
Honeycomb-like carbon with doping of a transition-metal and nitrogen for highly efficient zinc–air battery and zinc-ion battery†
Qian
Huang‡
a,
Shuxian
Zhuang‡
b,
Xin
You
b,
Jinpeng
Zhang
a,
Ao
Xie
b,
Yu
Chen
c,
Yang
Tang
 *a,
Yongmei
Chen
b,
Mingfei
Shao
d,
Xiao Jin
Yang
b and
Pingyu
Wan
*b
*a,
Yongmei
Chen
b,
Mingfei
Shao
d,
Xiao Jin
Yang
b and
Pingyu
Wan
*b
aInstitute of Applied Electrochemistry, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: tangyang@mail.buct.edu.cn
bNational Fundamental Research Laboratory of New Hazardous Chemicals Assessment & Accident Analysis, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: pywan@mail.buct.edu.cn
cInstitute of Future Technology, Ningbo Ronbay New Energy Technology Co., Ltd., Ningbo 315000, China
dState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China
First published on 15th November 2021
Abstract
The aqueous zinc-based batteries such as the Zn–air battery and the Zn-ion battery, featured with low-cost, high safety, high specific capacity and environmental-friendliness, have attracted intensive interest for their energy storage capability. For high performance zinc-based batteries, electrocatalysts with good conductivity, convenient mass transfer path and adequate accessible active sites are required. Herein, the fabrication of transition-metal/nitrogen co-doped honeycomb-like carbon materials with a hierarchical porous structure using a freeze-drying and one-step pyrolysis method is described. The as-prepared FeZnN–C is used as the cathode material of the zinc–air battery, which displays a high peak power density of 257 mW cm−2, a superior discharging voltage platform of about 1.36 V and a large specific capacity of 785 mA h gZn−1. The prepared Zn/ZnN–C was used as the anode of a Zn-ion battery which exhibits great cycle stability and a high charging/discharge rate. This work provides a novel and simple method for the construction of highly efficient electrocatalysts with hierarchical macro–meso–micro-porous structure and multi-active sites, which could be used for potential applications involving aqueous zinc-based batteries.
1 Introduction
The environmental pollution and resource depletion caused by fossil fuel dominated energy systems are becoming increasingly serious.1,2 The urgent requirement for high-efficiency, low-cost and environmental-friendly energy conversion and storage technologies have generated the investigation of electrochemical devices, such as electrolysis cells,3 fuel cells,4 batteries,5,6 supercapacitors7 and so on.8 Recently, aqueous zinc-based batteries, such as a Zn–air battery9,10 and a Zn-ion battery,11 have attracted increased attention due to their advantages of high specific capacity of zinc (820 mA h g−1), low reduction potential (−0.76 V vs. RHE), high stability, low cost (3 $ per kg) and good safety.12 For the Zn–air battery, high-cost, scarce reserves and poor-durability of the precious metal catalysts (such as platinum (Pt)-based materials)13 and especially the sluggish kinetics of the oxygen reduction reaction (ORR),14 have seriously hindered its performance and commercial applications.13,15 Specific to the Zn-ion battery, the primary problems of dendrite formation and side reactions (Zn corrosion and hydrogen evolution reaction) during charging and discharging cycles result in the low utilisation efficiency of the Zn anode, poor cycle life and short circuiting of batteries.16,17Currently, the transition metals (TM = Co, Cu, Fe, Ni, and Zn) and heteroatoms (X = B, F, N, P, S) of the co-doped carbon material (TMX-C) featuring low-cost, comparable catalytic activity and stability have been regarded as one of the most promising alternatives to Pt-based materials for ORR, especially in an alkaline medium.18–20 Among numerous TMX-Cs reported in the past decades, the FeNC-based electrocatalysts have attracted the most intensive study because of their good ORR performance.21,22 The high temperature pyrolysis of the TM, nitrogen and carbon precursors is the most common method to prepare TMN-Cs. However, the Fe atoms and carbon-based frameworks always aggregate during the high-temperature process, which decreases the active sites and hinders the porous structure formation, respectively.23 To improve the actual performance of the FeNC-based materials, it is essential to adopt two strategies: (1) fabricating of a hierarchical micro–meso–macro-porous structure with a large specific surface area and good conductivity to improve the apparent activity by increasing accessible active sites, optimizing electron conduction and providing a convenient path for electrolyte and oxygen diffusion,24,25 and (2) introducing multiple metal components to further adjust the electron state and O2 adsorption model, improve intrinsic activity and selectivity.26 It is disclosed that the coordination of multiple metals [(Cu, Fe)–N–CNT,27 (Fe, Co)/N–C,28 Ni–Co–Zn–N–PC29 and Fe–Zn–SA/NC30] and nitrogen atoms is conducive to easily breaking the O–O bonds.
In order to construct and maintain the highly porous structure, conventional templates (SiO2,31 MgO,32 F127 (ref. 33) and P123 (ref. 34)) are commonly used, which inevitably involve a high cost, use of toxic regents or tedious template-removal steps.35 In contrast, using NaCl as a low-cost, highly water soluble and chemically stable inorganic salt is convenient to operate, and it is easy to remove and recycle high purity NaCl by simple recrystallisation.36 Therefore, using crystalline NaCl as a template is a simple and environmental-friendly way to construct a porous structure. However, according to previous research,37 the porous structure carbon prepared by a method of rapid freezing of NaCl mainly consists of macro-pores, with only a few micro- and meso-pores. Wang et al.38 mixed poly(vinylpyrrolidone) and NaCl followed by rapid freezing, and then ground the freeze-dried mixtures with zinc chloride (ZnCl2). The mixture was pyrolyzed at 900 °C and then annealed again at 900 °C after washing the pyrolysis product with H2SO4. In addition to the macro-pores created by NaCl, as expected, the ZnCl2 enters the carbon framework during the pyrolysis and sublimation process because of capillary action, simultaneously forming a meso–micro-pore structure. However, this process is rather complicated not only because of the additional step to introduce Zn but also because of the second high temperature treatment.
In this work, honeycomb-like hierarchically porous FeZnN–C electrocatalysts were fabricated by freeze-drying precursors of carbon, nitrogen and metals, followed by a one-step pyrolysis process, in which the ZnCl2 and NaCl act as a co-template. The NaCl provides a framework for the precursor molecules and creates a macro-porous structure, whereas the ZnCl2 acts as a chemical activator to introduce abundant micro- and meso-pores and as a metal precursor to fabricate a Zn modified FeN–C material. The prepared FeZnN–C shows an excellent ORR performance with a high half-wave potential (E1/2) of 0.901 V (vs. RHE) and a diffusion-limiting current density (JL) of −6.04 mA cm−2, an extremely low average peroxide yield of only 1.25% and a high electron-transfer number (n) up to 3.97, which were superior to those of a commercial Pt/C catalyst (E1/2 of 0.851 V, JL of −5.27 mA cm−2, a H2O2 yield of 2.85% and a n of 3.94). The zinc–air battery using FeZnN–C as an electrocatalyst for cathode displays a high peak power density of 257 mW cm−2, high discharging potential of 1.36 V at 10 mA cm−2 and a large specific capacity of 785 mA h gZn−1. Moreover, the ZnN–C prepared by a similar method is further used to fabricate Zn/ZnN–C as a dendrite-free Zn containing anode for the advanced zinc-ion battery, which exhibits a high cycling stability and a high charging/discharging rate.
2 Experimental
2.1 Fabrication of catalysts
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50:50 were added. A uniform orange solution was obtained after adding 1.5 g of ZnCl2 under heating and stirring. This orange solution was fast frozen by liquid nitrogen and then placed in a lyophilizer for 24 h. The light-yellow powder obtained was placed in a porcelain boat and pyrolyzed at 550 °C for 1 h and then pyrolyzed at 900 °C for 2 h. The heating rate for both pyrolysis temperatures was 5 °C min−1 under an Ar atmosphere, meanwhile 1 M KOH solution was used to absorb the exhaust gas produced during the pyrolysis process. After cooling down to room temperature, the annealed product was etched with 0.5 M H2SO4 for 6 h followed by washing with ultrapure water until the pH was neutral. The black powder obtained was dried in a vacuum drying oven at 70 °C for 12 h, and the as-prepared sample was designated as FeZnN–C. Besides, the N–C, FeN–C and ZnN–C were prepared by a similar method without FeCl3 and ZnCl2, and with FeCl3 and with ZnCl2, respectively.
