
Activating atomically dispersed Co–N/C sites on g-C3N4 nanosheets via incorporating sulfur enables efficient visible light H2 evolution†
Fang
Wang
ab,
Yuan
Xue
b,
Weibing
Xu
 *a and
Shixiong
Min
*b
*a and
Shixiong
Min
*b
aSchool of Chemistry and Chemical Engineering, Hefei University of Technology, Hefei, 230009, P. R. China. E-mail: weibingxu@hfut.edu.cn
bSchool of Chemistry and Chemical Engineering, North Minzu University, Yinchuan, 750021, P. R. China. E-mail: sxmin@nun.edu.cn
First published on 15th November 2021
Abstract
Embedding atomically dispersed metal (M)–N/C sites within two-dimensional (2D) g-C3N4 nanosheets (CNs) is able to greatly improve charge separation efficiency; however, these sites are typically inert for catalyzing the photochemical H2 evolution reaction (HER). In this work, a simple room-temperature sulfurization strategy is proposed to activate the inert Co–N/C sites on CNs for efficiently catalyzing the dye-sensitized photocatalytic HER. By simply immersing cobalt-doped CNs (Co-CNs) into aqueous solution with high-concentration S2−, the coordinated O atoms at Co–N/C sites are partially replaced by S atoms at room temperature, generating ample highly active S-coordinated Co–N/C sites with excellent dispersion preservation. The as-prepared S-Co-CNs catalysts exhibit enhanced activity in catalyzing the HER in an Erythrosin B–triethanolamine (ErB–TEOA) system under visible light irradiation, while both the pristine CNs and Co-CNs show negligible activity under the same reaction conditions. The most efficient S-Co-CNs catalyst achieves a H2 evolution rate of 6.38 mmol h−1 g−1 and a quantum efficiency (QE) of 13.02% at 520 nm, and this S-Co-CNs catalyst shows excellent HER stability when sensitized with a more stable fluorescein (FL) dye. The enhanced catalytic performance originates from the favorable electronic structure of the active sites adjusted by S modulation, leading to reduced H2 evolution overpotential while maintaining the favorable electron transfer kinetics. This work provides an effective strategy to activate inert sites embedded in CNs for high-performance photocatalytic water splitting, organic transformation, and CO2 reduction.
Introduction
The solar photocatalytic H2 evolution from water splitting has been recognized as a promising route to solve the ever-increasing environmental issues and energy crisis.1–8 Among the photocatalysts developed so far, metal-free graphitic carbon nitride (g-C3N4) is a highly promising candidate for photocatalytic H2 evolution due to its suitable band gap of ca. 2.7 eV, good chemical and thermal stability, and low cost.9–15 However, pristine g-C3N4 shows low photocatalytic activity, attributed to the insufficient visible light absorption (λ < 470 nm), the fast recombination of charge carriers, and the lack of active sites for the H2 evolution reaction (HER).12–15 Several strategies, including dye-sensitization,16–21 metal and/or non-metal doping,22–32 nanostructuring,33–35 coupling with other inorganic semiconductors and conductive materials,36–41 and cocatalyst loading,9–12,42–46 have been proposed to enhance the photocatalytic activity of g-C3N4. Among these methods, dye-sensitization and loading cocatalysts have been proved to be facile yet effective methods for improving the photocatalytic HER performance of g-C3N4. Dye-sensitization of g-C3N4 could greatly extend the visible light absorption for maximizing the utilization efficiency of solar light,17 while loading a suitable cocatalyst on g-C3N4 can accelerate carrier transportation and reduce the barriers for the HER.9 Although noble metals, such as Pt-group metals, are the best cocatalysts for g-C3N4, their scarcity and high cost limit their practical applications. Therefore, it is still desirable to develop an alternative cocatalyst based on earth-abundant elements.47–55On the other hand, it has been found that introducing M–N/C sites (M = Fe, Co, Ni, Zn, etc.) at the atomic level within the framework of g-C3N4 could effectively tune the electronic structure of g-C3N4, and the resulting M-doped g-C3N4 (M-g-C3N4) materials typically possess reduced bandgap energy and enhanced charge separation efficiency,21–26,56,57 and thus improved photocatalytic performances. However, the confined M–N/C sites are hardly active for catalyzing the photocatalytic HER over M-g-C3N4, and an additional cocatalyst, such as Pt, still needs to be loaded on M-g-C3N4 to provide active sites for enhancing the photocatalytic HER activity. Fortunately, it has been found that the M-N/C sites could be activated to achieve reactivity for catalyzing the photocatalytic HER by rationally regulating the electronic structures of coordinatively unsaturated M–N/C sites via introducing an additional active ligand. In this context, Liu et al.58 reported that phosphidation of a single Co1-oxo site confined on g-C3N4 nanosheets led to a single Co1–P4 site that can act as an efficient reaction center for the overall water splitting reaction. Chen et al.59 discovered that single Ni active sites coordinated to a S-containing ligand chemically bonded to g-C3N4 can efficiently catalyze visible light H2 evolution with an activity comparable to that of 3 wt% Pt loaded g-C3N4 under the identical conditions. While these studies provided a new approach to construct active single transition metal-based active sites on the g-C3N4 matrix by deliberately choosing secondary active ligands, the synthesis of these materials typically involved multistep reactions using toxic reagents under harsh reaction conditions. Therefore, it is still highly desirable to develop a facile yet effective approach to activate inert M–N/C sites on g-C3N4 to obtain H2 evolution catalysts with high intrinsic activity.
Herein, we present a facile yet effective approach to activate inert Co–N/C sites on 2D g-C3N4 nanosheets (CNs) to highly active S-coordinated Co–N/C (S-Co–N/C) sites for catalyzing the H2 evolution reaction in a dye-sensitized system under visible light. The S-Co–N/C sites are generated in situ on CNs via a room-temperature sulfurization strategy by simply immersing Co-doped CNs (Co-CNs) precursor in a solution with a high concentration of S2−, and the O atoms (O2−) that are coordinated to Co–N/C sites are partially replaced by S2− according to the solubility equilibrium. The resulting S-Co–N/C sites can precisely retain the atomic-level dispersion of the Co–N/C sites and avoid the formation of aggregated nanoparticles of Co sulfides. Benefitting from the high dispersion and favorable electronic structure, the S-Co–N/C sites on CNs can not only lead to the efficient transportation of electrons but also greatly reduce the reaction barriers for the H2 evolution reaction. As a result, the best S-Co-CNs catalyst sensitized by Erythrosin B (ErB) exhibits a much higher H2 evolution rate of 6.38 mmol h−1 g−1 under visible light irradiation and an AQY of 13.02% at 520 nm, and greatly outperforms both pristine CNs and Co-CNs. In addition, when sensitized with a stable dye, the S-Co-CNs catalyst also shows an excellent long-term stability.
