
The role of nitrogen and sulfur dual coordination of cobalt in Co-N4−xSx/C single atom catalysts in the oxygen reduction reaction†
Asnake Sahele
Haile
 a,
Heine Anton
Hansen
*b,
Weldegebriel
Yohannes
c and
Yedilfana Setarge
Mekonnen
*a
a,
Heine Anton
Hansen
*b,
Weldegebriel
Yohannes
c and
Yedilfana Setarge
Mekonnen
*a
aCenter for Environmental Science, College of Natural and Computational Sciences, Addis Ababa University, P.O. Box 1176, Addis Ababa, Ethiopia. E-mail: yedilfana.setarge@aau.edu.et
bDepartment of Energy Conversion and Storage, Technical University of Denmark, Anker Engelunds Vej, 2800 Kgs. Lyngby, Denmark. E-mail: heih@dtu.dk
cChemistry Department, College of Natural and Computational Sciences, Addis Ababa University, P.O. Box 1176, Addis Ababa, Ethiopia
First published on 16th November 2021
Abstract
Single-atom catalysts (SACs) have been considered as a potential candidate for fuel cell application due to the fact that they exhibit good oxygen reduction reaction (ORR) activity. In this study, the ORR catalytic activities of Co-N4/C, Co-N3S/C, Co-N2S2/C, Co-NS3/C, and Co-S4/C catalysts are studied using density functional theory (DFT) calculations based on the BEEF-vdW functional. The reduction of *OH into H2O is found to be the potential determining step of the ORR on Co-N4/C catalysts. This implies that the activity of the Co-N4/C system could be improved by weakening the binding energy of the *OH intermediate. The DFT results revealed that the adsorption energy of the *OH intermediate bound on Co-N3S/C, Co-N2S2/C, Co-NS3/C, and Co-S4/C is weaker than that on the Co-N4/C catalyst. The results show that the overpotentials of Co-N4/C, Co-N3S/C, Co-N2S2/C, Co-N3S/C, and Co-S4/C catalysts are 0.57, 0.37, 0.41, 0.40, and 0.47 V, respectively. Thus, the Co-N3S/C catalyst revealed a lower overpotential pathway. A slightly smaller number of charges are transferred from the Co atom in Co-N3S/C to ORR intermediates as compared to Co-N4/C and the d-band center of the Co atom changes from −0.71 eV (Co-N4/C) to −0.91 eV (Co-N3S/C). This can explain the weaker adsorption energy of *OH on the Co-N3S/C catalyst. Therefore, Co-N3S/C is a promising non-precious single-atom catalyst for efficient ORR activity in acidic solutions in fuel cells.
1 Introduction
A proton exchange membrane fuel cell (PEMFC) is an energy conversion device that converts chemical energy stored in fuel like hydrogen into electrical energy. Due to its high efficiency, environmental friendliness, and non-toxic emission, this technology has been attracting much attention for applications in the automotive industry and portable appliances.1,2 However, the slow kinetics of the oxygen reduction reaction (ORR) which occurs at the cathode, and the price of platinum-based catalysts have hindered this device from being widely used. Therefore, the search for alternative catalysts should be focused on non-precious catalysts which are made from earth-abundant materials such as carbon, nitrogen, and non-precious transition metals.3–5Previous studies reported that nitrogen-coordinated metal atom (TM–N–C, TM = Fe, Co, Cr, Mn, Ni)6–12 materials have shown better performance for the ORR in fuel cells. Fe–N–C has gained considerable attention due to its remarkable activity as compared to other TM–N–C materials.13–16 The ORR of Fe–Nx–G (x = 2–4) catalysts also shows a nearly four-electron reduction mechanism and the energy barriers are smaller than those of the pure Pt energy barrier.17–19 Although the Fe–Nx catalysts exhibit excellent catalytic activity for the ORR, they suffer from poor stability in aqueous electrolytes and this has been considered a major challenge.20 In contrast, TM–N–C catalysts such as Co–N–C exhibited outstanding durability with high ORR and oxygen evolution reaction (OER) catalytic activity, which is mainly affected by the concentration of N and the doping configuration.21,22
Recently, it has been found that doping M–N–C materials with heteroatoms can further improve the ORR activity.23–26 For example, Jinqiang Zhang et al. developed a template-assisted method to synthesize a series of single metal atoms anchored on a porous N, S-codoped carbon (NSC) matrix as highly efficient ORR catalysts to investigate the correlation between their structure and catalytic performance.27 In addition, Kai Yuan et al. reported that both experimental and theoretical results suggested that the N and P dual-coordinated iron sites were favorable for oxygen intermediate adsorption/desorption, resulting in accelerated reaction kinetics and promising catalytic oxygen reduction activity.28 Further, J. Wang et al. reported that, remarkably, doping S atoms into P/GN-rich Co–N–C provides more oxygen adsorption sites and promotes the formation of OOH species due to altering the electron and spin densities of the adjacent carbon atom, and thus synergistically enhances the ORR activity in an acidic medium, in cooperation with efficient Co–N–C sites.29
Based on the previous related studies mentioned above, the ORR catalytic activity of sulfur-doped Co-N4/C catalysts, denoted as Co-N4−xSx/C (x = 0–4), is systematically studied via density functional theory calculations. The interaction of the ORR intermediates (*OOH, *O and *OH) with catalysts and the thermodynamics of the ORR process are also taken into account. Besides, electronic structure analysis is employed to investigate the source of the ORR activity, which will help to design promising ORR catalysts with high efficiency at a reasonable price.
2 Computational methods
In this study, spin-polarized DFT calculations are performed using the Vienna ab initio simulation package (VASP)30–33 coupled with the atomic simulation environment (ASE).34 All calculations are done with the BEEF-vdW exchange correlation-functional in which vdW-DF2 (ref. 35) non-local correlation is used to account for non-local van der Waals interactions. The self-consistent electron density is determined by the iterative diagonalization of the Kohn Sham Hamiltonian. The projector augmented wave (PAW) method is used to describe the core electrons, and the basis set for the electronic wavefunctions is plane waves below a 600 eV energy cutoff. A 4 × 4 × 1 Monkhorst–Pack k-point mesh is used to sample the Brillouin zone for structural optimization and a 5 × 5 × 1 k-point grid was sampled for the calculation of density of states (DOS). The structures are relaxed until the maximum force on each atom drops below 0.01 eV Å−1. Corrections for the surface dipole interaction are applied. Solvation of the surface and adsorbates is taken into account by using the implicit solvation model implemented in VASPsol.36,37 The dielectric constant was set to be 78.54 for H2O solvent.The Co-N4−xSx/C geometrical structure is modeled based on the experimentally observed configuration in which the sulfur atom forms a bond with the Co atom.27 The pristine structure of Co-N4/C is shown in Fig. 1a, and the structures of Co-N3S/C, Co-N2S2/C, Co-NS3/C, and Co-S4/C are shown in Fig. 1b–e, respectively. To construct the Co-N4−xSx/C systems, one C–C bond in pristine graphene was removed to create di-vacancy defective graphene and then C atoms around the di-vacancy are replaced by 4 − x (x = 0–4) nitrogen atoms and x sulfur atoms. Finally, the Co atom is incorporated at the center of the di-vacancy, as illustrated in Fig. 1a–e.