:1, then 1 mL of ethanol solution (1:1 in volume) was added. The mixture was stirred vigorously, pressed by a tablet machine into a catalyst flake with a thickness of 200 μm, and then laminated on a copper mesh and pressed tightly by a tablet press at 3 MPa. The Zn was electrodeposited on this catalytic flake at −10 mA cm−2 for 1 h in a 2 M ZnSO4 solution to obtain Zn/ZnN–C and this catalyst layer was used as a working electrode, with a commercial Zn plate as a counter electrode and a saturated calomel electrode (SCE) as a reference electrode.
:50:50 were added. A uniform orange solution was obtained after adding 1.5 g of ZnCl2 under heating and stirring. This orange solution was fast frozen by liquid nitrogen and then placed in a lyophilizer for 24 h. The light-yellow powder obtained was placed in a porcelain boat and pyrolyzed at 550 °C for 1 h and then pyrolyzed at 900 °C for 2 h. The heating rate for both pyrolysis temperatures was 5 °C min−1 under an Ar atmosphere, meanwhile 1 M KOH solution was used to absorb the exhaust gas produced during the pyrolysis process. After cooling down to room temperature, the annealed product was etched with 0.5 M H2SO4 for 6 h followed by washing with ultrapure water until the pH was neutral. The black powder obtained was dried in a vacuum drying oven at 70 °C for 12 h, and the as-prepared sample was designated as FeZnN–C. Besides, the N–C, FeN–C and ZnN–C were prepared by a similar method without FeCl3 and ZnCl2, and with FeCl3 and with ZnCl2, respectively.
:1, then 1 mL of ethanol solution (1:1 in volume) was added. The mixture was stirred vigorously, pressed by a tablet machine into a catalyst flake with a thickness of 200 μm, and then laminated on a copper mesh and pressed tightly by a tablet press at 3 MPa. The Zn was electrodeposited on this catalytic flake at −10 mA cm−2 for 1 h in a 2 M ZnSO4 solution to obtain Zn/ZnN–C and this catalyst layer was used as a working electrode, with a commercial Zn plate as a counter electrode and a saturated calomel electrode (SCE) as a reference electrode.
2.2 Characterisation
Scanning electron microscopy (SEM) measurements were performed on a Zeiss SUPRA 55 instrument. High-resolution transmission electron microscopy (HRTEM) measurements were carried out on a Jeol JEM-2100 electron microscope. The X-ray diffraction (XRD) patterns were examined with a Bruker D8 Advance diffractometer with Cu Kα radiation (hυ = 1486.6 eV) from 5°–90° at a scanning rate of 5° min−1. The X-ray photoelectron spectroscopy (XPS) measurement was carried out on an ThermoFisher ESCALAB 250Xi XPS spectrometer with a monochromated Al Kα X-ray source. The Raman spectra were measured using a Renishaw inVia Reflex spectroscope with a 532 nm laser. The adsorption and desorption isotherms of nitrogen was measured using a Quantachrome QUADRASORB evo/SI fully automatic specific surface area and pore size analyzer on a QUADRASORB evo/SI. The specific surface area and pore size distribution of the catalysts were determined by the BET and DFT methods.2.3 Electrochemical measurements
All the electrochemical measurements were carried out on a Pine Research electrochemical workstation at room temperature in a typical three-electrode system at 27 °C in O2- or Ar-saturated 0.1 M KOH solution with a SCE as a reference electrode, a Pt wire as the counter electrode and a rotating ring disk electrode (RRDE) as a working electrode in which the ring was a Pt electrode and always retained 0.2 V (vs. SCE) during testing. A portion (2.5 mg) of the prepared catalyst was dispersed in a mixed solution of 200 μL of absolute ethanol, 200 μL of 0.5 wt% Nafion and 100 μL of N,N-dimethylformamide (DMF), and then ultrasonically mixed for 30 min to obtain a uniform ink. A portion (10 μL) of the prepared ink was dripped onto the RRDE (5.56 mm in diameter and 0.25 cm2 in area), on which the catalyst loading was about 200 μg cm−2. All the potentials mentioned in this work are transformed into reversible hydrogen electrode scale (RHE). E(RHE) = E(SCE) + 0.2414 V + 0.0592 × pH = E(SCE) + 1.011 V. The measurements were carried out with 80% iR-compensation.The electron transfer number (n) and hydrogen peroxide yield (H2O2%) at various potentials were calculated as follows:
| n = 4Id/(Id + Ir/N) |
| H2O2% = (200Ir/N)/(Id + Ir/N) |
2.4 Assembly of zinc-based batteries
:water (1:1 volume) under vigorous stirring. The product was then placed in a tablet press and pressed into a uniform flake with a thickness of 200 μm. Finally, it was calcined in a muffle furnace at 130 °C for 40 min and then cooled to room temperature. The gas diffusion layer was prepared by adding 400 mg of acetylene black and 300 mg of ammonium nitrate (NH4NO3) in a mixed solution of 800 mg PTFE emulsion and 2.5 mg ethanol solution (1:1 in volume) under vigorous stirring. The product was then placed in a tablet press and pressed into a uniform flake with a thickness of 300 μm. Finally, it was calcined in a muffle furnace at 320 °C for 40 min and then cooled to room temperature. The zinc–air battery was assembled using a commercial Zn plate as the anode (2 × 2 cm2) and the prepared FeZnN–C as an oxygen diffusion cathode (2 × 2 cm2) which consisted of a catalytic layer and a diffusion layer separately pressed onto the two sides of a Ni foam at 10 MPa. The Pt/C oxygen diffusion cathode (2 × 2 cm2) was prepared by coating a commercial Pt/C with a loading of 1 mg cm−2. The discharging performance was tested in 6 M KOH.
