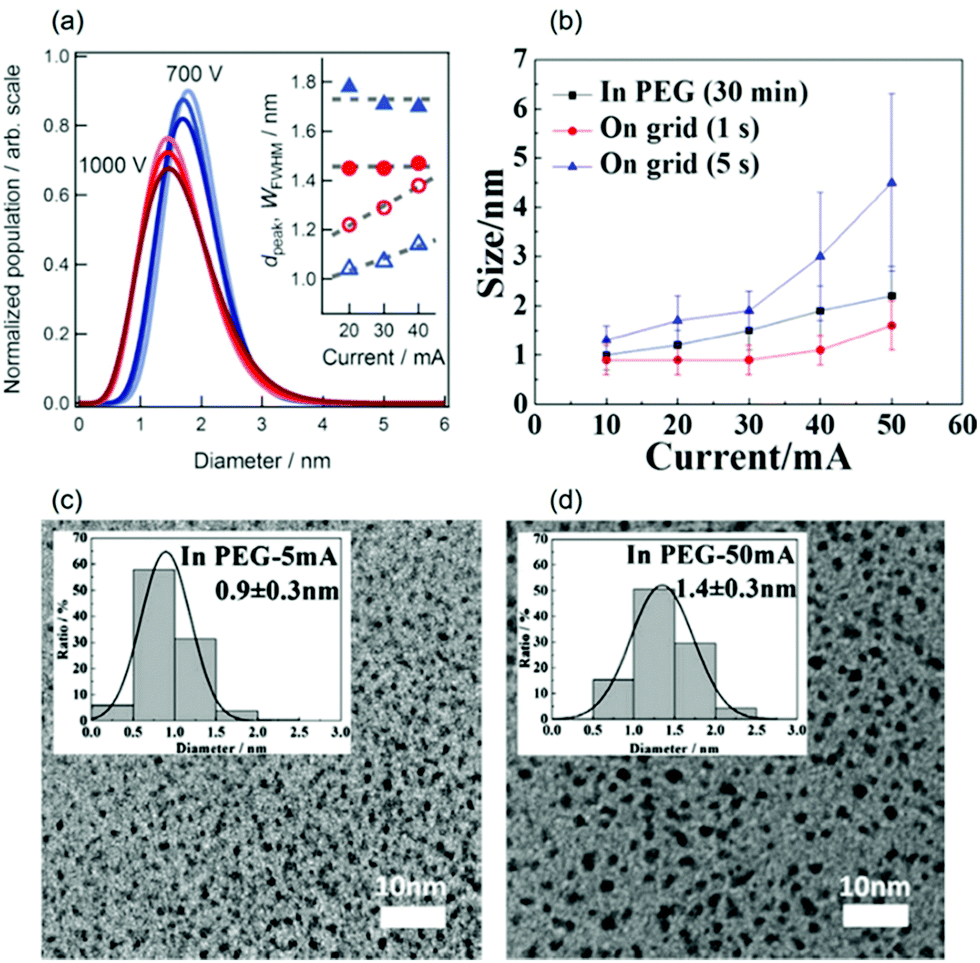
| | Fig. 6 (a) The size distributions (from SAXS) of the sputtered Au NPs at V = 700 V (blue) and V = 1000 V (red). The increase in the colour intensity corresponds to the increase of discharge current (I = 20–40 mA). The inset shows the average size (dpeak) and the width of the FWHM (WFWHM) of the size distribution against I.96 (b) The size vs. sputtering current of the Pt/Cu target onto PEG (t = 30 min) and TEM grids (t = 1–5 s).106 TEM images and size distribution (inset) of Pt NPs sputtered onto PEG (t = 30 min) with (c) I = 5 mA and (d) I = 50 mA.105 (Reproduced with permission from ref. 96. Copyright 2011 The Royal Society of Chemistry. Reproduced with permission from ref. 105 and 106. Copyright 2018 and 2019 The American Chemical Society.) | |
2.1.5. Applied current.
Sputtering current is related to the number of ejected atoms/clusters at a time. Low vacuum conditions of the deposition chamber can allow for negligible collision in a vacuum (gas phase) at a low sputtering current. On a solid substrate, faster growth of particles on the substrate can be expected with a higher sputtering current as more atoms/clusters are supplied to the growth at a time. In a liquid matrix, sputtered particles first land on the vacuum–liquid interface. Metal–liquid interaction, metal–metal interaction, and liquid physical properties (viscosity, surface tension) can affect how long sputtered particles can stay there and grow. If growth on the liquid surface occurs (e.g., high liquid viscosity79), similar to a solid substrate, one can also expect a larger particle size for a higher sputtering current. If growth on the liquid surface is insignificant, sputtered particles fall into the bulk liquid. The number of supplied atoms/clusters is related to their concentration in liquids, which in turn affects the collision, nucleation, and growth of particles in liquids. Depending on how effectively the liquid stabilizes the particles, the particle size can increase or negligibly change with a higher sputtering current. Therefore, we will review the impact of sputtering current on particle size for different liquids,90,96,102–106i.e., ILs and PEG of strong and weaker stabilization behavior to NPs, respectively, and for alloy NPs wherein metal–metal and metal–liquid interactions are important.108–113
(a) In ILs.
When Pd and AuPd alloy NPs were deposited onto multi-walled carbon nanotubes (MWCNTs) dispersed in C4mim+/BF4− using a single target, Liu et al. observed a negligible increase in particle size (Pd: 1.92 ± 0.37–2.03 ± 0.37 nm) for I = 20–40 mA (Table 2).90 For Au sputtered onto C4mim+/BF4−, despite many differences in the experimental parameters with Liu et al. (Ttarget, t, Tliquid, and matrix), Hatakeyama et al. also observed no obvious influence of the discharge current (I = 20–40 mA, Table 2 and Fig. 6a) on the average particle size of the Au NPs (by SAXS).96 These findings are opposite to the results from other groups,102,103 wherein larger particle size was observed for higher sputtering currents. For example, Wender et al. reported particle sizes of Au NPs of 3.2 ± 0.5 nm (20 mA, 299 V), 3.5 ± 0.6 nm (40 mA, 335V), and 4.6 ± 0.7 nm (110 mA, 410 V) (IL = C4mim+/NTf2−, Ar, P = 2 Pa, WD = 50 mm, t = 2.5 min).102 Suzuki et al. observed that the average size of Ag NPs increased from 5.6 to 11 nm for an increase of the sputtering current from 10 to 40 mA (IL: C4mim+/PF6−, Ar, P = 5 Pa, WD = 85 mm, t = 5 min, Tliquid = RT without temperature control during the sputtering, 0.6 cm3 IL/10 cm2).103 The noticeable difference is that Hatakeyama et al. had all the sputtering parameters under control (maintained constant) during sputtering among comparative experiments (e.g., V, Tliquid, and Ttarget), whereas others did not.96,102–104 Thus, an increase in the temperature of the target and the liquid, as suggested by Nishikawa, or an increase of the voltage coupled with the current, as suggested by Wender et al., could occur by increasing the discharge current, which causes the particle size to increase.96,102 The effect of the temperature of the target and liquid on the particle size was proved to be significant.96,104
(b) In PEG.
The Yonezawa group observed no obvious tendency in the particle size of Au deposited onto PEG (MW = 600, 10 g/31.2 cm2) against discharge currents of 20–40 mA (air, P = 2–30 Pa, WD = 25 mm, V = 200 V, t = 10–40 min) when sputtering was performed without control of Ttarget and Tliquid.100 To ensure that the temperatures of the target and the liquid were in control, a cool liquid cycled from a reservoir maintained at 0 °C was applied to the metal target and a Tliquid of 30 °C was maintained during sputtering (WD = 50 mm, Ar, P = 2 Pa, 10 cm3 PEG/31 cm2, vstir = 80 rpm) to investigate the impact of the sputtering current on the particle size. Fig. 6b–d shows an increase in particle size of Pt/Cu alloy NPs (from 0.9 ± 0.3 to 2.2 ± 0.6 nm) and Pt NPs (from 0.9 ± 0.3 to 1.4 ± 0.3 nm) when the discharge current increased from 5 to 50 mA. To exclude the effect of particle growth in the liquid, 1 s sputtering onto a TEM grid for the current range of 10–50 mA was performed. The results revealed that larger particle sizes were attained at a higher discharge current: that is, for Pt – 0.9 ± 0.3 nm (10 mA) to 1.6 ± 0.4 nm (50 mA) and for Pt/Cu – 0.9 ± 0.3 nm (10 mA) to 1.6 ± 0.5 nm (50 mA) as shown in Fig. 6b.105,106 Thus, the initial sizes of the particles formed upon landing on the surfaces of the PEG and TEM substrates can be different. This can be caused by the collision and growth of metal clusters in the gas phase, the probability of which depends on the number of sputtered atoms/clusters regulated by the discharge current. The collision is possible because at 2 Pa the mean free path (MFP), λ, of Ar at temperatures from 298 K (room temperature) to 373 K (100 °C) ranges roughly from 3.3 to 4.1 mm, which is one order of magnitude smaller than the WD (50 mm). The MFP of a gas is given in eqn (1):107where d is the diameter of the gas molecule (m) and nV = nNA/V is the number of molecules per unit volume wherein n is the number of moles of gas, NA is Avogadro's number, and V is the gas volume (m3). Using the relations kB = R/NA and PV = nRT for ideal gas, the MFP can be calculated as in eqn (2):where kB is the Boltzmann constant (1.38 × 10−23 J K−1), R is the gas constant, T is the temperature of the gas (K), and P is the gas pressure (Pa).
(c) Alloy NPs.
The effect of discharge current on the particle size of alloy NPs prepared by co-sputtering is different from that of single head sputtering of a monometallic target or of an alloy target. In particular, the size of co-sputtered alloy NPs in liquid PEG (MW = 600) did not always linearly increase with an increase in the discharge current of a metal target.108–112 There was a negligible effect of the sputtering current (generally 10–50 mA) on the particle size of co-sputtered Au/Ag, Au/Cu, Pd/Cu, and Pt/Ag alloy NPs.108–111 In contrast, the particle size was highly correlated with the Pt composition of the co-sputtered Au/Pt alloy NPs, but was not directly related to the sputtering current of only Au or Pt.112 For the same sputtering current of Pt, the particle size increased with an increase in sputtering current for Pt (Fig. 7, top to bottom of each column). For the same sputtering current of Au, the particle size of the Au/Pt NPs decreased with an increase in sputtering current for Pt, meaning that higher contents of Pt resulted in smaller size Pt (Fig. 7, left to right of each row).112 The finding is in good agreement with the decrease in particle diameter in the order of Au (2.9 nm) > Au/Pt (1.5 nm) > Pt (1.0 nm) previously reported for the Au, Au/Pt alloy, and Pt NPs deposited onto an IL, N,N,N-trimethyl-N-propylammonium bis(trifluoromethanesulfonyl)amide (TMPA-TFSA) using a single monometallic (Au, Pt) and binary Pt/Au target (Ar, P = 10 Pa, WD = 20 mm, V = 500 V, I = 40 mA, t = 5 min, Tliquid = RT).113 A decrease in the grain size of Au/Pt NPs for a higher Pt content was also detected (Fig. 8a).113 Wang and coworkers observed that Pd NPs/MWCNTs were smaller than Pd/Au 1/1 (mol/mol) NPs/MWCNTs using a single metal and an alloy target (Ar, P = 3 Pa, I = 40 mA, t = 5 min, C4mim+/BF4−).90 Further, for the same Pt content, the same particle sizes were found for various pairs of sputtering currents of Pt and Au (Fig. 7).112 Thus, the key size control factor for co-sputtered Au/Pt alloy NPs is Pt content (Fig. 8b), which is regulated by changing the relative sputtering current of the Pt and Au targets, not the sputtering current of a target itself.112 This is the result of the alloying effect, the stability of the particles in PEG, and the high formation energy of Pt during particle growth in PEG, which we will discuss later.
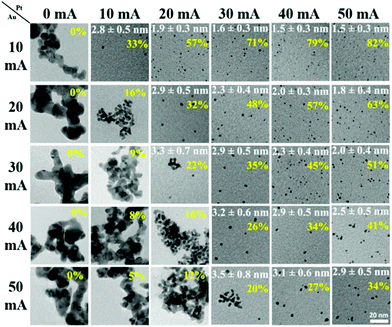 |
| | Fig. 7 TEM images of the Au/Pt NPs prepared by co-sputtering of Pt and Au targets onto PEG using various sputtering currents for the Pt (top row) and Au targets (left column). The mean particle size and standard deviation (white letters) and Pt at% (yellow letters) are shown. (Reproduced with permission from ref. 112. Copyright 2020 The American Chemical Society.) | |
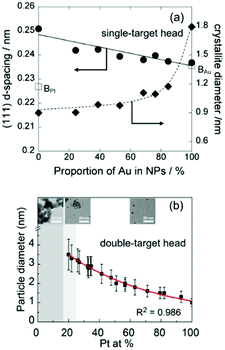 |
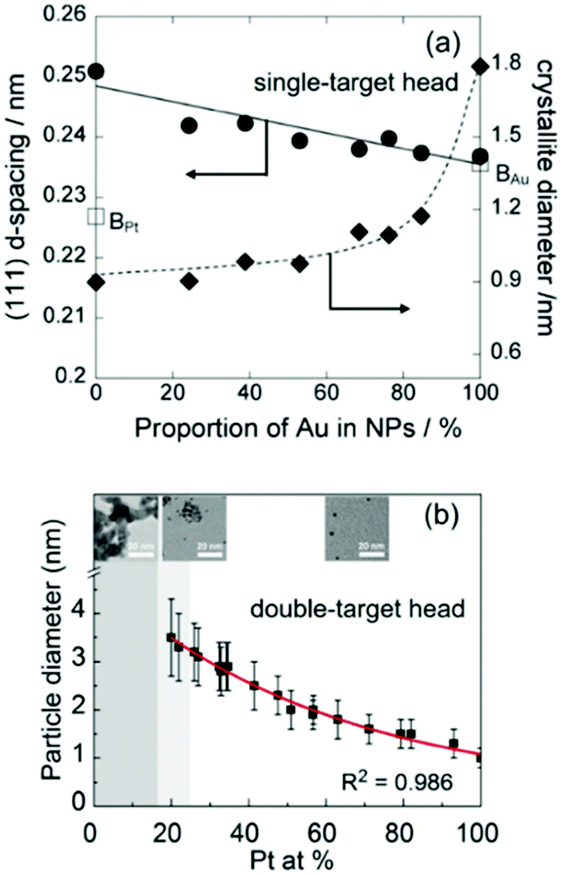
| | Fig. 8 (a) Relation between the d-spacing of (111) planes and crystallite diameters and Au at% in Pt/Au prepared in an IL using a binary target.113 (Reproduced with permission from ref. 113. Copyright 2012 The Royal Society of Chemistry.) (b) Relation between the particle size and Pt at% of Pt/Ag NPs co-sputtered onto PEG.112 (Reproduced with permission from ref. 112. Copyright 2020 The American Chemical Society.) | |
2.1.6. Metal targets.
Under the same deposition conditions, intuitively different sizes of NPs sputtered from targets of different metals can be expected, as the size of sputtered atoms/clusters and the sputtering yield depend on metals.114 In the presence of a liquid matrix, its interaction with metals adds a factor to regulate the particle size; for instance, stronger binding of the liquid to a metal often leads to a smaller and more uniform size.
For metals and alloys prepared by sputtering onto liquids, a general order in particle size from different metal elements is not readily available. This is because the particle sizes of different metals reported by different research groups and even by the same group are not always comparable, as more than one experimental condition was varied at a time. The following discussion is based on the comparison of the particle sizes of couples of metals under similar sputtering conditions (Table 3).
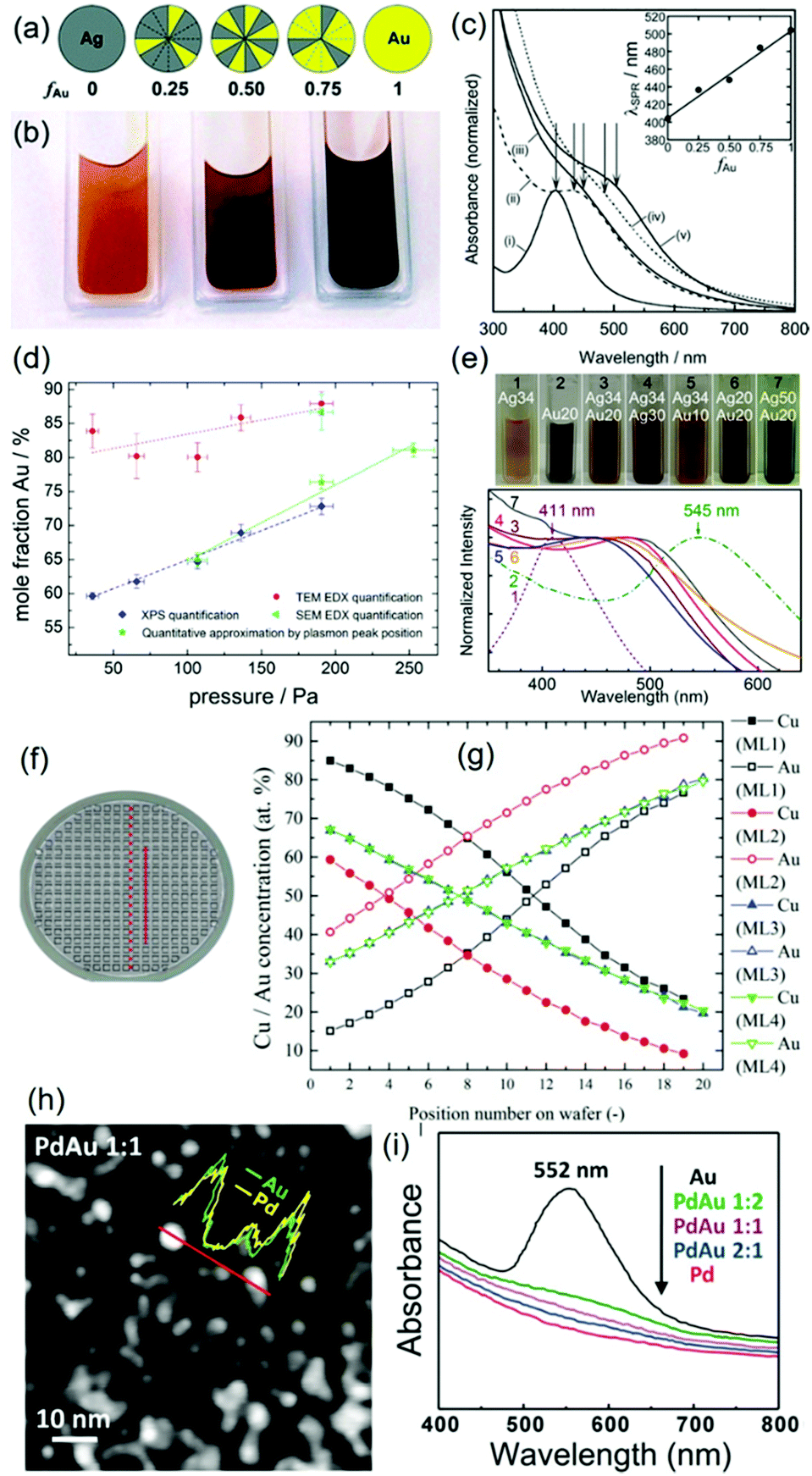
(a) Au, Ag, and Au/Ag.