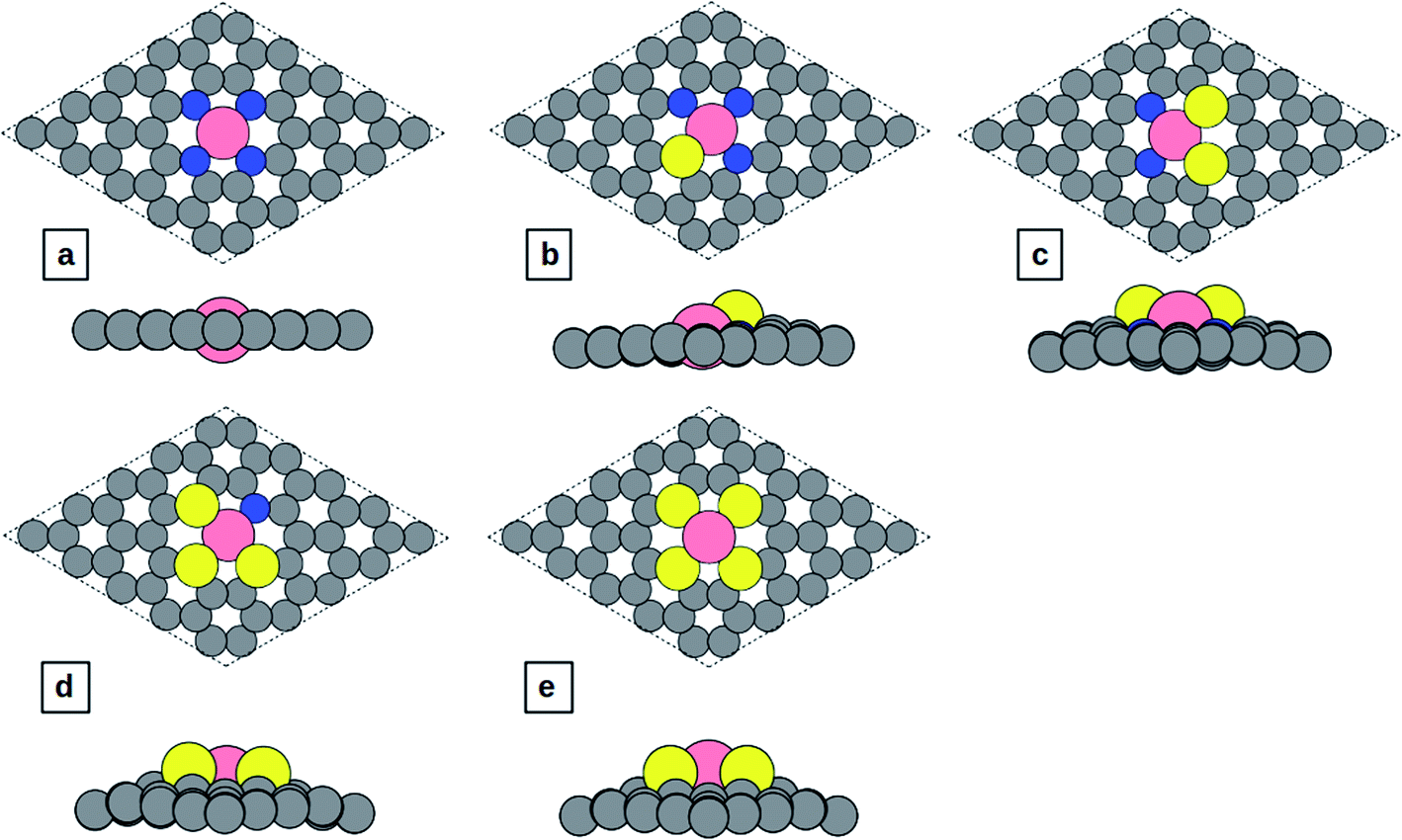 | ||
| Fig. 1 Top and side view of the structures: (a) Co-N4/C, (b) Co-N3S/C, (c) Co-N2S2/C, (d) Co-NS3/C, and (e) Co-S4/C. | ||
The binding energy of Co (ΔEb) in these catalysts can be calculated from ref. 10 and 38:
| ΔEb = ECo-N4−xSx/C − EN4−xSx/C − ECo | (1) |
The d-band center of the catalyst can be calculated from ref. 39:
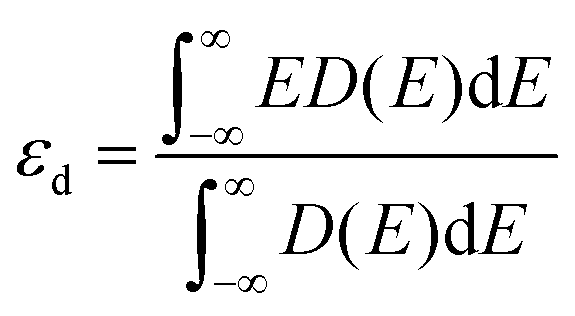 | (2) |
The DFT adsorption energies of OOH*, O*, and OH* are calculated relative to H2O and H2 molecular energies according to the following equations (eqn (3)–(5))
| ΔEads(O*) = EO* − E* − (EH2O − EH2) | (3) |
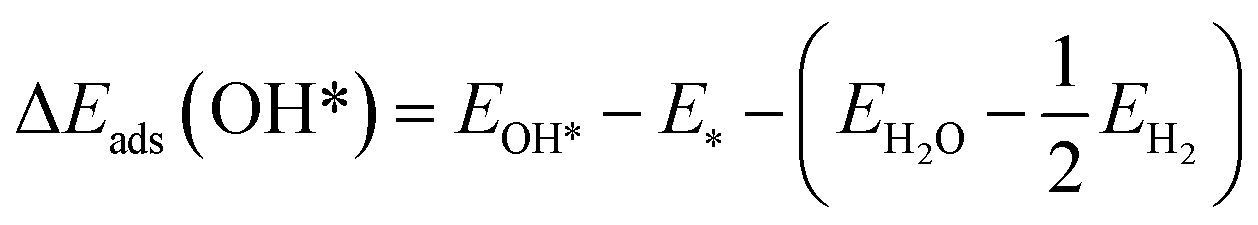 | (4) |
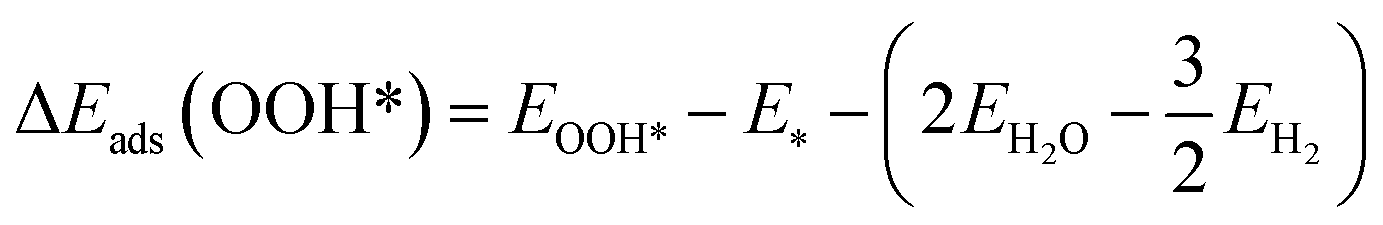 | (5) |
The Gibbs free energy of the intermediates can be calculated as:
| ΔG(U = 0 V) = ΔEads + ΔEZPE − TΔS | (6) |
The effect of applied potential (U) on the Gibbs free energy can be approximated by
| ΔG(U) = ΔG(U = 0 V) − neU | (7) |
In this paper, the associative 4e− pathway is considered to study the ORR mechanism as presented in eqn (8)–(11).
| * + O2 + H+ + e− → *OOH | (8) |
| *OOH + H+ + e− → *O + H2O | (9) |
| *O + H+ + e− → *OH | (10) |
| *OH + H+ + e− → H2O + * | (11) |
The Gibbs free energy changes (ΔG) of each ORR step on Co-N4−xSx/C are calculated as:
| ΔG1 = ΔGads(*OOH) − 4.92 eV | (12) |
| ΔG2 = ΔGads(*O) − ΔGads(*OOH) | (13) |
| ΔG3 = ΔGads(OH*) − ΔGads(*O) | (14) |
| ΔG4 = −ΔGads(*OH) | (15) |
The Gibbs free energy of O2 is set as 4.92 eV from the formation free energy of two water molecules: 2H2O → O2 + 2H2. We calculate ΔG1 − ΔG4 using the computational hydrogen electrode (CHE)44 to approximate the free energy of a proton and an electron by half the free energy of a hydrogen molecule at U = 0 V vs. RHE.