Experimental
Chemicals and reagents
All reagents including (NH4)2S solution (21 wt%), Co(NO3)2·6H2O, Ni(NO3)2·6H2O, CuSO4·5H2O, Zn(NO3)2·6H2O, and urea were of analytical grade and used without further purification. Triethanolamine (TEOA) was purchased from Xilong Scientific (>99.8%). Erythrosin B (ErB) and fluorescein (FL) were obtained from Tianjin Guangfu Fine Chemical Research Institute. All solutions in the experiments were prepared with deionized water (>15 MΩ) that was obtained from a YL-100B-D water-purification system.Synthesis of S-Co-CN catalyst
Co-doped g-C3N4 nanosheets (Co-CNs) precursor was first synthesized by a well-established thermal polymerization method.17 Typically, 20 g of urea and 40 mg of Co(NO3)2·6H2O were dissolved into 100 mL of water under stirring to form a homogeneous solution. Then, the water was evaporated to generate a solid mixture. After being ground into powders, the solid mixture was placed in a crucible with a cover and calcined in a muffle furnace at 550 °C for 2 h with a heating rate of 5 °C min−1. The obtained products were washed with water and ethanol several times, and dried in a vacuum oven at 60 °C overnight. The final product was labeled as Co-CNs. The pristine CNs was prepared by identical processes to Co-CNs without the addition of Co salt.S-Co-CNs catalyst was then synthesized via the room-temperature sulfurization of Co-CNs. Typically, 150 mg of Co-CNs was dispersed into 25 mL of water and ultrasonicated for 10 min to form a homogeneous suspension. After that, 500 μL of (NH4)2S solution was added into the above suspension and the resulting mixture was stirred for 6 h at room temperature. The obtained product, denoted as S-Co-CNs, was collected by centrifugation and then rinsed with water and ethanol several times, and finally dried in a vacuum oven at 60 °C overnight. A series of metal-doped (Ni, Cu, Zn, and Fe) CNs catalysts were also synthesized using the corresponding metal salts and sulfurized with the exact same procedures as for the synthesis of S-Co-CNs catalyst.
Characterization
Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) images of the samples were recorded using a FEI Tecnai-G2-F30 transmission electron microscope. X-ray diffraction (XRD) patterns of the samples were recorded with a Rigaku smartlab powder X-ray diffractometer with nickel filtered Cu Kα radiation from 5 to 80°. Fourier transform infrared spectroscopy (FTIR) spectra of the samples were collected by using a Thermo Nicolet Avatar 380 FT-IR spectrometer. X-ray photoelectron spectroscopy (XPS) measurements of the samples were performed on a Thermo Scientific Escalab-250Xi electron spectrometer and the C 1s peak at 284.8 eV was a signal-calibrating standard for all the binding energies. UV-vis absorption spectra of the reaction solutions were recorded by using a Thermo Scientific-Evolution 220 spectrophotometer. UV-vis diffuse reflectance spectra of the solid samples were recorded on a PerkinElmer Lambda-750 UV-vis-near-IR spectrometer. The textile structures of the samples were determined by using a Micromeritics ASAP 2020 HD88 adsorption apparatus. Photoluminescence (PL) spectra were collected on a Horiba Scientific FluoroMax-4 spectrofluorometer spectrometer. The PL decay profiles were measured using the Horiba Jobin Yvon Data Station HUB operating in time correlated single photon counting mode (TCSPC) (time resolution: 200 ps) with a nano LED diode emitting pulses at 369 nm with 1 MHz repetition rate as an excitation source. Light-scattering Ludox solution was used to obtain the instrument response function (prompt). Horiba Jobin Yvon DAS6 fluorescence decay analysis software was used to fit the model functions to the experimental data.Photocatalytic H2 evolution experiments
Photocatalytic H2 evolution experiments were performed in a sealed Pyrex reactor (340 mL) with a top flat quartz window (38.47 cm2) for light irradiation and a silicone rubber septum was fixed on its side for sampling the produced H2. In a typical procedure, the S-Co-CNs catalyst (50 mg) and ErB (0.2 mM) were added to a reaction cell containing 100 mL of 10 vol% TEOA (pH 8) aqueous solution under vigorous stirring. The reaction system was then thoroughly degassed for 10 min and backfilled with N2. After that, the reaction solution was irradiated by using a 30 W LED lamp equipped with a cut-off filter (λ ≥ 450 nm). The amount of H2 evolved was measured by using a gas chromatograph (Tech comp; GC-7900) with a thermal conductivity detector, a 5 A molecular sieve column (4 mm × 5 m), and with N2 as carrier gas. The apparent quantum yield (AQY) for the H2 evolution reaction was measured under light irradiation of different wavelengths. The photon flux of the incident monochromatic light was determined using a ray virtual radiation actinometer (Apogee MQ-500), and the AQY was calculated based on the following equation: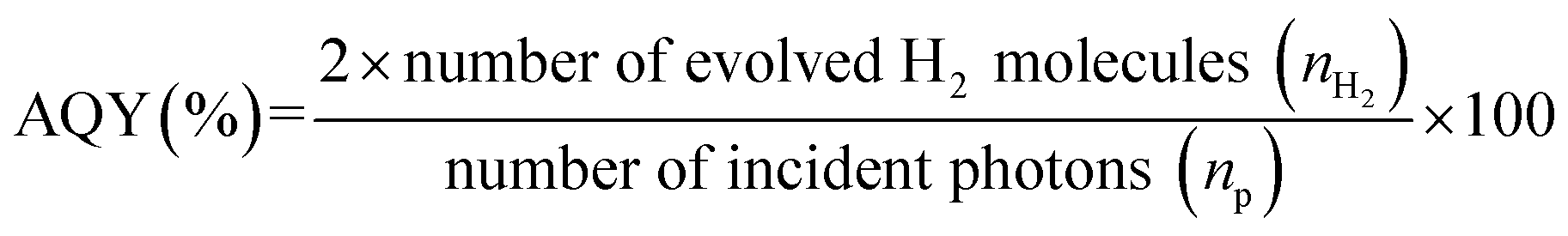
Electrochemical measurements
The electrochemical measurements were carried out in a standard three-electrode system with an electrochemical workstation (CHI760E, Shanghai, China). Saturated Ag/AgCl and a graphite rod were used as the reference and counter electrodes, respectively. The supporting electrolyte was degassed 10% TEOA (pH 8) solution containing 0.5 M Na2SO4. The working electrodes were prepared by drop-casting 0.12 mL of the catalyst (CNs, Co-CNs, or S-Co-CNs) suspension in ethanol (5 mg mL−1) onto both sides of carbon paper (HESEN, HCP030P, thickness: 0.3 mm, 1.5 cm × 0.5 cm) and dried at room temperature. The electrocatalytic H2 evolution activity was examined by measuring polarization curves using the linear sweep voltammetry (LSV) technique at a scan rate of 5 mV s−1. Electrochemical impedance spectroscopy (EIS) was carried out with an AC amplitude of 5 mV and a frequency range of 10 mHz to 100 kHz. The Mott–Schottky analysis was conducted at a frequency of 1 kHz and a scan rate of 50 mV s−1.Results and discussion
The S-Co-CNs catalyst was prepared by a simple liquid-phase anion-exchange reaction with Co-CNs and high-concentration S2− at room temperature. As schematically illustrated in Fig. 1, Co-CNs precursor was first prepared by pyrolyzing a mixture of urea and Co(NO3)2 in air, which produces numerous O-coordinated Co–N/C sites that are highly dispersed and firmly embedded in the frameworks of CNs. Then, the Co-CNs precursor was directly reacted with (NH4)2S in aqueous solution at room temperature, during which the coordinated O atoms at Co–N/C sites were partially replaced by S atoms, generating ample highly active S-coordinated Co–N/C sites. The successful anion-exchange along with S incorporation can be visually confirmed by the color change from light yellow for Co-CNs to grey for S-Co-CNs. Unlike conventional vapor-phase and hydrothermal sulfurization processes that are typically performed at high temperature involving the use of toxic chemicals (such as thiourea and thioacetamide), and in which the active sites tend to agglomerate to form larger isolated nanoparticles,52 the present developed synthetic route based on the solution-based sulfurization reaction allows the efficient activation of Co–N/C sites while maintaining the high dispersion, which would be beneficial to increase the catalytic activity for photocatalytic H2 evolution. | ||
| Fig. 1 Schematic illustration of the synthesis of S-Co-CNs catalyst via the room-temperature sulfurization strategy and their applications in the visible light H2 evolution. | ||
X-ray diffraction (XRD) patterns of pristine CNs, Co-CNs, and S-Co-CNs are shown in Fig. 2A. For pristine CNs, two characteristic peaks at around 13.0° and 27.5° are observed, which can be indexed to the (100) plane of the in-plane ordering of tri-s-triazine repeating units and the (002) plane of the graphitic stacking of C3N4 layers, respectively.9 The XRD pattern of Co-CNs does not differ significantly from that of pristine CNs, except for a decrease in the intensity of the (002) peak due to the interruption of polymeric condensation by Co incorporation.23,26 No diffraction peaks related to Co species, such as metallic Co, Co oxides, Co nitrides, and Co carbides, could be detectable. These results suggest that the Co species were homogeneously dispersed in the CN matrix, most likely via forming atomically dispersed Co–N/C species.26 After room-temperature sulfurization, the diffraction pattern of S-Co-CNs catalyst shows no significant difference compared to those of pristine CNs and Co-CNs, and no peaks originating from Co-based sulfides could be found, indicating that the coordination of S atoms to the Co atoms has no significant effect on the phase structure of CNs and the resulting Co species still preserve their high dispersion in the frameworks of CNs.