3 Results and discussion
3.1 Physical characterisation of FeZnN–C electrocatalysts
The honeycomb-like hierarchically porous FeZnN–C was prepared with a simple one-step pyrolysis method after rapid freeze-drying as shown in Scheme 1 by using glucose and DCDA as carbon and nitrogen sources, respectively, with FeCl3·6H2O as metal source and NaCl–ZnCl2 as co-template to create a porous structure. Similarly, three other electrocatalysts were also prepared without adding both FeCl3 and ZnCl2, and without adding either of them, denoted as N–C, FeN–C and ZnN–C. The SEM images of these four materials are shown in Fig. S1 (see ESI†). All of the images display honeycomb-like porous carbon frames, which are attributed to the carbonisation of the salt crystallite precursor used as a template during pyrolysis. The EDX mapping image of FeZnN–C is shown in Fig. S2 (see ESI†), and shows the uniform distribution of Fe, Zn, N and C elements on the surface. The normalised mass contents were 0.04% for Fe, 1.01% for Zn, 52.18% for N and 46.77% for C. It was observed that FeZnN–C and ZnN–C (Fig. S1b and f, see ESI†) prepared with the addition of ZnCl2 possessed many more pores on the ultra-thin carbon wall in comparison to the smooth surface of the carbon wall of FeN–C and N–C (Fig. S1d and h, see ESI†) without ZnCl2. As verified by the TEM images in Fig. 1a, b, d and e, FeZnN–C exhibits more abundant crosslinked pores than FeN–C. These comparisons reveal that the addition of ZnCl2 would produce more meso-/macro-pores on the carbon wall, further enlarging the specific surface area, creating more defect sites and providing more convenient mass-transfer paths. The comparison between ZnN–C and N–C (Fig. S1e–h, see ESI†) also confirmed the pore-forming effect of ZnCl2 as a chemical activator. Fig. 1c and f show the magnified cross section of the carbon walls of FeZnN–C and FeN–C, respectively. Both samples exhibit a discontinuous carbon lattice with a distance of about 0.34 nm and abundant defects on the ultra-thin and curved carbon walls.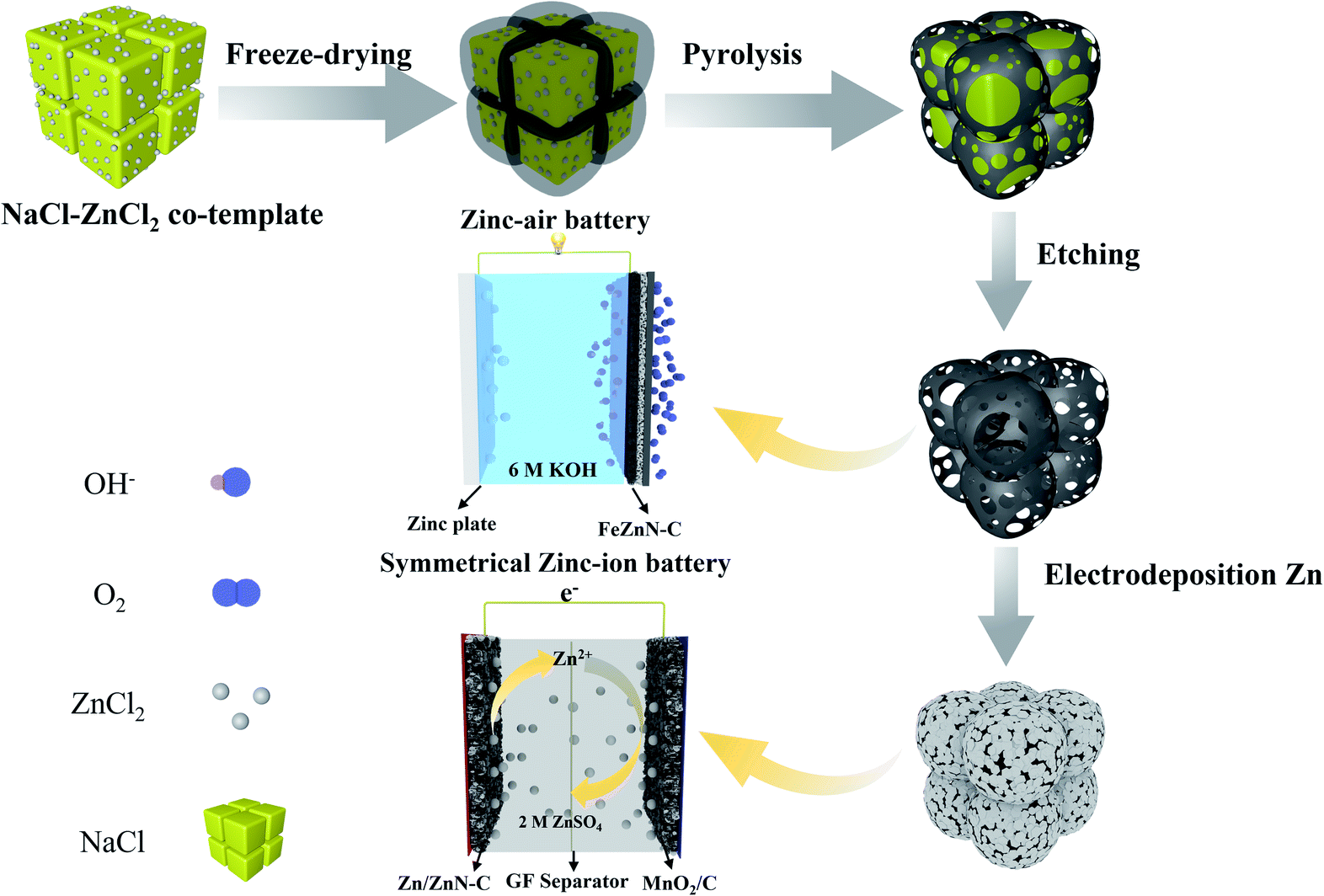 | ||
| Scheme 1 Schematic diagram of the synthetic route for honeycomb-like hierarchically porous carbon materials and their application in a zinc–air battery and a zinc-ion battery. | ||
 | ||
| Fig. 1 TEM and HRTEM images of (a–c) FeZnN–C and (d–f) FeN–C. | ||
The micro-crystal structure of carbon was further investigated by XRD and Raman spectroscopy. As shown in Fig. 2a, all these carbon materials exhibit similar and excessively broad diffraction peaks at 25° and 43°, belonging to the (0 0 2) and (1 0 0) planes of graphitic carbon, respectively, which was consistent with the unperfect carbon lattice with abundant defects in the HRTEM result. Moreover, FeZnN–C and ZnN–C exhibit relatively higher intensities at low angles below 10°, which can be attributed to the diffraction of micro-pores in the carbon matrix and indicate more micro-pores than those of FeN–C and N–C.39 The Raman spectra (Fig. 2b) of the four previously described materials show two peaks at about 1353 cm−1 and 1583 cm−1, which were attributed to the D band and the G band, respectively. The ID/IG values of FeZnN–C, ZnN–C, FeN–C and N–C were 0.93, 0.94, 0.96 and 0.98, respectively. The lower ID/IG values of FeZnN–C and ZnN–C proved that the presence of the Zn component improved the degree of graphitisation of the carbon structure, which would optimize electronic conductivity, and thus facilitate the ORR process.
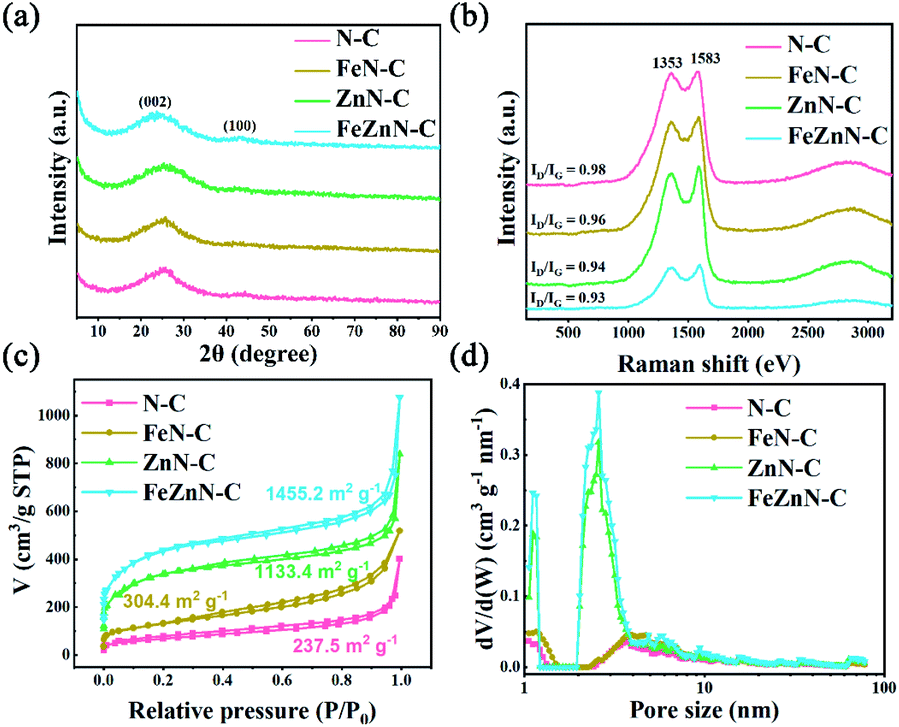 | ||
| Fig. 2 (a) The XRD patterns, (b) the Raman spectra, (c) the nitrogen adsorption–desorption isotherms, and (d) the corresponding pore size distribution of N–C, FeN–C, ZnN–C and FeZnN–C catalysts. | ||
The enriched porous structure benefitted from ZnCl2 was further confirmed by the N2 adsorption–desorption isotherms of the four catalysts (Fig. 2c). All these electrocatalysts exhibit type IV isotherm curves with a high N2 uptake in the medium and high relative pressure areas, which indicated the existence of meso- and macro-pores. In the comparison with the FeN–C and N–C, FeZnN–C and ZnN–C showed a much sharper uptake in the low relative pressure (P/P0 < 0.05) area, which suggested there were plentiful micro-pores inside the materials prepared with the addition of ZnCl2. The BET specific surface areas calculated from the isotherms of FeZnN–C and ZnN–C were as large as 1455.2 m2 g−1 and 1134.4 m2 g−1, respectively, which were significantly increased from the 304.4 m2 g−1 and 237.5 m2 g−1 for FeN–C and N–C, respectively. It was calculated that the specific surface areas of the catalysts with the addition of ZnCl2 (FeZnN–C, ZnN–C) were approximately 4.7 times higher than the results for the catalysts without ZnCl2 (FeN–C, N–C), respectively. Fig. 2d shows the corresponding pore size distribution of the four materials. The FeZnN–C and ZnN–C possessed an obviously intensive pore size distribution in the range of 1–1.5 nm (micro-pores) and 2–4 nm (small meso-pores), suggesting that the addition of ZnCl2 could increase the number of smaller meso-pores and give extra micro-pores. It was inferred that the FeZnN–C and ZnN–C possessed simultaneously, a hierarchical porous structure with abundant micro-, meso- and macro-pores, in which the micro-pores provided a larger specific surface area to facilitate the exposure of active sites whereas the meso-/micro-pores promoted the transmission of electrolytes during the ORR process. It was expected that the unique architecture with pores of various sizes and a large specific surface area would contribute to the high electrocatalytic performance, especially the high diffusion-limiting current densities for FeZnN–C or ZnN–C.