The activity of an ORR catalyst can be evaluated using the thermodynamic limiting potential (Ulim) and limiting overpotential (ηlim). Based on the computational hydrogen electrode (CHE) model,44 the limiting potential (Ulim) can be calculated from eqn (16) by determining the greatest free energy increase of all reaction steps.
| Ulim = −max{ΔG1,ΔG2,ΔG3,ΔG4}/e | (16) |
The limiting overpotential is calculated as the absolute deviation between the calculated limiting potential and the 4e− ORR equilibrium potential from eqn (17)
| ηlim = 1.23 − Ulim = 1.23 + max{ΔG1,ΔG2,ΔG3,ΔG4}/e | (17) |
The potential determining step (PDS), which is defined as the step with the largest ΔG value, is calculated. The catalyst with a smaller value of ΔG of the PDS can have a higher activity.
3 Results and discussion
3.1 Electronic properties and structural stability of Co-N4−xSx/C
The atomic charge transfer of the Co-N4−xSx/C system is investigated and the charge density difference is described in Fig. 2 for Co-N4/C and Co-N3S/C catalysts, where the cyan regions represent electron depletion and yellow represents electron accumulation. Few electrons are distributed around the Co atom because the coordinated N and S atoms (3.05 and 2.58) are more electronegative than the Co atom (1.88).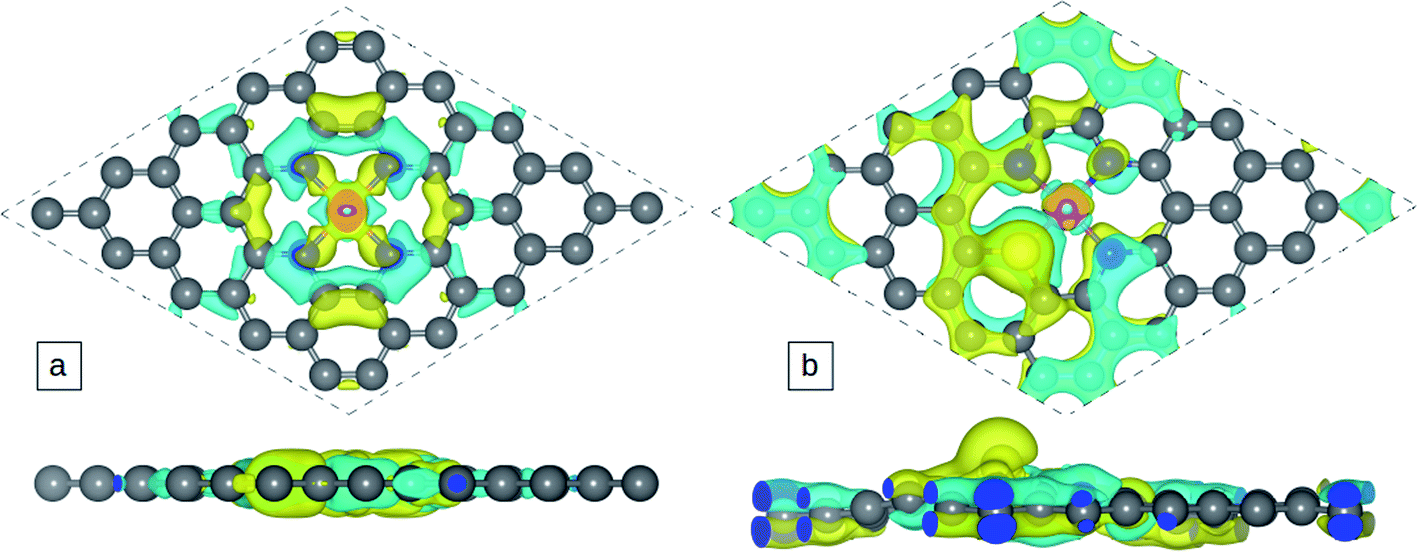 | ||
| Fig. 2 Charge density distributions of (a) Co-N4/C and (b) Co-N3S/C. | ||
The Bader charge method is used to calculate the Co-N4−xSx/C atomic charge transfer (Table 1), which can be used to judge the strength of the bond of Co-N4−xSx/C systems. We found that the Co atom is positively charged, and also C and S atoms around the Co atom are positively charged but N atoms are negatively charged. Co atom become less positively charged when the number of S atoms is increased which can be attributed to the weaker electronegativity of sulfur (2.58) than nitrogen (3.04). Significant charge transfer occurs between the Co atom and the surrounding four N atoms, which indicates the formation of covalent bonds between Co and the surrounding N atoms. The density of states (DOS) is displayed in Fig. 3a and b for Co-N4/C and Co-N3S/C, and the DOS of Co-N2S2/C, Co-NS3/C, and Co-S4/C is shown in Fig. S3a–c.† There is strong Co-3d and N-2p orbital hybridization below the Fermi level (Fig. 3), which is further indication of the formation of a covalent bond.
| Systems | d C–N | d Co–N | ΔEb | Q(Co) | Q(S) | Q(N) | Co d-band center |
|---|---|---|---|---|---|---|---|
| Co-N4/Cact | 1.376 | 1.879 | −9.38 | 1.06 | — | −1.21 | −0.71 |
| Co-N3S/C | 1.375 | 1.889 | −7.96 | 0.72 | 0.16 | −1.21 | −0.91 |
| Co-N2S2/C | 1.375 | 1.890 | −7.45 | 0.63 | 0.14 | −1.21 | −0.96 |
| Co-NS3/C | 1.385 | 1.940 | −5.97 | 0.47 | 0.14 | −1.18 | −1.06 |
| Co-S4/C | — | — | −5.97 | 0.37 | 0.12 | — | −1.24 |
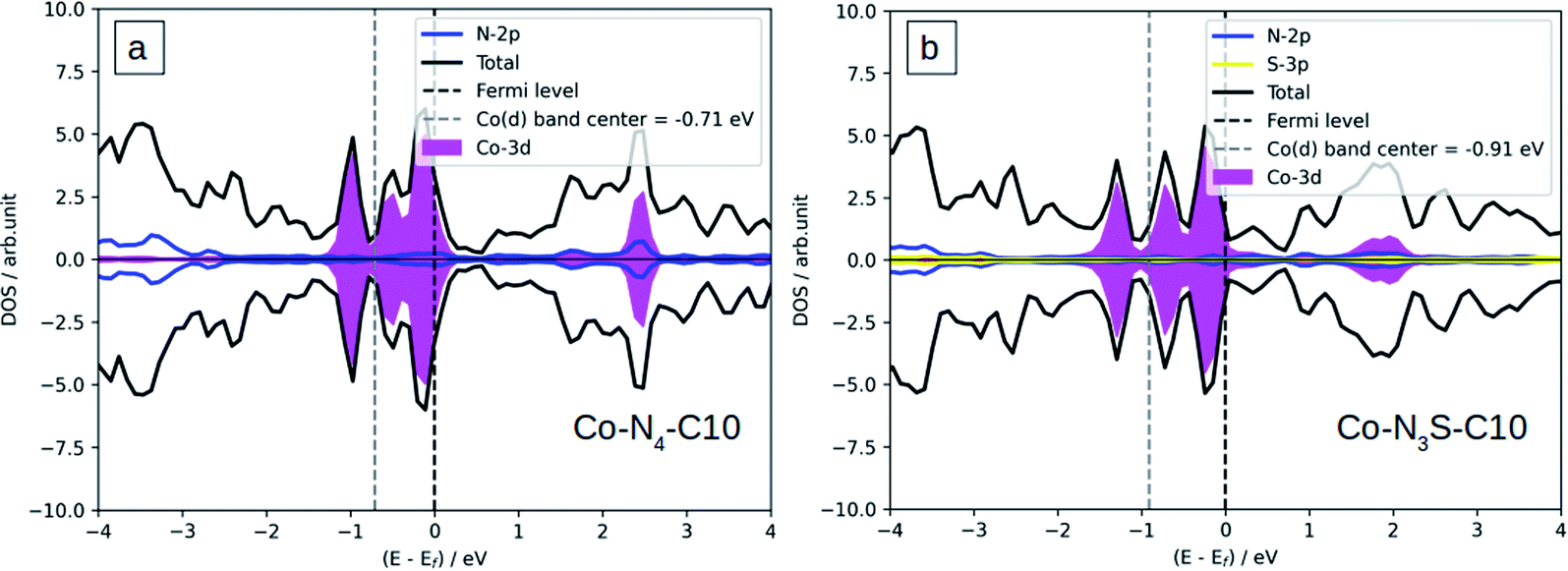 | ||
| Fig. 3 Density of states (DOS) of (a) Co-N4/C and (b) Co-N3S/C systems. | ||
To evaluate the stability of the structures the binding energy of Co in these catalysts is considered, since the stability of the structure is directly related to the binding energy, while the reaction activity has an indirect relationship with bonding.10,38 The Co atom exhibits a short bond length with N of 1.879 Å in Co-N4/C and the strongest bonding of −9.38 eV, which are much larger than those of Co-N3S/C, Co-N2S2/C, and Co-NS3/C (−7.96, −7.45, and −5.97 eV, respectively).