 | ||
| Fig. 2 (A) XRD patterns, (B) UV-Vis DRS, (C) FT-IR spectra, and (D) N2 adsorption–desorption isotherms of pristine CNs, Co-CNs, and S-Co-CNs. Insets in panel (A) and (D) show the digital images and pore size distribution curves of pristine CNs, Co-CNs, and S-Co-CNs, respectively. | ||
Fig. 2B shows the UV-visible diffuse reflectance spectra (UV-vis-DRS) of pristine CNs, Co-CNs, and S-Co-CNs. The pristine CNs exhibit a strong absorption from 200 to 380 nm with a sharp absorption edge at around 438 nm, corresponding to a band gap (Eg) of 2.83 eV (Table 1). Compared to the pristine CNs, it is clearly observed that the absorption band edge of Co-CNs is shifted to the red wavelength region (∼447 nm, Eg = 2.77 eV). In addition, obvious enhanced optical absorption was found in the visible light spectral range of 450–650 nm. This indicates that a ligand-to-metal charge transition (LMCT) similar to molecular systems occurs in solid transition metal-doped CNs,23–26 demonstrating the success of Co incorporation in CNs at the atomic level and the strong electronic interaction between Co and CNs. After sulfurization, the obtained S-Co-CNs shows a similar band gap to Co-CNs, but the absorption in the range of 450–800 nm is slightly enhanced along with a color change from light yellow to grey, indicating the formation of S-coordinated Co–N/C species on CNs. Fourier transform infrared (FTIR) spectroscopy characterization was performed to confirm the chemical structure of the samples. As shown in Fig. 2C, the FTIR spectrum of pristine CNs shows the vibrations of various C/N-containing groups, e.g., the characteristic mode of the triazine units (≈807 cm−1) and CN heterocycles (1200–1700 cm−1), as well as a broad peak in the range of 3000–3300 cm−1 corresponding to the stretching modes of the –OH/–NH2 groups.17 All these characteristic bands are well-preserved after the Co incorporation and subsequent sulfurization, and no new peaks attributed to the C–S/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bands typically at 900–1300 cm−1 can be observed in the spectrum of S-Co-CNs. These results suggest that the S atoms are coordinated to the unsaturated Co atoms in Co–N/C sites instead of incorporating into the frameworks of CNs.
S bands typically at 900–1300 cm−1 can be observed in the spectrum of S-Co-CNs. These results suggest that the S atoms are coordinated to the unsaturated Co atoms in Co–N/C sites instead of incorporating into the frameworks of CNs.
| Sample | S BET (m2 g−1) | Pore sizeb (nm) | V pore (cm3 g−1) | E g (eV) |
|---|---|---|---|---|
| a BET surface area is calculated from the linear part of the BET plot. b Adsorption average pore width (4 V A−1 by BET). c Single point total pore volume of the pores at P/P0 = 0.99. | ||||
| CNs | 48.1 | 21.0 | 0.252 | 2.83 |
| Co-CNs | 92.5 | 21.6 | 0.499 | 2.77 |
| S-Co-CNs | 94.7 | 22.8 | 0.541 | 2.77 |
The N2 adsorption–desorption isotherms measured at 77 K and pore size distribution curves of the samples are given in Fig. 2D, and the calculated textural data are summarized in Table 1. The isotherms for all samples are identified as typical type IV with a H3-type hysteresis loop, which are characteristics of mesoporous materials. The pore size distribution curves indicate the presence of ample mesopores with an average size of ∼20 nm for all the samples.17 The calculated Brunauer–Emmett–Teller (BET) specific surface area (SBET) for pristine CNs is 48.2 m2 g−1. Incorporating Co into CNs nearly doubles the SBET (92.5 m2 g−1) due to the generation of more micro- and mesopores, as evidenced by the increase of the total pore volume from 0.252 to 0.499 cm3 g−1. This is probably due to the disturbance of the CN network structure by the incorporated Co species or the stabilization of micropores via introducing Co species.26 After sulfurization, the S-Co-CNs catalyst shows slightly higher SBET, pore size, and pore volume than Co-CNs, indicating that introducing larger S atoms to replace smaller O atoms at Co–N/C sites will further expand the pore size and thus enhance the SBET but maintain the higher dispersion of in situ formed S-Co–C/N sites. The larger surface area and highly dispersed S-Co–C/N sites would be expected to provide a better H2 evolution activity.