The chemical states and composition of FeZnN–C, ZnN–C, FeN–C and N–C were investigated by XPS measurement. The XPS survey spectra for these samples are shown in Fig. 3a, and they verify the existence of C, N, O and Zn. The presence of a low content of Fe in FeZnN–C and FeN–C could be observed from magnification of the XPS spectra (Fig. S3a, see ESI†). The Zn mainly existed in the chemical state of Zn2+ as shown in Fig. S3b (see ESI†). Table S1 (see ESI†) and Fig. 3b show the atomic percentage of each element for the four catalysts. The FeZnN–C had a relatively high N content of 9.85% with 0.21% Fe and 0.60% Zn. The contents and chemical state of N and the metals have an important effect on the determination of the catalytic activity for the ORR. As shown in Fig. 3c, the N 1s spectrum of FeZnN–C was deconvoluted into five peaks, which were attributed to pyridinic-N (398.3 eV), metal-N (399.1 eV), pyrrolic-N (399.8 eV), graphitic-N (401 eV) and oxidized N (403 eV). According to the results given previous reports,40,41 pyridinic-N, graphitic-N and metal-N are three effective N species that have high ORR activity, whereas pyrrolic-N and oxidized N are ineffective N species. Thus, Fig. 3d and Table S1 (see ESI†) further compare the contents of different N species and the chemical states of these materials. Out of all the catalysts, the percentage of effective N species of FeZnN–C was the highest, not only with respect to all the nitrogen species (85.7%) but also with respect to all the elements' composition (8.44%). It was expected that among these materials, FeZnN–C would exhibit the highest ORR activity. In the comparison of these catalysts, it was observed that the introduction of ZnCl2 can not only enrich the porous structure, but also increase the content ratio of the effective nitrogen species, thereby simultaneously increasing the apparent and intrinsic catalytic activity.
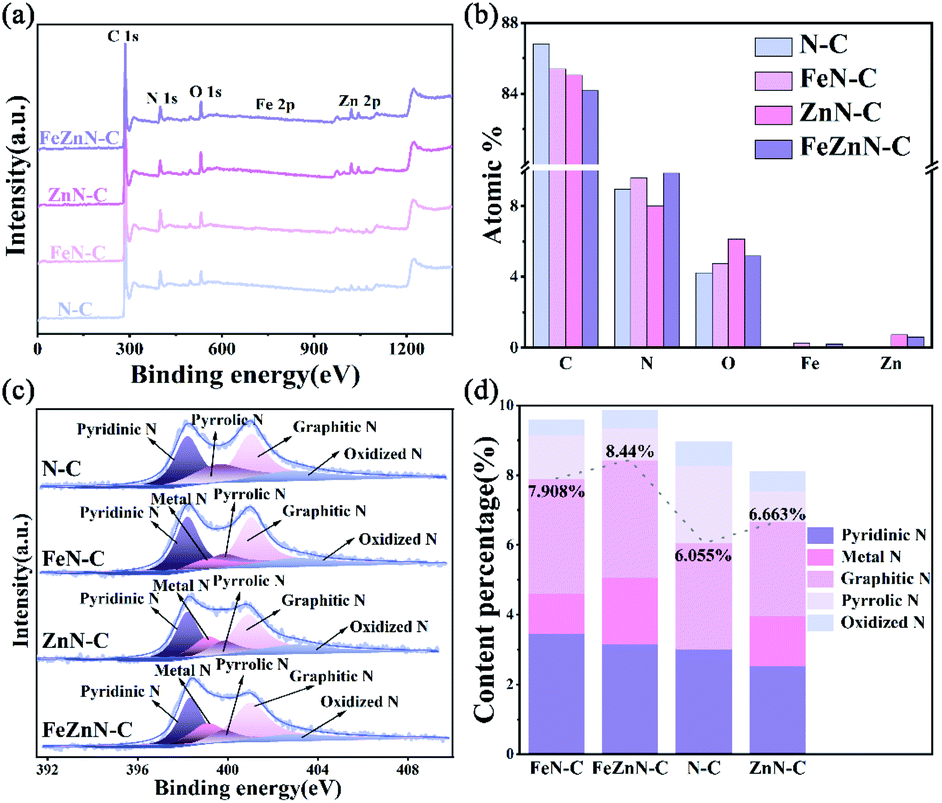 | ||
| Fig. 3 (a) The XPS survey spectra, (b) the atomic percentage content of different elements (C, N, O, Fe, Zn), (c) the N 1s spectra, and (d) the atomic percentage content of different nitrogen species of N–C, FeN–C, ZnN–C and FeZnN–C. | ||
3.2 The ORR and zinc–air battery tests
Cyclic voltammetry (CV) measurements were carried out on a rotating ring disk electrode (RRDE) in O2-saturated 0.1 M KOH at a scan rate of 50 mV s−1 and the results are shown in Fig. 4a. All of the catalysts, FeZnN–C, ZnN–C, FeN–C, N–C and 20% Pt/C, displayed obvious ORR peaks, in which FeZnN–C showed the highest peak current, the most positive peak potential and the largest area surrounded by a CV loop. Fig. 4b shows the classical linear sweep voltammetry (LSV) curves of the five materials in O2-saturated 0.1 M KOH at a scan rate of 5 mV s−1 and a rotating speed of 1600 rpm. The corresponding half-wave potential (E1/2) and diffusion-limiting current density (JL) obtained from the LSV curves are shown in Fig. 4c. The FeZnN–C exhibited the highest E1/2 of 0.901 V and a JL of −6.04 mA cm−2, both of which were superior to those of ZnN–C (0.856 V, −5.72 mA cm−2), FeN–C (0.810 V, −5.16 mA cm−2), N–C (0.789 V, −4.51 mA cm−2) and even the 20% Pt/C (0.851 V, −5.27 mA cm−2). The excellent ORR performance of FeZnN–C could be attributed to accessible active sites, the large specific surface area and the high content of effective nitrogen species in the hierarchical macro–meso–micro-porous structure.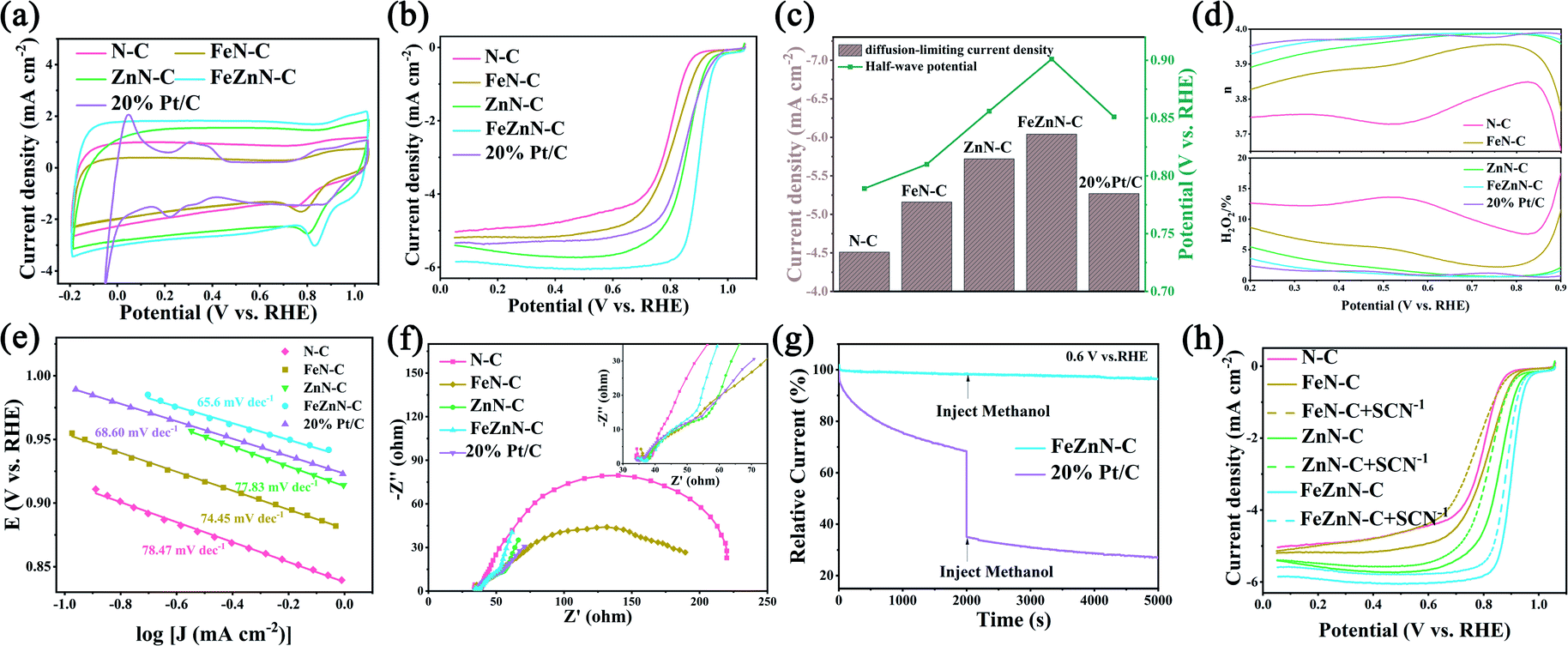 | ||
| Fig. 4 (a) The CV curves at a scan rate of 50 mV s−1, (b) the LSV curves at a scan rate of 5 mV s−1, (c) half-wave potentials and difussion-limiting current densities, (d) the electron transfer number and H2O2 yield, (e) the Tafel slopes, and (f) the electrochemical impedance spectroscopy spectra of N–C, FeN–C, ZnN–C, FeZnN–C and 20% Pt/C, measured at 0.811 V, (g) results of the methanol tolerance test of FeZnN–C and 20% Pt/C, and (h) the SCN− tolerance tests for the ORR performance of N–C, FeN–C, ZnN–C and FeZnN–C. | ||
To clarify the ORR kinetics, the LSV curves of FeZnN–C and 20% Pt/C at different rotating speeds were measured and the results are shown in Fig. S4a and b (see ESI†). Fig. S4c (see ESI†) shows the corresponding Koutecký–Levich curves obtained at 0.4 V and 0.6 V, from which it was derived that FeZnN–C undergoes a four-electron ORR process, similar to 20% Pt/C. As shown in Fig. 4d, both FeZnN–C and ZnN–C exhibited a low yield of H2O2 produced by a two-electron process, and their electron transfer numbers (n) were very close to 4. In particular, the H2O2 yield on FeZnN–C was as low as 1.25% and the average n was 3.97 in the potential range of 0.2–0.9 V. It was observed that the ORR performance of FeZnN–C surpassed that of 20% Pt/C (H2O2 yield of 2.85%, n of 3.94) and most of the non-noble metal electrocatalysts reported previously (Table S2, see ESI†).