The catalytic activity of catalysts can be described using the d-band center (average energy of d-states), so the d-band center of Co in Co-N4−xSx/C catalysts was calculated and is presented in Table 1. It can be seen from Table 1 that there is a decreasing trend in the d-band center from Co-N4/C to Co-S4/C. The effect of decreasing the number of N atoms does show a clear correlation between decreasing the number of N atoms and the d-band center, suggesting that the catalytic activity of Co-N4/C can be tailored through changing the number of N atoms by sulfur atoms.
The down-shift in the d-band center with increased S coordination correlates with a decrease in the Bader charge on Co. The Bader charges suggest that the expected Co oxidation state (II) is decreased as S replaces N around the Co. The decrease in Co Bader charge is consistent with the lower electronegativity of S than N mentioned above.
3.2 Adsorption energy of the ORR intermediates on model catalysts
To study the catalytic activity of a catalyst, there should be an interaction between reaction species, in this case ORR intermediates, and the catalyst surface. The adsorption energy of ORR intermediates is an indication of how the ORR intermediates interact with the catalyst surface. This interaction plays a very important role in investigating the catalytic activity. Previous studies have shown that the ORR potential-determining step (PDS) for Co-N4/C is the reduction of *OH into H2O.5,7,21,45–47 If the binding of *OH on Co-N4/C is too strong, it is not easy to desorb from the surface of the catalyst for further reduction. Due to this the binding strength of *OH can be considered as a descriptor of the activity for those catalysts which have similar structures to Co-N4/C. Therefore, we need to have a system on which the adsorption energy of *OH is weaker.The binding energies of the ORR intermediates (*OOH, *O, and *OH) on Co-N4/C as well as on Co-N3S/C, Co-N2S2/C, Co-NS3/C, and Co-S4/C are calculated using eqn (3)–(5).
Based on the calculation results, the most favorable adsorption site for the ORR intermediates is on the top of the central metal atom for all considered catalyst models. The optimized geometrical structures of ORR adsorbates adsorbed on catalyst surfaces are illustrated in Fig. 4 and S3.† The first step of the ORR process is adsorption of O2 at the active site. As clearly shown in Fig. S4a and b,† O2 prefers to adsorb on for example Co-N4S/C and Co-N3S/C with end-on configuration as shown in Fig. S4a and b.† The O–O bond lengths of the adsorbed O2 on Co-N4S/C and Co-N3S/C are elongated from the initial bond length 1.246 Å to 1.291 and 1.293 Å, respectively, which is due to the charge transfer from the metal atom to the O2-2π* orbital. The next step is the formation of *OOH by hydrogenation of adsorbed *O2. Then *OOH is dissociated into atomic oxygen and a free water molecule. Then *OH is formed via hydrogenation of adsorbed *O, which can be further reduced to the second H2O molecule.
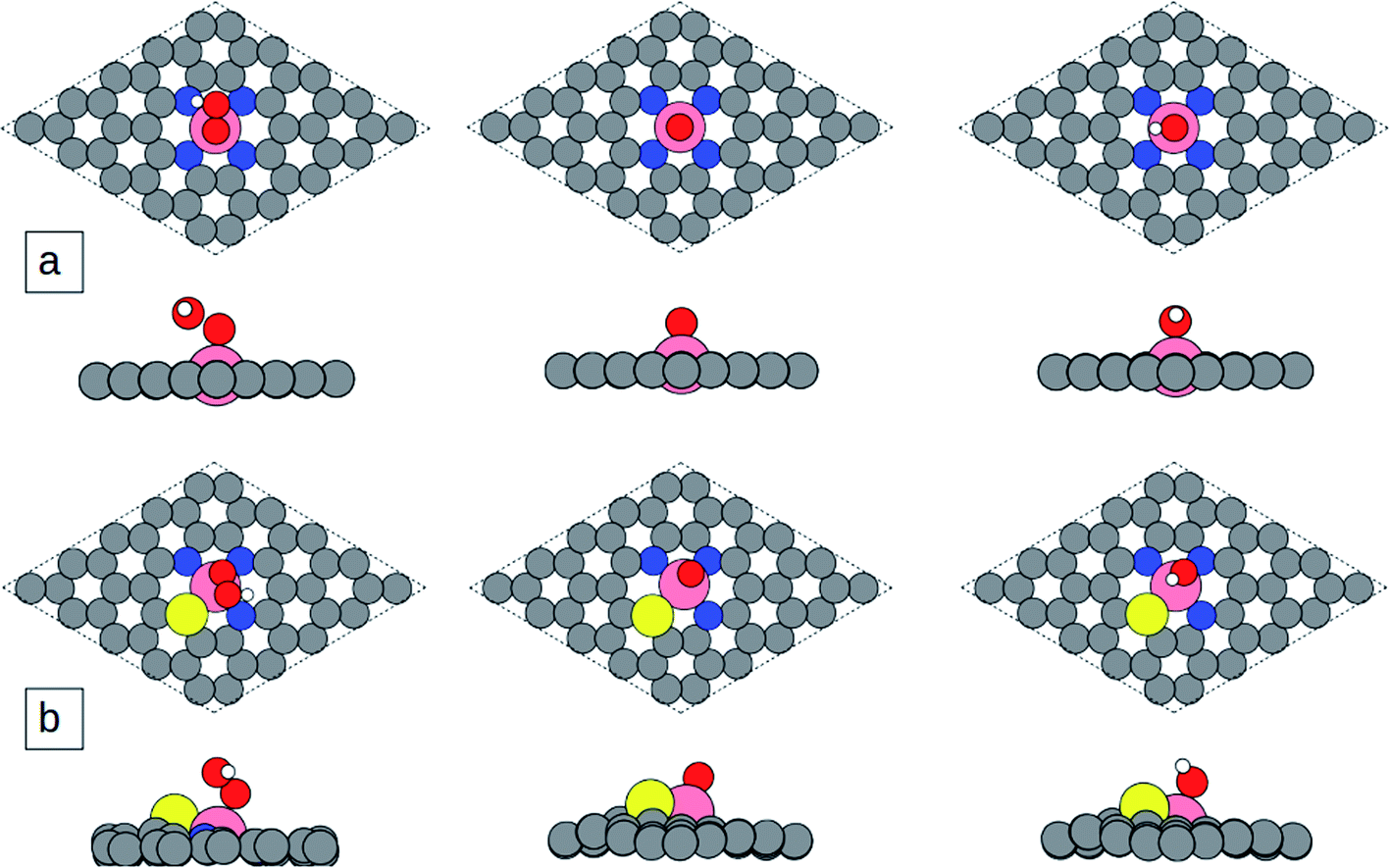 | ||
| Fig. 4 Optimized adsorption structures of ORR intermediates: (a) Co-N4/C and (b) Co-N3S/C. Pink, cobalt (Co); blue, nitrogen (N); yellow, sulfur (S); gray, carbon (C); red, oxygen (O); and white, hydrogen (H). | ||
As can be seen in Table 2, the adsorption energies of *OOH, *O, and *OH on Co-N4/C are 3.12, 2.17, and 0.32 eV, respectively. Meanwhile, the adsorption energies of *OOH, *O, and *OH on Co-N3S/C are 3.39, 2.05, and 0.52 eV; on Co-N2S2/C they are 3.42, 1.84, and 0.51 eV; on Co-NS3/C they are 3.43, 1.94, and 0.57 eV; and on Co-S4/C they are 3.50, 2.01, and 0.63 eV, respectively. The results reveal that the adsorption energy of *OH becomes weaker with the incorporation of S atoms. In other words, when one to three sulfur atoms are doped into Co-N4/C, its activity is substantially improved.