The Torimoto group reported the particle size increase in the order of Au (2.6 ± 0.3 nm) < Au50Ag50 (3.1 ± 0.6 nm) < Ag (6.0 ± 1.5 nm) in an IL, C4mim+/PF6− (0.6 cm3/10 cm2), when using a single metal target or a binary target with alternating areas of Au and Ag (Ar, P = 20 Pa, WD = 35 mm, I = 40 mA, t = 5 min, Tliquid = RT).65 The Wang group also reported smaller sizes of Au (0.5–2 nm, av. 1.5 nm) compared with Ag NPs (3–9 nm, av. 5.5 nm); both were embedded on graphene oxide (GO) which was dispersed in C4mim+/OTf− in DC sputtering (Ar, P = 1 Pa).86 For PEG MW = 600, Au NPs were bigger than Ag NPs (Ag:111 50 mA, 2.8 ± 0.7 nm; Au:112 20–40 mA, severe aggregation of particles ∼5 nm or more) under similar sputtering conditions (Ar, P = 2 Pa, Ttarget = 0 °C, 10 cm3 PEG/31 cm2, vstir = 80 rpm, Tliquid = 30 °C, t = 30 min, double target head system). Previously, aggregation and growth of Au in PEG to a larger extent compared with that of Au in IL was reported.72 Therefore, the stabilization ability of PEG and IL towards metals, i.e., Au and Ag, are possibly different; that is, PEG stabilizes Ag better than it does for Au, and hence, growth and aggregation of Au occurred more severely in PEG.
(b) Au, Pt, and Au/Pt.
A good agreement in particle size increase in the order of Pt < Au/Pt < Au was reported for both an IL and PEG as liquid substrates (Fig. 7 and 8).112,113 A single target113 and double-head target system was used for sputter deposition onto the IL and PEG, respectively.112 Yonezawa et al. suggested that the main reasons include the better stabilization behavior of the liquid towards Pt and the high formation energy of Pt which limits its growth in PEG.112
(c) Au, Pd, and Au/Pd.
Torimoto115 and Wang116,117 reported the particle size order of Pd < Au/Pd < Au for free NPs deposited in C4mim+/TFSA− and for NPs embedded on graphene dispersed in C4mim+/BF4−, respectively. Not much difference in the particle sizes of Pd (1.7–2.5 nm) was found among the reports, whereas the particle sizes of the free-standing Au NPs (2.2 nm115) and Au embedded in graphene (6.68 nm116 and 5.7 ± 1.2 nm117) were significant different. Perhaps, this is caused by the differences in sputtering conditions, such as the ILs and the presence of the solid support. In a previous report, Pd of slightly bigger size than Au NPs was observed when GO dispersed in C4mim+/OTf− was used as the matrix for sputtering.86
2.2.1. Sputtering time and period.
So far, a longer sputtering time has either negligibly affected the particle size or increased it. In the pioneering work by Torimoto et al. for sputtering of Au onto an IL, C2mim+/BF4−, they varied the deposition time from 5 to 120 min (air, P = 20 Pa, I = 4 mA). Based on the similar shapes of the UV-vis spectra of the dispersion, they reported that no significant change in particle size occurred for Au atom concentration values up to 0.33 mmol dm−3.63 They reported a similar phenomenon for Ag NPs (d ≈ 5.7 nm by TEM: t = 5–45 min, C4mim+/PF6−, I = 10 mA, Ag atom concentrations of 0.26–1.4 mmol dm−3).103 Wender et al. also observed no significant change in the size of Au NPs with deposition time (t = 2.5–5 min on castor oil71 and t = 5–10 min on C4mim+/NTf2− at 40 mA).102 Wang shared the same conclusion for the time-dependent particle size of Pd NPs sputtered onto MWCNTs dispersed in C4mim+/BF4− for t =1.7–10 min (Ar, P = 3 Pa, I = 40 mA).90 Raghuwanshi et al. also reported that prolonging the sputtering time (t = 0.5–5 min) at 20 mA has no effect on the size of the formed Au NPs deposited onto deep eutectic solvents (TC–urea), a class of IL analogues, which are eutectic mixtures of Lewis or Brønsted acids and bases containing varieties of anionic and/or cationic species.74
The Nishikawa group reported an increase in the particle size of Au NPs measured by SAXS for a longer time (t = 12–36 min) of sputtering onto three ILs, C2mim+/BF4−, C4mim+/BF4−, and C8mim+/BF4− (Ar, P = 10–15 Pa, V = 1000 V, I = 5 mA).104 However, later they suggested that the size increase was not ultimately caused by the increase in concentration of particles, but by the increase in Tliquid and Ttarget for a long sputtering. Following that, they conducted arrays of experiments by (i) maintaining the temperature of the IL (C4mim+/BF4−) at either 50 °C or 20 °C and varying the sputtering time (t = 30–90 min) or (ii) maintaining Ttarget = 20 °C and Tliquid = 50 °C and varying the metal concentration by changing the discharge current (I = 20–40 mA).96 As a result, the particle size clearly depends on Tliquid96,121 and there was no obvious change with sputtering time and current,96 thus proving their hypothesis. The collision of the particles correlated with Tliquid, which was suggested as a size-control factor.96,121
As presented above, the sputtering-time independent particle-size has been reported for the discharge current as high as 40 mA for up to 90 min in ILs. The Yonezawa group tested for longer deposition times onto PEG, up to t = 240 min, for higher sputtered atom concentrations of Cu, Pt, and Cu/Pt (Ar, P = 2 Pa, WD = 50 mm, Ttarget = 0 °C, I = 30 mA, Tliquid = 30 °C, vstir = 80 rpm).106 The size of Cu/Pt increased from 1.2 ± 0.4 (15 min) to 3.1 ± 1.7 nm (240 min) and after 240 min Cu/Pt NPs were bigger than Pt and smaller than Cu NPs.106Fig. 9a–e show the particle size increased rapidly from t = 120 min to t = 240 min and the large NPs were polycrystalline. Thus, secondary growth via NP collision, aggregation, adhesion, and attachment due to high particle concentration can account for the size increase. Hence, the diffusion velocity of metal NPs in PEG and the stabilization behavior of PEG towards NPs are different from those in the IL. Previously, at the same Tliquid, Nishikawa showed that NPs in PEG were bigger than those in C4mim+/BF4−.72
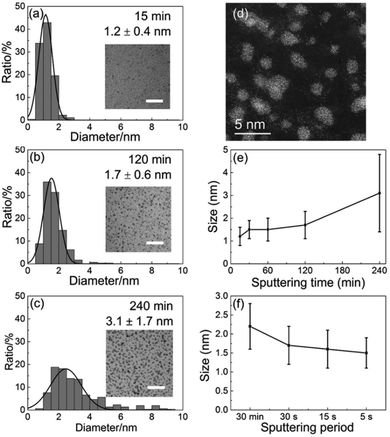 |
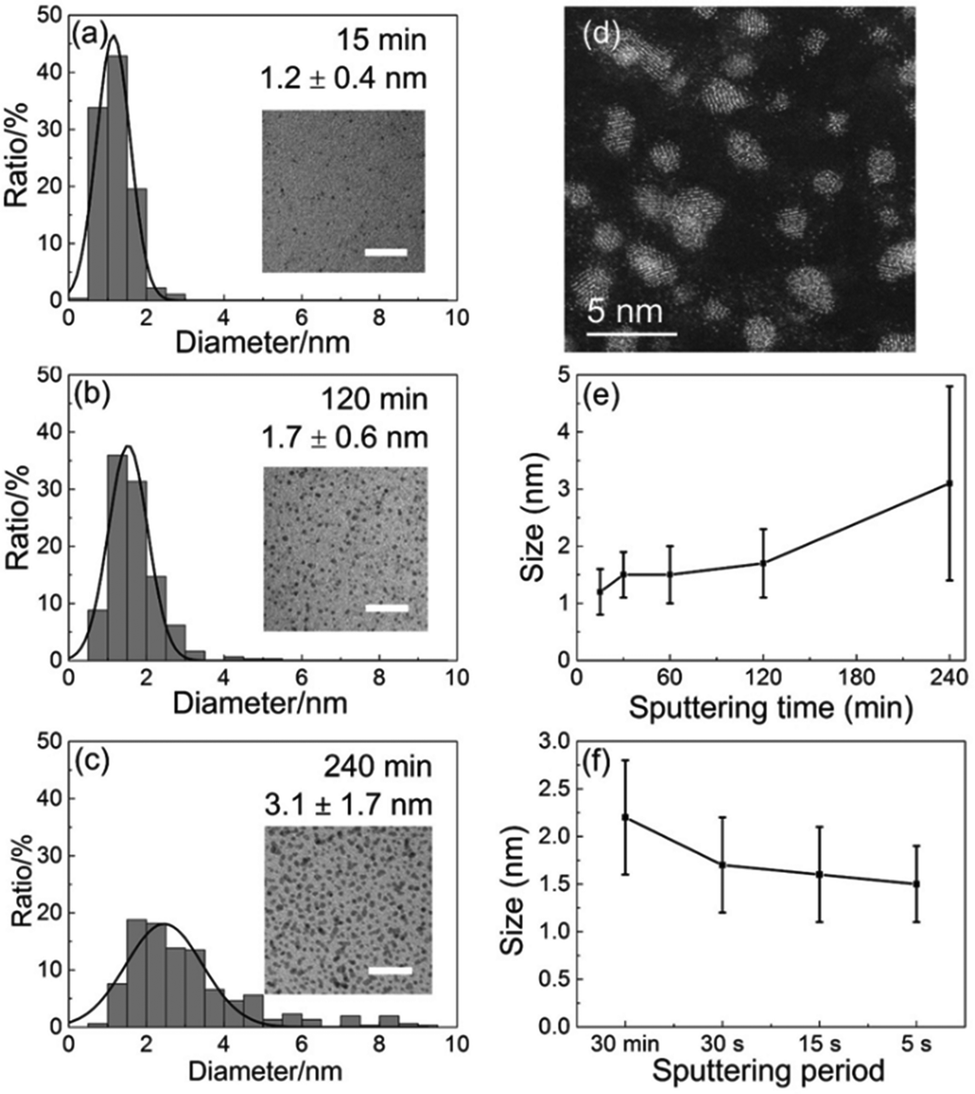
| | Fig. 9 Size histograms and the inset TEM images (scale bar of 20 nm) of the sputtered Pt/Cu NPs for deposition times of (a) 15 min, (b) 2 h, and (c) 4 h. (d) Scanning TEM-high-angle annular dark-field (STEM-HAADF) image of Pt/Cu NPs sputtered for 4 h. The NP size depends on the (e) sputtering time and (f) period. (Reproduced with permission from ref. 106. Copyright 2019 The American Chemical Society.) | |
In an attempt to detect the growth of Cu/Pt particles sputtered onto a PEG surface, Deng et al. also found that the sputtering period affected the particle size to a certain extent (Fig. 9f).106 In particular, when the total sputtering time (t = 30 min) was constant for having the same metal atom concentration in PEG, smaller NPs were obtained for a shorter sputtering period. This was explained by considering that the sputtering performed for a short period could allow for particle falling into PEG before the next arrival of sputtered species; thus, less growth occurred on the liquid surface.
2.2.2. Stirring speed.
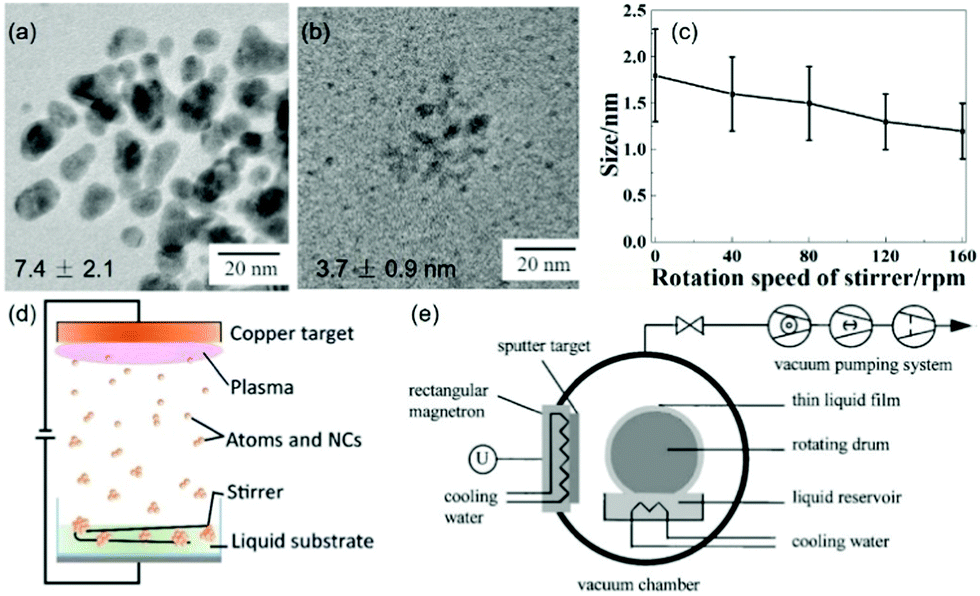
While NPs could be obtained by sputtering onto a liquid, rotation and/or stirring of capture medium was not always necessary or performed, particularly, when ILs and molten salts were used.63,65–68,71,72,74,79,81,82,86,87,89–91,93–96,99,102–104,113,115–119,121 However, it was found that when applying stirring at 100 rpm to PEG (Fig. 10a and b), the diameter of the sputtered Au NPs substantially decreased from 7.4 ± 2.1 (with aggregation) to 3.7 ± 0.9 nm (Ar, P = 2 Pa, WD = 50 mm, I = 20 mA, t = 20 min).122 Further, the particle size (Fig. 10c) decreased almost linearly with the rotation speed of the stirrer (vstir = 0–160 rpm) when a Pt/Cu alloy target was sputtered onto PEG (Ar, P = 2 Pa, WD = 50 mm, I = 30 mA, t = 30 min, Tliquid = 30 °C).106 In this case, the particle uniformity was improved with a higher stirring speed.106 Stirring helps refresh the PEG surface. It can also help the sputtered particles diffuse into the bulk liquid more quickly, and the collision/coalescence of particles during growth occurs more evenly. Thus, smaller and more uniform NPs could be attained. Prior to using a stirrer (Fig. 10d), refreshing the liquid capture medium was done using a rotating drum in contact with the liquid (Fig. 10e). This created a continuously running thin liquid film during sputtering which could capture the sputtered NPs and transport them to the liquid reservoir.62
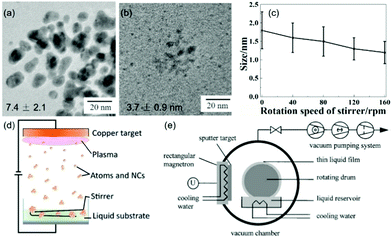 |
| | Fig. 10 TEM images of Au NPs (a) without and (b) with stirring PEG at 100 rpm.122 (Reproduced with permission from Ref. 122. Copyright 2016 Nature Publishing Group, England.) (c) Particle size and standard deviation of the size distribution of the sputtered Pt/Cu NPs decrease when increasing the rotation speed of PEG.106 (Reproduced with permission from ref. 106. Copyright 2019 The American Chemical Society.) (d) Stirring the liquid medium with a stirrer.77 (Reproduced with permission from ref. 77. Copyright 2016 The Royal Society of Chemistry.) (e) A rotating drum for making continuous flow of the liquid film in the vacuum chamber.62 (Reproduced with permission from ref. 62. Copyright 1999 Elsevier.) | |
2.2.3. Viscosity, surface tension, and temperature of the liquid.
(a) Surface tension and viscosity of the liquid.
The growth of the sputtered particles can occur on the liquid surface and/or inside the bulk liquid and/or in both places depending on the time they can stay on the surface and diffuse into the bulk liquid. In general, high surface tension can favour particle aggregation on the surface. This is because the energy (ΔG) for a particle to submerge into the liquid is proportional to the surface tension, as given by eqn (3):79| | ΔG = πr2γ(1 − cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θe)2 for 0 ≤ θe ≤ 90° θe)2 for 0 ≤ θe ≤ 90° | (3) |
where r is the particle radius, γ is the liquid–vapor surface tension, and θe is the equilibrium three phase contact angle.
In the bulk liquid, the liquid viscosity can affect the particle growth. High viscosity reduces particle collision by reducing the diffusion velocity of particles because the diffusion coefficient, D, of a particle in a quiescent liquid under Brownian motion is inversely proportional to the liquid viscosity, η, given by the Stokes–Einstein relationship:121
where
kB is the Boltzmann constant and
r is the hydrodynamic radius of the particles. Therefore, the final particle size and/or aggregation structures do not simply depend on a single factor. For example, particles grow less and thus have a smaller size in a high viscosity liquid. However, the highly viscous liquid can also slow down the time that particles can diffuse into the bulk liquid and to a certain extent can favour particle growth on the liquid surface to form big particles or a thin film.
Table 4 shows the particle sizes of sputtered NPs in correlation with the viscosity and surface tension of liquid media.72,79,80,104,121,124–126 To vary the surface tension and viscosity of ILs, the Nishikawa group used the cations with different lengths of the alkyl chain, that is, C2mim+, C4mim+, and C8mim+ with the same anion BF4−.104 The stabilization of NPs by these ILs was attributed mainly to the anion which adsorbs to the surfaces of the NPs; therefore, the effects of the viscosity and surface tension on particle size could be compared among the 3 ILs. With the increase of the alkyl chain length, the surface tension decreases and the viscosity of the liquid increases, whereas the sputtered Au NPs have particle sizes which decrease with narrower size distribution. This is to say, an IL with higher viscosity and lower surface tension favours the formation of well dispersed NPs with small size. Further, Vanecht et al. also observed in UV-vis spectra slower kinetics of the aggregation and sedimentation of sputtered Au NPs after sputtering in an IL of higher viscosity.123
Table 4 Surface tensions and viscosities of some liquid matrices
| Liquid |
Metal |
d/nm |
T
d/K |
γ/mN m−1 |
T
γ
/K |
η/cP |
T
η
/K |
Ref. |
|
Measured by SAXS; otherwise NPs were observed using a TEM or an optical microscope; Td, Tγ, and Tη: temperature of the capture medium for nanoparticle synthesis, for measuring surface tension, and for measuring viscosity, respectively.
|
| C2mim+/BF4− |
Au, 53 mmol dm−3 |
∼3.5a |
|
54.4 |
298.15 |
66.5 |
293 |
104
|
| C4mim+/BF4− |
∼3a |
44.81 |
293.15 |
154 |
293 |
| C8mim+/BF4− |
∼2a |
33.62 |
293.15 |
439 |
293 |
| C4mim+/BF4− |
Au |
∼2.8 |
333 |
44.81 |
293.15 |
∼25 |
333 |
104 and 121
|
| PEG MW 600 |
Au, 40 mmol dm−3 |
∼2a |
293 |
∼40 |
296.15 |
167 |
293 |
72 and 125
|
| ∼7.5a |
333 |
∼30 |
333 |
| PEG MW 400 |
Au |
Filmsurf, aggregatesl |
|
43.2 |
|
112 |
|
79
|
| Glycerol |
Filmsurf, NPl |
64.3 |
946 |
| Squalene |
|
31.2 |
12 |
| Silicone oils |
Au, Ag |
No film, aggregate + NPsl |
|
21.53 (100 cP) |
298 |
48, 96 |
298 |
79 and 126
|
| Filmsurf, NPsl |
23.95 (500 cP) |
339, 485 |
| PEMP |
Ag |
Film |
273 |
|
|
1279 |
273 |
124
|
| 2.3 |
293 |
658 |
293 |
| 1.8 |
373 |
∼8.5 |
373 |
| Oleic acid/oleylamine |
Au |
5 |
303 |
|
|
31 |
298 |
80
|
| 2.4 |
303 |
648 |
298 |
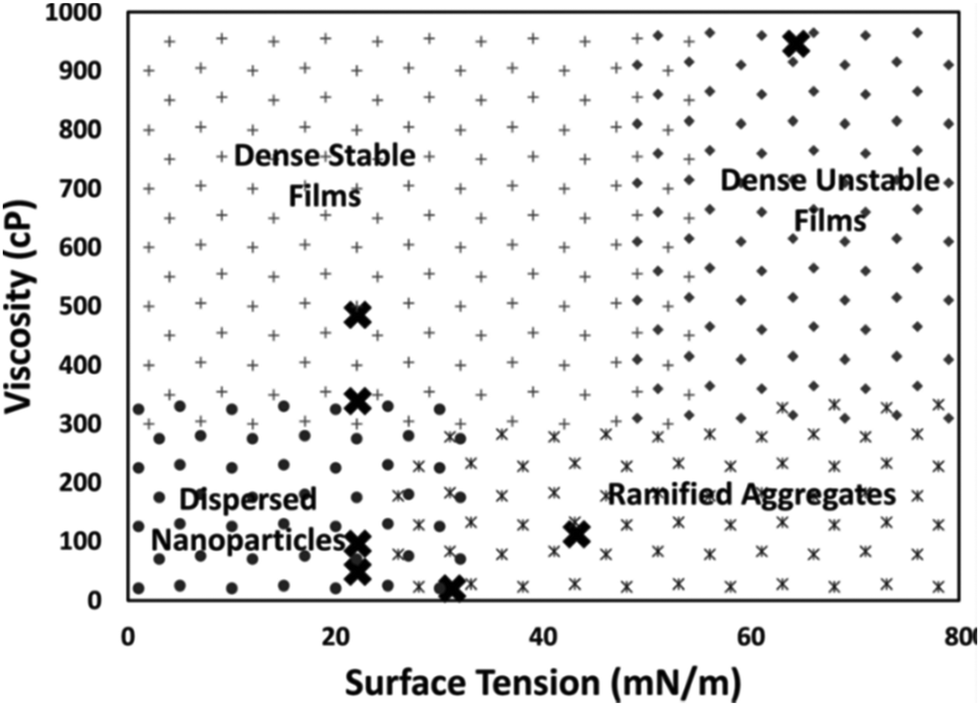
De Luna et al. proposed that surface tension and viscosity could take part in control of particle growth by regulating the rate that the liquid wets the particle surface. Particularly, the wetting rate increases with higher surface tension due to the decrease in the free energy of the system and decreases with higher liquid viscosity.79 To check the impact of viscosity on particle size, the authors deposited Au on silicone oils with viscosities of 48, 96, 339, and 585 cP (WD = 50 mm, P = 80 Pa, I = 20 mA, t = 30 s, 0.5 mL liquid/2.5 × 2.5 cm × cm glass slide). The energy required for submersion of Au into silicone oils (ΔG) is close to zero, suggesting that Au readily submerges into the oil. There was no Au film on the oil surface and the authors could observe Au NPs dispersed in the bulk silicone oils of 48 and 96 cP. The 48 cP silicone oil contained bigger Au NPs and some aggregates, whereas the latter had only dispersed NPs. However, at higher viscosities (339 and 585 cP), an Au film was formed on the liquid surface with a denser film on the higher viscosity liquid. The result (Table 4) supports their hypothesis that high viscosity could reduce the wetting rate of the particle surface, and thus, particles could accumulate and interact with and adhere to each other on the liquid surface to form a film structure. They also sputtered Au and Ag on liquids with different surface tensions, that is, squalene (12 cP, 31.2 mN m−1), PEG (MW = 400, 112 cP, 43.2 mN m−1), and glycerol (946 cP, 64.3 mN m−1). Metal films formed on all liquids and less stable films were formed on glycerol (highest surface tension). Inside the liquids, NPs were found in squalene, whereas ramified aggregates were found in the others. Note that silicone oil with viscosity (96 cP) similar to that of PEG and surface tension half of that of PEG allowed for NP dispersion without film formation. Therefore, liquids with low surface tension and not very high viscosity favoured dispersed NPs, liquids with high surface tension and low viscosity were for aggregates, and the others supported the formation of dense films on liquid surfaces. Fig. 11 shows the combined effect of surface tension and viscosity on the deposited NPs.79 It should be noted that the results may not be valid for liquids which bind strongly to Au and Ag and with electrostatically charged species which are involved in stabilizing NPs. For instance, using a strongly coordinating liquid, i.e., PEMP, with very low viscosity (∼8.5 cP), Yonezawa obtained well-dispersed and small Ag NPs (<2.5 nm) in liquids (Table 4).124
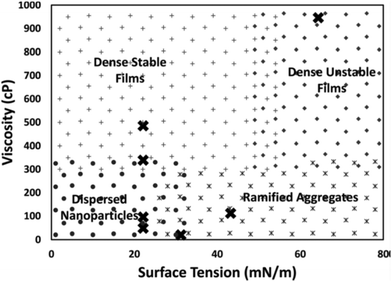 |
| | Fig. 11 Morphology diagram of the resulting structures in relation to the viscosity and surface tension of the liquids (PEG, glycol, squalene, and silicone oils). (Reproduced with permission from ref. 79. Copyright 2018, American Institute of Physics.) | |
| | | η = Aexp(B/Tliquid) | (5) |
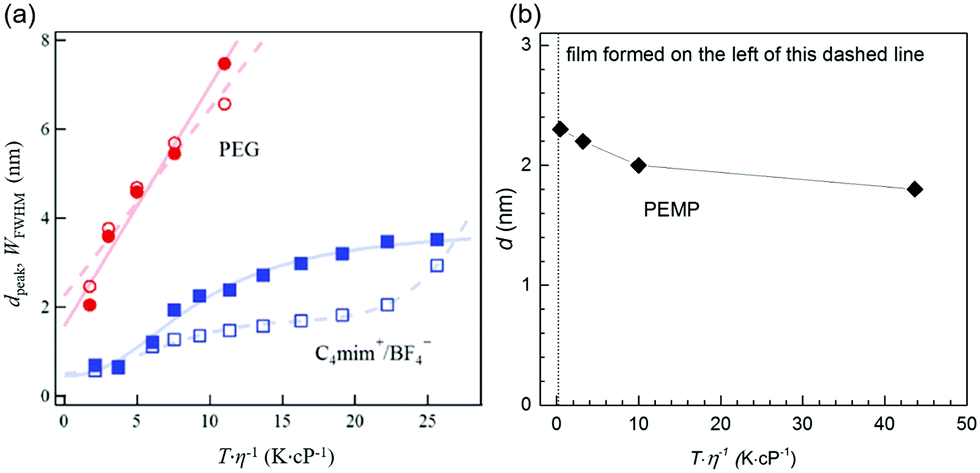
where A and B are constants for the liquid. Both smaller η and higher Tliquid promote a faster diffusion velocity of the sputtered particles into and in the liquid because the diffusion coefficient, D, depends linearly on Tliquid/η (eqn (4)). Hence, particles in the liquid undergo more collisions to grow bigger. The results of the Nishikawa group demonstrate this relation.72,121 They detected an increase in the particle size of Au (measured by SAXS) with increasing Tliquid of C4mim+/BF4− (20–80 °C) and PEG (20–60 °C) during sputtering. Further, heat treatment of the PEG containing Au NPs after sputtering (20–110 °C) also increased the particle size to even a larger extent.72 The authors used similar sputtering conditions for the IL and PEG (Ar, P = 12–13 Pa (IL) and 16–19 Pa (PEG), WD = 25 mm, t = 50 min, V = 1000 V, I = 20 mA, 2 cm3 liquid/15.9 cm2, with and without a cooling system for the target in the experiment with the IL and PEG, respectively).72,121 The increase of particle size with Tliquid/η for both the IL and PEG (Fig. 12a) indicates that diffusion velocity is one factor regulating the particle size. However, the size and size distribution of Au NPs in PEG varied linearly with Tliquid/η and increased more significantly compared with that in the IL. Thus, the authors suggested taking the stabilization ability of the liquid to NPs into account; in comparison to the IL, PEG insufficiently stabilized Au NPs. Besides, the stabilization ability of PEG varies with metals; therefore, the Tliquid-dependent particle size of NPs sputtered onto PEG also depends on the metals. For instance, different from Au, when varying the temperature of PEG, negligible change in the particle diameters was observed for the sputtered Pt/Cu NPs, that is, from 1.5 ± 0.4 to 1.6 ± 0.5 nm with Tliquid from 20 to 50 °C (Pt/Cu target, Ar, P = 2 Pa, WD = 50 mm, Ttarget = 0 °C, I = 30 mA, vstir = 80 rpm, t = 30 min, 10 cm3 PEG/31 cm2).106 The result can be ascribed (1) to the stirring of PEG for refreshing the liquid surface, faster particle mixing, and more uniform particle growth in the bulk liquid and (2) mainly to the better stabilization behavior of PEG to Pt/Cu alloy than to Au.106 The effect of sputtering time can be excluded in comparison to Nishikawa's report (50 min)72 because an increase in the sputtering time from 30 min to 1 h did not cause any obvious change in particle size.106 Stirring of PEG is not the main factor which causes the change of particle size because under similar experimental conditions Pt NPs and Pt alloy NPs with Au, Ag, and Cu in PEG were smaller than Au, Ag and Cu NPs, respectively (Table 3).105,106,112 Consequently, an increase of T/η (Tliquid = 20–50 °C) did not cause an increase in particle size for the Pt/Cu alloys as it did for Au.
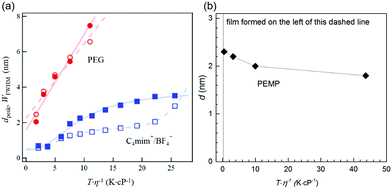 |
| | Fig. 12 (a) T/η dependences of dpeak (closed symbols) and WFWHM (open symbols) for Au NPs sputtered onto PEG (red circles) and onto the IL 1-butyl-3-methylimidazolium tetrafluoroborate, C4mim+/BF4− (blue squares), of different Tliquid where dpeak and WFWHM are the peak top and full-width at half maximum of the particle diameter distribution obtained by SAXS, respectively. (Reproduced with permission from ref. 72. Copyright 2011 The American Chemical Society.) (b) T/η dependence of the diameter (measured by TEM) of Ag NPs sputtered onto PEMP, which was composed using results given in ref. 124. | |
The nature of metal–liquid bonding and the stabilization ability of the liquid towards metal can change the effect of increasing Tliquid/η on particle size. Ishida et al. found that deposited Ag formed a film on the surface of liquid PEMP for Tliquid of 0 and 10 °C.124 At low temperatures, high viscosities of PEMP (η > 1200 cP at Tliquid = 0 °C) hindered the immediate penetration of the sputtered particles into PEMP, causing Ag to accumulate on the PEMP surface to form a film. This agrees with the finding of De Luna et al.79 Interestingly, well dispersed Ag NPs with smaller diameters were observed at higher Tliquid; that is, the particle size decreased from 2.3 to 1.8 nm when Tliquid increased from 20 (η ≈ 658 cP) to 100 °C (η ≈ 8.5 cP).124 Here, the particle size decreases with increasing Tliquid/η (Fig. 12b), which is opposite to the results for Au NPs in PEG (MW 600) and ILs.72 The results are also different from Ag deposited on silicone oils, PEG (MW 400), and glycerol with similar viscosity ranges.79 Particularly, well dispersed NPs below 2.5 nm were formed in PEMP, whereas a metal film formed on the liquid surface and ramified/aggregated structures were formed in the bulk liquid of the other liquids (Fig. 13).79 The key difference is that PEMP binds to the surfaces of the Ag NPs via 4 mercapto (–SH) functional groups much more strongly than the other liquids do. High temperature reduces the liquid viscosity, and allows sputtered particles to diffuse more rapidly into the bulk PEMP, where they can be capped with abundant PEMP, stabilized, and protected from growing. The capping capability of PEMP is so strong that at the lowest viscosity well dispersed and smallest Ag NPs were observed. Hence, the extent to which the liquid temperature and viscosity regulate the particle size by varying the diffusion velocity of the particles highly depends on the kind of liquid, and the binding and the stabilization ability of the liquid towards the NPs.
 |
| | Fig. 13 (a) Sputtered Ag NPs formed in PEMP at different temperatures: 20 °C (∼658 cP)–100 °C (∼8.5 cP). (Reproduced with permission from ref. 124. Copyright Elsevier 2016.) (b) Optical microscope images and (c) TEM images of the surface, and (d) TEM images of the bulk of the sputtered Ag onto (first column) squalene (12 cP, 31.2 mN m−1), (second column) PEG MW 400 (112 cP, 43.2 mN m−1), and (third column) glycerol (946 cP, 64.3 mN m−1). (Reproduced with permission from ref. 79. Copyright American Institute of Physics 2018.) | |
2.2.4. Kind of liquid.
ILs, viscous polymers, and various ligands have been mainly used as capture media in sputter deposition to create NPs. Liquid matrices are involved in regulating particle growth and stabilizing the sputtered particles, and thus in control of the particle size. This capability of the liquids will be viewed from their chemical structure, binding functionality, and stabilizing mechanism aside from the resulting macroscopic physical properties (e.g., surface tension, viscosity) discussed in Section 2.2.3.
(a) Ionic liquids: anion/cation effect and surface composition.
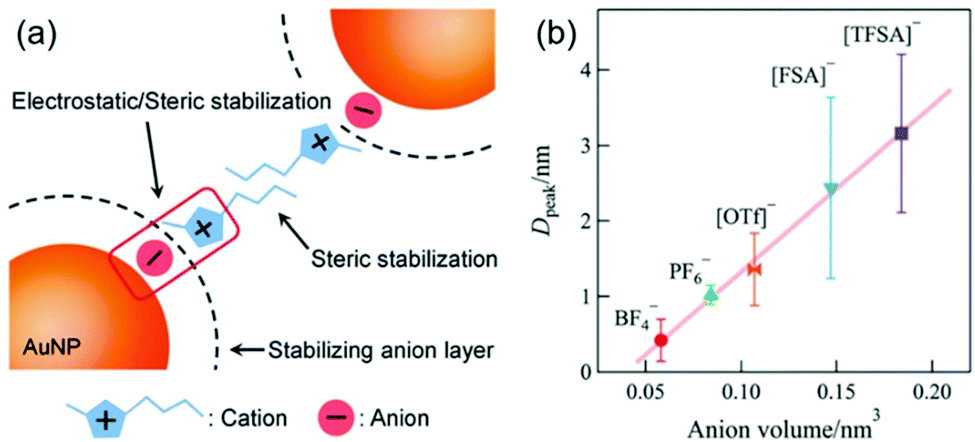
The effect of anions and cations of ILs on the particle size of deposited NPs has been found in early reports in this field.63,68,86,102,104,123 However, Nishikawa suggested that non-regulation of some sputtering parameters,104 such as Tliquid and Ttarget, could complicate the evaluation of the effect on particle size.128 The surfaces of small sputtered NPs are considered to be electron deficient;97,129,130 therefore, the anions of ILs can preferably bind to the NPs’ surfaces. The cations, on the other hand, can form the second shell on the NPs’ surfaces (Fig. 14a).128 This resembles a double layer structure, which stabilizes NPs via electrostatic repulsion. Besides, the alkyl chain of the cation can involve in steric stabilization. The chemically synthesized NPs in ILs by Janiak and Dupont support the electrostatic stabilization and steric hindrance.130–132 For instance, Janiak found bigger NPs formed in ILs with larger anion volume.131,132 This is because bigger anions (higher bulkiness) hinder packing into layers and result in smaller surface charge density. This lowers the surface potential, and thus, particles grow bigger. Hatakeyama et al. used the above model to verify and explain the anion-cation effect. Under controlled experiments (WD = 25 mm, Ttarget = 20 °C, V = 1000 V, I = 20 mA, t = 50 min, Tliquid = 20 °C), Hatakeyama et al. obtained a linear relationship between the anion volume and the size of the sputtered Au NPs measured by SAXS (Fig. 14b).128 The size increases in the order of increasing anion volume: BF4− < PF6− < OTf− <FSA− < TFSA−. The anion effect overcomes the impact of viscosity for at the same temperature, i.e., 25 °C; the viscosity of C4mim+/BF4− is almost 3 times as small as that of C4mim+/PF6−.128 Further, to weigh the effect of the alkyl chain length of cations Cnmim+ (n = 2–8) and the kind of anion (BF4−, PF6−, OTf−, FSA−, TFSA−) on particle size, the authors varied the temperature of the ILs (20–80 °C) during sputtering. They found that up to 40 °C the particle size negligibly changes with the alkyl chain length of the cations, whereas it depends on the anion; that is, the particle size increases in the anion order of BF4− ∼ PF6− <OTf− < FSA− < TFSA−. This confirms the dominant effect of the anions compared with the cations on the size of the sputtered NPs. Above 50 °C, a decrease in the stabilization capability of anions towards NPs and an increase of diffusion velocity can be expected, and thus, cohesion of the sputtered NPs occurred more significantly compared with that at low temperature. As a result, Hatakeyama et al. observed the size dependence on the alkyl chain length in the cations; smaller NPs were correlated with the cations of a longer alkyl chain as this restricts particle cohesion. This result demonstrated the stabilization mechanism and size regulation properties due to the cationic part of the ILs.
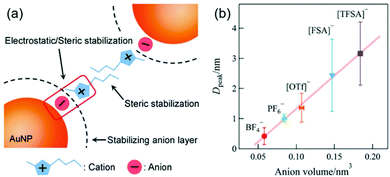 |
| | Fig. 14 (a) Model based on a double layer structure for the stabilization of NPs in imidazolium-based ILs via electrostatic repulsive force and steric stabilization. (b) Relationship between the volume of anions of ILs and particle diameter at the peak extracted from the particle size distribution of Au NPs measured by SAXS. (Reproduced with permission from ref. 128. Copyright 2016 The Royal Society of Chemistry.) | |
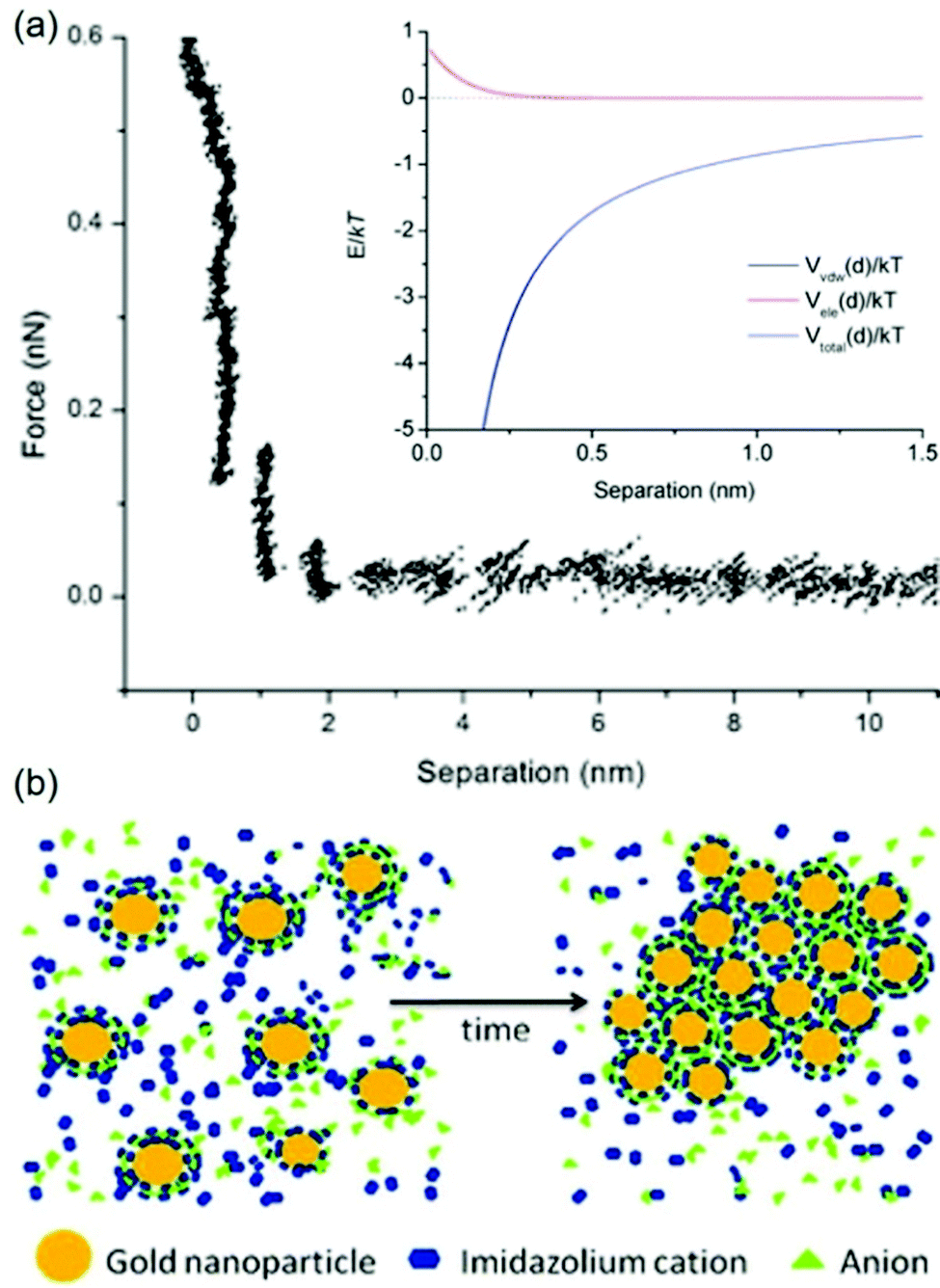
The stabilization mechanisms of NPs in ILs based on the organization of ILs in the form of supermolecules, the formation of carbine, the alkyl chain of cations as the inner shell, and cation–anion interaction have been also considered elsewhere.133–137 Also it should be noted that some previous experimental results did not completely follow the anion-size dependent particle-size trend.128 For instance, Wender et al. reported that the size of Au NPs increased in the order of NTf2− (3.5 ± 0.6 nm) ∼ BF4− (3.6 ± 0.4 nm) ∼ PF6− (3.7 ± 0.4 nm) < FAP−:F3P−(CF2CF3)3 (4.9 ± 0.9 nm), whereas the anion size increased in the order of BF4− < PF6− < NTf2− < FAP−.102 Based on DLVO theory for the repulsive force from a double layer structure and the van der Waals attraction, Vanecht et al. estimated the total negative potentials over the whole distance from the particle surface (Fig. 15a, inset). Thus, other stabilization mechanisms should be taken into account. Further, Vanecht et al. observed a step-wise repulsive force in the AFM force–distance profile between an Au-coated AFM tip and an Au-coated mica surface in C4mim+/N(CN)2− (Fig. 15a). Thus, they suggested that anions and cations of ILs form layer structures on the particle surface and change the electronic structure and local viscosity near particles, which reduces the effective collision of particles (Fig. 15b).
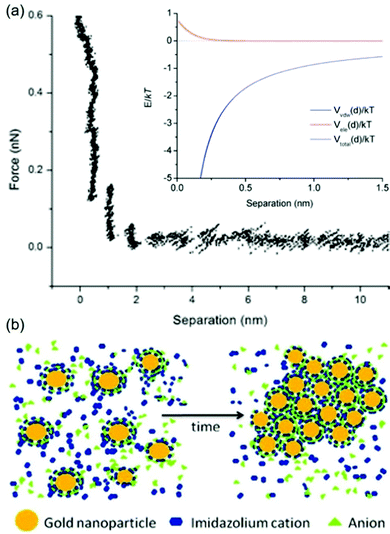 |
| | Fig. 15 (a) The measured AFM force–distance profile in C4mim+/N(CN)2− of an Au-coated AFM tip and an Au-coated mica surface. The inset shows the simulated total potential, Vtotal(d), the sum of van der Waals attraction, Vvdw(d), and electrostatic repulsion, Vele(d), as functions of distance between two spherical Au NPs of 6 nm. (b) Proposed layer structure of anions and cations of the IL on the Au surface for stabilizing Au. (Reproduced from ref. 123 with permission from the PCCP Owner Society. Copyright 2012.) | |
The liquid surface composition (at the vacuum–liquid interface) is another factor in regulating particle growth, particularly, when using functionalized ILs.102,138–140 Wender et al. suggested that the surface composition of Cmim+ ILs varies with anions (BF4−, PF6−, NTf2−:N(SO2CF3)2, and FAP−:PF3(CF2CF3)3).102 The alkyl side chain of Cmim+ cations tends to head to the vacuum, whereas the charged parts of the anions and cations stay in the polar liquid. However, when FAP− anions are present, their alkyl side chain also heads to the vacuum and forms a rich fluorinated surface moiety. The alkyl and perfluorinated region of the surface possibly favours the gathering and growth of bigger particles as observed.102 Thus, particle growth is governed by the anions and the surface composition. Additionally, Wender et al. obtained spherical and ellipsoid Au NPs by sputter deposition onto a nitrile functionalized IL, 1-(butyronitrile)-3-methylimidazolium bis(trifluoromethylsulfonyl)imide (BCN)MI+/N(Tf)2−.138 The ellipsoid Au NPs (together with spherical NPs) were formed in experiments conducted at relatively low discharge voltages (V = 275–340 V), whereas the spherical ones were observed at high voltages (V = 365–400 V). The authors proposed that on the liquid surface the highly coordinating group, i.e., the nitrile group of the alkyl chain of the cation, moved to the vacuum (Fig. 16b). The sputtered Au particles of lower kinetic energy (lower discharge voltage) interacted with the nitrile group, decreased the diffusion into the bulk ILs, and thus interacted with other Au particles on the liquid surface. These Au particles underwent 2D growth to form ellipsoid NPs. In contrast, sputtered particles of high kinetic energy (high voltage) could penetrate the surface more easily and be stabilized by the anion in the liquid to experience 3D growth into spherical NPs. However, the atomic density profiles of the IL surface obtained by molecular dynamics simulations did not support the protrusion of the side chain (–(CH2)3–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N in the cation) of the IL (BCN)MI+/N(Tf)2− into the vacuum (Fig. 16a), unlike the non-polar end groups (e.g., aliphatic group) in other ILs or surfactants.141 Hence, experimental data are needed to confirm the hypothesis of the surface composition of the functionalized ILs and its contribution to the 2D growth of Au NPs.
N in the cation) of the IL (BCN)MI+/N(Tf)2− into the vacuum (Fig. 16a), unlike the non-polar end groups (e.g., aliphatic group) in other ILs or surfactants.141 Hence, experimental data are needed to confirm the hypothesis of the surface composition of the functionalized ILs and its contribution to the 2D growth of Au NPs.
 |
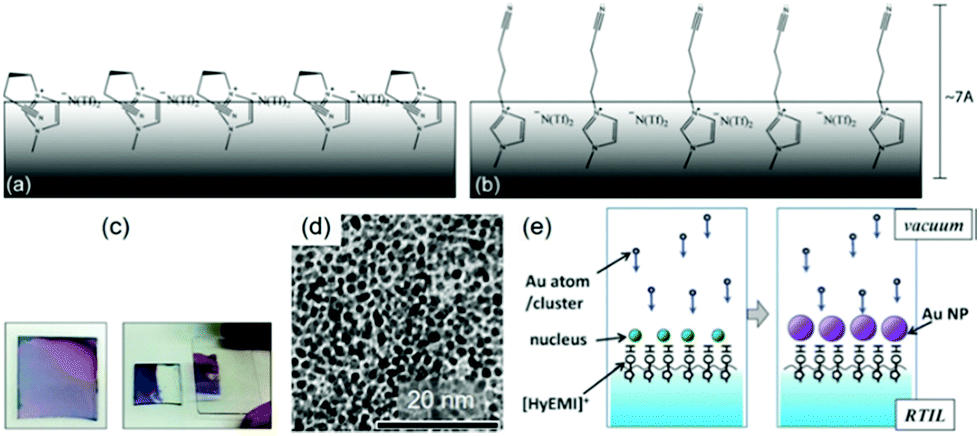
| | Fig. 16 Possible surface configuration of the IL (BNC)MI+/N(Tf)2−: (a) nitrile functional groups interact with cationic regions and (b) orient out of the surface. (Reproduced with permission of ref. 138. Copyright 2011 The Royal Society of Chemistry.) (c) Photographs of an Au film deposited on the HyEMI+/BF4− surface for 300 s (left) before and (right) after transferring half of the film onto a glass plate. (d) TEM image of the Au NPs in the film shown in (c). (e) Formation of Au NPs floating on the IL functionalized with the –OH group heading out of the liquid surface, which supports the formation of a monolayer of Au NPs obtained in (c) and (d). (Reproduced with permission of ref. 139. Copyright 2015 The Royal Society of Chemistry.) | |
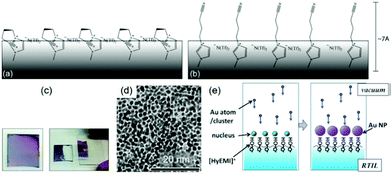
The impact of the surface composition of ILs is more obvious in the reports of Torimoto.139,140 A hydroxyl functionalized IL, 1-(2-hydroxyethyl)-3-methylimidazolium tetrafluoroborate (HyEMI+/BF4−), was used in sputtering to create a thin film of Au NPs (Fig. 16c–e), which can be readily transferred to a solid substrate (Ar, P = 20 Pa, WD = 35 mm, I = 10 mA, Tliquid = RT, t = 0.5–20 min).139 Under similar conditions and using a non-functionalized IL, they obtained a dispersion of Au NPs, not a film.139 This proves the role of the hydroxyl functional group of the cation which was proposed to head to the vacuum and bind to the deposited Au, thus allowing the NPs to grow on the liquid surface (Fig. 16e). A similar method was used to make a monolayer of Au@Pt core@shell NPs by step-wise sputtering.140 These results reveal that the functional group of ILs and its coordination with the sputtered NPs are important in locating and controlling the growth of NPs on the liquid surface.
(b) Viscous liquid polymers.
Liquid polymers used in sputtering such as silicone oils and PEG allow for obtaining NPs in liquids and/or thin films on liquid surfaces, which depend on the synthesis conditions. The high viscosity of the liquids can stabilize NPs via reduction of the diffusion velocity.62,72,79,81,82,94,105,106,108–112,120 PEG can further stabilize NPs via binding of O in the ether (–H2C–O–CH2–) and –OH groups with the particles’ surfaces. This binding offered by PEG is weaker than the electrostatic repulsion in ILs and more strongly coordinating capping ligands (e.g., thiol, carboxylic acid, amine). Thus, in general, the NPs (e.g., Au, Ag) deposited in these liquids are larger and less stable than those in ILs under relatively similar conditions.63,72,73,77,80,122,124 Because the viscosity of the polymers generally increases with their molecular weight, to a certain extent, the particle size are expected to be correlated with the molecular weight of the polymers.
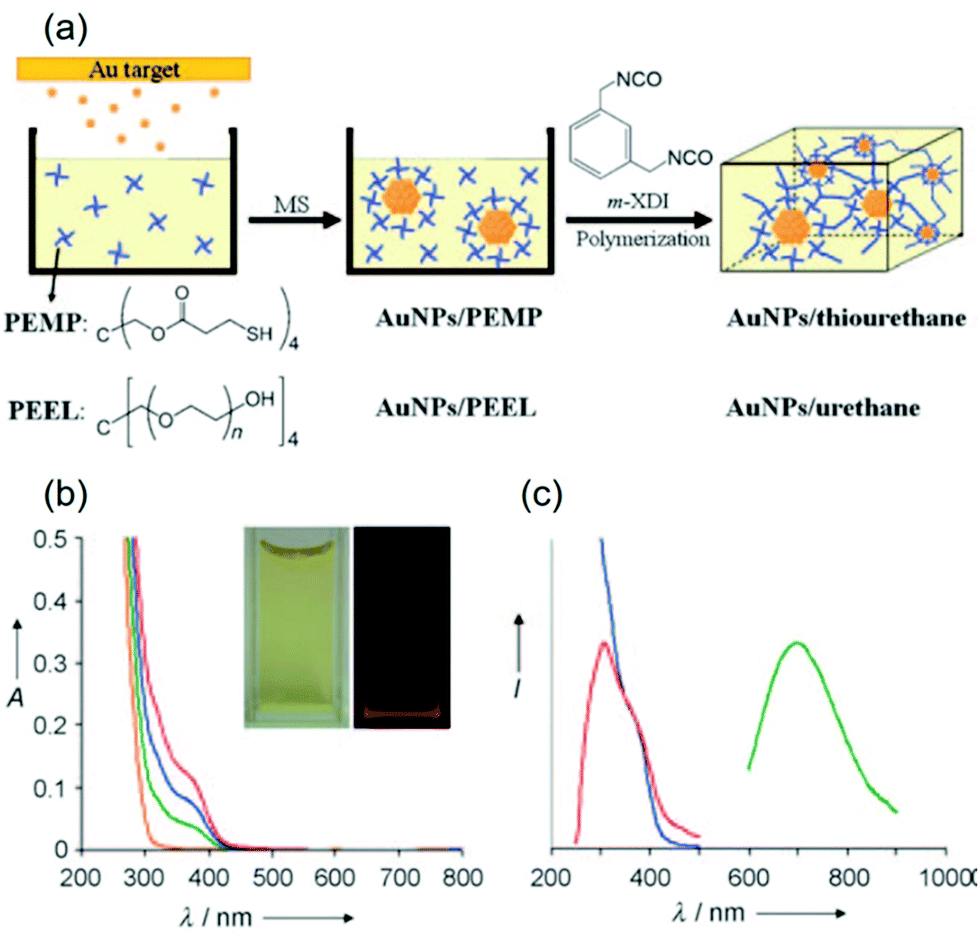
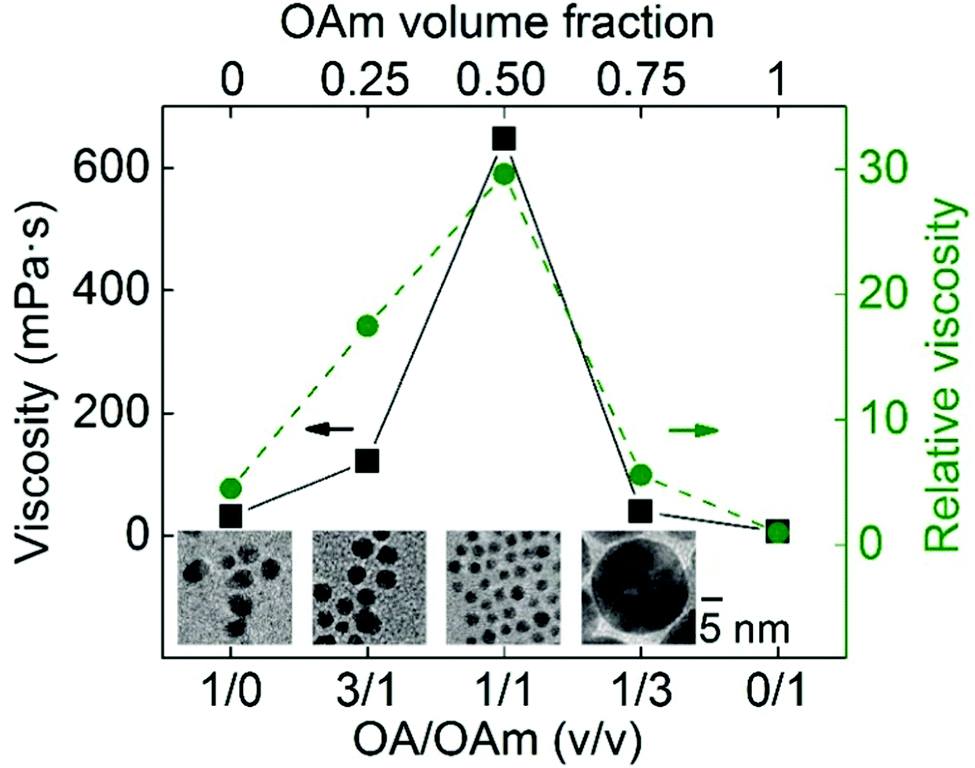
(c) Capping ligands.
In solution synthesis, capping ligands in the reaction solution help mediate particle growth and regulate the particle size and colloidal stability by binding with the particle surface.4,14,16,17,19,25–29,35,36 With the emerging of sputtering onto liquids, capping ligands have been used as substrates themselves,70,71,73,80,124,142 or introduced into the gas phase,100 or dissolved in liquid based substrates (ILs, DG, PEG)75–77,100,122,143–147 for stabilization and size control of the sputtered particles. Thiol molecules have been the most reported in sputter deposition for obtaining noble metal nanoclusters owing to their strong binding affinity. Other ligands with carboxylic acid, amine, hydroxyl, and ester functional groups with weaker bonding to NPs have also been used for size control of 2 nm or more. Ligands can have single, multiple, or mixed functional groups. The functional groups play a key role in regulating the size and properties of sputtered particles. Table 5 summarises the ligands and their functional groups most likely binding to the NPs, and the size and properties of the sputtered particles.
Table 5 Sputtered metal NPs and nanoclusters with ligands in liquid media
| Metal |
Ligand |
Functional group |
Base liquid |
Size (nm) |
λ
em or λab (nm) |
Ref. |
|
λ
em: wavelength of the fluorescent emission peak, λab: wavelength of the UV-vis absorption. |
| Au |
6-(Mercaptohexyl)trimethylammonium bromide (6-MTAB) (molten) |
–SH |
None |
1.3 ± 0.3 |
769em |
70
|
| Au |
Castor oil |
–COO− |
None |
∼2.4–3.6 |
525–550ab |
71
|
| Au |
Pentaerythritol ethoxylate, PEEL |
–OH |
None |
∼2.2 |
520ab |
73
|
| Au |
PEMP |
–SH |
None |
<1 |
690em |
73
|
| Au |
DG |
–OH |
DG |
6.7 ± 3.2 |
520ab |
75
|
| TC |
–SH |
DG |
2.0 ± 0.7 |
673em |
| Au |
11-Mercaptoundecanoic acid (MUA) |
–SH |
PEG |
2.2 |
440ab |
76
|
| Cu |
1.6 |
437em |
77
|
|
|
| Au |
Oleic acid, oleylamine |
–COO− |
None |
2.4 ± 0.4 |
515ab |
80
|
| –NH2 |
| Au |
α-Thioglycerol (volatile) |
–SH |
PEG |
2.4–3.3 |
705–778em |
100
|
| None |
PEG |
4.4–5.4 |
520ab |
| Au |
MUA (molten) |
–SH |
None |
1.6 ± 0.3 |
660em |
122
|
| MUA |
–SH |
PEG |
1.6–4.0 |
657–684em |
| Ag |
PEMP |
–SH |
None |
1.8–3.4 |
NA |
124
|
| Cu |
PEMP |
–SH |
None |
1.1 ± 0.2 |
445em |
142
|
| Cu–Sx |
1.6 ± 0.2 |
630em |
| 1.9 ± 0.3 |
| Ag |
HSCH2CH2COO−Na+ |
–SH |
PEG |
2.7–5.7 |
650em |
143
|
| Au |
11-Mercaptoundecyl-N,N,N-trimethyl ammonium bromide (MUTAB) |
–SH |
PEG |
1.2 ± 0.6 |
629em |
144
|
| Ag |
1.3 ± 0.6 |
432em |
| Cu |
1.0 ± 0.3 |
428em |
| Au–Ag |
MUTAB |
–SH |
PEG |
1.2 ± 0.3 |
444em |
145
|
| ∼1.5 ± 0.5 |
∼661em |
| Au |
Thiocholine hexafluorophosphate TC+·PF6− |
–SH |
PEG |
1.7 ± 0.6 |
755em |
146
|
| Au |
DG |
–OH |
DG |
6.7 ± 3.2 |
530ab |
147
|
| 1-Heptanoic acid |
–COO− |
4.6 ± 0.9 |
530ab |
| 6-Amino-1-hexanol |
–NH2 |
3.0 ± 0.8 |
509ab |
| 6-Mercapto-1-hexanol |
–SH |
2.1 ± 0.8 |
707em |
| Au |
1-Octadecanethiol (OD) |
–SH |
Silicone oil |
1.9 ± 0.4 |
677em |
148
|
| OD (molten) |
–SH |
None |
1.3 ± 0.3 |
664em |
Yonezawa et al. pioneered the use of a protecting agent in sputtering to obtain fluorescent light emitting metal nanoclusters.70,73 They introduced (6-mercaptohexyl)trimethylammonium bromide (6-MTAB) in the molten state called molten salt as the matrix for sputter deposition to achieve near infra-red (NIR) fluorescent Au nanoclusters of 1.3 nm.70 This approach excludes byproducts and purification and allows single-step production of water soluble Au nanoclusters. The authors also reported another thiol molecular capping agent, PEMP, as the liquid matrix for obtaining red fluorescent Au nanoclusters (Fig. 17).73 PEMP with Au was then polymerized to form solid thiourethane resin containing fluorescent Au nanoclusters.73 Later, by a similar synthesis method, blue emitting Cu nanoclusters of 1.6 nm size142 and size tunable Ag NPs of 1.8–3.4 nm124 were attained in PEMP.
 |
| | Fig. 17 (a) Preparation of resin containing Au NPs. (b) UV/vis absorption spectra of Au nanoclusters/PEMP generated with t = 0, 5, 10, and 15 min, which are indicated by orange, green, blue, and red curves, respectively; inset: photographs of AuNPs/PEMP with t = 15 min under room light (left) and UV light (right). (c) UV/vis absorption (blue), photoluminescence (green), and photoluminescence excitation (red) spectra of Au/PEMP with t = 15 min. (Reproduced with permission of ref. 73. Copyright 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.) | |
Yonezawa advanced the idea of using various thiol capping agents by adding them to the vacuum chamber in the form of volatile molecules100 and to liquid based media (e.g., PEG,76,77,122,143–146 DG,75,147 silicone oil148). The ligands can be either cationic70,75,144–146 or anionic,76,77,122,143,147 providing metal nanoclusters with positive or negative charges. In the presence of thiol capping agents, the obtained metal (Au, Ag, Cu, Au/Ag alloy) particles are substantially smaller and more uniform than those without ligands (Table 5). Further, the nanoclusters emit luminescent light dependent on the metal elements, the composition of the alloy nanoclusters, and the metal–ligand bond rather than the observed particle sizes (Table 5 and Fig. 18a).143 In some cases, particles of 2 nm or more (e.g., Au 2.0–2.1 nm, Ag 2.7 nm) also emit fluorescent light similar to small metal clusters (e.g., Au6, Ag8).75,143,146,147 Thus, it is also proposed that the actual size of the fluorophores can be smaller than the particle sizes and that the particles can be clusters/aggregates of the small fluorophores.143,146,147 This was revealed in the STEM-HAADF images of the sputtered Au NPs showing an unclear atomically ordered feature in contrast to the ordered structure of chemically synthesized Au NPs of similar size (Fig. 18b).146 For the same time of sputtering, the emission intensity was proportional to the amount of the ligands in liquid based media to a certain extent.77,122,143 This suggests the role of the ligand in capturing, stopping cluster growth, and stabilizing the formed nanoclusters.77,122,143 Besides, reactive metal nanoclusters such as Cu could react with ligands (PEMP) under storage or UV-irradiation, causing changes in the emission wavelength and intensity over time.142 Freshly prepared Cu nanoclusters emitting blue light could be converted to CuSx nanoclusters with red light emission.142 Yonezawa et al. addressed the structures of nanoclusters, the fluorophore, the origin of the fluorescence, and the fluorescence stability. Ishida et al. reviewed in-depth various aspects of fluorescent nanoclusters created by sputtering.149
 |
| | Fig. 18 (a) Fluorescence emission spectra of the sputtered TC capped Au nanoclusters in acetonitrile after size-fractionation with reversed-phase high-performance liquid chromatography (HPLC) into 5 fractions (I–V) of different particle diameters shown in the figure. (b) STEM-HAADF images of (left) a sputtered Au nanocluster and (right) a chemically synthesized Au NP, both protected with TC. (Reproduced with permission of ref. 146. Copyright 2018 The American Chemical Society.) | |
Ligands containing other functional groups have been compared with a thiol ligand. The size of the sputtered Au NPs decreased from 6.7 to 2.1 nm with an increase in the binding strength of the ligands to Au, i.e., in the order of –OH (DG) < –COOH (1-heptanoic acid) < –NH2 (6-amino-1-hexanol) < –SH (6-mercapto-1-hexanol).147 Further, light emitting Au nanoclusters were observed only with the thiol ligand. In another approach, by mixing the ligands to vary the viscosity and the binding strength, such as oleic acid and oleylamine, Au NPs with tailorable particle size (2.4–30 nm), size uniformity, and colloidal stability (more than a year) were attained (Fig. 19).80 These results demonstrate the capability of fine control of particle sizes in sputter deposition by choosing ligands of suitable binding strength and stabilizing behavior towards NPs as in the bottom-up chemical synthesis.
 |
| | Fig. 19 Mixing oleic acid (OA) and oleylamine (OAm) ligands at different volume ratios (v/v) for control of the size and uniformity of sputtered Au NPs by tailoring the viscosity of the liquid matrix. (Reproduced with permission of ref. 80. Copyright 2020 The American Chemical Society.) | |
Other organic molecules with weak coordination with metal NPs and in the form of viscous and low volatile liquids such as glycerol and its esters (castor oil, canola oil) have been also used as capture media.71,79,150 Wender et al. suggested that the oil surface composition combined with the kinetic energy of sputtered particles could help direct particle growth on the vacuum–liquid interface or inside the liquid.150 For example, low voltages can favour the particles staying on the oil surface, while weak liquid–metal coordination supports the metal–metal interaction/collision across the liquid surface for metal film formation. As observed by Wender et al., in the order of decrease in coordination strength of castor > canola > caprylic/capric triglyceride, Ag NPs, a thin film of many cracks, and a thin film of fewer cracks were formed respectively.150
3. Composition of NPs
Composition control is important for alloy NPs. A recent review discusses various approaches to attain a precise and tuneable metal composition of alloy NPs deposited in liquids.151 Here some representative strategies for composition control and their pros and cons are presented (Fig. 20).
 |
| | Fig. 20 Composition control of sputtered bimetal alloy NPs, control parameters, and complicating factors. (Illustrations a-1, b, c and d are adopted with permission from ref. 151. Copyright 2018 Taylor & Francis Group. Illustration a-2 was redrawn from ref. 152. Copyright 2017 IOP Publishing.) | |
3.1. Using a single target
Torimoto et al. was the first to achieve the composition control of alloy NPs deposited onto ILs by using a binary target (Fig. 20a-1) comprising alternating and radial sections of two metals, i.e., Au and Ag.65 The sputtered Au/Ag alloy NPs in the IL (Fig. 21b, middle) were of different colour from those of pure Ag and Au NPs (Fig. 21b, left and right photos). The final metal ratios of the resulting alloy NPs in the ILs varied linearly with the surface area ratios of the two metals on the target (Fig. 21c).65,113,115,119 However, it is noted that the metal ratios in alloy NPs, e.g., Au/Cu, Au/Pt, and Au/Pd, are not as the same as the area ratios of the metals in the target for the metals having different sputtering rates.113,115,119 Vahl et al. introduced a configuration of a binary target for making Au/Ag alloy NPs which allows for composition control by varying the sputtering gas pressure (Fig. 20a-2).152 In the Ag target, a radially symmetric trench was created for putting a concentric ring of Au wires. The position of the Au wires was the focus of the bombarding Ar ions. The sputtering profile became narrower and more focused on Au wires with a shorter MFP for an increase in Ar gas pressure; thus, this increased the Au content in the sputtered Au/Ag NPs (Fig. 21d). Alloy targets, e.g., AuPd and PtCu, were also used to create alloy NPs with a composition similar to that of the metal target (Fig. 20b).90,106,116 The synthesis and composition control are straightforward in a simple set-up, whereas variation of the NPs’ composition can require the fabrication of various binary or alloy targets with different metal compositions. Besides, when using an alloy target made of two metals with a significant difference in the sputtering rates (yield), the metal composition of the target, and hence the composition of the sputtered NPs, varies with sputtering time.
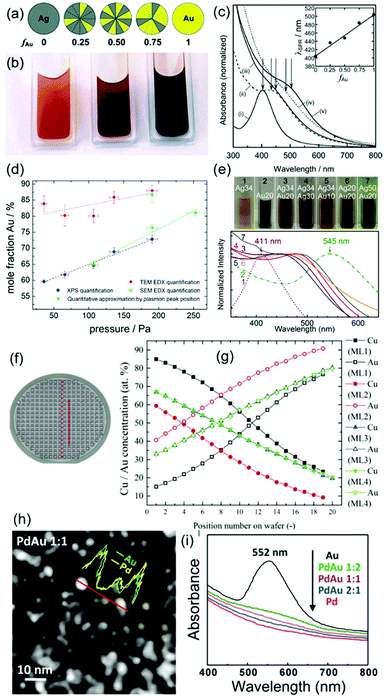 |
| | Fig. 21 Sputtered alloy NPs with composition control using various methods. (a–c) Sputtering using a binary target: (a) binary targets of various surface area fractions of Au (fAu), (b) photos (left to right) of the sputtered Ag, Au/Ag (fAu = 0.5), and Au NPs in an IL, and (c) UV-vis absorption spectra of the obtained Au, Ag, and Au/Ag NPs and the relation between the peak top and fAu.65 (d) Ar-gas-pressure dependent Au composition in Au/Ag NPs by single head sputtering (shown in Fig. 20a-2).152 (e) Co-sputtering of a double-target head with the composition control by sputtering currents of the Au and Ag targets shown in white letters in the inset of the photos (top row) and UV-vis spectra of the Au/Ag alloy NPs (bottom row).108 (f) Co-sputtering of a double-target head of Au and Cu with composition control via positioning of capture media: position of the cavities containing an IL in the vacuum chamber and (g) position dependent composition (by EDX) of Au/Cu NPs.118 Sequential sputtering of Pd followed by Au for different sputtering times for composition control of Au/Pd alloy NPs: (h) HAADF image with the inset of elemental line scans of equimolar Au/Pd and (i) composition-dependent UV-vis spectra of the Au/Pd NPs.156 (Reproduced with permission from ref. 65, 108, 118, 152 and 156 with Copyright 2008 The Royal Society of Chemistry, 2017 IOP Publishing, 2016 Elsevier, 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, and 2015 The Royal Society of Chemistry, respectively.) | |
3.2. Co-sputtering of two or more targets
The problems in Section 3.1 can be solved by using co-sputtering of two or more metal targets wherein varying the sputtering current applied to each target allows for fine control of the alloy composition and the targets themselves can be used constantly until running out (Fig. 20c and 21e). This method was utilized to produce bimetal alloy NPs in liquids with a wide range of compositions for Au/Ag,108 Au/Cu,109,118,119 Ag/Pt,111 Au/Pt,112 Pt/Ni,91 and Au/Pd.110,115 The dependence of the bimetal composition on the relative position of the liquid matrix to the target was reported (Fig. 21f and g).118 Co-sputtering is able to produce solid solution alloy NPs with tunable compositions in various liquid media, e.g., ILs and low volatile liquid polymers (PEG).65,90,106,108–113,115,116,118,119,153–155 Further, Ludwig et al. successfully created high-entropy alloy NPs by co-sputtering of multiple target onto ILs.153–155
3.3. Sequential sputtering
Some success in sequential sputtering (Fig. 20d) for obtaining bi- and tri-metallic alloy NPs of noble metals with well controlled composition has been reported wherein the sputtering time and the applied current of metal targets well correlated with the resulting composition of NPs (Fig. 21h and i).87,117,140,156,157 However, the sequential sputtering and mixing of monometallic NP dispersions do not always form alloy NPs or result in good control of alloy composition and size even with heat treatment.87,112,158 For example, Meischein et al. attempted to achieve high Cu content in AuCu alloy NPs by mixing dispersions of sputtered monometallic Cu and Au NPs with twice the amount of Cu compared with that of Au.158 However, the resulting Au/Cu alloy NPs had Au:Cu of 75:25 (mol/mol) which is close to the Au3Cu phase. The desired composition with higher Cu content could not be reached by the subsequent heat treatment at 100 °C for 1–5 h, which resulted in less Cu in the alloy NPs for longer heating times. This was attributed to the formation of an energetically stable Au3Cu phase and dissolution of Cu atoms into the ionic liquid. Therefore, the control of composition is challenging by physical mixing and heat treatment of mixtures of the deposited monometallic NPs after sputtering.
4. Morphological structures of sputtered NPs
4.1. Particle shape
Not only can particle size and composition be tailored, but sputtering onto liquids also allows for control of NP morphologies, i.e., shape and nanostructure. As mentioned in Section 2.2.4a, Wender et al. attained anisotropic and spherical Au NPs by varying the sputtering discharge voltage of Au onto a nitrile functionalized IL, (BCN)MI+/N(Tf)2−.138 The lower kinetic energy of the sputtered Au is beneficial for Au staying and Au–Au interaction on the IL liquid–vacuum interface. In addition, the nitrile group heading to the vacuum allows for Au–IL interaction on the surface. These factors favour the growth of 2D Au nanodiscs. On the other hand, Au particles with high kinetic energy can penetrate through the liquid surface to a larger distance, and preferably undergoes 3D isotropic growth via interaction with the anion in the internal region of the IL surface as they do with non-functionalized ILs. Thus, spherical Au NPs were formed.
4.2. Core@shell structures
Core@shell structures have been achieved by sequentially sputtering different metals onto liquid substrates.87,140,159 For example, Wang synthesized Ag@Au and Ag@Pd core@shell NPs by successive sputtering onto ILs; that is, Ag was first sputtered, then the target was switched to Au or Pd (Fig. 22a).87 Torimoto et al. used a similar approach to synthesize Au@Pt core@shell NPs.140 However, their approach has a unique feature of regulating the formation of core@shell NPs to occur locally on the IL surface, not in the entire bulk liquid (Fig. 22b). To do that, an IL with a hydroxyl functionalized cation, that is, 1-(2-hydroxyethyl)-3-methylimidazolium tetrafluoroborate (HyEMI-BF4), was used. The hydroxyl group on the liquid surface heading towards the vacuum supported the formation of a monolayer of Au NPs as the core on the liquid surface before a Pt shell was deposited. Besides, Torimoto et al. demonstrated that monolayers of other metal NPs, e.g., Pt and AuPd, could be attained by the same method.158 Further, the metal shells were oxidized to form metal@metal oxide core@shell NPs such as Au@In2O3, AuPd@In2O3, and Pt@In2O3.159 Oxidation of the metal core can also be used to create metal@metal oxide core@shell NPs.160
 |
| | Fig. 22 ILs support the formation of NPs with core@shell structures (a) underneath and (b) on the liquid surface under sequential sputtering using (a) a nonfunctionalized IL and (b) an –OH functionalized IL, respectively. | |
4.3. Hollow and porous structures
Sputtering onto liquids together with post-treatment is able to produce hollow or porous NPs. Suzuki et al. created hollow In2O3 and Cu2O NPs via oxidation of sputtered metallic In and Cu NPs. The diffusion of the metal core toward the shell, growth of a thicker oxide shell, and the formation of a hollow core can be due to the Kirkendall effect under oxidation (Fig. 23a–c).160 In another approach, de-alloying of bimetal alloy NPs obtained by sputtering resulted in porous NPs. This process is related to the removal or dissolution of the more active metal from the alloy NPs. Chauvin et al. dipped co-sputtered Au/Cu alloy NPs into concentrated HNO3 to etch away Cu, leaving 10–20 nm, porous Au NPs with residual Cu (Fig. 23d–f).161 The method can be utilized to create porous nanostructures of other metals or alloys such as porous Pt, which is a highly active catalyst. In control of hollow or porous nanostructures, sputtering onto liquids is advantageous to form small NPs with good dispersibility as the template.
 |
| | Fig. 23 (a) Strategy to form hollow nanostructures of Cu2O NPs via post-sputtering oxidation of the as-sputtered Cu NPs and TEM images of (b) the as-sputtered Cu NPs and (c) the resulting hollow Cu2O NPs.160 (d) De-alloying of co-sputtered alloy Au/Cu NPs to create porous Au NPs. (e and f) TEM images with the insets showing FFTs of the porous Au NPs.161 (Reproduced from ref. 160 and 161. Copyright 2021 and 2020 IOP Publishing.) | |
5. Crystal structure of sputtered NPs
5.1. Fine structure of solid solution alloys
One basic aspect associated with sputter deposition onto liquids is that the crystal structure of deposited NPs is different from that of the bulk counterpart and that of NPs obtained using other synthesis methods. It is well documented that sputtering of two metals (in a single target or in two different targets) resulted in binary solid solution alloy NPs at room temperature of sizes typically below 5 nm for almost the entire range of compositions even within the miscible gaps or for the compositions of intermetallic compounds in the bulk state.103,108–113,115–119 Similar results have also been reported for chemically synthesized NPs wherein the synthesis conditions were varied largely for different metal pairs and the particle size could be bigger than 5 nm.162–164 Thus, the advantages of the former method compared with the latter include the feasibility, reproducibility, and general applicability for almost all metal pairs, which comes from simultaneous generation of atoms of two or more metal elements for alloying at room temperature. The negative enthalpy mixing and small particle size can account for the formation of solid solution alloys. Although the solid solution structure in sputtered NPs is well acceptable, the degree of the complete alloying and composition/structure homogeneity down to each NP may need to be considered. Deng et al. found that the XRD results of sputtered Au/Pt NPs indicated the NPs are a face-centred cubic (fcc) solid solution alloy without any obvious phase segregation (Fig. 24a and b).112 In particular, a single (111) XRD peak between that of Au and Pt was observed for the sputtered Au/Pt NPs wherein the peak shift from Au to Pt was observed for samples with higher Pt at%. Further, the lattice parameter calculated from the XRD peak position was linearly proportional to Pt at%, following Vegard's law for solid solution alloys. These facts suggest a solid alloy structure for Au/Pt NPs. However, in high resolution STEM-EDS line scan and mapping, alloy NPs (∼3 nm) with slightly richer Pt on the surface than that inside were observed, suggesting some local phase segregation or differences in crystal structure (Fig. 24c and d). Recently, Zhu et al. found that the lattice spacing and the composition of Ag/Pt solid solution alloy NPs for a single pair of the applied sputtering currents distribute over a range (Fig. 24e and f) rather than a single number.111 Hence, fine control of the crystal structure of the sputtered NPs in the form of a solid solution is challenging, whereas the formation of the complete alloy of high uniformity among NPs and evaluation of the fine structure are also difficult. In the next sections, we discuss some findings regarding the differences in the crystal structure of the deposited NPs and further crystal transformation under heat treatment or during TEM observation.
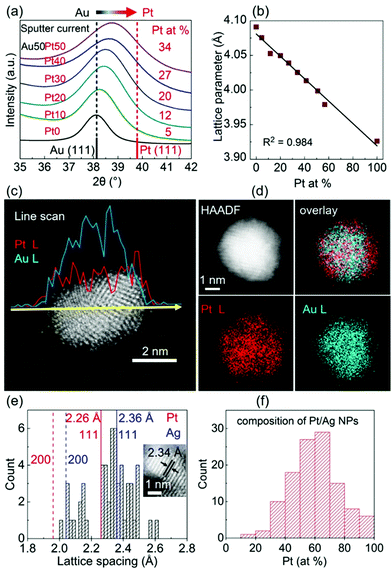 |
| | Fig. 24 Solid solution alloy Au/Cu NPs synthesized by co-sputtering onto liquid PEG: (a) XRD patterns with a peak shift from (111)Au to (111)Pt for sputtered Au/Pt samples with higher Pt at% obtained by keeping the sputtering current for the Au target constant at 50 mA and increasing the sputtering current for the Pt target from 10 to 50 mA, (b) linear relation between the lattice parameter (calculated from the XRD (111) peak) and Pt at% (measured by EDS), (c) STEM-EDS line scan, and (d) mappings of the Au/Pt alloy NPs.112 Co-sputtered solid solution Ag/Pt NPs sputtered at 50 mA for each target onto PEG: (e) distribution of the lattice spacings measured in HAADF images (the inset shows an example) and (f) STEM-EDS composition distribution of single NPs.111 (Reproduced with permission of ref. 111 and 112. Copyright 2021 and 2020 The American Chemical Society.) | |
5.2. Solid solutions and chemically ordered structures
Suzuki et al. reported the crystal structure change of fcc Au/Cu alloy NPs sputtered in the C2mim+/BF4− IL. They conducted comparative experiments without and with heating during sputtering followed by 1 h heat treatment (323–573 K) after sputtering, called Procedures X and Y, respectively. In both cases, they observed that the fcc Au/Cu alloy NPs converted to a chemically ordered structure, L10.119 The key differences are the particle size and the heat treatment temperature for forming the L10 structure. Procedure X resulted in a large particle size (>20 nm at 473 K or more) and the L10 structure appeared at a temperature of >473 K. In contrast, Procedure Y with heat treatment during sputtering (323–473 K) enabled a lower phase transition temperature (423 K) compared with Procedure X. As a result of the lower heat treatment temperature, the particle size was smaller than 20 nm. The authors confirmed that the heat treatment at 323–423 K during sputtering itself did not convert the fcc Au/Cu NPs to an L10 structure. Thus, they proposed that the first heating during sputtering helped form L10 Au/Cu seeds, which could then grow during post-sputtering heat treatment (Fig. 25). Thus, this allowed the lower phase transition temperature in Au/Cu NPs while maintaining small particle size and avoiding aggregation.
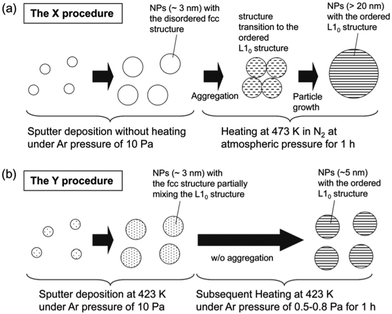 |
| | Fig. 25 Heat treatment of the sputtered fcc AuCu alloy NPs to form an L10 structure by (a) Procedure X at 473 K and (b) Procedure Y at 423 K. Procedure X did not include a 1st step heating under Ar at 423 K during 1 h sputtering, whereas Procedure Y did. The second step heating at 473 K (Procedure X) and 423 K (Procedure Y) is for complete conversion from fcc solid solution alloy NPs to a chemically ordered structure, L10. (Reproduced with permission from ref. 119. Copyright 2015 The Royal Society of Chemistry Publishing.) | |
5.3. Amorphous–crystalline structure transition
Garzón-Manjón et al. reported an amorphous structure for high-entropy alloy NPs, that is, Cr/Mn/Fe/Co/Ni, deposited in an IL by DC co-sputtering of 5 targets.154 They found the transformation from the amorphous to a body-centered cubic (bcc) structure after in situ electron beam irradiation by means of a TEM (Titan, 300 kV, 1.2 × 104 e− nm−2 s−1, 40 min irradiation) or after heat treatment under vacuum (30 Pa) of the NP dispersion in the IL at 100 °C for 2–10 h (Fig. 26a–c). However, an fcc structure was also found after a longer heating time (Fig. 26d). It should be noted that the fcc structure was reported for the bulk Cr/Mn/Fe/Co/Ni high-entropy alloy. Besides, the as-sputtered NPs had a low content of Mn (1–3 at%). However, in the presence of bcc and fcc crystal structures, an increase in particle size and more Mn (5–8 at%) were detected by TEM and EDS, respectively. In contrast, a significant number of the as-deposited alloy NPs had a bcc structure with a more equimolar metal composition (Mn of 10–19 at%) when high-power impulse magnetron sputtering (HiPIMS) was used. HiPIMS can generate sputtered species with higher energies compared with DC sputtering. Therefore, the sputtered species can penetrate the liquid surface and form an alloy in the bulk liquid with more uniformity in the composition. Thus, the authors suggested that the crystal structure transformation perhaps is associated with the change in the Mn content of the deposited multinary alloy NPs and the energy of the sputtered NPs by different deposition methods (Fig. 26e).
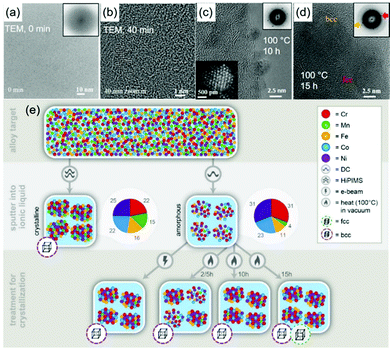 |
| | Fig. 26 Sputtered CrMnFeCoNi alloy NPs: (a) amorphous structure as observed in TEM, (b) bcc structure after 40 min electron beam irradiation in TEM, (c) bcc structure after ex situ heat treatment (100 °C, 10 h), and (d) bcc and fcc structures after heat treatment (100 °C, 15 h). (e) Crystal transformation of CrMnFeCoNi alloy NPs in relation to the Mn content (by ICP), deposition method, and post-treatment. (Reproduced with permission of ref. 154. Copyright 2018 MDPI.) | |
Chau et al. also revealed the formation of disordered or amorphous Pd/Cu alloy NPs aside from alloy NPs with an fcc crystal structure in the as-deposited sample by magnetron co-sputtering of Pd and Cu targets. They observed the transition from disorder/amorphous Pd/Cu NPs to an fcc crystal structure under a scanning electron probe (ARM, 200 kV) for a short period of time (∼60 s) during STEM-EDS analysis (Fig. 27a).110 A chemically ordered structure was not obvious. The increase in particle size was not obvious but joining of NPs and crystal reorientation were observed (Fig. 27b). Hence, the focused electron probe can have an impact on the tiny NPs, triggering the atom re-arrangement and crystallite formation.
 |
| | Fig. 27 Pd/Cu alloy NPs before (t = 0 s) and after STEM-EDS mapping (t = 60 s): (a) improved crystal structure and (b) joining of NPs and crystal reorientation.110 (Reproduced with permission of ref. 110. Copyright 2020 The Royal Society of Chemistry.) | |
6. Particle growth and formation mechanisms
In sputter deposition, the formation of particles can occur in the gas phase (vacuum) and after landing on a substrate. On a solid substrate, e.g., an amorphous Al2O3 film, RF (13.56 MHz) sputtered Pt particles nucleated, then diffused and coalesced, and finally agglomerated.85 In contrast, in high frequency (100 MHz) plasma DC sputtering of Pt on amorphous C or SiO2, the nucleation and island growth occurred from an early stage to complete substrate coverage.84 Using liquid substrates, the particle growth mechanism is not necessarily the same as that on solid substrates due to the mobility of the liquid and its binding to the particles. Where growth mainly occurs during sputtering often depends on many factors associated with the liquids and the deposition conditions used.72,79,96,97,102,106,128 Experimentally, growth was found to occur on the liquid surface to form NPs or thin films; they then could fall into or be stabilized in the bulk liquid.64,71,79,102,138–140,142,150,165 Alternatively, particles could grow in bulk liquids without significant collision and growth on the liquid surface.63,71,80,105,106,109–113,128,147,165 Moreover, the growth could proceed after sputtering, which varies with storage conditions, the kind of metal and the properties of the liquid matrices used.72,80,105,120,123,128,142,166 Below, we discuss the growth mechanism and the regulating factors of particle growth during and after sputtering for various liquids.
6.1. Particle growth and formation during sputtering
Generally, before landing on a liquid surface, the kinetic energy and size of sputtered species depend on V and Ttarget. At a higher kinetic energy, the particles can penetrate the liquid surface faster. Hence, fewer interactions between the particles with the liquid surface and with other particles on the liquid surface are expected. Upon landing on the liquid, γ, η, vstir, Tliquid, and surface functional groups can jointly determine particle growth on the liquid surface by influencing the time that the sputtered particles can stay there and collide with other particles. Meanwhile, the larger the number of particles that arrive at the liquid surface at a time the more the nucleation and growth on the surface possible.105,106 This is regulated by I and tp. Further, the stronger metal–metal interaction than metal–liquid interaction partially favours the surface film formation. In the bulk liquid, η and Tliquid regulate the frequency of the particle collisions. Meanwhile, the functional groups and the kind of liquid determine how well they cap and stabilize particles from the collision. Therefore, these factors affect the particle growth and stability of NPs in liquids during and after sputtering. The extent of the impact of each factor on particle growth varies with the kind of liquid and metal. Hence, the growth on the liquid surface or in the liquid can be tailored. Transfer of particles from the vacuum–liquid interface to the bulk liquid can be also viewed as a diffusion control process, which is faster for particles with higher kinetic energy, in a liquid medium of lower viscosity, at higher temperature, at a faster rotation speed of the liquid, and for weaker interaction of particles with the liquid surface. The more particles arriving at the surface at a time with a lower kinetic energy and stronger metal–metal interaction give more chance for particle collision and growth on the liquid surface than falling into the liquid. Particles in liquids can be protected from growth to different extents by decreasing the liquid temperature, increasing the liquid viscosity, decreasing the particle concentration, and choosing suitable capping agents and liquids. Fig. 28 and 29 depict general growth-control factors in ILs and other liquids, respectively.
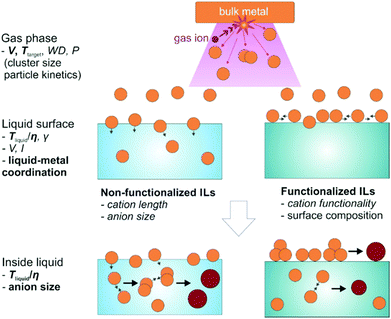 |
| | Fig. 28 Growth mechanism of sputtered particles onto ILs. Common control factors for each step are written on the left with the important ones in boldface. | |
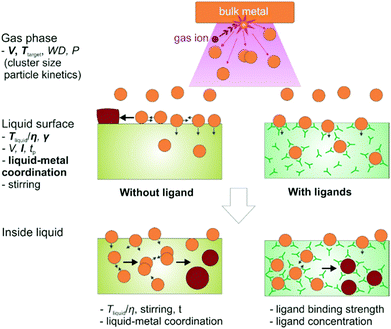 |
| | Fig. 29 Growth control of sputtered particles onto viscous liquids without and with ligands. Common control factors for each step are written on the left with the important factors in boldface. | |
6.1.1. Growth in non-functionalized ILs during sputtering.
Particle growth mainly occurs inside liquids for wide ranges of V (several hundreds to kV), I (several to hundred mA), t (tens of seconds to 50 min), and Tliquid values (Fig. 28).65,90,96,104,119,128 A certain extent of particle growth on surfaces was considered.104 However, interaction of sputtered species with the liquid surface is not likely significant in the sense of having the particle growth mainly on the surface. Thus, the main regulating factors are Tliquid/η (for the diffusion velocity) and the sizes of anions and cations (for stabilizing NPs via electrostatic repulsion and steric hindrance). Hatakeyama et al. demonstrated that the size of Au NPs is linearly proportional to the volume of anions.128 The NPs favourably adsorb anions to form a surface charged double layer. A higher surface charge density and better layer packing attained with smaller anions can stabilize NPs better via electrostatic repulsion and limit the particle collision and growth.
6.1.2. Growth in functionalized ILs during sputtering.
Particle growth can be adjusted to occur partially138 or mainly139,140 on the surface (Fig. 22b), which depends on the functional group of the cations heading to the vacuum–liquid interface and its binding power to the deposited NPs.97 This strategy was used to create anisotropic Au NPs by Wender et al.138 and to form a thin film of NPs by Torimoto.139,140 The applied voltage was adjusted (low or high) to further favour growth on surfaces (more or less, respectively). Besides, the functionalized anions in ILs could also support particle growth on surfaces to a certain extent.102 Wender et al. suggested that a richer fluorinated surface moiety of the surface composition of C4mim+ ILs was formed by using PF3(CF2CF3)3 anions compared with other anions (BF4−, PF6−, NTf2−:N(SO2CF3)2).102 The alkyl and perfluorinated region of the surface can favour the gathering and growth of NPs on the surface to form bigger NPs. The growth inside a liquid is also present when partial growth on the liquid surface occurs. The growth is regulated by Tliquid/η and anion size similar to non-functionalized ILs (Fig. 28, right).
6.1.3. Growth in weakly to moderately coordinating liquids during sputtering.
Liquids such as PEG, glycerol, PEEL, and vegetable oils can bind to particle surfaces, with their binding strength ranging from weak to moderate. Factors such as viscosity and liquid–metal coordination initially determine particle growth on or underneath the liquid surface.79 Static liquids with high viscosity and surface tension with weak coordination to metal favour particle growth on the liquid surface to form a metal film as the liquid wetting of the metal surface decreases and metal–metal interaction becomes more feasible.79 These liquids coordinate with metal NPs more weakly than ILs. This promotes film formation and less stable NP dispersions (Fig. 29, left). For instance, PEG (MW = 600) has a slightly higher viscosity than C4mim+/BF4− IL (e.g., at 20 °C PEG: 167 cP, C4mim+/BF4−: 154 cP72); however, bigger size and more severe size increase with the temperature of Au NPs were observed in PEG than in the IL. Ag thin films and dispersed NPs were formed on liquid PEEL and in C4mim+/TFSI− IL, respectively, due to the weaker coordination of PEEL to Ag than the IL to Ag.165 Due to the low discharge voltage combined with weak co-ordination of vegetable oils to Ag, a Ag film was observed. Otherwise, particles grew inside liquids.150 Once particles submerge into the liquid, their growth is governed by the diffusion velocity via Tliquid, η, and the stabilizing properties of the liquid towards NPs. PEG stabilizes Au NPs more likely more weakly than the electrostatic stabilization and steric hindrance of ILs. Thus, generally temperature accelerates collision, aggregation, and growth in these liquids to a larger extent than that in ILs.72
Yonezawa et al. proved a certain type of growth on both the surface of and inside PEG at 30 °C.105,106 This can explain for the increase in the particle size in PEG with discharge currents more obviously than that in non-functionalized ILs.96 The reason is that the discharge current controls the number of particles reaching the surface at a time, to which the growth on the surface is sensitive. The growth in liquids is also significant as an increase in sputtering time for higher concentrations of particles clearly results in bigger particle sizes and agglomeration or aggregation. Besides, the kind of metal has a clear impact on particle growth in liquids because the liquid–metal binding varies with metals. For example, Au and Ag are not colloidally stable in PEG, whereas Pt is well stabilized because PEG interacts with the latter much more strongly than with the former.
6.1.4. Growth in strongly coordinating liquids during sputtering.
PEMP, MTAB, molten MUA, etc., or liquids containing thiol ligands can be considered as strongly coordinating liquids. Like in weakly coordinating liquids, at high viscosity, particle growth on the liquid surface can take place and overcome the binding of the ligand to metal particle surface (Fig. 29). At relatively lower viscosity, particles were trapped in the liquid (Fig. 29, right), the growth could be well restricted, and thus, small and uniform particles (<2 nm) were formed and stabilized in liquids (Table 5).70,73,75–77,122,124,142–148 The decrease in particle size with an increase in the concentration of ligands has been observed, suggesting that particle growth inside the liquids is hindered by the ligands.75–77,122,143 However, the agglomeration of primary metal clusters can occur.146
6.1.5. Formation and growth of co-sputtered bimetal alloy NPs.
In co-sputtering of two or more metal targets onto a liquid to create alloy NPs, the formation of alloy NPs can occur in the vacuum and liquid phases.108–112,118,153–155,158,161 The particle size upon landing onto the liquid was estimated by deposition on a solid substrate for one second. The size is smaller than that obtained by sputtering onto the liquid for a long time (e.g., 30 min). Thus, it was believed that the growth of alloy NPs occurs in the liquid.111,112 Further, co-sputtered bimetal alloy NPs in PEG are generally more colloidally stable than NPs of one of the metal components.108–112 For example, Pt alloy NPs are smaller and more stable than Au, Ag, and Cu NPs;106,111,112 Au/Cu and Au/Ag alloy NPs are smaller than Au.108,109 This tendency was also observed in other liquid matrices (Table 3).103,113,118 The underlying reason was investigated by Deng et al. with Au/Pt as a case study.112 DFT calculation results suggest that Pt has a higher formation energy than Au, and PEG binds to Pt more strongly than to Au. This causes the Au/Pt alloy NPs with higher Pt content to be smaller and more stable than those with lower Pt content. Their experimental (Fig. 24c and d) and simulation results further revealed that by forming the Pt rich surface, particle growth can be stopped. This leads to the same size for NPs of the same composition despite using higher sputtering currents for both metal targets.112 Selective attachment growth, that is, the effective collision and sticking of Au–Au sites when two particles collide, was proposed for the formation of alloy NPs with a slightly rich Pt surface (Fig. 30).
 |
| | Fig. 30 Formation of Pt/Au alloy NPs in PEG via selective attachment at Au sites and growth to form a surface rich in Pt. The formation of the Pt-rich surface to a certain Pt content helps stop the particle growth. (Reproduced with permission of ref. 112. Copyright 2020 The American Chemical Society.) | |
6.2. Particle growth after sputtering
Monitoring particle size and aggregation after sputtering helps evaluate particle growth in liquid and dispersion stability during storage. Particle size can be measured with TEM, estimated by SAXS, or inferred from the UV-vis spectra of the samples. Localized plasmon resonance bands in the UV-vis range of plasmonic particles such as Au, Ag, and Cu are sensitive to particle size, shape, and inter-particle distance; hence, UV-vis spectra can be used for quick assessment of the NPs’ growth and dispersion stability (size, agglomeration state, precipitation).
6.2.1. Growth in ILs after sputtering.
Vanecht et al. traced the growth of Au NPs with TEM and UV-vis spectra (Fig. 31) after sputtering (Ar, P = 10 Pa, WD = 40 mm, I = 50 mA, t = 60 s) onto C4mim+/N(CN)2− (4 cm3/7 cm2).123,166 The plasmon absorption of the Au increased with time (10 min–4 h). The peak was initially broad (10 min) and then became well-defined, indicating the particle growth of small clusters to >2 nm NPs (Fig. 31a). The increase of the UV-vis peak absorbance was observed for other ILs of different anions (Fig. 31b) and the evolution speed increased in the order of PF6− < BF4− < TfO− < Tf2N− < N(CN)2−. This is also the order of decreasing liquid viscosity, suggesting a diffusion-limited growth. The growth was accompanied by an increase in particle size observed by TEM wherein the number of big NPs increased and that of small NPs decreased. Thus, Vanecht et al. suggested that Au underwent aggregative growth; that is, primary Au clusters aggregate and coalesce to form NPs (Fig. 32).166 The NPs can further aggregate to form precipitates. Heating (at 50 °C) and an increase of the water content in the ILs could accelerate the process by lowering the IL viscosity and loosening the IL layer on the particle surface, respectively.123,166,167 Consequently, the Au dispersions became less stable.123,167 Sedimentation of the thermal vapor deposited Au NPs occurred after the longest period of time for C4mim+/PF6− (9 days) followed by C4mim+/BF4− (5 days) and then C4mim+/Tf2N− (1 day).123 Nishikawa found the smallest particle size in the IL with the smallest anion (e.g., PF6− ∼ BF4− < OTf− < FSA− < TFSA−) as they stabilize NPs better by electrostatic repulsion.128 Though the extents of size increase and the times for having stable colloids are different, similar tendencies are observed regarding the stability effect of anions. This suggests that the growth of particles after sputtering is related to the stabilization ability of ILs. In Vanecht et al.'s reports, the liquid was kept at room temperature and the concentration of Au and sputtering time were significantly smaller than those in other reports.63,102,104 These conditions can help differentiate growth during and after sputtering.
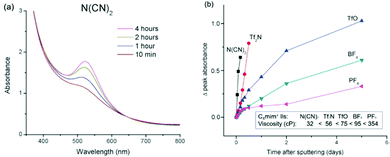 |
| | Fig. 31 (a) UV-vis spectra of Au NPs in C4mim+/N(CN)2− over time after sputter deposition. (b) Increase in peak absorbance as a function of time after sputtering onto different C4mim+ ILs of various anions suggests particle growth. The viscosities (at 25 °C) of the C4mim+ ILs with different anions are shown. (Reproduced with permission of ref. 166. Copyright 2011 The Royal Society of Chemistry.) | |
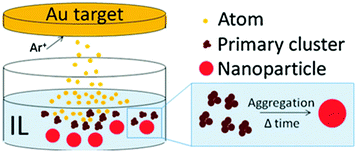 |
| | Fig. 32 Aggressive growth of Au in the IL after sputter deposition. (Reproduced with permission from ref. 166. Copyright 2011 The Royal Society of Chemistry.) | |
6.2.2. Growth in other liquids after sputtering.
(a) In liquid PEG.
Yonezawa et al. reported two-step growth of Pt after sputtering (Ar, P = 2 Pa, WD = 50 mm, I = 5–50 mA, t = 30 min, 10 cm3 PEG/31.2 cm2).105 In the first step, a slight increase in particle size for about 10 days was observed (Fig. 33a–c). Meanwhile, the number density of free Pt atoms (by STEM-HAADF imaging) decreased continuously (Fig. 33a and b).105 This reveals the consumption of the primary free atoms/clusters by ∼0.9–1.4 nm Pt NPs in the aggressive growth (Fig. 33d). In the second step, slight agglomeration of Pt NPs occurred without any obvious increase in the particle size. Meanwhile, precipitates were not observed, and the dispersions were stable for at least 5 months. The growth of Pt in PEG after sputtering shares some common features with that of Au in ILs; however, its kinetics is slower.166 In contrast, Au and Ag NPs agglomerated and formed precipitates more quickly than Pt (few weeks to months).112 Nishikawa reported that the size of Au increased by heating the PEG dispersion (30–110 °C) after sputtering, especially, above 80 °C.72 These results suggest that the growth is via particle diffusion and aggregation for metal NPs not stabilized well in PEG.
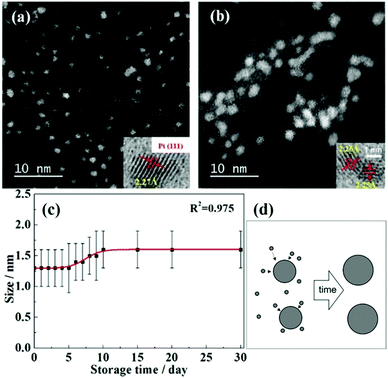 |
| | Fig. 33 STEM-HAADF images of (a) the as-sputtered Pt NPs and (b) after 30 days in PEG. (c) Particle size increase with time after sputtering. (d) Particle growth via consumption of small free Pt atoms/clusters. (Reproduced with permission from ref. 105. Copyright 2018 The American Chemical Society.) | |
Chau et al. reported fast growth of Cu NPs in the first 4 h after sputtering followed by a negligible size change as monitored by TEM and UV-vis spectroscopy.120 Meanwhile, Cu underwent oxidation obviously after a day. The particle growth in PEG containing a strong capping agent, e.g., MUA, can hardly proceed after sputtering. A negligible change in the size and oxidation stability of Cu is expected judging from the stable fluorescent emission intensity of the dispersion after a month.77 Here, the binding strength of the ligand determines the extent of the particle growth.
(b) In oleic acid and oleylamine.
The growth of Au NPs after sputtering in oleic acid mixed with oleylamine occurred slowly over a longer period of time (more than a year) compared with that in PEG and ILs.80 The most stable dispersion was attained with equal volumes of the two ligands. Both the viscosity and binding strength of the liquids, which vary with the ligand volume ratio, determine particle growth after sputtering. It took the longest time for size increase and the plasmon peak of Au NPs in the most viscous liquid (ligand ratio 1/1 (v/v)) to be observed (Fig. 34).80 The increase in plasmon peak absorbance continues after 2 weeks in the sample with a ligand ratio of 1/1 (v/v) though the particle size does not change significantly. This suggests the growth of Au NPs ∼2 nm via consumption of small Au clusters and atoms (Fig. 33d). Mixing two ligands can create carboxylate ions which stabilize Au NPs better than amine or carboxylic acid groups.80 The deposited Au aggregates in oleic acid underwent “dissolution” shortly after the sample had come into contact with air after sputtering. The process is ascribed to the oxidation of the π-bonding of oleic acid surrounding Au aggregates, which introduces the repulsive force for dissolution of Au NPs from the aggregates.168 This can also occur in post sputtering onto oleic acid and oleylamine.80 The longer term growth and better colloidal stability of Au NPs in oleic acid/oleylamine than in PEG after sputtering can be ascribed to the stronger binding of the liquids to NPs via their functional groups.
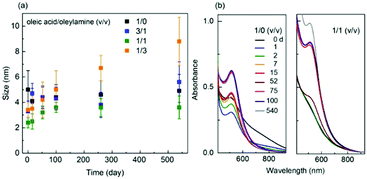 |
| | Fig. 34 (a) Particle size and (b) UV-vis spectra of Au increases as functions of time after sputtering onto oleic acid and oleylamine of various volume ratios (agglomeration and partial sedimentation occurred in samples with ligand ratios other than 1/1 v/v). (Reproduced with permission from ref. 80. Copyright 2021 American Chemical Society.) | |
7. Separation of sputtered NPs from liquid matrices
The obvious advantages of sputtering onto liquids to prepare NPs are it enables making liquid dispersions of NPs and its ability to tailor the size, composition, structure, and stability of NPs. The as-prepared NP dispersions and NP thin films (or metal films) on liquids can be applied in optical devices.62,64,72,169 They can be further transferred to other solid substrates or can be solidified before being utilized.73,78,139,140,170–172 For other applications, which take advantage of their small particle size, complexity in composition, and/or alloy structure, such as catalysts, it is required to separate NPs from liquid matrices. The separation of small and well dispersed NPs, however, is not always a simple task. Centrifugation of particles out of poor solvents,80,118,146 using solid supports (before or after sputtering) to collect NPs (graphite, O, TiO2, SiO2, etc.),67,86–93,95,111,112 and potential-assisted immobilization of NPs onto electrode surfaces153 have been used so far for NP isolation and purification. The anion (of ILs) adsorbed onto metal surfaces can help couple NPs to support materials (e.g., C) via electrical interaction.95 The catalytic properties have been evaluated with promising results, especially for solid solution alloy NPs.67,68,87–94,98,99,113,115,116,140,153,155–157,160,170–172 The sputtered NPs of small sizes and without a strongly coordinating ligand on the surface are good in catalysis for capping ligands can hinder their catalytic performance.173,174
8. Concluding remarks
In this review, we have introduced sputter deposition onto liquids for synthesis of NPs and its relation to the conventional solution approach and vacuum deposition technique. The impact of each experimental factor on the particle size, composition, and morphological and crystal structures of NPs has been discussed and evaluated for various kinds of liquids. Physical parameters can be tuned to regulate the particle size, crystal structure and alloy formation. On the other hand, “chemistry” associated with the liquid plays a bigger and bigger role when it comes to finer control of the particle size, morphological structure, and particle growth mechanism of NPs. The particle size can be tuned to the range below 10 nm to a single digit wherein selection of ligands and kind of liquid are critical. The particle growth mechanisms during and after sputtering, their stability, and where the growth mainly occurs have been categorized based on the liquids. Following that, the main growth-control factors has been discussed. Above all, the liquid viscosity, liquid functionality, and its interaction with the particles are the most important factors. A broad picture of the particle formation with details of the impact of the parameters involved has thus been presented.
The synthesis method and strategies discussed in this review enable the formation of alloys with a tailorable composition and number of metal elements, and crystal structure control in a reproducible and expandable manner. Creating multi-element alloys, crystal control over a small size range and well dispersed NPs are among the most significant advantages of this method. There remain challenges which include the fine control of alloy structure and uniformity in composition. Besides, the growth and the formation mechanism of alloy NPs are not fully understood yet. The applications of metal and metal alloy NPs are of interest and should be considered.
We hope this review not only provides an overview and background for people new to this field but also stimulate the interest in the area of fine control of the size, shape, composition, and structures of NPs. We also expect a lot of novel structures, and new understandings of structure formation of small NPs and their novel properties for advanced applications such as optics and catalysis.
Author contributions
MTN and LD composed the initial draft in consultation with TY. MTN and TY drew illustration and prepared data presentation. MTN and TY revised the draft and competed the manuscript. All authors reviewed the manuscript. TY conducted overall management.
Conflicts of interest
There is no conflict of interest to declare.
Acknowledgements
This work was supported by Hokkaido University. TY acknowledges the financial support by the Grants-in-Aid for Scientific Research (B) program (18H01820) and for Scientific Research on Innovative Areas (Research in a proposed research area) (19H05162 and 20H00138) from JSPS. MTN acknowledges financial support by the Grant-in-Aid for Young Researcher (B) program (17K14072) from JSPS. LD acknowledges the scholarship from the Japanese government for her stay in Sapporo.
Notes and references
- R. P. Feynman, Eng. Sci., 1960, 23, 22–36 Search PubMed . There's plenty of room at the bottom.
-
G. Schmid, Nanoparticles: From Theory to Application, Wiley-VCH Verlag GmbH & Co. KGaA, 2004 Search PubMed.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- A. Tao, P. Sinsermsuksakul and P. Yang, Angew. Chem., 2006, 118, 4713–4717 CrossRef.
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 405–408 CrossRef CAS.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS PubMed.
- M. S. Chen and D. W. Goodman, Science, 2004, 306, 252–255 CrossRef CAS PubMed.
- D. Astruc, Chem. Rev., 2020, 120, 461–463 CrossRef CAS PubMed.
- W. J. Stark, P. R. Stoessel, W. Wohlleben and A. Hafner, Chem. Soc. Rev., 2015, 44, 5793–5805 RSC.
- N. Geerts and E. Eiser, Soft Mater., 2010, 6, 4647–4660 RSC.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS PubMed.
- R. K. Ramamoorthy, E. Yildirim, E. Barba, P. Roblin, J. A. Vargas, L.-M. Lacroix, I. Rodriguez-Ruiz, P. Decorse, V. Petkov, S. Teychené and G. Viau, Nanoscale, 2020, 12, 16173–16188 RSC.
- C. B. Whitehead, S. Özkar and R. G. Finke, Mater. Adv., 2021, 2, 186–235 RSC.
- N. Toshima and T. Yonezawa, New J. Chem., 1998, 22, 1179–1201 RSC.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108(3), 845–910 CrossRef CAS PubMed.
- Y. Ma, W. Li, E. C. Cho, Z. Li, T. Yu, J. Zeng, Z. Xie and Y. Xia, ACS Nano, 2010, 4, 6725–6734 CrossRef CAS PubMed.
- C. Wang, W. Tian, Y. Ding, Y. Ma, Z. L. Wang, N. M. Markovic, V. R. Stamenkovic, H. Daimon and S. Sun, J. Am. Chem. Soc., 2010, 132, 6524–6529 CrossRef CAS PubMed.
- S. Mishra, K. Song, K. C. Ghosh and M. Nath, ACS Nano, 2014, 8, 2077–2086 CrossRef CAS PubMed.
- T. Bian, H. Zhang, Y. Jiang, C. Jin, J. Wu, H. Yang and D. Yang, Nano Lett., 2015, 15, 7808–7815 CrossRef CAS PubMed.
- K. D. Malviya and K. Chattopadhyay, J. Phys. Chem. C, 2016, 120, 27699–27706 CrossRef CAS.
- D. F. Swearer, R. K. Leary, R. Newell, S. Yazdi, H. Robatjazi, Y. Zhang, D. Renard, P. Nordlander, P. A. Midgley, N. J. Halas and E. Ringe, ACS Nano, 2017, 11, 10281–10288 CrossRef CAS PubMed.
- H. Shirai, M. T. Nguyen, D. Čempel, H. Tsukamoto, T. Tokunaga, Y.-C. Liao and T. Yonezawa, Bull. Chem. Soc. Jpn., 2017, 90, 279–285 CrossRef CAS.
- C. Wadell, A. Yasuhara and T. Sannomiya, J. Phys. Chem. C, 2017, 121, 27029–27035 CrossRef CAS.
- D. Čempel, M. T. Nguyen, Y. Ishida, T. Tokunaga and T. Yonezawa, New J. Chem., 2018, 42, 5680–5687 RSC.
- M. J. Saw, M. T. Nguyen, S. Zhu, Y. Wang and T. Yonezawa, RSC Adv., 2019, 9, 21786–21792 RSC.
- C. Song, A. Tayal, O. Seo, J. Kim, Y. Chen, S. Hiroi, L. S. R. Kumara, K. Kusada, H. Kobayashi, H. Kitagawa and O. Sakata, Nanoscale Adv., 2019, 1, 546–553 RSC.
- K. Kusada, D. Wu, T. Yamamoto, T. Toriyama, S. Matsumura, W. Xie, M. Koyama, S. Kawaguchi, Y. Kubota and H. Kitagawa, Chem. Sci., 2019, 10, 652–656 RSC.
- D. Wu, K. Kusada, T. Yamamoto, T. Toriyama, S. Matsumura, I. Gueye, O. Seo, J. Kim, S. Hiroi, O. Sakata, S. Kawaguchi, Y. Kubota and H. Kitagawa, Chem. Sci., 2020, 11, 12731–12736 RSC.
- D. Wu, K. Kusada, T. Yamamoto, T. Toriyama, S. Matsumura, S. Kawaguchi, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2020, 142, 13833–13838 CrossRef CAS PubMed.
- S. G. Jang, A. Khan, M. D. Dimitriou, B. J. Kim, N. A. Lynd, E. J. Kramer and C. J. Hawker, Soft Matter, 2011, 7, 6255–6263 RSC.
- N. Toshima, K. Kushihashi, T. Yonezawa and H. Hirai, Chem. Lett., 1989, 1769–1770 CrossRef CAS.
- T. Yonezawa and N. Toshima, J. Chem. Soc., Faraday Trans., 1995, 91, 4111–4119 RSC.
- A. Candreva, G. D. Maio and M. La Deda, Soft Matter, 2020, 16, 10865–10868 RSC.
- S. Abalde-Cela, B. Auguié, M. Fischlechner, W. T. S. Huck, R. A. Alvarez-Puebla, L. M. Liz-Marzán and C. Abell, Soft Matter, 2011, 7, 1321–1325 RSC.
-
T. Yonezawa, S. Zhu and M. T. Nguyen, Chapter 15. Miscellaneous reductants, in Reducing Agents in Colloidal Nanoparticle Synthesis, ed. S. Mourdikoudis, The Royal Society of Chemistry, 2021 Search PubMed.
- P. Pulkkinen, J. Shan, K. Leppänen, A. Känsäkoski, A. Laiho, M. Järn and H. Tenhu, ACS Appl. Mater. Interfaces, 2009, 1, 519–525 CrossRef CAS PubMed.
- I. Díez, H. Hahn, O. Ikkala, H. G. Börner and R. H. A. Ras, Soft Matter, 2010, 6, 3160–3162 RSC.
- S. Liu, R. Tokura, M. T. Nguyen, H. Tsukamoto and T. Yonezawa, Adv. Powder Technol., 2020, 31, 4570–4575 CrossRef CAS.
- M. Tomonari, K. Ida, H. Yamashita and T. Yonezawa, J. Nanosci. Nanotechnol., 2008, 8, 2468–2471 CrossRef CAS PubMed.
- T. Rao, X.-H. Dong, B. C. Katzenmeyer, C. Wesdemiotis, S. Z. D. Cheng and M. L. Becker, Soft Matter, 2012, 8, 2965–2971 RSC.
- I. Maity, D. B. Rasale and A. K. Das, Soft Matter, 2012, 8, 5301–5308 RSC.
- Z. Y. Li, N. P. Young, M. Di Vece, S. Palomba, R. E. Palmer, A. L. Bleloch, B. C. Curley, R. L. Johnston, J. Jiang and J. Yuan, Nature, 2008, 451, 46–48 CrossRef CAS PubMed.
- I. Lee, S. W. Han and K. Kim, Chem. Commun., 2001, 1782–1783 RSC.
- T. Seto, K. Koga, H. Akinaga, F. Takano, T. Orii and M. Hirasawa, J. Nanopart. Res., 2006, 8, 371–378 CrossRef CAS.
- S. Sato, K. Mori, O. Ariyada, H. Atsushi and T. Yonezawa, Surf. Coat. Technol., 2011, 206, 955–958 CrossRef CAS.
- Y. Hattori, S. Mukasa, H. Toyota, T. Inoue and S. Nomura, Mater. Chem. Phys., 2011, 131, 425–430 CrossRef CAS.
- A. Yamaguchi, Y. Mashima and T. Iyoda, Angew. Chem., Int. Ed., 2015, 54, 12809–12813 CrossRef CAS PubMed.
- M. Y. Rekha, N. Mallik and C. Srivastava, Sci. Rep., 2018, 8, 8737 CrossRef CAS PubMed.
- P. Gambardella, S. Rusponi, M. Veronese, S. S. Dhesi, C. Grazioli, C. Grazioli, I. Cabria, R. Zeller, P. H. Dederichs, K. Kern, C. Carbone and H. Brune, Science, 2003, 300, 1130–1133 CrossRef CAS PubMed.
- W. R. Grove, Philos. Trans. R. Soc. London, 1852, 142, 87–101 CrossRef.
- J. E. Greene, J. Vac. Sci. Technol., A, 2017, 35, 05C204 CrossRef.
- P. Davidse and L. Maissel, J. Appl. Phys., 1966, 37, 574–579 CrossRef.
- S. Terauchi, N. Koshizaki and H. Umehara, Nanostruct. Mater., 1995, 5, 71–78 CrossRef CAS.
- J.-H. Kwon, D.-Y. Kim and N.-M. Hwang, Coatings, 2020, 10, 443 CrossRef CAS.
- P. J. Kelly and R. D. Arnell, Vacuum, 2000, 56, 159–172 CrossRef CAS.
- J. T. Gudmundsson, Plasma Sources Sci. Technol., 2020, 29, 113001 CrossRef CAS.
- G. E. Johnson, R. Colby and J. Laskin, Nanoscale, 2015, 7, 3491–3503 RSC.
- R. Cai, P. R. Ellis, J. Yin, J. Liu, C. M. Brown, R. Griffin, G. Chang, D. Yang, J. Ren and K. Cooke, Small, 2018, 14, 1703734 CrossRef PubMed.
- P. Benzo, S. Combettes, C. Garcia, T. Hungria, B. Pécassou and M.-J. Casanove, Cryst. Growth Des., 2020, 20, 4144–4149 CrossRef CAS.
- J. Vernieres, S. Steinhauer, J. Zhao, P. Grammatikopoulos, R. Ferrando, K. Nordlund, F. Djurabekova and M. Sowwan, Adv. Sci., 2019, 6, 1900447 CrossRef PubMed.
- G.-X. Ye, Q.-R. Zhang, C.-M. Feng, H.-L. Ge and Z.-K. Jiao, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 14754 CrossRef CAS PubMed.
- M. Wagener and B. Günther, J. Magn. Magn. Mater., 1999, 201, 41–44 CrossRef CAS.
- T. Torimoto, K.-i. Okazaki, T. Kiyama, K. Hirahara, N. Tanaka and S. Kuwabata, Appl. Phys. Lett., 2006, 89, 243117 CrossRef.
- E. Borra, O. Seddiki, R. Angel, D. Eisenstein, P. Hickson, K. R. Seddon and S. P. Worden, Nature, 2007, 447, 979–981 CrossRef CAS PubMed.
- K.-i. Okazaki, T. Kiyama, K. Hirahara, N. Tanaka, S. Kuwabata and T. Torimoto, Chem. Commun., 2008, 691–693 RSC.
- T. Suzuki, K.-I. Okazaki, S. Suzuki, T. Shibayama, S. Kuwabata and T. Torimoto, Chem. Mater., 2010, 22, 5209–5215 CrossRef CAS.
- T. Tsuda, K. Yoshii, T. Torimoto and S. Kuwabata, J. Power Sources, 2010, 195, 5980–5985 CrossRef CAS.
- Y. Oda, K. Hirano, K. Yoshii, S. Kuwabata, T. Torimoto and M. Miura, Chem. Lett., 2010, 39, 1069–1071 CrossRef CAS.
- S. Kuwabata, T. Tsuda and T. Torimoto, J. Phys. Chem. Lett., 2010, 1, 3177–3188 CrossRef CAS.
- Y. Shishino, T. Yonezawa, K. Kawai and H. Nishihara, Chem. Commun., 2010, 46, 7211–7213 RSC.
- H. Wender, L. F. de Oliveira, A. F. Feil, E. Lissner, P. Migowski, M. R. Meneghetti, S. R. Teixeira and J. Dupont, Chem. Commun., 2010, 46, 7019–7021 RSC.
- Y. Hatakeyama, T. Morita, S. Takahashi, K. Onishi and K. Nishikawa, J. Phys. Chem. C, 2011, 115, 3279–3285 CrossRef CAS.
- Y. Shishino, T. Yonezawa, S. Udagawa, K. Hase and H. Nishihara, Angew. Chem., Int. Ed., 2011, 50, 703–705 CrossRef CAS PubMed.
- V. S. Raghuwanshi, M. Ochmann, A. Hoell, F. Polzer and K. Rademann, Langmuir, 2014, 30, 6038–6046 CrossRef CAS PubMed.
- Y. Ishida, C. Lee and T. Yonezawa, Sci. Rep., 2015, 5, 15372–15377 CrossRef CAS PubMed.
- Y. Ishida, R. Nakabayashi, M. Matsubara and T. Yonezawa, New J. Chem., 2015, 39, 4227–4230 RSC.
- M. Porta, M. T. Nguyen, Y. Ishida and T. Yonezawa, RSC Adv., 2016, 6, 105030 RSC.
- M. Porta, M. T. Nguyen, T. Yonezawa, T. Tokunaga, Y. Ishida, H. Tsukamoto, Y. Shishino and Y. Hatakeyama, New J. Chem., 2016, 40, 9337–9343 RSC.
- M. M. De Luna and M. Gupta, Appl. Phys. Lett., 2018, 112, 201605 CrossRef.
- M. T. Nguyen, K. Wongrujipairoj, H. Tsukamoto, S. Kheawhom, S. Mei, V. Aupama and T. Yonezawa, ACS Sustainable Chem. Eng., 2020, 8, 18167–18176 CrossRef CAS.
- H. Ge, C. Feng, G. Ye, Y. Ren and Z. Jiao, J. Appl. Phys., 1997, 82, 5469–5471 CrossRef CAS.
- G.-X. Ye, T. Michely, V. Weidenhof, I. Friedrich and M. Wuttig, Phys. Rev. Lett., 1998, 81, 622 CrossRef CAS.
-
K. Wasa and S. Hayakawa, Handbook of sputter deposition
technology, 1992 Search PubMed.
- P. Andreazza, C. Andreazza-Vignolle, J. Rozenbaum, A.-L. Thomann and P. Brault, Surf. Coat. Technol., 2002, 151, 122–127 CrossRef.
- B. Ramalingam, S. Mukherjee, C. J. Mathai, K. Gangopadhyay and S. Gangopadhyay, Nanotechnology, 2013, 24, 205602 CrossRef PubMed.
- C.-H. Liu, B.-H. Mao, J. Gao, S. Zhang, X. Gao, Z. Liu, S.-T. Lee, X.-H. Sun and S.-D. Wang, Carbon, 2012, 50, 3008–3014 CrossRef CAS.
- C.-H. Liu, X.-Q. Chen, Y.-F. Hu, T.-K. Sham, Q.-J. Sun, J.-B. Chang, X. Gao, X.-H. Sun and S.-D. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 5072–5079 CrossRef CAS PubMed.
- L. Luza, A. Gual, D. Eberhardt, S. R. Teixeira, S. S. Chiaro and J. Dupont, ChemCatChem, 2013, 5, 2471–2478 CrossRef CAS.
- I. Y. Cha, M. Ahn, S. J. Yoo and Y.-E. Sung, RSC Adv., 2014, 4, 38575–38580 RSC.
- C.-H. Liu, J. Liu, Y.-Y. Zhou, X.-L. Cai, Y. Lu, X. Gao and S.-D. Wang, Carbon, 2015, 94, 295–300 CrossRef CAS.
- Y.-Y. Zhou, C.-H. Liu, J. Liu, X.-L. Cai, Y. Lu, H. Zhang, X.-H. Sun and S.-D. Wang, Nano-Micro Lett., 2016, 8, 371–380 CrossRef PubMed.
- L. Luza, C. P. Rambor, A. Gual, F. Bernardi, J. B. Domingos, T. Grehl, P. Bruner and J. Dupont, ACS Catal., 2016, 6, 6478–6486 CrossRef CAS.
- K. Yoshii, K. Yamaji, T. Tsuda, H. Matsumoto, T. Sato, R. Izumi, T. Torimoto and S. Kuwabata, J. Mater. Chem. A, 2016, 4, 12152–12157 RSC.
- M. W. Chung, I. Y. Cha, M. G. Ha, Y. Na, J. Hwang, H. C. Ham, H.-J. Kim, D. Henkensmeier, S. J. Yoo and J. Y. Kim, Appl. Catal., B, 2018, 237, 673–680 CrossRef CAS.
- I. Y. Cha, H. T. Kim, M. Ahn, J. H. Jang, Y. G. Kim, Y.-E. Sung and S. J. Yoo, Appl. Surf. Sci., 2019, 471, 1083–1087 CrossRef CAS.
- Y. Hatakeyama, K. Onishi and K. Nishikawa, RSC Adv., 2011, 1, 1815–1821 RSC.
- H. Wender, P. Migowski, A. F. Feil, S. R. Teixeira and J. Dupont, Coord. Chem. Rev., 2013, 257, 2468–2483 CrossRef CAS.
- I. Y. Cha, S. J. Yoo and J. H. Jang, J. Electrochem. Sci. Technol., 2016, 7, 13–26 CrossRef CAS.
- T. Tsuda, T. Kurihara, Y. Hoshino, T. Kiyama, K.-I. Okazaki, T. Torimoto and S. Kuwabata, Electrochemistry, 2009, 77, 693–695 CrossRef CAS.
- T. Sumi, S. Motono, Y. Ishida, N. Shirahata and T. Yonezawa, Langmuir, 2015, 31, 4323–4329 CrossRef CAS PubMed.
- P. Asanithi, S. Chaiyakun and P. Limsuwan, J. Nanomater., 2012, 963609 Search PubMed (8 pages).
- H. Wender, L. F. de Oliveira, P. Migowski, A. F. Feil, E. Lissner, M. H. Prechtl, S. R. Teixeira and J. Dupont, J. Phys. Chem. C, 2010, 114, 11764–11768 CrossRef CAS.
- T. Suzuki, K.-i. Okazaki, T. Kiyama, S. Kuwabata and T. Torimoto, Electrochemistry, 2009, 77, 636–638 CrossRef CAS.
- Y. Hatakeyama, M. Okamoto, T. Torimoto, S. Kuwabata and K. Nishikawa, J. Phys. Chem. C, 2009, 113, 3917–3922 CrossRef CAS.
- L. Deng, M. T. Nguyen and T. Yonezawa, Langmuir, 2018, 34, 2876–2881 CrossRef CAS PubMed.
- L. Deng, M. T. Nguyen, S. Mei, T. Tokunaga, M. Kudo, S. Matsumura and T. Yonezawa, Langmuir, 2019, 35, 8418–8427 CAS.
-
D. Halliday, R. Resnick and J. Walker, Fundamentals of Physics, John Wiley & Sons, Inc., 8th edn, 2008, p. 660 Search PubMed.
- M. T. Nguyen, T. Yonezawa, Y. Wang and T. Tokunaga, Mater. Lett., 2016, 171, 75–78 CrossRef CAS.
- M. T. Nguyen, H. Zhang, L. Deng, T. Tokunaga and T. Yonezawa, Langmuir, 2017, 33, 12389–12397 CrossRef CAS PubMed.
- Y.-t. R. Chau, M. T. Nguyen, M. Zhu, A. Romier, T. Tokunaga and T. Yonezawa, New J. Chem., 2020, 44, 4704–4712 RSC.
- M. Zhu, M. T. Nguyen, Y.-T. R. Chau, L. Deng and T. Yonezawa, Langmuir, 2021, 37, 6096–6105 CrossRef CAS PubMed.
- L. Deng, M. T. Nguyen, J. Shi, Y.-T. R. Chau, T. Tokunaga, M. Kudo, S. Matsumura, N. Hashimoto and T. Yonezawa, Langmuir, 2020, 36, 3004–3015 CrossRef CAS PubMed.
- S. Suzuki, T. Suzuki, Y. Tomita, M. Hirano, K.-I. Okazaki, S. Kuwabata and T. Torimoto, CrystEngComm, 2012, 14, 4922–4926 RSC.
-
K. Wasa, 2 - Sputtering Phenomena, in Handbook of Sputtering Technology, ed. K. Wasa, I. Kanno and H. Kotera, William Andrew Publishing, 2nd edn, 2012, pp. 41–75 Search PubMed.
- M. Hirano, K. Enokida, K.-I. Okazaki, S. Kuwabata, H. Yoshida and T. Torimoto, Phys. Chem. Chem. Phys., 2013, 15, 7286–7294 RSC.
- C. Liu, X. Cai, J. Wang, J. Liu, A. Riese, Z. Chen, X. Sun and S.-D. Wang, Int. J. Hydrogen Energy, 2016, 41, 13476–13484 CrossRef CAS.
- C. Liu, N. Chen, J. Li, X. Gao, T.-K. Sham and S.-D. Wang, J. Phys. Chem. C, 2017, 121, 28385–28394 CrossRef CAS.
- D. König, K. Richter, A. Siegel, A. V. Mudring and A. Ludwig, Adv. Funct. Mater., 2014, 24, 2049–2056 CrossRef.
- S. Suzuki, Y. Tomita, S. Kuwabata and T. Torimoto, Dalton Trans., 2015, 44, 4186–4194 RSC.
- Y.-T. R. Chau, L. Deng, M. T. Nguyen and T. Yonezawa, MRS Adv., 2019, 4, 305–309 CrossRef CAS.
- Y. Hatakeyama, S. Takahashi and K. Nishikawa, J. Phys. Chem. C, 2010, 114, 11098–11102 CrossRef CAS.
- Y. Ishida, I. Akita, T. Sumi, M. Matsubara and T. Yonezawa, Sci. Rep., 2016, 6, 29928 CrossRef CAS PubMed.
- E. Vanecht, K. Binnemans, S. Patskovsky, M. Meunier, J. W. Seo, L. Stappers and J. Fransaer, Phys. Chem. Chem. Phys., 2012, 14, 5662–5671 RSC.
- Y. Ishida, S. Udagawa and T. Yonezawa, Colloids Surf., A, 2016, 498, 106–111 CrossRef CAS.
- F. Beiranvand, H. Najibi and B. H. Shahraki, Iran. J. Oil Gas Sci. Technol., 2020, 9(3), 26–43 Search PubMed.
-
A. M. Al-Shareef, Wetting kinetics in forced spreading, Doctoral Dissertations, 2016, p. 2612. https://scholarsmine.mst.edu/doctoral_dissertations/2612.
- E. N. da and C. Andrade, Nature, 1930, 125, 309–310 Search PubMed.
- Y. Hatakeyama, K. Judai, K. Onishi, S. Takahashi, S. Kimura and K. Nishikawa, Phys. Chem. Chem. Phys., 2016, 18, 2339–2349 RSC.
- S. Özkar and R. G. Finke, J. Am. Chem. Soc., 2002, 124, 5796–5810 CrossRef PubMed.
- P. Migowski, G. Machado, S. R. Texeira, M. C. M. Alves, J. Morais, A. Traverse and J. Dupont, Phys. Chem. Chem. Phys., 2007, 9, 4814–4821 RSC.
- E. Redel, R. Thomann and C. Janiak, Inorg. Chem., 2008, 47, 14–16 CrossRef CAS PubMed.
- E. Redel, R. Thomann and C. Janiak, Chem. Commun., 2008, 1789–1791 RSC.
- L. S. Ott, M. L. Cline, M. Deetlefs, K. R. Seddon and R. G. Finke, J. Am. Chem. Soc., 2005, 127, 5758–5759 CrossRef CAS PubMed.
- A. Mele, G. Romano, M. Giannone, E. Ragg, G. Fronza, G. Raos and V. Marcon, Angew. Chem., Int. Ed., 2006, 45, 1123–1126 CrossRef CAS PubMed.
- T. Gutel, J. Garcia-Antõn, K. Pelzer, K. Philippot, C. C. Santini, Y. Chauvin, B. Chaudret and J. M. Basset, J. Mater. Chem., 2007, 17, 3290–3292 RSC.
- P. S. Campbell, C. C. Santini, D. Bouchu, B. Fenet, K. Philippot, B. Chaudret, A. A. H. Pádua and Y. Chauvin, Phys. Chem. Chem. Phys., 2010, 12, 4217–4223 RSC.
- J. Dupont and J. D. Scholten, Chem. Soc. Rev., 2010, 39, 1780–1804 RSC.
- H. Wender, P. Migowski, A. F. Feil, L. F. de Oliveira, M. H. Prechtl, R. Leal, G. Machado, S. R. Teixeira and J. Dupont, Phys. Chem. Chem. Phys., 2011, 13, 13552–13557 RSC.
- D. Sugioka, T. Kameyama, S. Kuwabata and T. Torimoto, Phys. Chem. Chem. Phys., 2015, 17, 13150–13159 RSC.
- D. Sugioka, T. Kameyama, S. Kuwabata, T. Yamamoto and T. Torimoto, ACS Appl. Mater. Interfaces, 2016, 8, 10874–10883 CrossRef CAS PubMed.
- A. Kauling, G. N. Ebeling, J. Morais, A. Pádua, T. Grehl, H. H. Brongersma and J. Dupont, Langmuir, 2013, 29, 14301–14306 CrossRef CAS PubMed.
- M. Porta, M. T. Nguyen, T. Tokunaga, Y. Ishida, W.-R. Liu and T. Yonezawa, Langmuir, 2016, 32, 12159–12165 CrossRef CAS PubMed.
- Y. Ishida, R. Nakabayashi, R. D. Corpuz and T. Yonezawa, Colloids Surf., A, 2017, 518, 25–29 CrossRef CAS.
- R. D. Corpuz, Y. Ishida and T. Yonezawa, New J. Chem., 2017, 41, 6828–6833 RSC.
- R. D. Corpuz, Y. Ishida, M. T. Nguyen and T. Yonezawa, Langmuir, 2017, 33, 9144–9150 CrossRef CAS PubMed.
- Y. Ishida, A. Morita, T. Tokunaga and T. Yonezawa, Langmuir, 2018, 34, 4024–4030 CrossRef CAS PubMed.
- I. Akita, Y. Ishida and T. Yonezawa, Bull. Chem. Soc. Jpn., 2016, 89, 1054–1056 CrossRef CAS.
- Y. Ishida, T. Sumi and T. Yonezawa, New J. Chem., 2015, 39, 5895–5897 RSC.
- Y. Ishida, R. D. Corpuz and T. Yonezawa, Acc. Chem. Res., 2017, 50, 2986–2995 CrossRef CAS PubMed.
- H. Wender, R. V. Gonçalves, A. F. Feil, P. Migowski, F. S. Poletto, A. R. Pohlmann, J. Dupont and S. R. Teixeira, J. Phys. Chem. C, 2011, 115, 16362–16367 CrossRef CAS.
- M. T. Nguyen and T. Yonezawa, Sci. Technol. Adv. Mater., 2018, 19, 883–898 CrossRef CAS.
- A. Vahl, J. Strobel, W. Reichstein, O. Polonskyi, T. Strunskus, L. Kienle and F. Faupel, Nanotechnology, 2017, 28, 175703 CrossRef PubMed.
- T. Löffler, H. Meyer, A. Savan, P. Wilde, A. Garzón Manjón, Y.-T. Chen, E. Ventosa, C. Scheu, A. Ludwig and W. Schuhmann, Adv. Energy Mater., 2018, 1802269 CrossRef.
- A. Garzón-Manjón, H. Meyer, D. Grochla, T. Löffler, W. Schuhmann, A. Ludwig and C. Scheu, Nanomaterials, 2018, 8, 903 CrossRef PubMed.
- A. G. Manjón, T. Löffler, M. Meischein, H. Meyer, J. Lim, V. Strotkötter, W. Schuhmann, A. Ludwig and C. Scheu, Nanoscale, 2020, 12, 23570–23577 RSC.
- C.-H. Liu, R.-H. Liu, Q.-J. Sun, J.-B. Chang, X. Gao, Y. Liu, S.-T. Lee, Z.-H. Kang and S.-D. Wang, Nanoscale, 2015, 7, 6356–6362 RSC.
- X.-L. Cai, C.-H. Liu, J. Liu, Y. Lu, Y.-N. Zhong, K.-Q. Nie, J.-L. Xu, X. Gao, X.-H. Sun and S.-D. Wang, Nano-Micro Lett., 2017, 9, 48 CrossRef PubMed.
- M. Meischein, A. Garzón-Manjón, T. Frohn, H. Meyer, S. Salomon, C. Scheu and A. Ludwig, ACS Comb. Sci., 2019, 21, 743–752 CrossRef CAS PubMed.
- T. Torimoto, Y. Ohta, K. Enokida, D. Sugioka, T. Kameyama, T. Yamamoto, T. Shibayama, K. Yoshii, T. Tsuda and S. Kuwabata, J. Mater. Chem. A, 2015, 3, 6177–6186 RSC.
- S. Suzuki, A. Morimoto, S. Kuwabata and T. Torimoto, Jpn. J. Appl. Phys., 2021, 60, SAAC01 CrossRef CAS.
- A. Chauvin, A. Sergievskaya, A.-A. El Mel, A. Fucikova, C. A. Corrêa, J. Vesely, E. Duverger-Nédellec, D. Cornil, J. Cornil, P.-Y. Tessier, M. Dopita and S. Konstantinidis, Nanotechnology, 2020, 31, 455303 CrossRef CAS PubMed.
- Q. Zhang, K. Kusada, D. Wu, N. Ogiwara, T. Yamamoto, T. Toriyama, S. Matsumura, S. Kawaguchi, Y. Kubota, T. Honma and H. Kitagawa, Chem. Sci., 2019, 10, 5133–5137 RSC.
- E. R. Essinger-Hileman, D. DeCicco, J. F. Bondi and R. E. Schaak, J. Mater. Chem., 2011, 21, 11599–11604 RSC.
- K. Kusada, M. Yamauchi, H. Kobayashi, H. Kitagawa and Y. Kubota, J. Am. Chem. Soc., 2010, 132, 15896–15898 CrossRef CAS PubMed.
- X. Carette, B. Debièvre, D. Cornil, J. Cornil, P. Leclère, B. Maes, N. Gautier, E. Gautron, A.-A. El Mel, J.-M. Raquez and S. Konstantinidis, J. Phys. Chem. C, 2018, 122, 26605–26612 CrossRef CAS.
- E. Vanecht, K. Binnemans, J. W. Seo, L. Stappers and J. Fransaer, Phys. Chem. Chem. Phys., 2011, 13, 13565–13571 RSC.
- K. Richter, A. Birkner and A.-V. Mudring, Phys. Chem. Chem. Phys., 2011, 13, 7136–7141 RSC.
- A. Fujita, Y. Matsumoto, M. Takeuchi, H. Ryuto and G. H. Takaoka, Phys. Chem. Chem. Phys., 2016, 18, 5464–5470 RSC.
- S. C. Hamm, S. Basuray, S. Mukherjee, S. Sengupta, J. C. Mathai, G. A. Baker and S. Gangopadhyay, J. Mater. Chem. A, 2014, 2, 792–803 RSC.
- T. Kameyama, Y. Ohno, T. Kurimoto, K.-I. Okazaki, T. Uematsu, S. Kuwabata and T. Torimoto, Phys. Chem. Chem. Phys., 2010, 12, 1804–1811 RSC.
- K. Yoshii, T. Tsuda, T. Torimoto and S. Kuwabata, ECS Trans., 2010, 33, 127 CrossRef CAS.
- K. Matsuyama, T. Tsubaki, T. Kato, T. Okuyama and H. Muto, Mater. Lett., 2020, 261, 127124 CrossRef CAS.
- D. Li, C. Wang, D. Tripkovic, S. Sun, N. M. Markovic and V. R. Stamenkovic, ACS Catal., 2012, 2, 1358–1362 CrossRef CAS.
- Z. Niu and Y. Li, Chem. Mater., 2014, 26, 72–83 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *,
Lianlian
Deng
and
Tetsu
Yonezawa
*,
Lianlian
Deng
and
Tetsu
Yonezawa