As shown in Fig. 4e, the Tafel slopes of FeZnN–C, ZnN–C, FeN–C, N–C and 20% Pt/C were 65.6 mV dec−1, 77.8 mV dec−1, 74.5 mV dec−1, 78.5 mV dec−1 and 68.6 mV dec−1, respectively. The smallest Tafel slope of FeZnN–C indicated that the rate-determining step of the ORR process was the second electron transfer step and this demonstrated the optimal intrinsic catalytic activity.42Fig. 4f shows the electrochemical impedance spectroscopy (EIS) of the five catalysts at 0.811 V. In the high frequency area, the same initial value represents the solution resistance (Rs) of 42 Ω for all the materials in the same test system. In the low frequency area, the diameter of the semicircle represents the charge transfer resistance (Rct). The lowest Rct of FeZnN–C indicated that it had the best electronic conductivity of ORR among all the materials. Both the smallest Tafel slope and lowest Rct show the improved ORR kinetics obtained with FeZnN–C.
On the other hand, the double-layer capacitance (Cdl) was also measured by CV in the non-Faraday response region at different scan rates (Fig. S5a–d, see ESI†) to evaluate the electrochemical surface area (ECSA) of these catalysts. The Cdl for FeZnN–C, ZnN–C, FeN–C and N–C were 20.60 mF cm−2, 15.97 mF cm−2, 12.52 mF cm−2 and 10.80 mF cm−2 (Fig. S5e, see ESI†), and the corresponding ECSA estimated from the Cdl were 515 cm2, 399 cm2, 313 cm2 and 270 cm2, respectively. The largest Cdl and ECSA values of FeZnN–C also revealed the largest charge storage capacity and specific surface area, which would provide the most sites of activation, thus presenting the highest apparent catalytic activity towards the ORR. The ECSA normalised LSV curves of N–C, FeN–C, ZnN–C and FeZnN–C in the electrochemical polarization-controlled region are shown in Fig. S6 (see ESI†), which verified that FeZnN–C showed the best intrinsic ORR activity. Furthermore, the methanol tolerance tests and thiocyanate (SCN−) poison tests were also performed, and the results are shown in Fig. 4g and h, respectively. The relative ORR current at 0.6 V on a 20% Pt/C electrode decreased sharply and immediately when methanol was added to the electrolyte. In contrast, there was almost no decay for the relative ORR current on FeZnN–C, which showed its higher resistance to methanol poisoning than that of 20% Pt/C (Fig. 4g), and this means that it is a promising ORR catalyst for use in fuel cells. SCN− is always used to evaluate the active sites of TMN-C because most non-noble metals are susceptible to SCN−. The results of the SCN− poison test showed that the E1/2 of FeZnN–C, ZnN–C and FeN–C negatively shifted by 21 mV, 38 mV and 44 mV, respectively, indicating that both Fe- and Zn-containing sites in FeZnN–C were simultaneously effective for ORR.
A zinc–air battery was assembled using the prepared FeZnN–C as an electrocatalyst for an oxygen diffusion cathode (2 × 2 cm2) with a commercial Zn plate (2 × 2 cm2) as anode and 6 M KOH as electrolyte. A similar zinc–air battery with commercial Pt/C as a cathodic electrocatalyst was also assembled for comparison. The polarization and power density curves of these two zinc–air batteries are shown in Fig. 5a. It was observed that the FeZnN–C battery achieved a peak power density of 257 mW cm−2 at a current density of 310 mA cm−2, which was superior to that of the Pt/C battery (191 mW cm−2 at 248 mA cm−2). Table S3 (see ESI†) also shows that FeZnN–C was much better than most of the previously reported ORR catalysts. Fig. 5b shows the discharge voltages of these two batteries at different constant current densities of 1 mA cm−2, 10 mA cm−2, 50 mA cm−2 and 100 mA cm−2. The cell voltage of the FeZnN–C battery was 60–70 mV higher than that of Pt/C at same current densities (Table S4, see ESI†). Typically, at 10 mA cm−2, the discharge voltage of the FeZnN–C battery could achieve 1.36 V with a specific energy density of 785 mA h gZn−1, which was higher than that of the Pt/C battery (1.30 V with 698 mA h g−1) as shown in Fig. 5c. In addition, the FeZnN–C battery only had a slight decline in discharge voltage at 10 mA cm−2 during each 30 h cycle with a total of six cycles in the durability test during which the zinc plate was replaced regularly, indicating the great durability of FeZnN–C. In contrast, the discharge voltage of the Pt/C battery was not only lower than that of the FeZnN–C battery, but also obviously fluctuated during the durability test with six cycles at the current density of 10 mA cm−2, which indicated that the FeZnN–C battery had better durability than the Pt/C one. These zinc–air battery tests strongly verified that FeZnN–C was a promising electrocatalyst for improving discharge voltage, power density and stability.