| System | *OOH | *O | *OH |
|---|---|---|---|
| Co-N4/C | 3.12 | 2.17 | 0.32 |
| Co-N3S/C | 3.39 | 2.05 | 0.52 |
| Co-N2S2/C | 3.42 | 1.84 | 0.51 |
| Co-NS3/C | 3.43 | 1.94 | 0.57 |
| Co-S4/C | 3.50 | 2.01 | 0.63 |
3.3 Thermodynamics of the ORR
The Gibbs free energy (ΔG) of each ORR step can be calculated by using eqn (12)–(15) in which the ΔGads(*OOH), ΔGads(*O), and ΔGads(*OH) are the adsorption free energies (ΔGads) of *OOH, *O, and *OH, respectively. And the ΔGads can be calculated using the equations ΔG(*OOH) = ΔEads(*OOH) + 0.67, ΔG(*O) = ΔGads(*O) + 0.11, and ΔGads(*OH) = ΔEads(*OH) + 0.34.As can be seen in Table 3, even after replacing N atoms by S atoms, the ΔG of each ORR step remains negative in all the Co-N4−xSx/C systems, which means that the modified catalysts can catalyze the whole ORR spontaneously at U = 0 V vs. RHE. In addition, except for Co-NS3/C and Co-S4/C, in which the formation of *OOH is the PDS which is in agreement with a previous study,48 the PDS of all the Co-N4−xSx/C samples is the reduction of *OH into H2O step, that is, *OH + H+ + e− → * + H2O. This shows once again that the ORR catalytic activity of this type of catalyst is indeed restricted by *OH adsorption. The calculated overpotential of Co-N4/C, Co-N3S/C, Co-N2S2/C, Co-NS3/C, and Co-S4/C is 0.57, 0.37, 0.41, 0.40, and 0.47 V, respectively.
Compared with the original Co-N4/C system, all sulfur-containing catalysts have lower ηORR which indicates better ORR catalytic activity. For example, the ηORR of Co-N3S/C is reduced to a very small value of 0.37 V, which is 0.2 V smaller than that of Co-N4/C. In addition, the ηORR of Co-N3S/C is also 0.04 V smaller than that of Co-S4/C. Further, these values of overpotential are all smaller than those of the Pt(111) surface (0.65 V),49 Pt3V (0.64 V),54 and other heteroatoms doped M–N–C materials such as phosphorus-doped Fe–N–C (0.75 V),26 S-doped Fe–N–C (0.58 V),27 and S doped Cu–N–C (0.39 V).24 This suggests that the doping with S atoms is an effective strategy to improve the catalytic activity of Co-N4.
Considering that the S atom couldn't form a bond directly with Co, S-doping in the graphene sheet was taken into account for simulation. Additional models with different S atom positions are constructed. For simplification, only two S positions around the Co-N4 species are considered (Fig. S1†). The ORR activity of the most stable structure (Fig. S1b†) is then investigated. The DOS and optimized geometry of ORR intermediates adsorbed on Co-N4S/C are displayed in Fig. S2d and S5.† The Co-N4S/C catalyst exhibited lower overpotential (0.51 V) than the Co-N4/C one but higher than that of Co-N3S/C. This implies better ORR catalytic activity than that of Co-N4/C. However, the catalytic performance of Co-N4S/C is lower than that of Co-N3S/C. The PDS of the ORR on Co-N4S/C also lies in the reduction of *OH.
The activity volcano plot (Fig. 5) based on the scaling relation (ΔadsG(*OOH) = ΔadsG(*OH) + 3.2 eV), which is obtained from the data for the structures considered in this study (Table S3†), represents the theoretical UL predicted as a function of the *OH adsorption free energy (ΔadsG(*OH)) calculated at the most active surface site for each of the model structures. The most active sites are found to lie at the top of the volcano. Thus, based on Fig. 5, the ORR activity can be ranked as Co-N3S/C > Co-NS3/C > Co-N2S2/C > Co-S4/C > Co-N4/C.
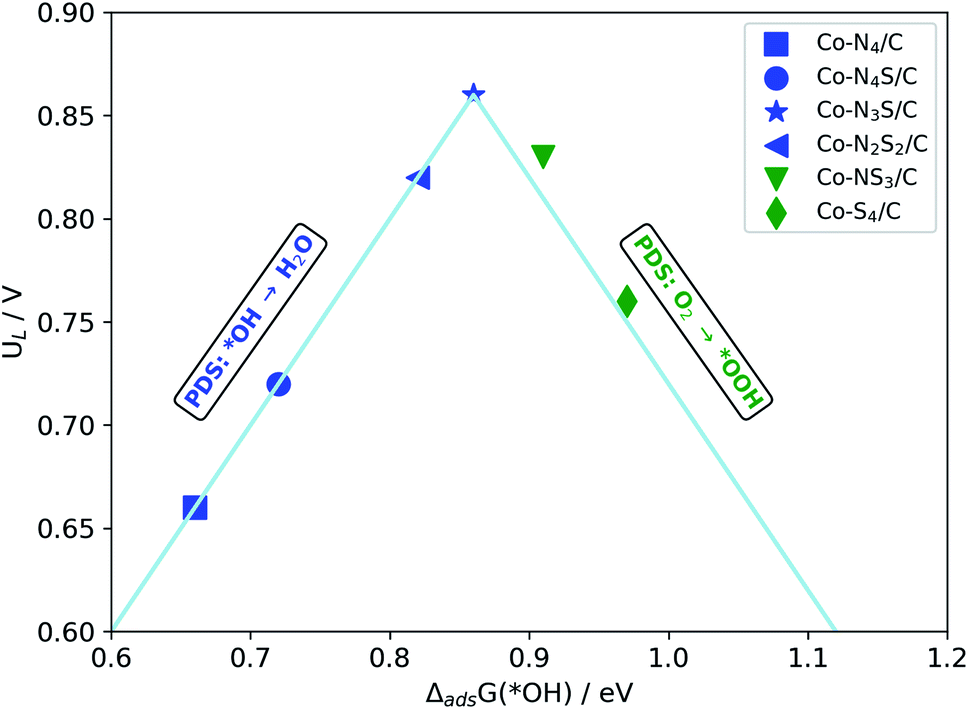 | ||
| Fig. 5 Volcano plot for the ORR limiting potential (UL) against the Gibbs free energy of ΔadsG(*OH). | ||
Fig. 6 displays the free energy diagram for the ORR via the four-electron reductions on Co-N4/C and Co-N3S/C systems and Fig. S6a–d† for Co-N2S2/C, Co-NS3/C, Co-S4/C, and Co-N4S/C at different electrode potentials. At U = 0, all ORR steps are downhill, which demonstrates that the reactions from O2 to H2O can spontaneously happen as an exothermic process. When U = UL, the free energy of *OH to H2O is isoenergetic except in the case of Co-NS3/C and Co-S4/C in which the free energy of *O2 to *OOH is isoenergetic. At U = Ueq = 1.23 V the formation of *OOH and H2O is uphill in all systems which implies that these processes are endothermic.