The microstructures of Co-CNs and S-Co-CNs were further investigated by transmission electron microscopy (TEM). The pristine CNs have a thin sheet structure with abundant pores (20–40 nm) on its surface (Fig. S1, ESI†), consistent with the result of N2 adsorption–desorption analysis, and the thin sheets are folded at their edges due to the high surface energy. As shown in Fig. 3A, the Co-CNs exhibits a similar sheet structure to pristine CNs, indicating that the basic morphology does not change after Co incorporation. Additionally, no aggregated nanoparticles or clusters can be found on the entire surface of CNs from its high-resolution TEM images (Fig. 3B and S2A, ESI†), revealing that the Co dopants are distributed in the conjugated frameworks of CNs at the atomic level. This is further confirmed by the high angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and the corresponding energy-dispersive X-ray spectroscopy (EDX) mapping analysis (Fig. 3C), revealing that C, N, Co, and O are homogeneously dispersed in the CN matrix. According to the EDX analysis (Table S1, ESI†), the loading amount of Co in the Co-CNs catalyst is determined to be 0.18 at%. Fig. 3D shows the TEM image of S-Co-CNs, revealing that the thin layer sheet structure is well preserved after room-temperature sulfurization. As shown in the HRTEM images in Fig. 3E and S2B, ESI,† it can also be seen that no particles are formed, which indicates that the high dispersion of Co-N/C sites remains unchanged after sulfurization. Furthermore, the HAADF-STEM image and the relevant EDX mapping images in Fig. 3F further confirm a homogeneous distribution of the S element in the S-Co-CNs, while other elements maintain their original distributions without any aggregation. In addition, the EDX analysis indicates that the S content increased from zero to 0.19 at% for the resulting S-Co-CNs catalyst. These results show that the S atoms have been successfully coordinated to the Co-N/C sites and that the in situ formed S-Co–N/C maintains the high dispersion of original Co–N/C sites.
 | ||
| Fig. 3 (A) TEM and (B) HRTEM images of Co-CNs. (C) HAADF-STEM image of Co-CNs and the corresponding elemental maps (C, N, Co, and O). (D) TEM and (E) HRTEM images of S-Co-CNs. (F) HAADF-STEM images of S-Co-CNs and the corresponding elemental maps (C, N, Co, O, and S). | ||
The electronic structures of Co-CNs and S-Co-CNs were investigated by using X-ray photoelectron spectroscopy (XPS). As shown in Fig. 4A, compared to Co-CNs that are composed of C, N, O, and Co elements, an additional S 2p signal can be found in the spectrum of the S-Co-CNs. This result is consistent with the observation of EDX analysis, further confirming the successful S incorporation via facile room-temperature sulfurization. For the Co 2p XPS spectrum of Co-CNs (Fig. 4B), the two Co 2p3/2 peaks centered at 779.8 and 781.5 eV can be assigned to the Co–O and Co–N species, respectively. The appearance of Co–O species indicates the coordination of O atoms to Co–N/C sites as the Co-CNs was prepared in air.25 The other two peaks at 784.5 and 788.1 eV are the satellite peaks of both Co–O and Co–N species.60 In contrast to Co-CNs, besides the Co–O and Co–N species, the Co 2p XPS spectrum of S-Co-CNs shows a new Co 2p3/2 peak at 778.9 eV, which can be identified as the peak of Co–S species.61,62 In addition, the binding energies of Co–O (780.2 eV) and Co–N (781.8 eV) species for S-Co-CNs reveal a shift to higher binding energies due to S incorporation. The high-resolution S 2p XPS spectrum of S-Co-CNs further confirms the formation of S-coordinated Co species. As shown in Fig. 4C, the two peaks observed at 163.2 and 161.9 eV are assigned to 2p1/2 and 2p3/2 of Co–S species, and the two peaks at 167.4 and 168.7 eV correspond to 2p1/2 and 2p3/2 of surface-adsorbed S oxides.63 These firmly confirm that O2− coordinated to Co–N/C sites was partly replaced by S2−. In the N 1s spectra (Fig. 4D), the Co-CNs shows three main peaks at 397.7, 399.0 and 400.1 eV that correspond to the C–NCC, N–C3, and C–NHx functional groups, respectively.17 All these peaks can also be observed for S-Co-CNs but slightly shift to lower binding energies. This is attributed to the fact that the S atoms are coordinated to Co and the electron transfer from CNs to in situ generated S-Co–N/C sites.
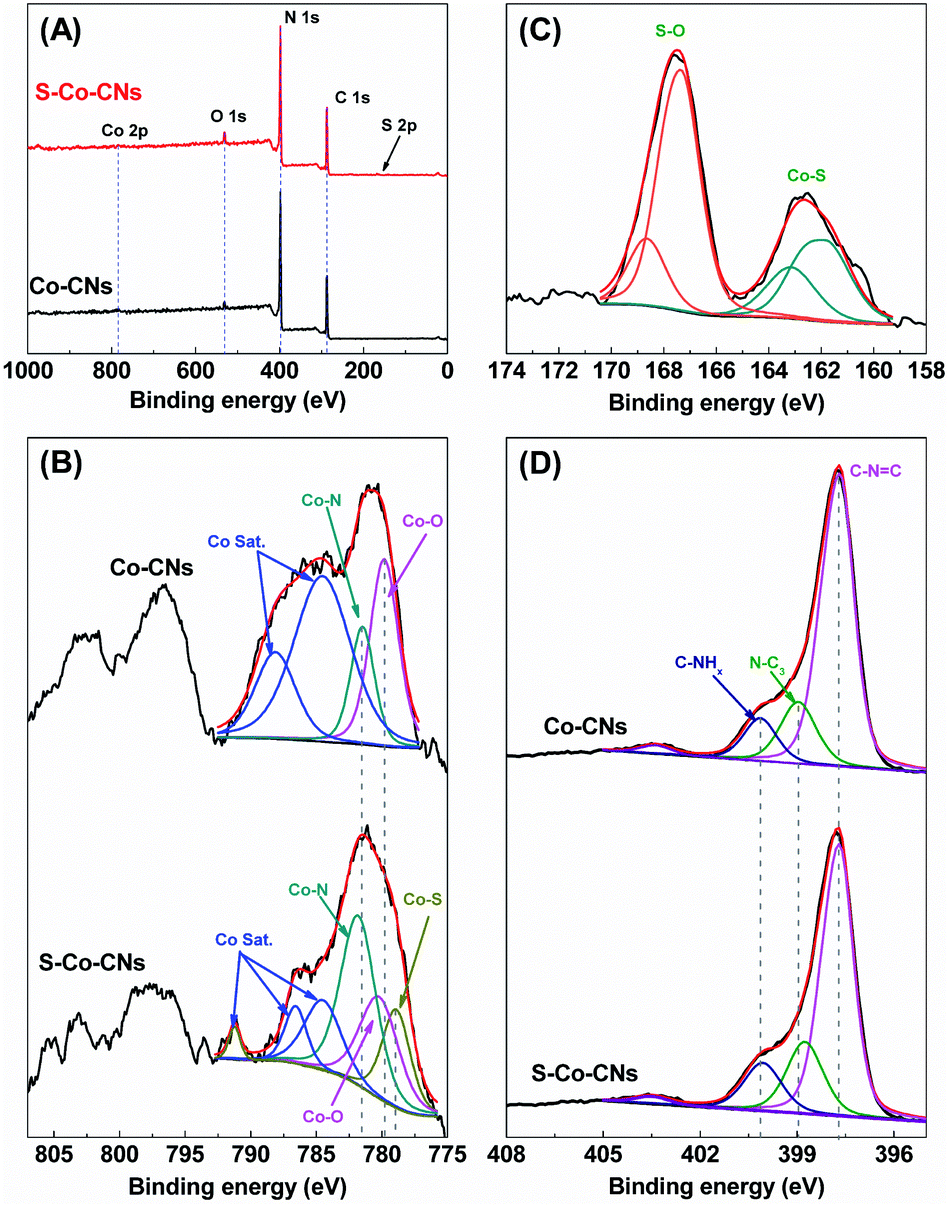 | ||
| Fig. 4 (A) XPS survey spectra of Co-CNs and S-Co-CNs. (B) Co 2p XPS spectra of Co-CNs and S-Co-CNs. (C) S 2p XPS spectrum of S-Co-CNs. (D) N 1s XPS spectra of Co-CNs and S-Co-CNs. | ||
The activities of the samples towards photocatalytic H2 evolution were evaluated in a dye-sensitized system composed of xanthene dye Erythrosin B (ErB) as the photosensitizer and triethanolamine (TEOA) as the sacrificial reagent under visible light irradiation (λ ≥ 450 nm). As shown in Fig. 5A, the ErB– TEOA system shows no activity towards H2 evolution in the absence of a catalyst. When CNs were added as the catalyst into the above system, only trace amounts of H2 were detected. The low activity of CNs is attributed to the lack of active sites. Similarly, the Co-CNs also exhibits a very low H2 evolution activity, demonstrating that the Co–N/C sites are inert for the H2 evolution reaction though they have been reported to effectively facilitate the charge separation.25,57 This result is consistent with previously reported results that an additional cocatalyst is still needed to be loaded on metal-doped CNs to promote the H2 evolution reaction.25,57 In strong contrast, the S-Co-CNs catalyst exhibits superior H2 evolution activity to CNs and Co-CNs under the same reaction conditions. In a 5 h reaction, 616.5 μmol of H2 is produced over S-Co-CNs catalyst in the ErB–TEOA system, corresponding to a turnover number (TON) of 62 based on ErB. Since there is no significant change in the textural properties (surface area, pore size, and pore volume) for the Co-CNs and S-Co-CNs, the significantly enhanced catalytic H2 evolution activity of S-Co-CNs over Co-CNs can be attributed to the activation of Co–N/C sites by S incorporation, and the in situ formed S-Co–N/C sites are highly active for H2 evolution. The time courses of H2 evolution over ErB-sensitized S-Co-CNs catalysts prepared with different amounts of Co salt are given in Fig. S3, ESI.† It can be noted that H2 evolves rapidly in the first 1 h of reaction over all of the S-Co-CNs catalysts. After 1 h of reaction, H2 evolution becomes slow probably due to the degradation of ErB. Fig. 5B shows the initial H2 evolution rates on S-Co-CNs catalysts sensitized by ErB. It is clear that the H2 evolution rate increases linearly with increasing Co content from 5 to 40 mg. As shown in the XRD patterns of Co-CNs and S-Co-CNs catalysts (Fig. S4, ESI†), it is clear that no peaks of crystalline Co species for Co-CNs and Co sulfides for S-Co-CNs can be observed even at higher Co contents, indicating that the enhanced H2 evolution activity with Co content is due to the formation of more S-Co–N/C active sites on CNs. Further increasing the Co content to 50 mg and higher will in turn deteriorate the activity most likely due to the aggregation of S-Co–N/C sites. In addition, the light-shielding caused by deep-colored S-Co-CNs catalysts at higher Co contents (inset in Fig. 5B) would also limit the light utilization of the reaction system and thus reduce the H2 evolution activity. This facile room-temperature sulfurization strategy was also extended to prepare various transition metal-doped CNs (S-M-CNs, M = Cu, Zn, Ni) catalysts aiming to develop more active catalysts. The XRD patterns in Fig. S5, ESI† indicate that Cu and Zn can be homogeneously distributed on CNs, but the Ni dopant will aggregate to form Ni7S6 particles in the final S-Ni-CNs catalyst. Their activities towards H2 evolution in the ErB–TEOA system were also tested. As shown in Fig. S6, ESI,† the as-prepared S-Cu-CNs and S-Zn-CNs show no H2 evolution activity in the ErB–TEOA system, while the S-Ni-CNs exhibit a H2 evolution rate of 1.81 mmol h−1 g−1, which is much lower than that obtained on S-Co-CNs (6.38 mmol h−1 g−1). These results further emphasize the importance of constructing highly active sites on CNs via simultaneously regulating the central metal and coordination structure.
 | ||
| Fig. 5 (A) Time course of H2 evolution over different catalysts in the ErB–TEOA system. (B) Average H2 evolution rates over S-Co-CN catalysts prepared with different masses of Co(NO3)2·6H2O (5 to 60 mg) in the ErB–TEOA system. The inset shows the pictures of the S-Co-CNs catalysts. (C) Dependence of the H2 evolution rate and of AQY on an ErB-sensitized S-Co-CNs catalyst as a function of wavelength of incident light. (D) Time course of cyclic H2 evolution from the ErB-sensitized S-Co-CNs system. The inset shows the H2 evolution from the Fl-sensitized S-Co-CNs system. Reaction conditions: catalyst, 50 mg; 100 mL of TEOA solution, 10%, pH 8; dye, 0.2 mM; light source, 30 W LED lamp; λ ≥ 450 nm. | ||
The high catalytic activity of the S-Co-CNs catalyst was further demonstrated by measuring the apparent quantum yield (AQY) of H2 evolution under visible light irradiation of different wavelengths. As displayed in Fig. 5C, the H2 evolution rate and the corresponding AQY first increase and then decrease with the increase of wavelength of incident light from 450 to 650 nm, which is fully consistent with the absorption spectrum of ErB. This indicates that ErB is a photosensitizer for capturing light and the S-Co-CNs act as a H2 evolution catalyst. At 520 nm, both the H2 evolution rate and AQY reach the highest values of 129.1 μmol h−1 and 13.02%, respectively. These values are among the best obtained on dye-sensitized g-C3N4-based catalysts under visible light irradiation (Table S2, ESI†), further demonstrating that our S-Co-CNs catalyst, albeit with very low Co content, is a highly efficient cocatalyst for photocatalytic H2 evolution in a dye-sensitized system.
The catalytic stability of S-Co-CNs catalyst towards H2 evolution in the ErB–TEOA system was also evaluated by performing a continuous repeated reaction over 24 h, as shown in Fig. 5D. As can be seen the system exhibits a steady H2 evolution activity in the first 6 h, after which the activity decreases with time, which is due to the gradual degradation of dye ErB (Fig. S7, ESI†). After the first run reaction, the S-Co-CNs catalyst was collected, washed, and redispersed into fresh ErB–TEOA solution for the next run reaction. Although the activity of the system decreases more significantly in the following runs compared to the first run, the S-Co-CNs catalyst is still active. The results of XRD (Fig. 6A) and FTIR (Fig. 6B) analyses reveal that the chemical structures of CNs and the high dispersion nature of S-Co–N/C sites are well retained in the used S-Co-CNs catalyst. XPS analysis (Fig. 6C and D) shows that the chemical states of the S-Co–N/C catalyst are also well maintained after the stability test. Specifically, the Co–S species can still be observed in the Co 2p and S 2p XPS spectra of the used S-Co-CNs catalyst. In addition, TEM analysis reveals that the microstructure of the used S-Co-CNs catalyst is similar to that of the fresh one, and no particle aggregation can be observed (Fig. S8, ESI†). These results reveal that the S-Co-CNs catalyst is robust with well-retained highly dispersed S-Co–N/C active sites during the long-term H2 evolution in the ErB–TEOA system. To improve the stability of the dye-sensitized S-Co-CNs system for H2 evolution, a more stable dye fluorescein (FL) was used to replace ErB. As shown in the inset of Fig. 5D, a steady H2 evolution activity is obtained during a continuous reaction for a period of 24 h, further demonstrating the good catalytic stability of the S-Co-CNs catalyst.
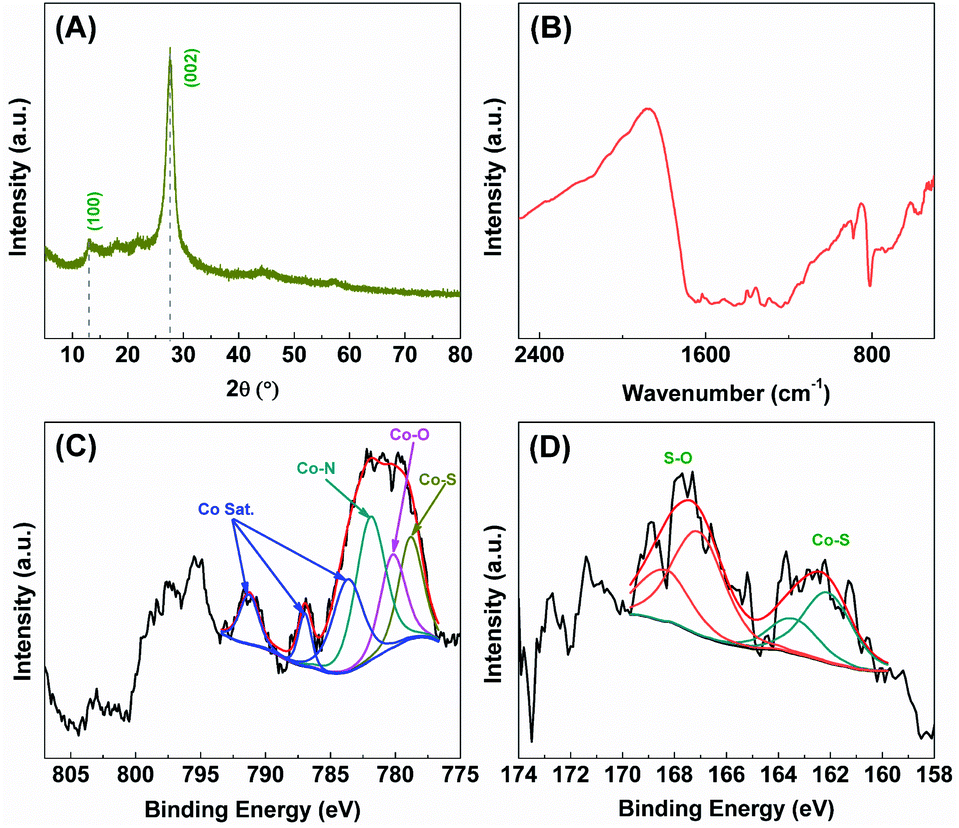 | ||
| Fig. 6 (A) XRD pattern, (B) FTIR spectrum, and high-resolution XPS spectra of (C) Co 2p and (D) S 2p of the S-Co-CNs catalyst after the stability test. | ||
To elucidate the origin of the high activity of the S-Co-CNs catalyst, the electrocatalytic H2 evolution experiments were first carried out by linear sweep voltammetry (LSV). As shown in the LSV plots in Fig. 7A, the overpotential to achieve a current density of 10 mA cm−2 on S-Co-CNs catalyst is 0.56 V, much lower than those of Co-CNs (0.63 V) and pristine CNs (0.86 V). This result indicates that the S incorporation is effective to activate the Co–N/C sites for enhancing the H2 evolution activity, which is consistent with previous observations that the Co sulfides are typically highly active catalysts for electrocatalytic and photocatalytic H2 evolution reactions. Electrochemical impedance spectroscopy (EIS) measurements were then performed to further disclose the origin of the activity enhancement on the S-Co-CNs catalyst (Fig. 7B). It can be seen that the Co-CNs catalyst shows a much smaller semicircle diameter and a much lower interfacial charge-transfer resistance (Rct = 38.0 kΩ) than those of pristine CNs (Rct = 113.3 kΩ), indicating that doping of Co into the CNs will significantly reduce the electron transfer resistance.25 After sulfurization treatment, the charge transfer resistance of S-Co-CNs is further reduced (Rct = 21.3 kΩ), which is beneficial for improving electron transfer from the excited dye to S-Co–N/C active sites in the ErB–TEOA system and thus leading to a better H2 evolution activity. The efficient charge separation promoted by Co–N/C and S-Co–N/C active sites was further revealed by PL analysis. As demonstrated in Fig. 7C, the strong PL emission of CNs in the wavelength range of 400–650 nm can be significantly quenched after the Co–N/C sites were embedded in the CNs, indicating that the charge recombination rate on CNs is greatly restricted due to the presence of Co–N/C sites.58 Time-resolved PL (TRPL) spectra (Fig. 7D) recorded at the corresponding steady-state emission peaks further reveal the average lifetimes (τave) of approximately 12.5 and 5.8 ns for pristine CNs and Co-CNs, respectively. This greatly decreased lifetime for charge carriers firmly reveals significantly enhanced charge separation in the Co-CNs due to the presence of Co–N/C sites.58 These results further confirm that embedding of Co–N/C sites is an effective strategy to promote the photoexcitation dissociation. After the S incorporation, the resulting S-Co-CNs catalyst, however, shows a slightly higher PL emission intensity compared to Co-CNs. Moreover, the PL emission lifetime of the S-Co-CNs catalyst (6.0 ns) is nearly the same as that of the Co-CNs catalyst. These results reveal that the efficient charge separation nature of Co-N/C sites is well-retained after the S incorporation, and the enhanced H2 evolution activity achieved on S-Co-CNs with respect to Co-CNs can be attributed to the enhanced intrinsic catalytic activity of S-Co–N/C towards the H2 evolution reaction, as evidenced by the LSV results. Furthermore, the Mott–Schottky (MS) measurements were performed to determine the possible change of the CB (ECB) and VB (EVB) potentials of CNs after Co doping and subsequent room-temperature sulfurization (Fig. S9, ESI†). The ECB of CNs, Co-CNs, and S-Co-CNs is determined to be −0.91, −0.86, and −0.83 V vs. Ag/AgCl, respectively. Based on the above results and the bandgap energy obtained from the UV-vis-DRS in Table 1, the VB potentials (EVB) were calculated to be +1.92, +1.91, and +1.94 V vs. Ag/AgCl for CNs, Co-CNs, and S-Co-CNs, respectively. These results clearly confirm that the embedding of Co-N/C sites and subsequent sulfurization will slightly reduce the ECB of CNs due to the strong interactions between the CN matrix and embedded S-Co–N/C active sites, which would be beneficial for the fast electron transfer from excited ErB to S-Co-CNs for further enhancing the photocatalytic H2 evolution activity. Based on the above results, the efficient photocatalytic H2 evolution catalyzed by S-Co-CNs in ErB–TEOA can be illustrated in Fig. 8. Upon visible light irradiation, ErB is excited to be ErB*. Owing to the estimated reduction potential of ErB of −0.91 V vs. NHE (1.15 V vs. SCE),64 and the ECB of S-Co-CNs of −0.63 V vs. NHE (−0.83 V vs. Ag/AgCl), the electrons will efficiently transfer from ErB* to the CB of S-Co-CNs catalyst. Owing to the efficient electron separation nature of S-Co-CNs, the accepted electrons can be rapidly transferred to the embedded S-Co–N/C sites and effectively catalyze the H2O/H+ reduction to H2, while the formed ErB+ can be regenerated to ErB by TEOA to complete the catalytic cycle.
 | ||
| Fig. 7 (A) H2 evolution LSV plots and (B) Nyquist plots of pristine CNs, Co-CNs, and S-Co-CNs recorded in 10% TEOA (pH 8) solution containing 0.5 M Na2SO4. The inset shows the equivalent circuit used for data fitting. (C) Steady-state PL spectra (excitation wavelength: 380 nm) and (D) time-resolved PL decay profiles of pristine CNs, Co-CNs, and S-Co-CNs. | ||
 | ||
| Fig. 8 Schematic illustration of the band alignment and the electron transfer from ErB to CNs and the H2 evolution on the S-Co–N/C sites under visible light irradiation. | ||
Conclusions
In summary, we have proposed a facile and effective strategy to activate inert Co–N/C sites on CNs to highly active S-Co–N/C sites for catalyzing the photochemical H2 evolution reaction via a facile and effective room-temperature sulfurization method. Attributed to the mild sulfurization conditions, the high dispersion of active sites can be well preserved, and the obtained S-Co-CNs catalysts render excellent catalytic activity for H2 evolution in an ErB-sensitized system under visible light irradiation (λ ≥ 450 nm). The most active S-Co-CNs catalyst shows a high H2 evolution rate of 6.38 mmol h−1 g−1 and a highest AQY of 13.02% at 520 nm, while both pristine CNs and Co-CNs are totally inactive. The S incorporation is found to effectively enhance the intrinsic activity of Co–N/C sites while maintaining the efficient charge separation nature of the active sites. This work affords a simple but effective method for the fabrication of highly dispersed and highly efficient active sites for the H2 evolution reaction on the 2D g-C3N4 matrix and is expected to provide new insights into the materials design of transition metal-based catalysts and their applications in various energy-conversion-related photo/electrocatalysis fields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22162001), the Natural Science Foundation of Ningxia Province (2021AAC03201), the Foundation of Academic Top-notch Talent Support Program of North Minzu University (2019BGBZ08), the Leading Talents Program of Science and Technology Innovation in Ningxia Province (2020GKLRLX14), the West Light Foundation of the Chinese Academy of Sciences (XAB2018AW13), and the Innovation and Entrepreneurship Projects for Returnees of Ningxia Province.References
- X. B. Chen, S. H. Shen, L. J. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS.
- Y. Ma, X. L. Wang, Y. S. Jia, X. B Chen, H. X Han and C. Li, Chem. Rev., 2014, 19, 9987–10043 CrossRef.
- L. Lei, W. J. Wang, Z. F. Xie, X. B. Wu, A. K. Yadav, P. M. Buschbaum and H. Q. Fan, Sustainable Energy Fuels, 2021, 5, 5227–5235 RSC.
- Y. Q. Xing, Z. R. Tan, J. Z. Cheng, Z. Q. Shen, Y. J. Zhang and S. Y. Liu, Sustainable Energy Fuels, 2021, 5, 5166–5174 RSC.
- S. Wageh, A. A. Al-Ghamdi, R. Jafer, X. Li and P. Zhang, Chin. J. Catal., 2021, 42, 667–669 CrossRef CAS.
- R. C. Shen, D. D Ren, Y. N. Ding, Y. T. Guan, Y. H. Ng, P. Zhang and X. Li, Sci. China Mater., 2020, 63, 2153–2188 CrossRef CAS.
- Z. Z. Liang, R. C. Shen, Y. H. Ng, P. Zhang, Q. J. Xiang and X. Li, J. Mater. Sci. Technol., 2020, 56, 89–121 CrossRef.
- R. C. Shen, J. Xie, Q. J. Xiang, X. B. Chen, J. Z. Jiang and X. Li, Chin. J. Catal., 2019, 40, 240–288 CrossRef CAS.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2008, 8, 76–80 CrossRef.
- X. C. Wang, K. Maeda, X. F. Chen, K. Takanabe, K. Domen, Y. D. Hou, X. Z. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680–1681 CrossRef CAS PubMed.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 RSC.
- X. C. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS.
- Y. Wang, X. C. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–69 CrossRef CAS.
- S. W. Cao and J. G. Yu, J. Phys. Chem. Lett., 2014, 5, 2101–2107 CrossRef CAS PubMed.
- Z. Chen, S. Zhang, J. Yang, C. Chen, Y. C. Song, C. L. Xu, M. Q. Wu and J. X. Liao, Sustainable Energy Fuels, 2021, 5, 4973–4980 RSC.
- K. Takanabe, K. Kamata, X. C. Wang, M. Antonietti, J. Kubota and K. Domen, Phys. Chem. Chem. Phys., 2010, 12, 13020–13025 RSC.
- S. X. Min and G. X. Lu, J. Phys. Chem. C, 2012, 116, 19644–19652 CrossRef CAS.
- J. Y. Xu, Y. X. Li, S. Q. Peng, G. X. Lu and S. B. Li, Phys. Chem. Chem. Phys., 2013, 15, 7657–7665 RSC.
- W. J. Wang, H. Xu, G. X. Yu, D. M. Chen, S. Q. Sun, M. Zhou and J. Chen, Sustainable Energy Fuels, 2021, 5, 5247–5256 RSC.
- Y. N. Liu, C. C. Shen, N. Jiang, Z. W. Zhao, X. Zhou, S. J. Zhao and A. W. Xu, ACS Catal., 2017, 7, 8228–8234 CrossRef CAS.
- Y. Xue, Y. G. Lei, X. Y. Liu, Y. N. Li, W. A. Deng, F. Wang and S. X. Min, New J. Chem., 2018, 42, 14083–14086 RSC.
- Y. F. Li, M. Zhang, L. Zhou, S. J. Yang, Z. S. Wu and Y. H. Ma, Acta Phys. -Chim. Sin., 2021, 37, 2009030 Search PubMed.
- X. C. Wang, X. F. Chen, A. Thomas, X. Z. Fu and M. Antonietti, Adv. Mater., 2009, 21, 1609–1612 CrossRef CAS.
- X. F. Chen, J. S. Zhang, X. Z. Fu, M. Antonietti and X. C. Wang, J. Am. Chem. Soc., 2009, 131, 11658–11659 CrossRef CAS PubMed.
- G. G. Zhang, C. J. Huang and X. C. Wang, Small, 2015, 11, 1215–1221 CrossRef CAS PubMed.
- G. G. Zhang, S. H. Zang, L. H. Lin, Z. A. Lan, G. S. Li and X. C. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 2287–2296 CrossRef CAS PubMed.
- G. Liu, P. Niu, C. H. Sun, S. C. Smith, Z. G. Chen, G. Q. Lu and H. M. Cheng, J. Am. Chem. Soc., 2010, 132, 11642–11648 CrossRef CAS PubMed.
- J. H. Zhang, J. H. Sun, K. Maeda, K. Domen, P. Liu, M. Antonietti, X. Z. Fu and X. C. Wang, Energy Environ. Sci., 2014, 39, 15373–15379 Search PubMed.
- C. H. Ng, S. H. Teo, N. Mansir, A. Islam, C. G. Joseph, S. Hayased and Y. H. Taufiq-Yap, Sustainable Energy Fuels, 2021, 5, 4457–4511 RSC.
- Y. P. Zhu, T. Z. Ren and Z. Y. Yuan, ACS Appl. Mater. Interfaces, 2015, 7, 1685–1686 CrossRef.
- X. J. Shen, L. Liu, H. Y. Ji, Z. Mo, Y. P. Li, L. Y. Huang, D. L. Du, H. Xu and H. M. Li, Appl. Catal., B, 2016, 187, 144–153 CrossRef.
- Z. Z. Lin and X. C. Wang, Angew. Chem., Int. Ed., 2013, 52, 1735–1738 CrossRef CAS.
- D. J. Martin, K. P. Qiu, S. A. Shevin, A. D. Handoko, X. W. Chen, Z. X. Guo and J. W. Tang, Angew. Chem., Int. Ed., 2014, 53, 9240–9245 CrossRef CAS PubMed.
- Y. Yu, W. Yan, X. F. Wang, P. Li, W. Y. Gao, H. H. Zou, S. M. Wu and K. J. Ding, Adv. Mater., 2018, 30, 1705060 CrossRef.
- J. H. Sun, J. S. Zhang, M. W. Zhang, M. Antonietti, X. Z. Fu and X. C. Wang, Nat. Commun., 2012, 3, 1139–1146 CrossRef.
- C. Han, Y. D. Wang, Y. P. Lei, B. Wang, N. Wu, Q. Shi and Q. Li, Nano Res., 2015, 8, 1199–1209 CrossRef CAS.
- R. Q. Ye, H. B. Fang, Y. Z. Zheng, N. Li, Y. Wang and X. Tao, ACS Appl. Mater. Interfaces, 2016, 8, 13879–13889 CrossRef CAS.
- H. J. Yan and Y. Huang, Chem. Commun., 2011, 47, 4168–4170 RSC.
- J. J. Duan, S. Chen, M. Jaroniec and S. Z. Qiao, ACS Nano, 2015, 9, 931–940 CrossRef CAS PubMed.
- L. Xu, W. Q. Huang, L. L. Wang, Z. A. Tian, W. Y. Hu, Y. M. Ma, X. Wang, A. L. Pan and G. F. Huang, Chem. Mater., 2015, 27, 1612–1621 CrossRef CAS.
- T. Ishaq, M. Yousaf, L. A. Bhattia, M. Ahmadc, M. Ikramd, M. UsmanKhanef and A. Qayyuma, Int. J. Hydrogen Energy, 2020, 45, 31574–31584 CrossRef CAS.
- D. D. Ren, W. N. Zhang, Y. N. Ding, R. C. Shen, Z. M. Jiang, X. Y. Lu and X. Li, Solar PRL, 2020, 4, 1900423 CAS.
- C. Liu, Y. Feng, Z. T. Han, Y. Sun, X. Q. Wang, Q. F. Zhang and Z. G. Zou, Chin. J. Catal., 2021, 42, 164–174 CrossRef CAS.
- H. T. Xu, R. Xiao, J. R. Huang, Y. Jiang, C. X. Zhao and X. Yang, Chin. J. Catal., 2021, 42, 107–114 CrossRef CAS.
- F. H. Li, R. R. Zhao, B. Y. Yang, W. Wang, Y. Liu, J. P. Gao and Y. L. Gong, Int. J. Hydrogen Energy, 2019, 44, 30185–30195 CrossRef CAS.
- L. Ge, C. C. Han, J. Liu and Y. F. Li, Appl. Catal., A, 2011, 409, 215–222 CrossRef.
- R. C. Shen, K. L. He, A. P. Zhang, N. Li, Y. H. Ng, P. Zhang, J. Hu and X. Li, Appl. Catal., B, 2021, 291, 120104 CrossRef CAS.
- M. Wang, J. J. Cheng, X. F. Wang, X. K. Hong, J. J. Fan and H. G. Yu, Chin. J. Catal., 2021, 42, 37–45 CrossRef CAS.
- D. D. Zheng, G. G. Zhang, Y. D. Hou and X. C. Wang, Appl. Catal., A, 2016, 521, 2–8 CrossRef CAS.
- D. Q. Zeng, T. Zhou, W. J. Ong, M. D. Wu, X. G. Duan, W. J. Xu, Y. Z. Chen, Y. A. Zhu and D. L. Peng, ACS Appl. Mater. Interfaces, 2019, 11, 5651–5660 CrossRef CAS.
- M. Ghaemmaghami and R. Mohammadi, Sustainable Energy Fuels, 2019, 3, 2176–2204 RSC.
- H. T. Li, M. Wang, Y. P. Wei and F. Long, J. Colloid Interface Sci., 2019, 534, 343–349 CrossRef CAS PubMed.
- R. C. Zhen, J. Xie, X. Y. Lu, X. B. Chen and X. Li, ACS Sustainable Chem. Eng., 2018, 6, 4026–4036 CrossRef.
- R. C. Zhen, J. Xie, H. D. Zhang, A. P. Zhang, X. B. Chen and X. Li, ACS Sustainable Chem. Eng., 2018, 6, 816–826 CrossRef.
- N. Zhao, L. G. Kong, Y. M. Dong, G. L. Wang, X. M. Wu and P. P. Jiang, ACS Appl. Mater. Interfaces, 2018, 10, 9522–9531 CrossRef CAS.
- L. F. Gao, T. Wen, J. Y. Xu, X. P. Zhai, M. Zhao, G. W. Hu, P. Chen, Q. Wang and H. L. Zhang, Appl. Surf. Sci., 2017, 392, 608–615 CrossRef.
- P. W. Chen, K. Li, Y. X. Yu and W. D. Zhang, Appl. Surf. Sci., 2017, 392, 608–615 CrossRef CAS.
- W. Lin, L. L. Gao, W. R. Cheng, Y. J. Cao, X. K. Liu, W. Zhang, X. L. Mou, L. L. Jin, X. S. Zheng, W. C. Prof, T. Yao and S. Q. Wei, Angew. Chem., Int. Ed., 2017, 56, 9312–9317 CrossRef.
- Y. Chen, B. Lin, W. L. Yu, Y. Yang, H. Huang, K. Z. Takanabe and J. M. Basset, Chem.–Eur. J., 2015, 21, 10290–10295 CrossRef CAS PubMed.
- X. R. Wang, J. Y. Liu, Z. W. Liu, W. C. Wang, J. Luo, X. P. Han, X. W. Du, X. Z. Qiao and J. Yang, Adv. Mater., 2018, 30, 1800005 CrossRef PubMed.
- P. W. Cai, J. H. Huang, J. X. Chen and Z. H. Wen, Angew. Chem., Int. Ed., 2017, 56, 4858–4861 CrossRef CAS PubMed.
- S. H. Chang, M. D. Lu, Y. L. Tung and H. Y. Tuan, ACS Nano, 2013, 7, 9443–9451 CrossRef CAS.
- C. B. Ouyang, X. Wang and S. Y. Wang, Chem. Commun., 2015, 51, 14160–14163 RSC.
- J. Y. Xu, Y. X. Li and S. Q. Peng, Int. J. Hydrogen Energy, 2015, 40, 353–362 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures. See DOI: 10.1039/d1se01702k |
| This journal is © The Royal Society of Chemistry 2022 |