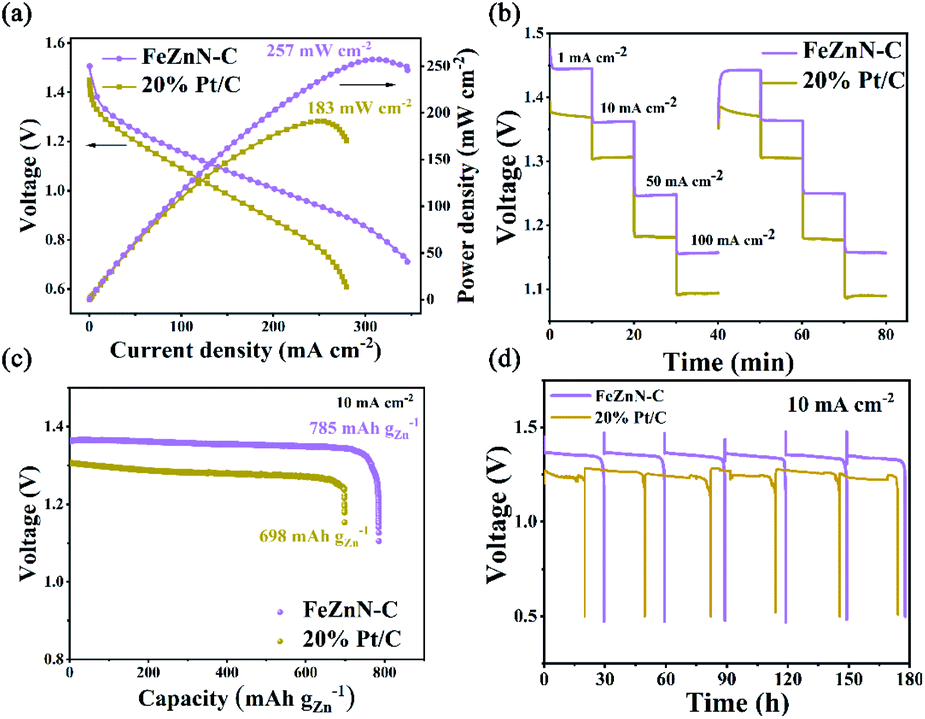 | ||
| Fig. 5 (a) Polarization curves and the corresponding power density plots, (b) discharge voltages at multi-current densities (1, 10, 50 and 100 mA cm−2), (c) discharge curves at 10 mA cm−2, and (d) constant galvanostatic discharge (CGD) curves at 10 mA cm−2 for Zn–air batteries employing FeZnN–C and 20% Pt/C as the cathodic catalysts. | ||
3.3 Zinc-ion battery test
For rechargeable zinc-based batteries, the limited cycle life and low utilization efficiency of the zinc anodes hinder their practical application. This section describes how the Zn and N co-doped ZnN–C was fabricated by a similar method to that used for FeZnN–C. The effect of preparation temperature was firstly evaluated so that an optimized zinc-ion battery with high efficiency could be constructed. The ZnN–Cx prepared at different calcination temperatures were employed as anode catalysts in the assembly of the series coin cell ZnN–Cx‖Zn plate. As shown in Fig. S7a (see ESI†), the coulombic efficiency (CE) of the ZnN–C550‖Zn plate was 99.76% after 100-cycles of plating/stripping tests at 1 mA cm−2 to 1 mA h cm−2, and the CE gradually decreased to 98.97% and 97.62% with temperature increases to 650 °C and 700 °C, respectively. In contrast, the ZnN–C800 and ZnN–C900 batteries had a short circuit after 70 cycles of testing because of the formation of dendritic Zn in the cell. Moreover, the CEs of them were almost 0 which means that the zinc affinity of ZnN–C800 and ZnN–C900 were very poor and Zn metal could not be deposited on their surface under these conditions. Fig. S7b (see ESI†) shows the charging and discharging of various coin cells based on ZnN–Cx. At the 50th cycle, the difference between the discharging voltage and the charging voltage (voltage gap) of ZnN–C550 was 51.4 mV. The smallest voltage gap achieved was 43.8 mV on ZnN–C650. Afterwards, as the preparation temperature increased, the charge–discharge voltage difference continuously increased. Therefore, 650 °C was found to be the optimal temperature for the anode catalyst in a zinc-ion battery.Therefore, the ZnN–C prepared at 650 °C, featuring a honeycomb-like hierarchically porous structure, a low voltage gap, a high CE and great Zn affinity, was used as the substrate for zinc deposition. The Zn/ZnN–C catalyst was obtained with an area capacity of 10 mA h cm−2, and the SEM images and schematic diagram are shown in Fig. 6a–c. It can be seen that the surface of the ZnN–C becomes uniform and flat, and notably no dendritic Zn was observed on the Zn/ZnN–C surface even for the high area capacity electrode. Two symmetrical coin cells, Zn/ZnN–C‖Zn/ZnN–C and Zn plate‖Zn plate, were assembled and tested in 2 M ZnSO4 to examine the zinc plating/stripping reversibility of Zn/ZnN–C and commercial Zn plate. The zinc-ion battery performances using Zn/ZnN–C and commercial Zn plate as zinc anodes were firstly tested at the same current density of 0.5 mA cm−2 (Fig. 6d). The polarisation voltage of Zn plate‖Zn plate was 45.3 mV and 92.7 mV in the first and 50th cycle, respectively (Fig. 6e and f). Comparatively, that of Zn/ZnN–C‖Zn/ZnN–C was as low as 15.5 mV in the first cycle (Fig. 6e), and only slightly increased to 18 mV after 50 cycles (Fig. 6f) and to 35 mV after 300 cycles (Fig. 6g). Moreover, the durability of the Zn/ZnN–C‖Zn/ZnN–C was much longer than that of Zn plate‖Zn plate. Short circuiting and large fluctuations of voltage did not happen to the former cell. The large specific surface area with the abundant active sites of ZnN–C containing Zn–N would provide sufficient sites for zinc deposition,43 which was conducive to the formation of a uniform and flat Zn layer, which further improving the cell performance. At the same time, the abundant porous structure of ZnN–C facilitated mass transfer and avoided the formation of Zn dendrites during the plating/stripping process, thereby ensuring the stability of the symmetrical coin cells.