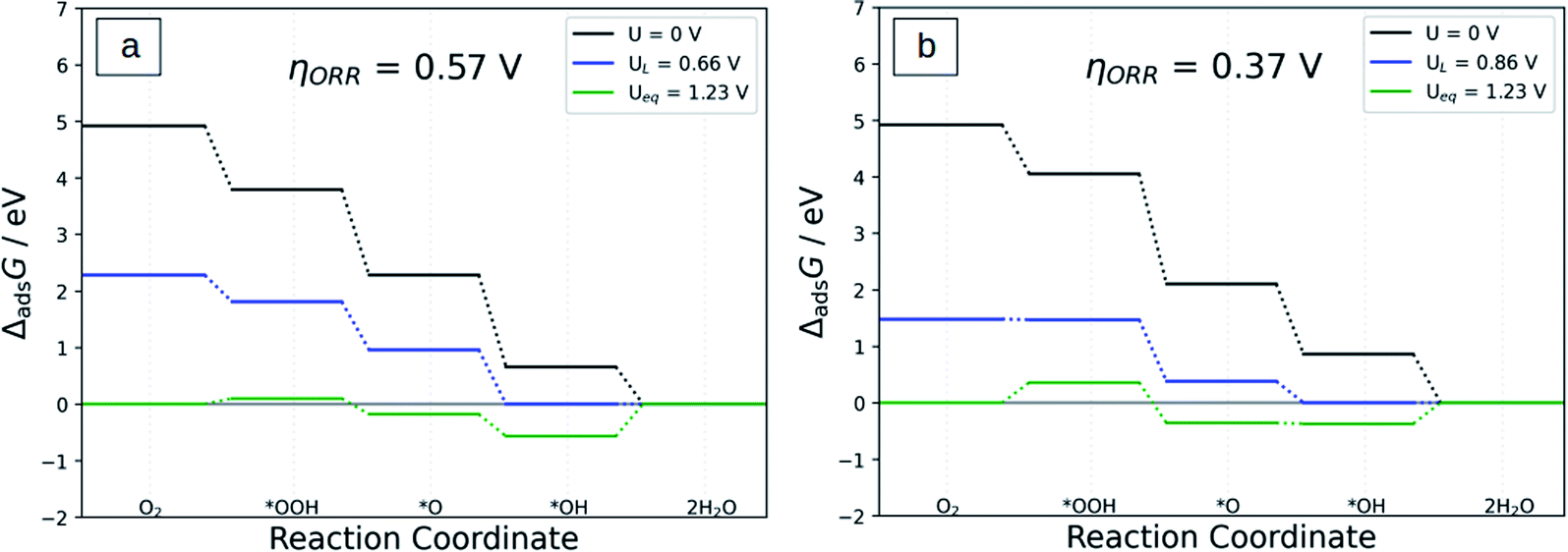 | ||
| Fig. 6 The Gibbs free energy diagram of the ORR: (a) Co-N4/C and (b) Co-N3S/C catalysts. | ||
3.4 Origin of the ORR activity
The electronic structure analysis approach plays a crucial role in investigating the source of the ORR activity because it can clearly show the electron-withdrawing/donating properties of the active sites during the ORR process.50–52 The Bader charge of the atoms at the active sites of the Co-N4/C and Co-N3S/C systems during the ORR process is shown in Fig. 7. Co-N3S/C is chosen due to its higher ORR activity as compared to the other studied systems.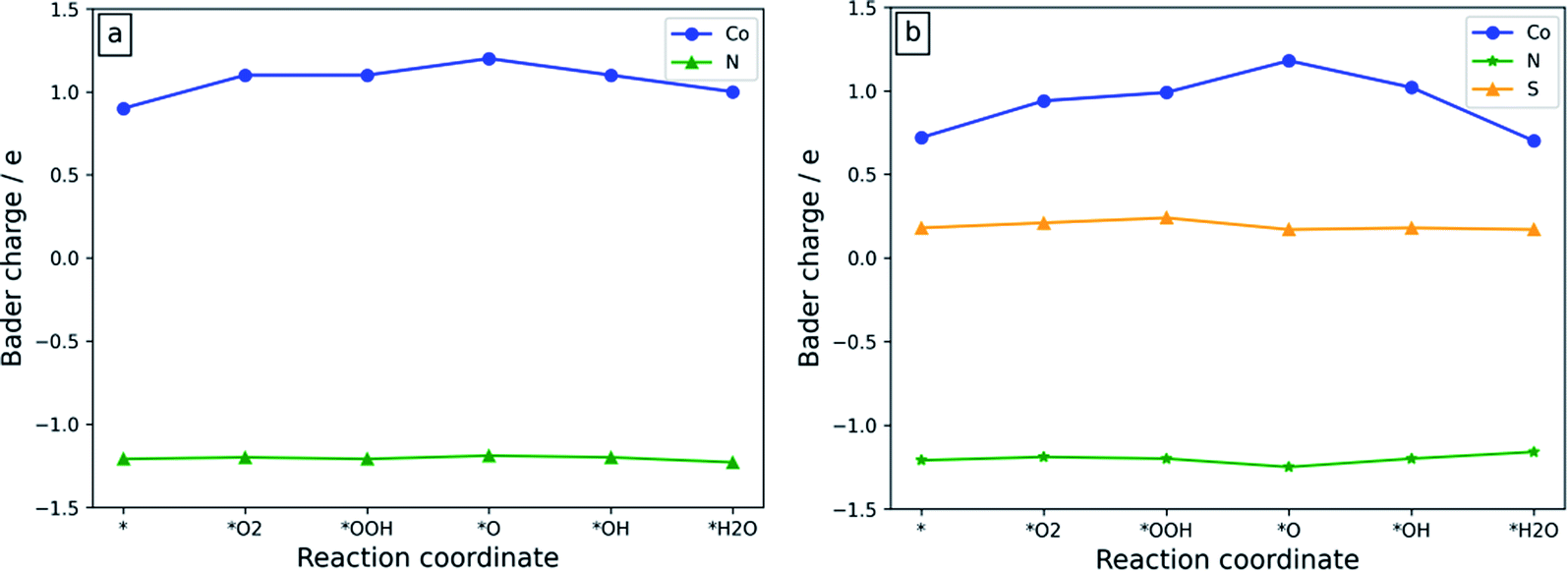 | ||
| Fig. 7 The Bader charge of the atoms at the active sites of (a) Co-N4/C and (b) Co-N3S/C catalysts. The charge of N is the average charge of the existing N atoms. The data in Table S1† are used to plot the graphs. | ||
As illustrated in Fig. 7a, for Co-N4/C, during the whole ORR process, the charge fluctuation of the Co atom is obvious, while after oxygen containing ORR intermediates are adsorbed on the active site the charge on N atoms remains almost constant. This suggests that the ORR activity is mainly due to the Co atom. As depicted in Fig. 7b, when the S atom is introduced near the Co atom, the Co atom shows a very strong change of charge throughout the whole ORR process, indicating that it is indeed an active site for catalyzing the ORR. The Bader charges of S and N atoms remain almost unchanged during the whole ORR process, suggesting that they are not contributing to the charge transfer. Therefore, the Co atom could be considered as the origin of the ORR activity.
To explain the origin of the activity we use the density of states (DOS).53 It is clearly shown that the d-band center of the Co atom is changed from −0.71 eV on Co-N4/C to −0.91 eV on Co-N3S/C (Fig. 3), contributing to the weaker binding strength of *OH. Therefore, this is another reason that the ORR activity of Co-N4 is greatly improved after S doping, owing to the fact that the binding strength of *OH as the activity descriptor is tuned.
4 Conclusion
In summary, the ORR activities of Co-N4/C, Co-N3S/C, Co-N2S2/C, Co-NS3/C, and Co-S4/C systems were investigated using density functional theory. The binding energy of the ORR species, the free energy of each ORR step, overpotential of the whole ORR, and the electronic structure were calculated. The results suggest that doping of S atoms is an effective strategy to improve the catalytic activity of Co-N4/C. The d-band center of the Co atom is down-shifted from the Fermi level due to the dual coordination with sulfur and nitrogen atoms. This is responsible for weaker adsorption of ORR intermediates, especially *OOH and *OH, on systems that contain both sulfur and nitrogen than that of the system with nitrogen alone (Co-N4/C). Among the studied systems, Co-N3S/C possesses enhanced ORR activity. The overpotential of Co-N3S/C is 0.37 V which is smaller than that of the Co-N4/C (0.57 V) catalyst. The formation of H2O is found to be the potential determining step of the ORR on the Co-N3S/C catalyst. Therefore, we can conclude that modifying the electronic structure of the central metal atom could enhance the ORR catalytic activity of the Co-N4/C system.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the thematic research project (No: TR/036/2020 and VPRTT/PY-026/2018/10) funded by Addis Ababa University. A. S. Haile acknowledges Assosa University for financial support. H. A. Hansen acknowledges funding from the Villum Foundation through the research center V-Sustain (grant 9455). Y. S. Mekonnen acknowledges support from the World Academy of Sciences (TWAS) (research grant 19-202 RG/CHE/AF/AC_G).References
- G. W. Crabtree and M. S. Dresselhaus, The hydrogen fuel alternative, MRS Bull., 2008, 33, 421–428 CrossRef CAS.
- J. Shin, W.-S. Hwang and H. Choi, Can hydrogen fuel vehicles be a sustainable alternative on vehicle market? Comparison of electric and hydrogen fuel cell vehicles, Technol. Forecast. Soc. Change, 2019, 143, 239–248 CrossRef.
- M. K. Debe, Electrocatalyst approaches and challenges for automotive fuel cells, Nature, 2012, 486, 43–51 CrossRef CAS.
- A. S. Haile, H. A. Hansen, W. Yohannes and Y. S. Mekonnen, Pyridinic-Type N-Doped Graphene on Cobalt Substrate as Efficient Electrocatalyst for Oxygen Reduction Reaction in Acidic Solution in Fuel Cell, J. Phys. Chem. Lett., 2021, 12, 3552–3559 CrossRef CAS PubMed.
- K. L. Svane, H. A. Hansen and T. Vegge, A comparison of single and double Co sites incorporated in N-doped graphene for the oxygen reduction reaction, J. Catal., 2021, 393, 230–237 CrossRef CAS.
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Identification of catalytic sites for oxygen reduction in iron-and nitrogen doped graphene materials, Nat. Mater., 2015, 14, 937–942 CrossRef CAS PubMed.
- Q. Cheng, L. Yang, L. Zou, Z. Zou, C. Chen, Z. Hu and H. Yang, Single Cobalt Atom and N Codoped Carbon Nanofibers as Highly Durable Electrocatalyst for Oxygen Reduction Reaction, ACS Catal., 2017, 7, 6864–6871 CrossRef CAS.
- J. Li, M. Chen, D. A. Cullen, S. Hwang, M. Wang, B. Li, K. Liu, S. Karakalos, M. Lucero and H. Zhang, et al., Atomically dispersed manganese catalysts for oxygen reduction in proton-exchange membrane fuel cells, Nat. Catal., 2018, 1, 935–945 CrossRef CAS.
- Z. Cai, P. Du, W. Liang, H. Zhang, P. Wu, C. Cai and Z. Yan, Single-atom-sized Ni–N4 sites anchored in three-dimensional hierarchical carbon nanostructures for the oxygen reduction reaction, J. Mater. Chem. A, 2020, 8, 15012–15022 RSC.
- M. Chen, M. Luo, C. Liu, X. Qi, S. G. Peera and T. Liang, Transition metal-Nx doped graphene as an efficient oxygen reduction reaction catalyst: a theoretical perspective, Comput. Theor. Chem., 2020, 1187, 112945 CrossRef CAS.
- E. Luo, H. Zhang, X. Wang, L. Gao, L. Gong, T. Zhao, Z. Jin, J. Ge, Z. Jiang, C. Liu and W. Xing, Single-Atom Cr−N4 Sites Designed for Durable Oxygen Reduction Catalysis in Acid Media, Angew. Chem., Int. Ed., 2019, 58, 12469–12475 CrossRef CAS.
- L. Li, R. Huang, X. Cao and Y. Wen, Computational screening of efficient graphene supported transition metal single atom catalysts toward the oxygen reduction reaction, J. Mater. Chem. A, 2020, 8, 19319–19327 RSC.
- Y. Wang, Q. Li, L.-c. Zhang, Y. Wu, H. Chen, T. Li, M. Xu and S.-J. Bao, A gel limiting strategy for large-scale fabrication of Fe–N–C single-atom ORR catalysts, J. Mater. Chem. A, 2021, 9, 7137–7142 RSC.
- M. Sun, D. Davenport, H. Liu, J. Qu, M. Elimelech and J. Li, Highly efficient and sustainable non-precious-metal Fe–N–C electrocatalysts for the oxygen reduction reaction, J. Mater. Chem. A, 2018, 6, 2527–2539 RSC.
- X. Ao, W. Zhang, Z. Li, L. Lv, Y. Ruan, H.-H. Wu, W.-H. Chiang, C. Wang, M. Liu and X. C. Zeng, Unraveling the high-activity nature of Fe–N–C electrocatalysts for the oxygen reduction reaction: the extraordinary synergy between Fe–N4 and Fe4N, J. Mater. Chem. A, 2019, 7, 11792–11801 RSC.
- J. Weiss, H. Zhang and P. Zelenay, Recent progress in the durability of Fe-N-C oxygen reduction electrocatalysts for polymer electrolyte fuel cells, J. Electroanal. Chem., 2020, 875, 114696 CrossRef CAS , Special Issue in Memory of Andrzej Wieckowski..
- W. Liang, J. Chen, Y. Liu and S. Chen, Density-Functional-Theory Calculation Analysis of Active Sites for Four-Electron Reduction of O2 on Fe/N-Doped Graphene, ACS Catal., 2014, 4, 4170–4177 CrossRef CAS.
- Y. Meng, K. Li, Y. Yang, Y. Wang and Z. Wu, The Oxygen Reduction Reaction Mechanism on FeN3 Doped Divacancy Graphene: A Theoretical Perspective, J. Electrochem. Soc., 2018, 165, F145–F151 CrossRef CAS.
- Y. Yang, K. Li, Y. Meng, Y. Wang and Z. Wu, A density functional study on the oxygen reduction reaction mechanism on FeN2-doped graphene, New J. Chem., 2018, 42, 6873–6879 RSC.
- K. Liu, Z. Qiao, S. Hwang, Z. Liu, H. Zhang, D. Su, H. Xu, G. Wu and G. Wang, Mn and N-doped carbon as promising catalysts for oxygen reduction reaction: theoretical prediction and experimental validation, Appl. Catal. B Environ., 2019, 243, 195–203 CrossRef CAS.
- X. Sun, K. Li, C. Yin, Y. Wang, M. Jiao, F. He, X. Bai, H. Tang and Z. Wu, Dualsite oxygen reduction reaction mechanism on CoN4 and CoN2 embedded graphene: theoretical insights, Carbon, 2016, 108, 541–550 CrossRef CAS.
- X. Sun, K. Li, C. Yin, Y. Wang, F. He, H. Tang and Z. Wu, CoN3 embedded graphene, a potential catalyst for the oxygen reduction reaction from a theoretical perspective, Phys. Chem. Chem. Phys., 2017, 19, 17670–17676 RSC.
- H. Shang, Z. Jiang, D. Zhou, J. Pei, Y. Wang, J. Dong, X. Zheng, J. Zhang and W. Chen, Engineering a metal–organic framework derived Mn–N4–CxSy atomic interface for highly efficient oxygen reduction reaction, Chem. Sci., 2020, 11, 5994–5999 RSC.
- H. Shang, X. Zhou, J. Dong, A. Li, X. Zhao, Q. Liu, Y. Lin, J. Pei, Z. Li and Z. Jiang, et al., Engineering unsymmetrically coordinated Cu-S1N3 single atom sites with enhanced oxygen reduction activity, Nat. Commun., 2020, 11, 1–11 Search PubMed.
- Y.-C. Wang, Y.-J. Lai, L. Song, Z.-Y. Zhou, J.-G. Liu, Q. Wang, X.-D. Yang, C. Chen, W. Shi, Y.-P. Zheng, M. Rauf and S.-G. Sun, S-Doping of an Fe/N/C ORR Catalyst for Polymer Electrolyte Membrane Fuel Cells with High Power Density, Angew. Chem., Int. Ed., 2015, 54, 9907–9910 CrossRef CAS.
- J.-C. Li, H. Zhong, M. Xu, T. Li, L. Wang, Q. Shi, S. Feng, Z. Lyu, D. Liu and D. Du, et al., Boosting the activity of Fe-Nx moieties in Fe-NC electrocatalysts via phosphorus doping for oxygen reduction reaction, Sci. China Mater., 2020, 63, 965–971 CrossRef CAS.
- J. Zhang, Y. Zhao, C. Chen, Y.-C. Huang, C.-L. Dong, C.-J. Chen, R.-S. Liu, C. Wang, K. Yan, Y. Li and G. Wang, Tuning the Coordination Environment in Single-Atom Catalysts to Achieve Highly Efficient Oxygen Reduction Reactions, J. Am. Chem. Soc., 2019, 141, 20118–20126 CrossRef CAS PubMed.
- K. Yuan, D. Lützenkirchen-Hecht, L. Li, L. Shuai, Y. Li, R. Cao, M. Qiu, X. Zhuang, M. K. H. Leung, Y. Chen and U. Scherf, Boosting Oxygen Reduction of Single Iron Active Sites via Geometric and Electronic Engineering: Nitrogen and Phosphorus Dual Coordination, J. Am. Chem. Soc., 2020, 142, 2404–2412 CrossRef CAS.
- J. Wang, Q. Wang, W. She, C. Xie, X. Zhang, M. Sun, J. Xiao and S. Wang, Tuning the electron density distribution of the Co-N-C catalysts through guest molecules and heteroatom doping to boost oxygen reduction activity, J. Power Sources, 2019, 418, 50–60 CrossRef CAS.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Ab initio molecular-dynamics simulation of the liquid-metal amorphous-semiconductor transition in germanium, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer and C. Hargus, et al., The atomic simulation environment—a Python library for working with atoms, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- J. Wellendorff, K. T. Lundgaard, A. Møgelhøj, V. Petzold, D. D. Landis, J. K. Nørskov, T. Bligaard and K. W. Jacobsen, Density functionals for surface science: exchange-correlation model development with Bayesian error estimation, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 235149 CrossRef.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. Arias and R. G. Hennig, Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- K. Mathew and R. G. Hennig, Implicit self-consistent description of electrolyte in planewave density-functional theory, arXiv preprint arXiv:1601.03346, 2016.
- M. Sun, Q. Ren, Y. Zhao, J.-P. Chou, J. Yu and W. Tang, Electronic and magnetic properties of 4d series transition metal substituted graphene: a first-principles study, Carbon, 2017, 120, 265–273 CrossRef CAS.
- B. Hammer and J. K. Nørskov, Theoretical surface science and catalysis—calculations and concepts, Adv. Catal., 2000, 45, 71–129 CAS.
- R. C. Weast, CRC Handbook of Chemistry and Physics : a Ready-Reference Book of Chemical and Physical Data, CRC Press, Boca Raton, Fla, 1986 Search PubMed.
- NIST computational chemistry comparison and benchmark database, NIST standard reference database number 101, ed. R. D. Johnson III, 21, August 2020 Search PubMed.
- R. Christensen, H. A. Hansen and T. Vegge, Identifying systematic DFT errors in catalytic reactions, Catal. Sci. Technol., 2015, 5, 4946–4949 RSC.
- R. Christensen, H. A. Hansen, C. F. Dickens, J. K. Nørskov and T. Vegge, Functional independent scaling relation for ORR/OER catalysts, J. Phys. Chem. C, 2016, 120, 24910–24916 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, Origin of the overpotential for oxygen reduction at a fuel-cell cathode, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- Z. Lu, G. Xu, C. He, T. Wang, L. Yang, Z. Yang and D. Ma, Novel catalytic activity for oxygen reduction reaction on MnN4 embedded graphene: a dispersion-corrected density functional theory study, Carbon, 2015, 84, 500–508 CrossRef CAS.
- K. Liu, S. Kattel, V. Mao and G. Wang, Electrochemical and Computational Study of Oxygen Reduction Reaction on Nonprecious Transition Metal/Nitrogen Doped Carbon Nanofibers in Acid Medium, J. Phys. Chem. C, 2016, 120, 1586–1596 CrossRef CAS.
- Y. Han, Y.-G. Wang, W. Chen, R. Xu, L. Zheng, J. Zhang, J. Luo, R.-A. Shen, Y. Zhu, W.-C. Cheong, C. Chen, Q. Peng, D. Wang and Y. Li, Hollow N-Doped Carbon Spheres with Isolated Cobalt Single Atomic Sites: Superior Electrocatalysts for Oxygen Reduction, J. Am. Chem. Soc., 2017, 139, 17269–17272 CrossRef CAS.
- P. Zhang, X. Hou, J. Mi, L. Liu and M. Dong, Oxygen reduction reaction on M-S4 embedded graphene: a density functional theory study, Chem. Phys. Lett., 2015, 641, 112–116 CrossRef CAS.
- M. Reda, H. A. Hansen and T. Vegge, DFT Study of the oxygen reduction reaction on carbon-coated iron and iron carbide, ACS Catal., 2018, 8, 10521–10529 CrossRef CAS.
- X. Chen, F. Ge and N. Lai, Cobalt-based coordination polymer as high activity electrocatalyst for oxygen reduction reaction: catalysis by novel active site CoO4N2, Int. J. Energy Res., 2020, 44, 2164–2172 CrossRef CAS.
- A. Legarreta-Mendoza, N. Flores-Holguín and D. Lardizabal-Gutiérrez, A proposal based on quantum phenomena for the ORR mechanism on nitrogen-doped carbon-based electrocatalysts, Int. J. Hydrogen Energy, 2019, 44, 12374–12380 CrossRef CAS , XVII Mexican Hydrogen Society Congress Special Issue..
- G. Zhu, F. Liu, Y. Wang, Z. Wei and W. Wang, Systematic exploration of N, C coordination effects on the ORR performance of Mn–Nx doped graphene catalysts based on DFT calculations, Phys. Chem. Chem. Phys., 2019, 21, 12826–12836 RSC.
- Y. Sun, J. Wang, Q. Liu, M. Xia, Y. Tang, F. Gao, Y. Hou, J. Tse and Y. Zhao, Itinerant ferromagnetic half metallic cobalt–iron couples: promising bifunctional electrocatalysts for ORR and OER, J. Mater. Chem. A, 2019, 7, 27175–27185 RSC.
- A. S. Haile, W. Yohannes and Y. S. Mekonnen, Oxygen reduction reaction on Pt-skin Pt3V(111) fuel cell cathode: a densoty functional theory study, RSC Adv., 2020, 10, 27346 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1se01654g |
| This journal is © The Royal Society of Chemistry 2022 |