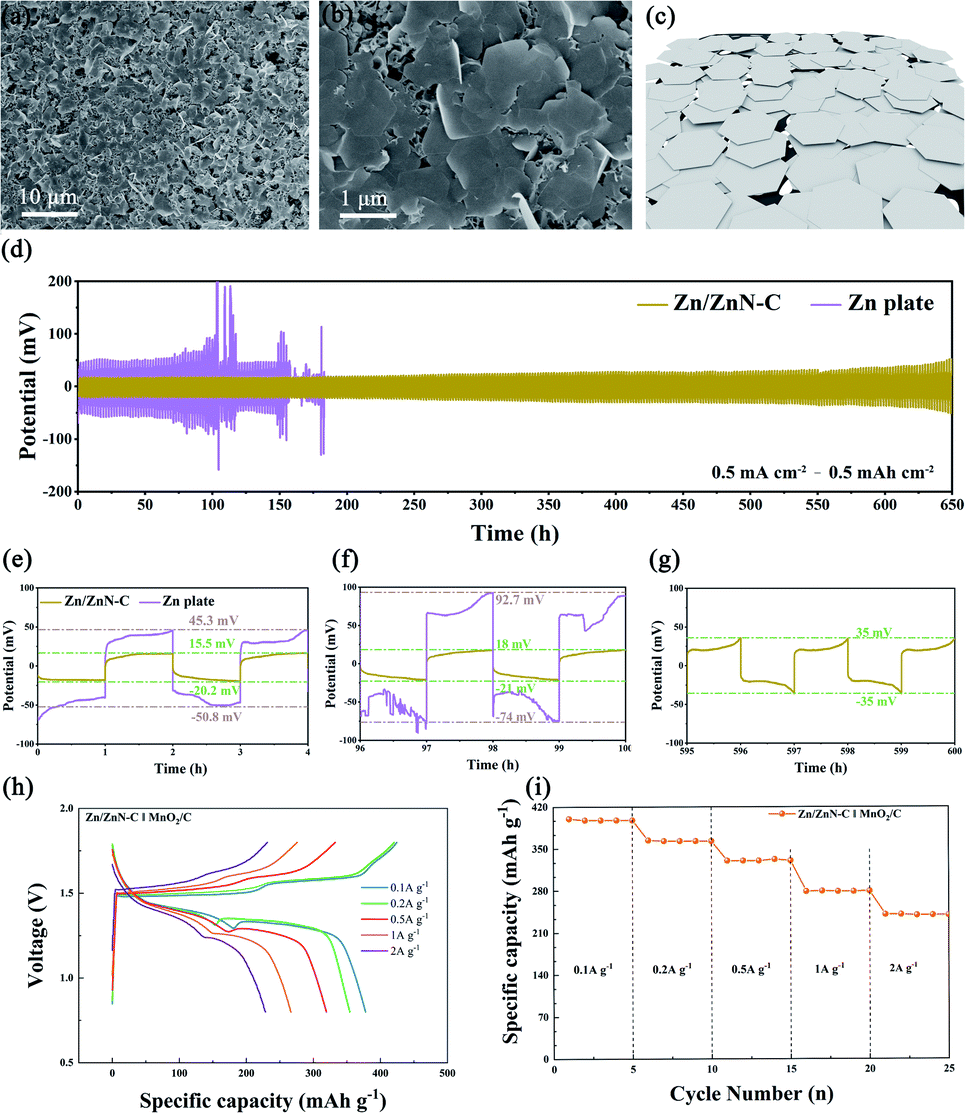 | ||
| Fig. 6 (a and b) The SEM images of Zn deposition on ZnN–C at 10 mA cm−2 to 10 mA h cm−2, (c) a schematic diagram of Zn/ZnN–C, (d) the electrochemical stability (Zn plating–stripping) tests of symmetrical cells based on Zn/ZnN–C and Zn plate at 0.5 mA cm−2 to 0.5 mA h cm−2, and (e–g) the amplified images at different cycles, (h) the GCD curves, and (i) the rate capability of a Zn/ZnN–C‖MnO2/C battery at different current densities (0.1, 0.2, 0.5, 1 and 2 A g−1). | ||
The rate performances of the two batteries described previously are shown in Fig. S8 (see ESI†). Compared with commercial Zn plate‖Zn plate, Zn/ZnN–C‖Zn/ZnN–C exhibited a better rate performance at all the current densities of 0.5, 1, 2, 5 and 10 mA cm−2, which was reflected in the low polarization voltage. Moreover, the Zn/ZnN–C‖Zn/ZnN–C showed a much more stable charging/discharging voltage than commercial Zn plate‖Zn plate. Fig. S9 (see ESI†) further showed that Zn/ZnN–C‖Zn/ZnN–C was superior to Zn plate‖Zn plate in the long-term charging/discharging process at different current densities – area capacities of 1 mA cm−2 to 1 mA h cm−2, 2 mA cm−2 to 2 mA h cm−2 and 5 mA cm−2 to 2 mA h cm−2. The Zn plate‖Zn plate showed a large vibration of voltage after dozens of hours of testing, suggesting that it suffers from short-circuiting, whereas the prepared Zn/ZnN–C‖Zn/ZnN–C displayed a more stable charging and discharging voltage in addition to the lower polarization voltage. Moreover, the Zn/ZnN–C still maintained a uniform and flat surface after repeated Zn stripping/deposition processes, because of the porous structure which is beneficial to mass transfer (Fig. S10a and b, see ESI†). On the other hand, there are dense and sharp Zn dendrites on the surface of the commercial Zn plate (Fig. S10c and d, see ESI†) after a long-term test with 50 cycles at a current density – area capacity of 1 mA cm−2 to 1 mA h cm−2.
Furthermore, the zinc-ion battery employing Zn/ZnN–C as the anode and MnO2/C as the cathode was assembled. The battery performance was evaluated in 2 M ZnSO4 to 0.2 M MnSO4. As shown in Fig. 6h, the Zn/ZnN–C‖MnO2/C battery exhibited typical charging and discharging curves with the platform voltage at 1.33–1.39 V and 1.50–1.60 V, respectively. The Zn/ZnN–C‖MnO2/C battery delivered discharge capacities of 397.7, 362.8, 330.2, 279.1 and 279.5 mA h g−1 at different current densities (0.1, 0.2, 0.5, 1 and 2 A g−1), respectively, displaying its good rate capability. The specific capacity of this battery hardly declined during each test at various current densities as shown in Fig. 6i. These results demonstrate that the Zn/ZnN–C is a highly efficient, dendrite-free anode.
4 Conclusions
In conclusion, the transition-metal/nitrogen co-doped honeycomb-like carbon materials with hierarchical porous structure were successfully synthesized using a simple freeze-drying and one-step pyrolysis method. As a co-template, ZnCl2 and NaCl created abundant macro–meso–micro pores which provided a convenient path for mass and electron transfer, which exposed more active sites. Furthermore, because of the high effective N content and the abundant Fe/Zn bi-active sites, the as-prepared FeZnN–C shows an excellent ORR performance with a high half-wave potential of 0.901 V and a limiting current density of −6.04 mA cm−2, which were superior to those of commercial Pt/C (0.851 V and −5.27 mA cm−2). The zinc–air battery using FeZnN–C as a cathodic electrocatalyst displayed a high peak power density of 257 mW cm−2, a superior discharging voltage platform of about 1.36 V and a large capacity of 785 mA h gZn−1. Moreover, the prepared Zn/ZnN–C possessing a hierarchical porous structure could effectively avoid the formation of Zn dendrites, therefore the zinc-ion battery employing Zn/ZnN–C exhibited a high cycle stability and charging/discharge rate.Author contributions
Qian Huang: methodology, formal analysis, investigation, data curation and writing – original draft, Shuxian Zhuang: methodology, formal analysis, investigation and writing – review and editing, Jinpeng Zhang: visualisation, Xin You: writing – review and editing, Ao Xie: methodology and investigation, Yu Chen: methodology and investigation, Yang Tang: conceptualisation, writing – review and editing, project administration and funding acquisition, Yongmei Chen: supervision and funding acquisition, Mingfei Shao: validation, Xiao Jin Yang: validation and supervision, Pingyu Wan: conceptualisation, supervision and project administration.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors greatly appreciate the financial support from the National Natural Science Foundation of China (No. 22075012, 21506010) and the Zhongguancun Frontier Technology and Industrial Service Alliance. The authors would like to thank staff at the Anqing Research Institute of Beijing University of Chemical Technology for their help with XRD testing. The authors especially thank Prof. Xiaoguang Liu for important discussions and suggestions of this work.Notes and references
- F. J. Kelly and T. Zhu, Science, 2016, 352, 934 CrossRef CAS PubMed.
- L. Yue, H. Zhao, Z. Wu, J. Liang, S. Lu, G. Chen, S. Gao, B. Zhong, X. Guo and X. Sun, J. Mater. Chem. A, 2020, 8, 11493–11510 RSC.
- R. d'Amore-Domenech and T. J. Leo, ACS Sustainable Chem. Eng., 2019, 7, 8006–8022 CrossRef.
- X. Tian, X. F. Lu, B. Y. Xia and X. W. Lou, Joule, 2020, 4, 45–68 CrossRef CAS.
- H.-F. Wang and Q. Xu, Matter, 2019, 1, 565–595 CrossRef CAS.
- W. Zhao, X. Wang, X. Ma, L. Yue, Q. Liu, Y. Luo, Y. Liu, A. M. Asiri and X. Sun, J. Mater. Chem. A, 2021, 9, 15807–15819 RSC.
- J. Yin, W. Zhang, N. A. Alhebshi, N. Salah and H. N. Alshareef, Small Methods, 2020, 4, 1900853 CrossRef CAS.
- H. Sun, J. Zhu, D. Baumann, L. Peng, Y. Xu, I. Shakir, Y. Huang and X. Duan, Nat. Rev. Mater., 2019, 4, 45–60 CrossRef.
- H. Xu, D. Wang, P. Yang, A. Liu, R. Li, L. Xiao, J. Zhang, Z. Qu and M. An, Sustainable Energy Fuels, 2021, 5, 2695–2703 RSC.
- J. Ma, J. Li, R. Wang, Y. Yang, P. Yin, J. Mao, T. Ling and S. Qiao, Mater. Today Energy, 2021, 19, 100624 CrossRef CAS.
- J. S. Ko, M. B. Sassin, J. F. Parker, D. R. Rolison and J. W. Long, Sustainable Energy Fuels, 2018, 2, 626–636 RSC.
- Z. Yi, G. Chen, F. Hou, L. Wang and J. Liang, Adv. Energy Mater., 2021, 11, 2003065 CrossRef CAS.
- M. R. Karim, M. Ferrandon, S. Medina, E. Sture, N. Kariuki, D. J. Myers, E. F. Holby, P. Zelenay and T. Ahmed, ACS Appl. Energy Mater., 2020, 3, 9083–9088 CrossRef.
- L. Chen, Y. Chen, C. Xu, W. Wang, W. Fu, W. Hu, M. Zhou, B. He, Q. Chen, Z. Hou and W. Xu, Mater. Today Energy, 2021, 20, 100670 CrossRef CAS.
- R. Zhang, M. Tahir, S. Ding, M. A. Qadeer, H. Li, Q.-X. Zeng, R. Gao, L. Wang, X. Zhang, L. Pan and J.-J. Zou, ACS Appl. Energy Mater., 2019, 3, 2323–2330 CrossRef.
- W. Du, E. H. Ang, Y. Yang, Y. Zhang, M. Ye and C. C. Li, Energy Environ. Sci., 2020, 13, 3330–3360 RSC.
- G. Xu, Y. Zhang, Z. Gong, T. Lu and L. Pan, J. Colloid Interface Sci., 2021, 593, 417–423 CrossRef CAS.
- T. Sharifi, E. Gracia-Espino, A. Chen, G. Hu and T. Wågberg, Adv. Energy Mater., 2020, 10, 1902084 CrossRef CAS.
- A. Bin Yousaf, J. R. Monnier, J. W. Weidner, M. K. Hassan, S. J. Zaidi and P. Kasak, Sustainable Energy Fuels, 2020, 4, 5050–5060 RSC.
- L. Yue, J. Liang, Z. Wu, B. Zhong, Y. Luo, Q. Liu, T. Li, Q. Kong, Y. Liu, A. M. Asiri, X. Guo and X. Sun, J. Mater. Chem. A, 2021, 9, 11879–11907 RSC.
- J. Cao, X. Jia, M. Guo, Y. Du, J. Xu and Z. Chen, Sustainable Energy Fuels, 2018, 2, 169–174 RSC.
- C. Xu, L. Chen, Y. Wen, S. Qin, H. Li, Z. Hou, Z. Huang, H. Zhou and Y. Kuang, Mater. Today Energy, 2021, 21, 100721 CrossRef CAS.
- Z. X. Mao, M. J. Wang, L. Liu, L. Peng, S. Chen, L. Li, J. Li and Z. Wei, Chin. J. Catal., 2020, 41, 799–806 CrossRef CAS.
- W. Tian, H. Zhang, X. Duan, H. Sun, G. Shao and S. Wang, Adv. Funct. Mater., 2020, 30, 1909265 CrossRef CAS.
- Y. Wu, H. Zhao, Z. Wu, L. Yue, J. Liang, Q. Liu, Y. Luo, S. Gao, S. Lu, G. Chen, X. Shi, B. Zhong, X. Guo and X. Sun, Energy Storage Materials, 2021, 34, 483–507 CrossRef.
- Z. Lu, B. Wang, Y. Hu, W. Liu, Y. Zhao, R. Yang, Z. Li, J. Luo, B. Chi, Z. Jiang, M. Li, S. Mu, S. Liao, J. Zhang and X. Sun, Angew. Chem., Int. Ed., 2019, 58, 2622–2626 CrossRef CAS PubMed.
- M. Kato, N. Fujibayashi, D. Abe, N. Matsubara, S. Yasuda and I. Yagi, ACS Catal., 2021, 11, 2356–2365 CrossRef CAS.
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS.
- K. Huang, C. Rong, W. Zhang, X. Yang, Y. Fan, L. Liu, Z. Yang, W. Chen and J. Yang, Mater. Today Energy, 2021, 19, 100579 CrossRef CAS.
- J. Xu, S. Lai, D. Qi, M. Hu, X. Peng, Y. Liu, W. Liu, G. Hu, H. Xu, F. Li, C. Li, J. He, L. Zhuo, J. Sun, Y. Qiu, S. Zhang, J. Luo and X. Liu, Nano Res., 2021, 14, 1374–1381 CrossRef CAS.
- Y. Tsuji, K. Yamamoto, K. Yamauchi and K. Sakai, Angew. Chem., Int. Ed., 2018, 57, 2 CrossRef CAS.
- T. Zhang, Z. Li, L. Wang, P. Sun, Z. Zhang and S. Wang, ChemSusChem, 2018, 11, 2730–2736 CrossRef CAS PubMed.
- L. Peng, C.-T. Hung, S. Wang, X. Zhang, X. Zhu, Z. Zhao, C. Wang, Y. Tang, W. Li and D. Zhao, J. Am. Chem. Soc., 2019, 141, 7073–7080 CrossRef CAS.
- J.-G. Wang, H. Liu, H. Sun, W. Hua, H. Wang, X. Liu and B. Wei, Carbon, 2018, 127, 85–92 CrossRef CAS.
- X. Huang, T. Shen, T. Zhang, H. Qiu, X. Gu, Z. Ali and Y. Hou, Adv. Energy Mater., 2020, 10, 1900375 CrossRef CAS.
- H. Zeng, W. Wang, J. Li, J. Luo and S. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 8721–8729 CrossRef CAS.
- Y. Chen, I. Kone, Y. Gong, A. Xie, H. Hu, D. Kong, J. Liu, Y. Tang, X. Yang, R. Pang and P. Wan, Carbon, 2019, 152, 325–334 CrossRef CAS.
- W. Wang, M. Tang, Z. Zheng and S. Chen, Adv. Energy Mater., 2019, 9, 1803628 CrossRef.
- V. Selvamani, R. Ravikumar, V. Suryanarayanan, D. Velayutham and S. Gopukumar, Electrochim. Acta, 2015, 182, 1–10 CrossRef CAS.
- Y. Deng, G. Wang, K. Sun, B. Chi, X. Shi, Y. Dong, L. Zheng, J. Zeng, X. Li and S. Liao, J. Power Sources, 2019, 417, 117–124 CrossRef CAS.
- S. G. Patnaik, R. Vedarajan and N. Matsumi, ACS Appl. Energy Mater., 2018, 1, 1183–1190 CrossRef CAS.
- C. H. Choi, H.-K. Lim, M. W. Chung, G. Chon, N. Ranjbar Sahraie, A. Altin, M.-T. Sougrati, L. Stievano, H. S. Oh, E. S. Park, F. Luo, P. Strasser, G. Dražić, K. J. J. Mayrhofer, H. Kim and F. Jaouen, Energy Environ. Sci., 2018, 11, 3176–3182 RSC.
- Y. Zeng, X. Zhang, R. Qin, X. Liu, P. Fang, D. Zheng, Y. Tong and X. Lu, Adv. Mater., 2019, 31, e1903675 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1se01427g |
| ‡ These co-first authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |