
Unravelling the chemistry of catalyst surfaces and solvents towards C–C bond formation through activation and electrochemical conversion of CO2 into hydrocarbons over micro-structured dendritic copper†
Nusrat
Rashid
 a,
Mohsin Ahmad
Bhat
b and
Pravin P.
Ingole
*a
a,
Mohsin Ahmad
Bhat
b and
Pravin P.
Ingole
*a
aDepartment of Chemistry, Indian Institute of Technology Delhi, New Delhi, India 110016. E-mail: ppingole@chemistry.iitd.ac.in; ppingole@gmail.com; Fax: +91(11)26581102; Tel: +91(11)26597547
bDepartment of Chemistry, University of Kashmir, Srinagar, Jammu and Kashmir, India 190006
First published on 8th November 2021
Abstract
Herein, we report the results from our study towards understanding the role of electrocatalyst surfaces and solvents (aqueous versus wet organic media) in C–C bond formation on microstructured spheres, polygons, aggregates and dendrites of copper, which is a crucial aspect for the selective and sustainable conversion of CO2 into high energy density hydrocarbons like ethylene. Our results establish that apart from morphology, the crystal orientations and characteristic electrode/electrolyte interfacial physico-chemical aspects significantly affect the electrochemical carbon dioxide reduction (ECR) reaction activity and selectivity of copper electrocatalysts. Moreover, we report that an appropriate combination of catalyst properties and solvent/electrode interface ensures selective electroreduction of CO2 into C1 and C2 hydrocarbons, especially ethylene, over dendritic copper electrocatalysts. We demonstrate that for a given solvent system, the morphology of the electrocatalyst is the sole determinant of its catalytic activity, with elaborate dendritic shape enhancing the ECR reaction irrespective of the solvent used. A detailed account of the electrocatalytic performance, product selectivity, and faradaic efficiency of Cu-electrodeposits for ECR in aqueous medium and wet-organic (DMSO) electrolyte is presented. In aqueous 0.2 M KHCO3 solution, the catalyst exhibits a faradaic efficiency of 43% for ethylene production at −1.0 V vs. RHE and a selectivity of 74% among all the hydrocarbon products. Importantly, the Cu-based electrocatalysts fabricated for the present study inhibit the parasitic hydrogen evolution reaction and production of parallel products, viz. CO, CH4, and C2H6, during the ECR. Our results further establish that C–C coupling for the formation of ethylene is affected by the nano-morphology of the electrocatalyst and the composition of the electrolyte system employed for the electroreduction.
1. Introduction
The sustainability of planet Earth is largely determined by energy transduction and 80% of this comes from fossil fuel utilization that primarily releases CO2, a greenhouse gas responsible for global warming and the deterioration of climate.1 CO2 capture and utilization is a promising means to extend the usage span for fossil fuels and close the CO2 cycle.2–4 Tremendous efforts have been made toward CO2 capture and utilization5–9 to mitigate some of the current energy and climate challenges brought about by CO2 emissions. Among the several technologies proposed for CO2 utilization, the electrochemical route seems to be a promising and eco-friendly approach.9,10 However, electrochemical CO2 reduction (ECR) into multicarbon, high energy density fuels such as ethylene, ethane and similar hydrocarbons has proven to be highly sluggish. The selective conversion of CO2 into specific hydrocarbons poses another steep challenge for the commercial application of ECR based CO2 utilization technology. Since the first report by Hori10 on the production of hydrocarbons over copper electrodes via ECR, several research groups have demonstrated the selective reduction of CO2 into a variety of hydrocarbon fuels over differently engineered copper and other metal based electrodes.9,11–13 The selectivity in these reports has been achieved via engineering of the morphology, surface area,12–15 and size11,16 of the metal nanomaterials.Mechanistically, ECR is an inner sphere heterogeneous electron transfer reaction associated with many solvent phase/solvent induced reactions. The electrode and the electrolyte employed in the CO2 electrolyser, because of their impact over the stability and reactivity of electro-generated intermediates, are expected to play a critical role in deciding the overall mechanism, and hence the product profile for ECR. The solvents/electrolytes are reported to not only affect the CO2 availability but are also demonstrated to influence the ECR mechanistic pathway.17 While in dry organic solvents, CO18,19 or oxalates20 or carbonates21 are the major ECR products, wet organic solvents yield a variety of hydrocarbons.17,22–24 Thus, the mechanistic and kinetic aspects of ECR are dictated by the solvent employed for the process25–27 as different solvents/electrolytes exhibit differing structural organization, and have significantly different mechanistic and kinetic implications across the electrode/electrolyte interface.26,27 Besides, for ECR, the electrode kinetics and the stability of intermediates are also very sensitive toward the electrocatalyst's morphology, surface structure28 and composition.19 For instance, compared to the electro-polished polycrystalline Cu-electrodes, the roughened Cu-electrode surfaces (e.g. covered with Cu-nanoparticles or sputtered surfaces) have been found to exhibit better ECR selectivity for hydrocarbons. Similarly, compared to their bulk and smooth counterparts the nano/micro-structured catalysts with preferential orientation/growth along a particular direction are demonstrated to be more active and to selectively favour the C–C coupling process.
Overall, besides its role in dictating the effective concentration of CO2,17,29 the solvent/electrolyte system plays a critical role in deciding the ECR mechanism.26,27 Due to their ability to limit the availability of reducible protons, the use of organic solvents has been reported to be very effective in facilitating the ECR while mitigating the competitive hydrogen evolution reaction (HER).22 However, organic media lack availability of protons for efficient production of hydrocarbons at a high yield. To overcome this limitation, deliberate addition of a small quantity of water into organic media has been attempted, which is found to alter the product distribution from only CO in organic solvents to hydrocarbons in wet organic media, which could be due to the altered mechanism. However, it will be interesting to study the process of ECR using the same catalyst but in these totally different electrolytes, i.e. aqueous and organic media, in order to gain better insight into the chemistry of the interplay between the electrode material and solvent employed for the ECR process. To the best of our knowledge, no such systematic study has ever been reported. Motivated by this, we attempted to explore and compare the electrocatalytic performance of a single catalyst with varying physicochemical properties like morphology and crystal orientations for ECR in different electrolytes, i.e. aqueous and organic solutions.
For this, copper electrodeposits obtained under varying electrodeposition conditions have been chosen as a model system. Special attention has been given to the catalyst with roughened surfaces. In general, the type of hydrocarbon produced depends upon the asymmetry of the structure, with roughened surfaces favouring the production of higher hydrocarbons. This is attributed to the higher surface area offered by such surfaces which ensures ample time for *CO dimerization in contrast to smaller sized particles that favour the production of methane. Among the various nano-dimensional electrocatalysts, dendritic materials with a self-assembled hierarchical structure consisting of a central stem and many side branches are reported to be very fascinating for both fundamental studies and practical applications. Recently, we have reported22 a Cu-electrodeposit based electrocatalyst with controlled morphologies and crystallinity for efficient and selective electrochemical conversion of CO2 into CO along with traces of C1 and C2 hydrocarbons from CO2-saturated moistened di-methyl sulfoxide (DMSO) electrolyte solution. The selectivity for C1 or C2 hydrocarbons as a major product was found to be dependent on the extent of branching in dendrite microstructures as well as the extent of exposure of (111) and (200) facets. Importantly, the production of C1 and C2 hydrocarbons along with CO was ascribed to the availability of protons from residual water (<0.2% v/v or ca. 1000 ppm) and hygroscopic nature of DMSO. The current study was therefore designed with the aim to assess, explore, and quantify the electrocatalytic performance of the same Cu-nano electrodeposits for ECR in 0.2 M KHCO3 aqueous solution. The results from these investigations in terms of activity and product selectivity are compared with those obtained with wet DMSO (0.1 M TBATBF4) as an ECR medium to propose a case for choice of electrode/electrolyte combination for the desired C2 hydrocarbons.
Through the present study, we report the results from our comprehensive electrochemical investigations regarding ECR activity of Cu-electrodeposits that clearly establish the significance of surface morphology, crystal orientation and role of solvent systems in dictating the mechanistic and kinetic aspects of ECR. The results presented herein suggest that the presence of ca. 1000 ppm moisture in organic solvent results in production of ethane along with CO during ECR over Cu-surfaces having dendritic kinks and branches. In contrast, for the ECR in aqueous medium over these surfaces, production of alkenes (ethylene) is favoured over alkanes. The effect of solvent is thus highlighted not only on immensely branched dendrites but also on spherical as well as polygonal shapes, with all studies leading to the conclusion that selectivity for a particular ECR product is a manifestation of electrode and electrolyte interplay and providing a comprehensive overview of different copper structures in different electrolytes. We foresee that the results from these studies could help us to unravel the effect of CO2 activation pathways on the product profile and selectivity of ECR. This study, we believe, should open avenues for designing electrode/electrolyte interfaces that combine the catalyst and electrolyte properties for commercially viable ECR feedstock.
2. Experimental
2.1 Materials and chemicals
All chemicals used in this study were of analytical reagent grade and used as received without further purification, unless stated otherwise. A copper metal rod (99.9% pure, metal basis) was procured from Alfa Aesar. Ascorbic acid (AsH) (99%) and potassium hydrogen carbonate (99%) (KHCO3) were procured from Fisher Scientific. Ultrapure CO2 and argon gases were passed through a filter equipped with an oxygen trap, gas purifier, and moisture trap to remove the traces of impurities and moisture (Sigma Gases, New Delhi).2.2 Synthesis of dendritic Cu-micro-structures
The synthesis of dendritic Cu-micro-structure samples was performed following our earlier reported electro-deposition protocol; for more details, please refer to ref. 22. Typically, L-ascorbic acid (AsH) was used as a stabiliser cum reducing agent in an electro-deposition bath with a copper rod (6 mm diameter) as a sacrificial anode and platinum wire (2 mm diameter) as a cathode. Both the electrodes were pre-cleaned in 0.1 M HNO3 and rinsed thoroughly with deionised water before immersing in a conventional glass electrochemical cell containing AsH aqueous solution. A Keithley 2231A-30-3 triple channel DC power supply (Tektronix Inc. USA) was used for applying a constant output DC voltage for the stipulated time intervals. The brownish mass of copper particles accumulated at the Pt cathode at a later part of the electrolysis was tapped off and the obtained suspension from electrolysis was collected, centrifuged for 10 minutes, washed with Milli-Q water and ethanol, and then dried in a Petri dish under vacuum at 50 °C for ca. 3 hours. Several samples were prepared following a similar approach but varying the electrolysis parameters, viz. applied voltage, AsH concentration, electrolysis time and temperature, one at a time.2.3 ECR measurements and analysis
All the electrochemical measurements were carried out on a Metrohm PGSTAT-302N potentiostat/galvanostat in a custom designed 2-chambered Teflon cell with 50 mL electrolyte capacity and 18 mL of headspace in both chambers. A Nafion 117 (Sigma Aldrich) proton exchange membrane, pre-activated by treating with H2SO4 and H2O2,30 was used as a separator. A Pt foil counter electrode was taken in the anodic chamber, while a Ag/AgCl (sat. KCl) reference electrode and a catalyst modified glassy carbon plate electrode (GCPE, 2 × 1 cm2) serving as a working electrode were placed together in the cathodic chamber. Prior to catalyst loading, the GCPE was first cleaned with a slurry of alumina powder (1 μm followed by 0.3 μm), washed thoroughly with Milli-Q water, and then sonicated in ethanol for a few seconds to get rid of adsorbed moieties, if any. Finally, a catalyst (ca. 30 μL) prepared by suspending 3 mg of a catalyst sample in a solution of 600 μL iso-propyl alcohol, 300 μL water and 10 μL Nafion, followed by ultra-sonication for ca. 30 minutes, was drop-cast on the GCPE and dried under ambient conditions before its use for ECR. The electrolyte (0.2 M KHCO3) was pre-electrolysed using another set of pre-cleaned carbon plates as both the cathode as well as anode to electro-deposit ionic/metallic impurities, if any. LSV polarization curves were recorded in CO2 saturated 0.2 M KHCO3 electrolyte at a scan rate of 20 mV s−1 from 0.6 to −1.1 V vs. the reversible hydrogen electrode (RHE). CO2 saturation was performed for 30 min at a 20 mL min−1 flow rate regulated by a mass flow controller (MFC) from Alicat, India. All the electrochemical data reported were corrected for IR drop by impedance measurements, and all the potentials are reported vs. RHE after conversion from the Ag/AgCl reference electrode scale using the following eqn (1),| ERHE = EAg/AgCl + 0.197 V + (0.059 × pH) | (1) |
The pH of CO2 and Ar-saturated 0.2 M KHCO3 solutions was 7.3 and 8.9, respectively. To understand the charge transfer dynamics, EIS analysis was carried out within a frequency range of 10 kHz to 0.1 Hz with a 10 mV AC voltage amplitude. The gas phase ECR products were collected in Tedlar bags at different applied potentials ranging from −0.3 V to −1.0 V vs. RHE, and were analysed using a Thermo Scientific Trace 1110 gas chromatography (GC) instrument by manually feeding 1 mL of the collected gaseous sample into the GC. Faradaic efficiency (FE) of the gaseous products was calculated using the following eqn (2),
| FE = (nx × ∅x × F × fm)/I | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), ∅x is the volume fraction of the gaseous products (gases are considered as ideal), fm is the molar flow rate (mol s−1), and I is the corresponding current (in amperes, A).
485 C mol−1), ∅x is the volume fraction of the gaseous products (gases are considered as ideal), fm is the molar flow rate (mol s−1), and I is the corresponding current (in amperes, A).
The liquid products were collected using a glass syringe and were analysed with a 500 MHz NMR spectrometer (Bruker Ascend™). Typically, 0.5 mL of catholyte (electrolyte solution from the cathode chamber) was mixed with 0.1 mL of D2O and 0.5 μL of di-methyl sulphoxide (DMSO) (as an internal standard), and the solvent suppression method was followed to suppress the overwhelming solvent peaks to make the small CO2 reduction product peaks observable. FEs for liquid samples were calculated using the following set of equations.31,32 First, the R factor for different concentrations of liquid products was calculated using eqn (3),
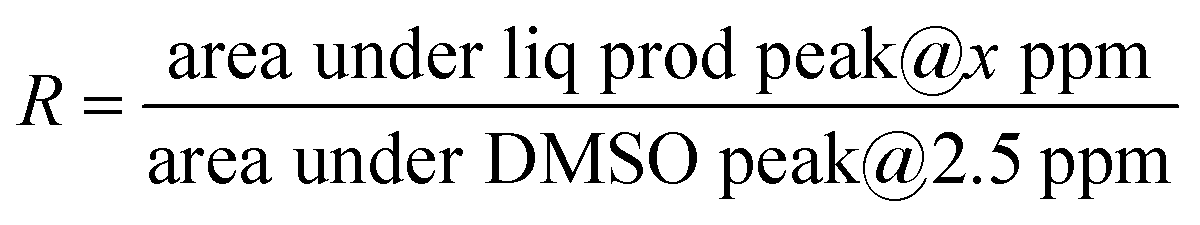 | (3) |
A calibration curve was plotted with R vs. concentration of the concerned liquid product, and was further used for calculation of FE for liquid products.
| Liq. product conc. = R/slope, M−1 | (4) |
The total catholyte amounted to 50 mL in the cathodic chamber; therefore, the total concentration of product produced was
| Nliq. product = 0.05L × liq. product conc. | (5) |
The number of moles of electrons used to produce n moles of liquid product is
| eoutput = Nliq. product × ne− | (6) |
| einput = Q/F | (7) |
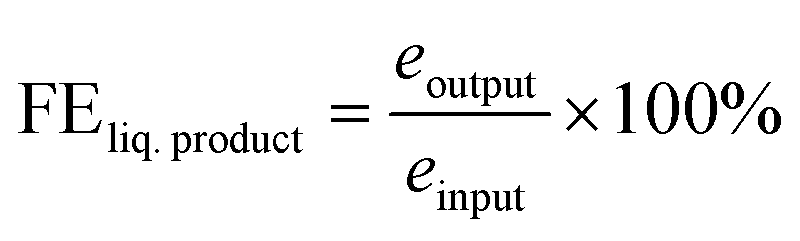 | (8) |
Total selectivity for C2H4 among all of the CO2 reduction products was calculated as:
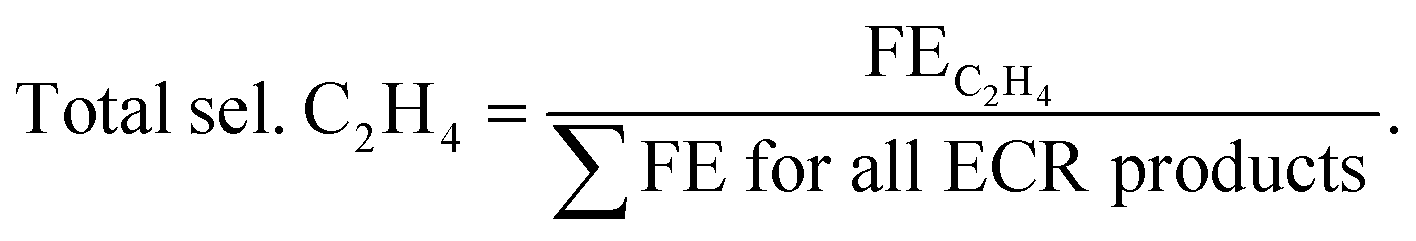 | (9) |
And, selectivity for ethylene among all hydrocarbons was calculated as:
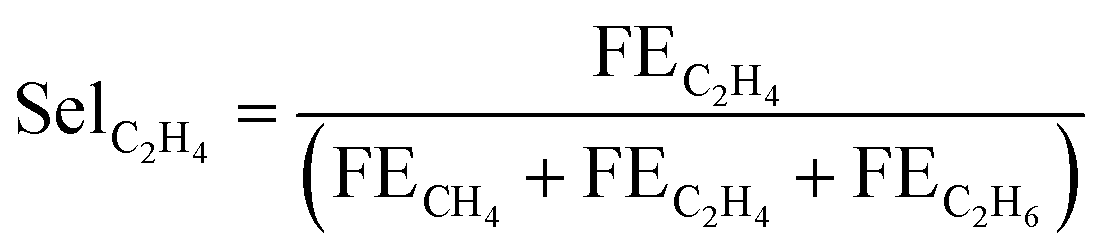 | (10) |
3. Results and discussion
3.1 ECR on dendritic Cu-micro-structured catalysts
Several samples of dendritic micro-structured Cu-electrodeposits were fabricated under varying experimental conditions, viz. AsH concentrations, applied potentials, electro-deposition bath temperatures, and electro-deposition time intervals. All the samples were well characterized for their crystalline and morphological properties, and the corresponding XRD patterns and FESEM images are given in the ESI.† The XRD analysis suggests Cu(0) as the prevalent phase in most of the samples except for a few that contain some Cu-oxides. A microscopic inspection of the surface topography suggests that optimum electro-deposition parameters, viz. AsH concentration, applied voltage, reaction temperature, and electrodeposition duration, affect the resultant catalyst morphology. The lower AsH concentration (25 mM) results in the formation of a mixed morphology (both polygons and small dendrimers), while for 75 mM AsH, the polygons are transformed into aggregates of small asymmetrical structures. For 100 mM AsH, well-defined and extensively branched dendrimers (ESI-1†) are obtained. In the applied voltage optimization set (keeping AsH concentration fixed at 100 mM), both 7.5 V and 15 V DC potentials lead to dendrite formation to some extent (ESI-2†), but at 7.5 V dendrites are not extensively branched, and at 15 V, independent dendrites are not observed (only a coral type of morphology is displayed). However, at 30 V, well-branched dendrites are noted. In the temperature control set, at 60 °C or at 70 °C (keeping the AsH concentration and DC potential fixed at 100 mM and 30 V, respectively), a mixed morphology of polygons and small dendrites is observed, while at 90 °C, the broken fragments and agglomerated branches of dendrites are observable (ESI-3†), but at 80 °C, extended dendrite structures are found. Similarly, in the study of the effect of electrodeposition time (at a fixed optimized AsH concentration, applied bias and temperature, viz. 100 mM, 30 V and 80 °C), a dendrite structure with extensive branches is observed at 60 min of electrodeposition, while at 15 min we have only polygons, at 30 min, the edges of the polygons are covered with Cu-adatoms, and at 45 min, most of the polygons have been covered with Cu-deposits (ESI-4†). The thus-obtained Cu-microstructures with varying morphologies were cast on the GCPE and tested for ECR.The ECR current density recorded under similar conditions was dependent on the morphological features of the Cu-electrodeposits obtained under different electrodeposition parameters, indicating the tuneable ECR catalytic activity (details in succeeding sections). Amongst these, the electrodeposits prepared at 60 minutes demonstrate the best ECR performance. For the sake of convenience, henceforth, the best-performing Cu-electrodeposits will be denoted as b-Cu-dendrites indicating that the said sample possesses the best dendritic features as well as ECR performance. Fig. 1(a) presents typical LSV polarization curves recorded over b-Cu-dendrites in Ar and CO2 saturated 0.2 M KHCO3 aqueous solutions, for which the cathodic current density in CO2-saturated 0.2 M KHCO3 was much higher than that in the Ar-saturated solution, especially after the onset potential of −0.324 V vs. RHE. This is in good accordance with our previous study with the same catalysts in wet DMSO, the Cu-electrodeposits showing the best activity in wet-DMSO, and also showing the highest activity in 0.2 M KHCO3, which can be attributed to the high capacity of dendritic structures to transfer electrons in electrochemical reactions. However, for the same electrocatalyst, the qualitative and quantitative distribution of ECR products was quite different in 0.2 M KHCO3 and wet DMSO, implying completely different reaction pathways, owing to different electrode/electrolyte interfacial structures. The b-Cu-dendrites in 0.2 M KHCO3 produce ethylene with an average FE of 43% and a selectivity of 74% among hydrocarbons at −1.0 V vs. RHE, while ethane was the major product in wet DMSO at the same overpotential.
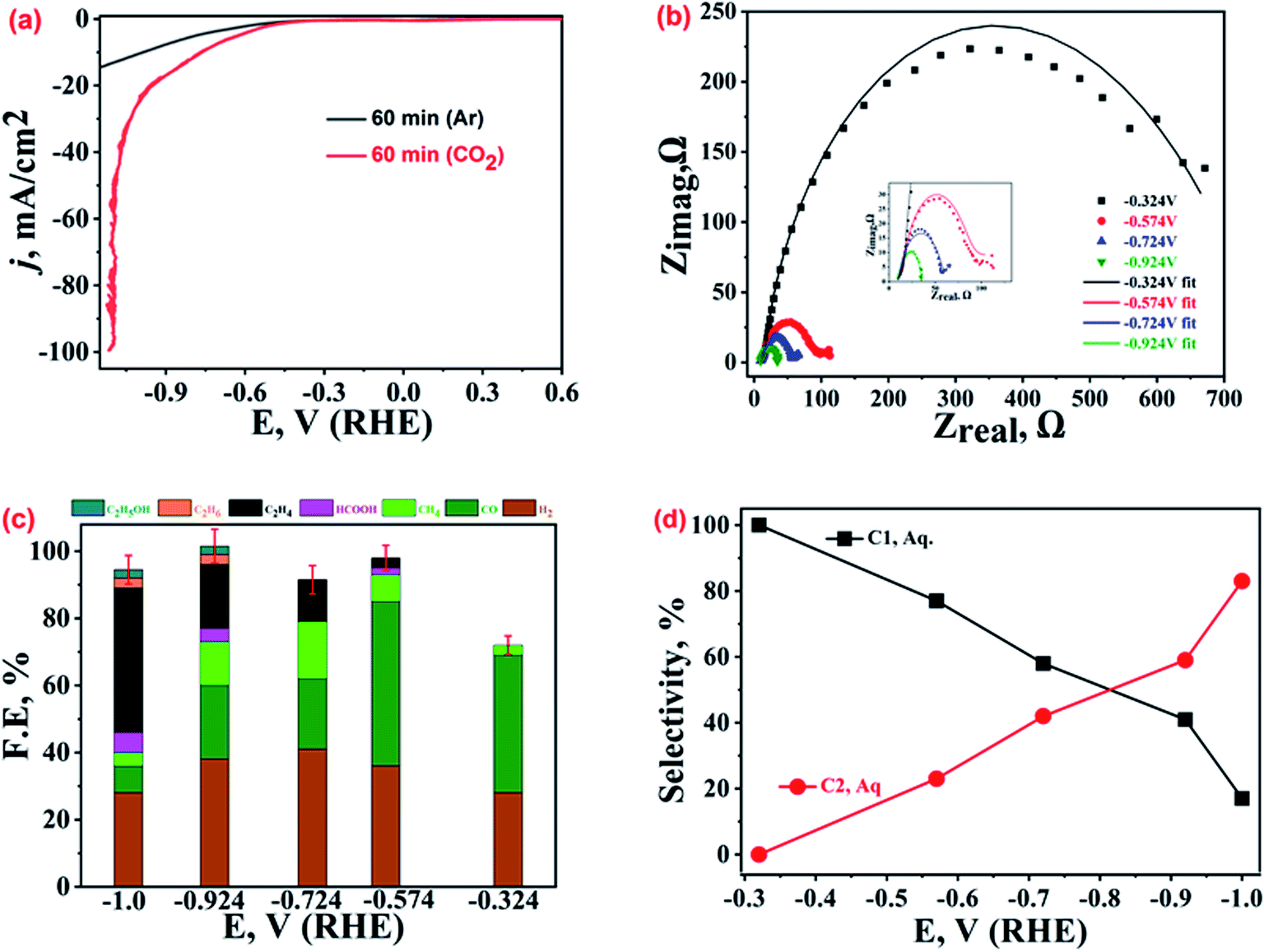 | ||
| Fig. 1 (a) Comparative LSV polarization curves for b-Cu-dendrites in Ar and CO2 saturated 0.2 M KHCO3. (b) Nyquist plots, and (c) product distributions with their FEs at different applied potentials. (d) Comparative selectivity for C1 and C2 hydrocarbons in water at different CO2 reduction potentials. | ||
Further, to quantify the ECR-kinetic performance of b-Cu-dendrites, EIS spectra (Nyquist plots) were recorded at five different applied potentials starting from an onset potential (Eonset) of −0.324 V up to −0.924 V, as shown in Fig. 1(b). The scatter of the data points at higher negative potentials indicates the evolution of gaseous products, making the EIS spectra noisy. The Nyquist plots fitted to equivalent circuits (Fig. SI-5†) showed a decrease in charge transfer resistance at high negative potentials, which confirms the applied potential dependent ECR mechanism. Therefore, to understand the potential dependent ECR mechanism, the ECR products at five different applied potentials, more negative than Eonset, were collected and analysed through GC and NMR spectroscopy. Averaged values from at least five repeatedly similar FE values observed during these investigations are shown in Fig. 1(c). ECR at Eonset preferentially yields CO (41%), followed by H2 (28%) along with traces of CH4 (<3%); the values in parentheses represent the corresponding FE values. At a more negative potential, −0.57 V, the production of small portions of HCOOH and C2H4 is also noted, with CO still being the major product (49%) followed by H2 (36%) and CH4 (8%). Further increasing the potential to −0.73 V, the generation of C2H4 increases (12.5%) while that of CO decreases (21%). The trend of increasing FE for ethylene and decrease for CO, H2, and CH4 continues with the more negative potentials, which is as predicted from theoretical investigations33 and is reported in numerous earlier studies.34–36 C2H4 production reaches a FE of 43% at −1.0 V vs. RHE, while for CO and H2, FEs were observed to drop down to 8% and 28%, respectively. Production of trace amounts of ethanol and ethane during ECR was also noticed at this potential. At peak potential for ethylene production (−1.0 V vs. RHE), the carbon balance for all products for b-Cu-dendrites was 94.5% ± 4%.
A comparison of the potential dependent selectivities towards the production of C1 and C2 hydrocarbons (calculated by dividing the FE of C1 and C2 hydrocarbons with the total FE for C1 and C2 added together, excluding the FE of H2 and CO) is shown in Fig. 1(d). The best-performing catalyst shows higher selectivity toward C1 hydrocarbons at low overpotentials, but at high overpotentials the selectivity is higher for C2 hydrocarbons. The formation of ethylene and other C2 hydrocarbons at ca. 400 mV or potentials more negative than the ECR onset potential has also been theoretically predicted.37 As per the established literature,37–40 the formation of ethylene along with traces of ethanol and methane suggests the prevalence of the *CO–*CO coupling mechanism, where surface adsorbed *CO species interact with each other. The current plateau region of CO adsorption noted at ca. −0.6 V to −0.9 V in the LSV curve (Fig. 1(a)) also indicates ethylene generation by the *CO–*CO coupling mechanism.41,42
Further, to confirm this mechanism, the presence of the *CO intermediate on the catalyst surface was probed through a chronopotentiometric approach proposed by Hori et al.34 The chronopotentiograms were recorded on Cu-dendrites (60 min. sample) in Ar, CO, and CO2 saturated 0.1 M KHCO3 as shown in Fig. 2. In Ar-saturated KHCO3 solution, the potentials remain constant, but for the CO saturated buffer there is a negative shift in potentials especially at higher applied currents due to CO adsorption on surfaces that hinders the HER.34 For instance, a negative shift of 0.13 V was observed at an applied current of 5 mA in CO-saturated compared to Ar-saturated 0.2 M KHCO3 solution. Moreover, for the same catalyst in CO2 saturated 0.2 M KHCO3 solution, the chronopotentiogram appears similar to that of the Ar-saturated 0.2 M KHCO3 solution at low applied currents, but at higher applied currents it starts reaching the potentials observed in the case of CO-saturated 0.2 M KHCO3 solution. These observations, similar to those reported by Hori et al.,34 support the prevalence of the *CO–*CO coupling mechanism in 0.2 M KHCO3 over the crafted catalysts. The negative shift of the potential in the presence of the CO-saturated solution as compared to the Ar-saturated one is ascribed to the *CO adsorption that prevents the cathodic reduction of H+ in CO-saturated electrolyte solutions. Thus, the adsorption of an intermediate *CO on the catalyst surface supports the prevalence of the *CO–*CO coupling mechanism.
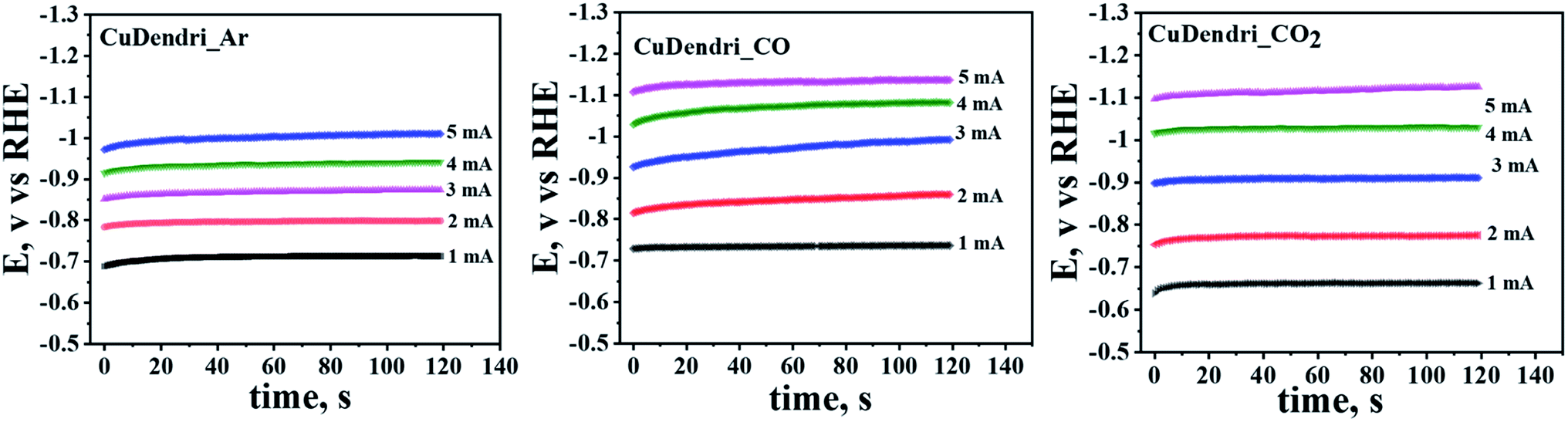 | ||
| Fig. 2 Chronopotentiograms of Cu-dendrites (60 min sample) in Ar, CO and CO2 saturated 0.2 M KHCO3 solutions. | ||
The large increase in local pH at more negative potentials is known to cause alkalization around sharp and/or dendritic surfaces, thereby reducing the chances of the parasitic HER while facilitating the ECR.40 Ma et al.43 showed that the changes in the local pH can be controlled by regulated diffusion of ionic and neutralizing species like HCO3− around the nano-morphologies. The elaborate structures show a larger reduction in the HER due to the higher pH changes around the kinks and corners which is similar to what we found in the present study. A detailed account of our observation vis-à-vis the electrocatalytic performance of differently crafted Cu-electrodeposits toward ECR in DMSO and aqueous bicarbonate solutions is presented in the following sections.
3.2 ECR on Cu-microstructures prepared with varying AsH concentrations
The Cu-electrodeposits obtained with three different AsH concentrations (25, 75, and 100 mM) were characterized by various techniques as discussed in an earlier section. More details can be found in ref. 22. As mentioned earlier, the variation in the AsH concentrations results in differently shaped Cu-microstructures; at 100 mM AsH, well-developed dendrites with spiky ends were noted suggesting the preferential growth of the Cu-microstructures along the (111) facets, while for 25 and 75 mM AsH, mixed morphologies were obtained. Fig. 3(a) shows the LSV polarization curves in CO2 saturated 0.2 M KHCO3 solution for these three samples. For the 100 mM AsH sample, the LSV curve (blue trace) shows the lowest onset potential (−0.45 V vs. RHE) and highest current density, reaching 100 mA cm−2 at −1.1 V followed by the 25 mM (black trace) and 75 mM (red trace) samples. The Nyquist plots (Fig. 3(b)) recorded at open circuit potential (EOCP) show the minimum charge transfer resistance for the 100 mM AsH sample. The equivalent circuits for these plots are shown in Fig. SI-6.†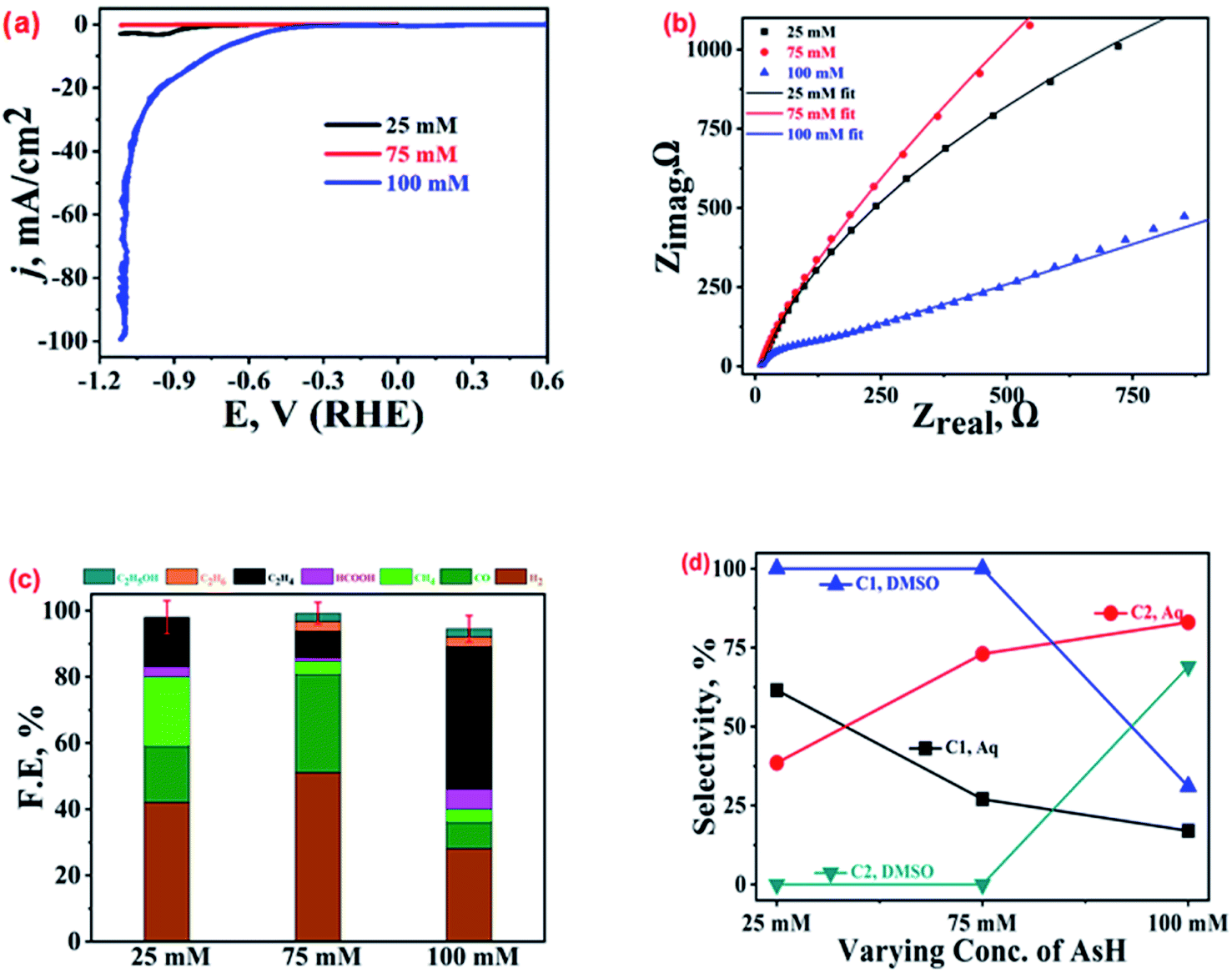 | ||
| Fig. 3 (a) LSV polarization curves, (b) Nyquist plots at EOCP, and (c) product profiles with FE values at −1.0 V vs. RHE for Cu-electrodeposit modified GCPEs in CO2 saturated 0.2 M KHCO3. (d) Selectivities of the C1 and C2 hydrocarbons in aqueous and DMSO electrolytes. | ||
The differences noted in activity for the different catalysts were further confirmed from GC and NMR spectroscopic characterization of the ECR products. Fig. 3(c) suggests the selective production of ethylene with selectivity of 43% among the total ECR products and 74% among only hydrocarbons for the 100 mM AsH sample as compared to those of the 25 mM (15%) and 75 mM (8%) AsH samples that show better selectivity towards CO and H2. Importantly, the parasitic HER was more suppressed over the 100 mM AsH sample. The enhanced ECR activity is attributed to the distinct morphology (highly branched and homogeneous) of the catalyst (Fig. SI-1†), which is expected to offer a higher number of active sites. The presence of these sites improves the charge transfer kinetics and stabilises the *CO intermediate (by increasing its residence time), and increases the local pH at the catalyst/electrolyte interfaces resulting in lower HER and increased ECR performance.38,39,44
A comparison of the ECR activity in aqueous medium and wet DMSO (Fig. SI-7†) suggests the best performance by the 100 mM AsH sample, irrespective of the medium. At −1.0 V vs. RHE, the 100 mM AsH electro-deposit produced ethylene (FE ∼ 43%) in aqueous medium, but ethane (FE ∼ 29%) in DMSO. No ethane was found on the catalysts prepared with 25 and 75 mM AsH samples in DMSO.22 The distribution of ECR products is tabulated in Table SI-1.† The production of ethane in DMSO was ascribed to its hygroscopic nature and presence of moisture in its commercially available supplies which becomes a proton source required for the hydrocarbon formation.22 These observations vis-a-vis the product profile for ECR in water and DMSO clearly establish the vital role of solvents in dictating the mechanistic aspects of CO2 electro-reduction.
Interestingly, for ECR over the 25 mM AsH sample, the production of CO and CH4 in both DMSO (17.7% and 21%, respectively) and 0.2 M KHCO3 (17% and 28%, respectively) was similar. However, for the 75 mM AsH sample, a solvent specific shift in the selectivity of the ECR product was noticed. For CO and CH4, FEs of 30% and 4% in 0.2 M KHCO3, respectively, were noted in comparison to 13% and 25.4% in DMSO. For the 100 mM AsH sample, the highest selectivity in DMSO was for CO (FE ∼ 32%) at −1.0 V vs. NHE, while in 0.2 M KHCO3, it exhibited the highest selectivity towards ethylene (FE ∼ 43%) at the same potential. However, for all these catalysts, the HER was observed to be more prevalent in 0.2 M KHCO3 electrolyte in comparison to moist DMSO as shown in Fig. 3(c) and SI-7.† The comparative selectivities for C1 and C2 hydrocarbon products in aqueous and DMSO electrolytes (Fig. 3(d)) further prove the best activity and selectivity towards C2 hydrocarbons (rather than C1) by the 100 mM AsH sample, both in DMSO and aqueous medium. The 25 and 75 mM samples are capable of exhibiting good selectivity towards C1 hydrocarbons, but their FE is very poor (less than 24%). The preferential selectivity observed for a particular product in both the media suggests that the shape and morphology of the electrocatalyst play a key role in preferential stabilization of particular intermediates and hence higher selectivity.
3.3 ECR on Cu-microstructures electrodeposited with different applied voltages
Similarly, Cu-electrodeposits obtained with different biases of 7.5 V, 15 V and 30 V were also characterized by various techniques; the SEM images and the XRD patterns recorded on these catalysts are shown in Fig. SI-2.† Comparison of the LSV polarization curves (Fig. 4(a)) suggests the best performance, i.e. lowest onset potential and highest current density, by the 30 V sample (blue curve), followed by the 7.5 V (olive curve) and 15 V bias (brown curve) samples. The corresponding Nyquist plots at EOCP shown in Fig. 4(b) and the corresponding equivalent circuits in Fig. SI-8† show the minimum charge transfer resistance and hence the best activity for the 30 V sample. The ECR product profiles shown in Fig. 4(c) suggest that the 30 V sample exhibits a high production of ethylene with selectivity of 74% and FE of ca. 43% followed by the 15 V (FE ∼ 20%) and 7.5 V (FE ∼ 10%) samples. Interestingly, this is in contrast to the trend observed for the current density noted in the LSV polarization curves, which clearly supports the view that the overall current density for an electrolytic setup is not always a direct measure of its activity for production of a particular reaction product. The parasitic HER was more suppressed on the 30 V sample as compared to the 15 V and 7.5 V samples. On the other hand, the 7.5 V sample exhibited selective production of methane with FE of 24%. The comparison of ECR activity in aqueous and wet-DMSO media (Fig. SI-9†) reveals that the most active catalyst selectively produces ethylene in aqueous medium, but exhibits the best performance for CO and ethane production in DMSO. The 15 V sample shows FE of 15% for ethane while no ethane was found for the ECR in DMSO for the 7.5 V sample.22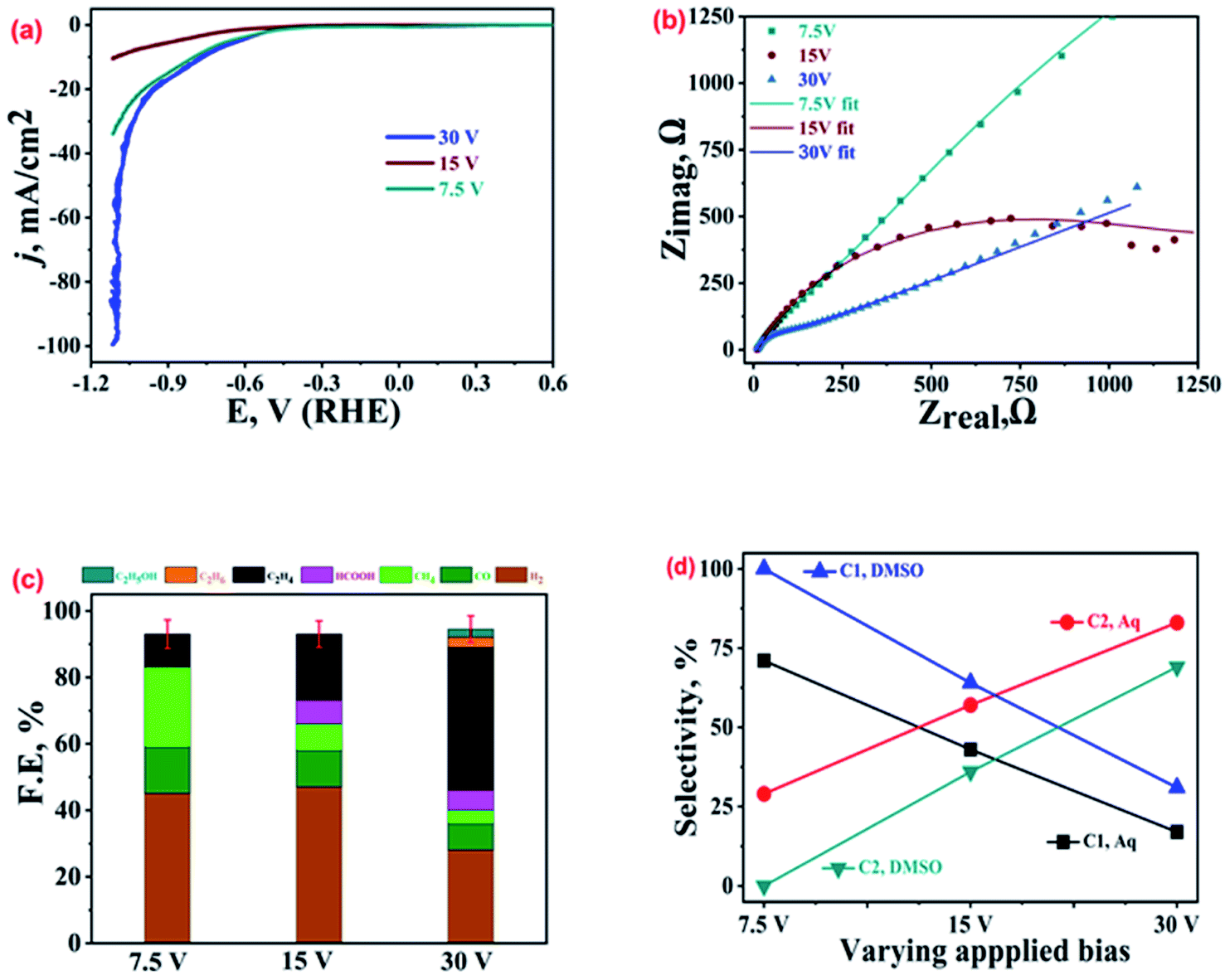 | ||
| Fig. 4 (a) LSV polarization curves, (b) Nyquist plots at EOCP, and (c) product profiles with FE values at −1.0 V vs. RHE for Cu-electrodeposit (at varying electro-deposition biases) modified GCPEs in CO2 saturated 0.2 M KHCO3. (d) Selectivities of the C1 and C2 hydrocarbons in 0.2 M KHCO3 and DMSO. | ||
The highest selectivity for a product (C2H6 in DMSO and C2H4 in KHCO3) was noticed for the 30 V sample, which supports our conclusion from the preceding section that the shape and morphology of the Cu-electrodeposits play a key role in selective stabilization of the intermediate and hence preferential production of a particular product. Another intriguing observation was that the production of CO and CH4 whether in DMSO or 0.2 M KHCO3 was similar (for CO, 14% FE in 0.2 M KHCO3 and 15% FE in DMSO; for CH4, 24% FE in 0.2 M KHCO3 and 29% FE in DMSO). As shown in Fig. 4(d), for the 30 V sample, the overall FE for C1 products (CH4 + HCOOH) from ECR adds up to only 10%, corresponding to a selectivity of ca. 17%, while for C2 products (C2H4 + C2H6 + C2H5OH), FE adds up to 49%, corresponding to a selectivity of 83% in 0.2 M KHCO3. In comparison, for the same catalyst in DMSO, the overall FE for C1 hydrocarbons adds up to only 13%, corresponding to a selectivity of 31%, while for C2 hydrocarbons it adds up to 29%, corresponding to a selectivity of 69%. Again, as shown in Fig. 4(d), the selectivity of the best-performing catalyst is higher for C2 hydrocarbons than C1 hydrocarbons, both in DMSO and aqueous electrolytes.
3.4 ECR on Cu-microstructures electrodeposited at varying bath temperatures
After studying the effect of AsH concentration and applied bias on the electrocatalytic performance of Cu-electrodeposits, we explored the impact of electro-deposition bath temperature on their activity and selectivity for ECR. The samples prepared at four different reaction bath temperatures, viz. 60 °C, 70 °C, 80 °C, and 90 °C, were characterised22 and their characteristic XRD and FESEM analysis results are shown in Fig. SI-3.† The LSV polarization curves recorded over these samples (Fig. 5(a)) reveal that the 80 °C sample (blue curve) exhibits the highest current density as well as the lowest ECR onset potential. The other catalysts to follow in a decreasing order of activity are the 60 °C (black curve) and 70 °C (red curve) samples, while the 90 °C sample (magenta curve) shows the lowest current density. These voltammetric inferences were further supported by the analysis of the corresponding Nyquist plots (Fig. 5(b) and SI-10†) which indicated the lowest charge transfer resistance for the 80 °C sample.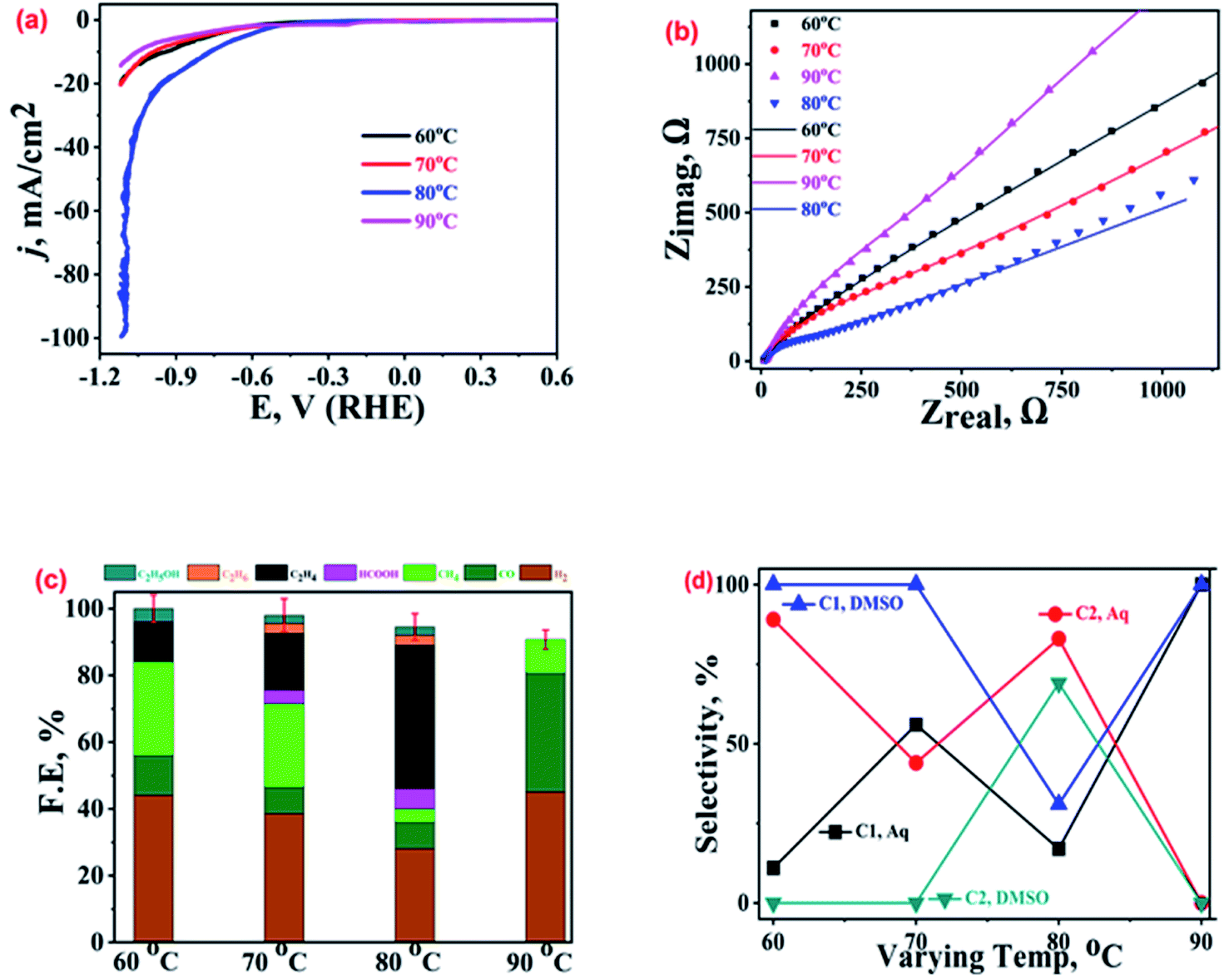 | ||
| Fig. 5 (a) LSV polarization curves, (b) Nyquist plots at EOCP, and (c) product profiles with FE values at −1.0 V vs. RHE recorded on dendritic Cu-microstructure (prepared at different electrodeposition bath temperatures) modified GCPEs in CO2 saturated 0.2 M KHCO3. (d) Selectivities of the C1 and C2 hydrocarbons in aqueous and DMSO electrolytes. | ||
The ECR product profiles in Fig. 5(c) suggest the highest ethylene production by the 80 °C sample followed by the 70 °C (FE ∼ 17%) and 60 °C (FE ∼ 12%) samples, while no ethylene was observed for the 90 °C sample. CH4 was the main ECR product for the 60 °C and 70 °C samples with FEs of 28% and 25%, respectively. Importantly, the parasitic HER was found to be the most suppressed over the electrodeposit prepared at 80 °C while the catalyst obtained at 90 °C was found to be the most HER active and also produces CO (FE ∼ 36%) alongside. As shown in Fig. SI-3,† the morphology of the 90 °C sample consists of smaller dendritic fragments but the 80 °C sample exhibits extensive branching in its dendritic structures which is responsible for the high activity and selectivity towards ethylene.39,45,46 Thus, the ECR selectivity of the catalysts is attributed to their distinct morphologies and structures.39
Once again, the comparison of ECR activity in aqueous and DMSO electrolytes suggests a complete change in the product profiles (Fig. SI-10†). For ECR in DMSO,22 CH4 is a major product for the 60 °C (FE ∼ 22%), 70 °C (FE ∼ 25%), and 90 °C (FE ∼ 28%) samples (refer to Table SI-1†); however, CO (FE ∼ 32%) and ethane (FE ∼ 29%) were the major products for the 80 °C sample. The comparison of selectivities for C1 and C2 hydrocarbons over the electrodeposits crafted at different temperatures, both in aqueous and DMSO electrolytes, is shown in Fig. 5(d). The most active catalyst, the 80 °C sample, exhibits higher selectivity towards C2 hydrocarbons than C1 hydrocarbons, both in DMSO and aqueous medium. Moreover, the highest selectivity for a product (ethane in DMSO and ethylene in KHCO3) for the same sample in both the media indicates a key role of catalyst morphology in stabilising the intermediate. Again, it is pertinent to mention here that the 90 °C sample exhibits a very good selectivity towards C1 hydrocarbons albeit with a poor FE of ca. 10%.
3.5 ECR over Cu-microstructures prepared with varying electro-deposition times
The samples prepared with varying electro-deposition times were characterised and their representative XRD and FESEM images are presented in Fig. SI-4.† The samples were tested for their ECR performance in CO2 saturated 0.2 M KHCO3, and the recorded LSV polarization traces are shown in Fig. 6(a). The 60 min sample has an extended dendritic structure (Fig. SI-4†), and exhibits the highest current density and lowest ECR onset potentials. This was further supported by the analysis of the impedance data (Fig. 6(b) and SI-12†) recorded over these samples at EOCP. The product profiles in Fig. 6(c) also suggest the best ECR activity and selectivity towards ethylene (FE 43%) for the 60 min sample. The 15 min and 30 min samples did not even show traces of ethylene production, though some ethylene generation (FE, 12%) was observed for the 45 min sample. The 15 min sample was composed of polygons with smooth surfaces, and ECR over this sample generated CO as a major product (FE, 49%) along with traces of HCOOH (FE, 5%). No methane, ethane or ethanol formation was observed over this catalyst. The 30 min sample consisted of polygons with its edges covered with the particulate deposits (Fig. SI-4†), and the ECR over this sample generated methane, CO, and HCOOH.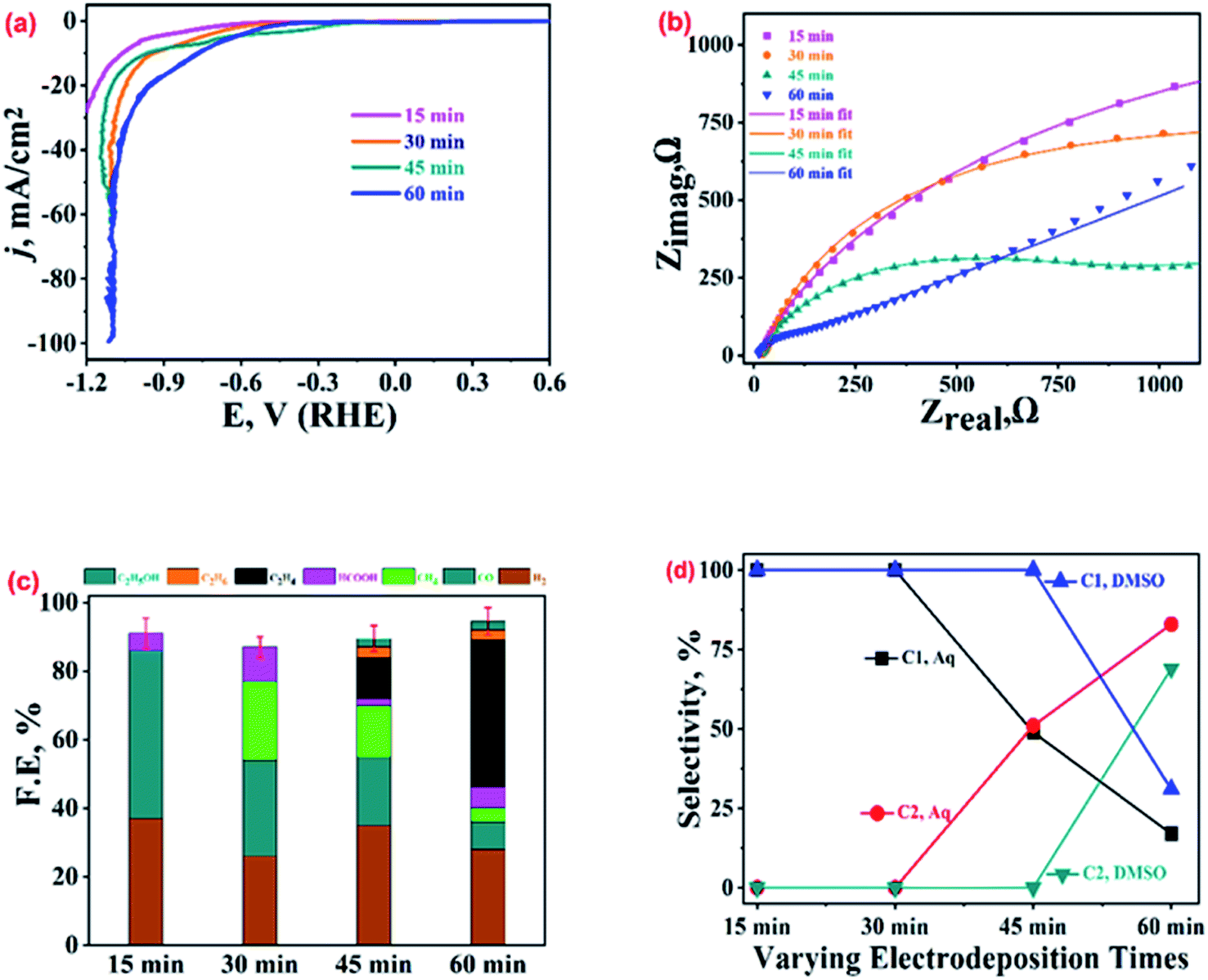 | ||
| Fig. 6 (a) LSV polarization curves, (b) Nyquist plots at EOCP, and (c) product profiles with FE values at −1.0 V vs. RHE recorded on dendritic Cu-microstructure (prepared with different electro-deposition time durations) modified GCPEs in CO2 saturated 0.2 M KHCO3. (d) Selectivities of the C1 and C2 hydrocarbons in aqueous and DMSO electrolytes. | ||
Fig. SI-13† presents a comparison of the ECR product profiles obtained over Cu-electrodeposits prepared with different electrodeposition times in both 0.2 M KHCO3 and DMSO electrolytes. For ECR in DMSO, CH4 and CO were the major products for the 15 min, 30 min, and 45 min samples (with FE values of 26%, 24%, and 25%, respectively, for CH4, and 19%, 17%, and 15% for CO, respectively) (refer to Table SI-1†). Meanwhile, for the 60 min sample, CO (FE, 32%) and ethane (FE, 29%) were the major products in DMSO. In contrast, in 0.2 M KHCO3, CO was observed to be the main product over the 15 min, 30 min, and 45 min samples, and ethylene (FE, 43%) was the main product over the 60 min sample. The comparison of the selectivity towards C1 and C2 hydrocarbons as shown in Fig. 6(d) suggests that in 0.2 M KHCO3, the 60 min sample exhibits a selectivity of 17% for C1 hydrocarbons (CH4 and HCOOH) and 83% for C2 hydrocarbons (C2H4, C2H6, and C2H5OH). The same catalyst exhibited a selectivity of 69% for C2 hydrocarbons and 31% for C1 hydrocarbons in DMSO. Although the 15 min and 30 min samples showed the highest selectivity for C1 hydrocarbons, their FEs were very low.
These observations regarding the electrocatalytic performance of Cu-electrodeposits crafted at varying electrodeposition parameters suggest the pivotal role of the catalyst's morphology and the solvent in determining the activity and product selectivity of ECR, respectively.
3.6 Discussion on the ECR mechanistic aspects in DMSO and aqueous medium
Improving and tuning the selectivity of Cu-based electrocatalysts for the production of different hydrocarbons in ECR is one of the most challenging tasks for materials electrochemists. As compared to other metal surfaces that can form not more than 5% of C2 products, the specific and very high affinity of Cu-surfaces towards C2 hydrocarbons is a very puzzling question. Several theoretical and experimental studies regarding the mechanistic aspects of ECR over Cu based electrocatalysts have underlined that the ECR to hydrocarbons over Cu-electrode surfaces occurs mainly via two pathways: (i) a C1 pathway leading to C1 hydrocarbons like methane,39 and (ii) a C2 pathway leading to C2 hydrocarbons such as ethane and ethylene.39 It has been reported by Koper and co-workers47 that in the C1 pathway, CHOads is the key intermediate towards the breaking of the C–O bond in CO2 leading to the formation of methane, while in the C2 pathway, the first step is the formation of a *CO dimer that is followed by the formation of a surface-bonded enediol or enediolate, or the formation of a surface oxametallacycle. Furthermore, as per this report,47 a *CO dimerization reaction is the key to explain the observed trend in ECR products. In general, ECR over Cu-surfaces simultaneously proceeds via a mix of C1 and C2 mechanisms. However, under certain circumstances, as dictated by the catalyst surface and the nature of electrolyte, there can be the dominance of one of these pathways over the others resulting in a preferential selectivity for C1 or C2 products. The mechanistic basis for the impact of solvent and catalyst morphology over ECR product distribution and selectivity in the present work can be explained as follows.Our observations vis-a-vis the product profile and selectivity for ECR over different Cu-electrodeposits are in good agreement with the above-stated morphology–activity relationship. In the present study, the dominance of C1 products was noted over electrodeposits obtained with the 25 mM and 75 mM AsH concentrations, and that of C2 hydrocarbons over the 100 mM sample, which can be attributed to the differing catalyst morphologies (Fig. SI-1†). Similarly, the catalysts electrodeposited with varying applied biases suggest that the 7.5 V and 15 V samples consist of a large number of polygons and a colony of corals emerging from a single base, while the 30 V sample appears as spiky dendrites (Fig. SI-2†). Accordingly, irrespective of the ECR medium used, the products generated on the 7.5 V and 15 V samples are predominantly C1 products, while the 30 V sample favours C2 products. The electrodeposits obtained at varying bath temperatures follow a similar trend; the 80 °C sample possesses uniform dendrites and shows the highest selectivity for C2 products, while the electrodeposits obtained at other temperatures are either highly agglomerated or consist of polygons (Fig. SI-3†) and favour the production of C1 products. The inference regarding morphology-dependent ECR product selectivity is also supported by the catalytic performance of Cu-microstructures obtained with varying electrodeposition time durations. As shown in Fig. 6(d), for the catalysts crafted with short electrodeposition times (15 and 30 min), the selectivity towards C2 products is very low, almost negligible. However, with longer electrodeposition times, catalysts with enhanced dendritic extensions (Fig. SI-4†) show higher selectivity. Therefore, in view of these observations it is concluded that the morphology of the electrocatalysts plays a pivotal role in deciding the ECR product profile, where the spiky ends of Cu-dendrites demonstrate significantly higher selectivity for the C2 pathway than the C1 mechanism, and smooth surfaces favour the C1 pathway over the C2 pathway. This observation and interpretation sounds obvious in light of the residence time of intermediates over the catalyst surfaces; the roughened surfaces provide higher residence time favourable for the C–C coupling process while smooth surfaces mainly produce C1 products.
In addition to the morphology, the crystalline features of the catalysts, especially the exposed facets, exhibit a profound effect on the ECR activity as well as selectivity. Hori et al.48 demonstrated that while ECR over Cu(100) favours the formation of C2H4, the Cu(111) surfaces favour the production of CH4. Koper and co-workers47 with their online electrochemical mass spectrometry (OLEMS) measurements confirmed that ethylene production is favoured on Cu(100) surfaces at lower (or less negative) potentials, but for Cu(111) surfaces, more negative potentials are needed. As per Nørskov et al.,49 the activation barrier for CO dimerization on Cu(100) is lower than that on Cu(111), probably because of a more exergonic *CO adsorption energy on the former. Overall, these studies pinpoint the profound effect of crystalline facets on the catalytic performance of Cu-based materials for ECR. While analysing our results in light of the crystallinity of the Cu-based electrodeposits crafted and tested in the present work, we noted that the higher the peak intensity ratio of (111)/(200) planes, the greater the formation of C2 products, in both electrolytes. Thus, the formation of ethylene or ethane seems to be determined by the nature of the electrolyte employed for the ECR, more than the type of crystalline facets exposed at the electrocatalyst/electrolyte interface.
According to the mechanistic pathway proposed by Yeo and co-workers52 for ECR over thick oxide-derived copper catalysts, the preferential formation of ethane follows the *CH3–*CH3 dimerization route while the predominating formation of ethylene follows the *CO–*CO dimerization pathway. They have also suggested that the higher selectivity for ethane and ethanol formation can be ascribed to the stabilization of –CH3 intermediates by the Cu+ sites in the thick oxide-derived Cu-films. It is pertinent to mention here that the XRD studies have clearly ruled out the possibility of substantial presence of Cu+ sites on well-developed dendrites crafted for the present study. Therefore, in the absence of any profound evidence for Cu+ sites on the Cu based electrodeposits crafted and tested for ECR in the present work, we ascribe the selective formation of ethane in wet DMSO and ethylene in aqueous medium to *CO–*CO stabilization by the electrolyte media. This is in accordance with the analysis of chronopotentiometry curves recorded on the most active catalyst in Ar, CO, and CO2 saturated 0.2 M KHCO3 solutions (Fig. 2).
Nørskov and co-workers,49 based on their DFT simulations and theoretical calculations, have suggested that the presence of a charged layer of water molecules on Cu-surfaces plays an important role in the stabilization of the CO dimer configuration. In particular, they have noted that while the solvent stabilizes the CO dimer by ca. 0.6 eV, the applied electric field stabilizes the same dimer by 0.2 eV. Without the charged water layer, the formation of CO dimers has been noted to be inhibitory/endergonic. Thus, a combination of the solvation effects and the resulting fields has been found to significantly reduce both the thermodynamic and kinetic energy requirements for CO dimerization from the adsorbed *CO. So, considering the ample presence of the water layer in case of the aqueous medium especially as compared to the wet DMSO, it can be concluded that the CO2 reduction follows the CO–CO dimerization route in an aqueous electrolyte, whereas it would follow the *CH3–*CH3 dimerization route in wet DMSO. Therefore, the preferential formation of ethylene in aqueous medium along with the preferential formation of ethane in wet DMSO in the current work seems to be in good agreement with this mechanism. The prevalence of the *CO–*CO dimerization mechanism in 0.2 M KHCO3 was also supported by the chronopotentiometric analysis shown in Fig. 2.
Overall, it was observed that the dendritic copper micro-structures having an elaborate nano-morphology, and synthesized under specific electro-synthesis conditions, selectively electroreduce the CO2 into ethylene and other C1 and C2 hydrocarbons in aqueous 0.2 M KHCO3 solution. The b-Cu-dendrites show a much higher selectivity of 83% for C2 over C1 hydrocarbons, generating ethylene with a selectivity of 74% among all the ECR products. Importantly, the formation of the most common parallel reaction products, viz. CO and H2, seems to be very minimal for ECR over extensively branched dendritic Cu-electrodeposits. These observations support the general conclusion that the key to the C2H4 formation via C–C coupling lies in having a distinct nano-morphology. Moreover, the comparison of the ECR efficiency and selectivity in DMSO and aqueous medium suggests that in the formation of C2 hydrocarbons through C–C coupling, the nature of the reaction medium is also indispensable. Thus, the present work suggests that both the catalyst surface characteristics and the nature of the ECR electrolyte play a pivotal role in determining the selectivity for C1 and C2 hydrocarbons. Table 1 shows the comparative catalytic activity of similar dendritic catalysts with the b-Cu dendritic catalyst, studied here.
| Material | Solvent | Voltage (V) | FE (%) C2H4 | FE (%) H2 | Ref. |
|---|---|---|---|---|---|
| Plasma-modified dendritic Cu | 0.1 M KHCO3 | −0.9 (RHE) | ∼28 | 48 | 50 |
| Electrodeposited Cu dendrites | 0.1 M KHCO3 | −1.7 (Ag/AgCl) | 26 | 38 | 53 |
| Ag embedded Cu nanoporous arrays | 0.5M KHCO3 | −1.2 (RHE) | 43.1 | 18 | 54 |
| Cu nanodendrites | 0.1 M KHCO3 | −1.2 (RHE) | 22.3 | ∼23 | 55 |
| 4H Au@Cu nanoribbons | 0.1 M KHCO3 | −1.11 (RHE) | 44.9 | ∼31 | 56 |
| Cu-coated Cu3N NPs | 0.1 M KHCO3 | −0.95 (RHE) | 39 | — | 57 |
| Cu NP ensemble | 0.1 M KHCO3 | −0.86 (RHE) | 33.2 | — | 58 |
| Twinned Cu NWs | 0.1 M KHCO3 | — | <5 | — | 59 |
| Cu dendrites | 0.2 M KHCO3 | −1.0 (RHE) | 43 | 28 | This work |
4. Conclusion
In summary, we demonstrate that the shape and morphology of the Cu electrodeposit and the nature of the solvent system employed play a critical role in deciding the efficiency and selectivity for production of various ECR products. Importantly, the dendritic structures with spikey ends and edges are more selective for ethylene than other structures. The higher activity of the fully exposed branches of dendrites can be attributed to the nano-hot spots, diffusion control of ionic species involved in lowering the HER, and an increase in *CO adsorption. The prevalence of the *CO–*CO dimerization route is also supported by the chronopotentiometry curves recorded for the most active electrocatalysts in Ar, CO and CO2 saturated 0.2 M KHCO3 electrolyte solutions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
NR acknowledges CSIR-UGC for the fellowship. PPI thanks DST-SERB, Govt. of India for the financial support, vide SB/EMEQ-339/2014 under the “Empowerment and Equity Opportunities for Excellence in Science”. Authors are thankful to the Central Research Facility (CRF) and Nanoscale Research Facilities (NRF) of IIT Delhi for assistance with material characterization.References
- H. Ritchie and M. Roser, Fossil Fuels, https://ourworldindata.org/fossil-fuels.
- T. Wang, A. H. A. Park, Y. Shi and G. Gadikota, Carbon Dioxide Capture and Utilization – Closing the Carbon Cycle, 2019, vol. 33, DOI: DOI:10.1021/acs.energyfuels.8b04502.
- M. Gross, Closing the Carbon Cycle, Curr. Biol., 2014, 24(13), 583–585, DOI:10.1016/j.cub.2014.06.051.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, New Insights into the Electrochemical Reduction of Carbon Dioxide on Metallic Copper Surfaces, Energy Environ. Sci., 2012, 5(5), 7050–7059, 10.1039/c2ee21234j.
- P. Gabrielli, M. Gazzani and M. Mazzotti, The Role of Carbon Capture and Utilization, Carbon Capture and Storage, and Biomass to Enable a Net-Zero-CO2 Emissions Chemical Industry, Ind. Eng. Chem. Res., 2020, 59(15), 7033–7045, DOI:10.1021/acs.iecr.9b06579.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, The Teraton Challenge. A Review of Fixation and Transformation of Carbon Dioxide, Energy Environ. Sci., 2009, 3, 43–81, 10.1039/b912904a.
- S. Chaemchuen, N. A. Kabir, K. Zhou and F. Verpoort, Metal–Organic Frameworks for Upgrading Biogas via CO2 Adsorption to Biogas Green Energy, Chem. Soc. Rev., 2013, 42(24), 9304–9332, 10.1039/c3cs60244c.
- S. Zeng, X. Zhang, L. Bai, X. Zhang, H. Wang, J. Wang, D. Bao, M. Li, X. Liu and S. Zhang, Ionic-Liquid-Based CO2 Capture Systems: Structure, Interaction and Process, Chem. Rev., 2017, 117(14), 9625–9673, DOI:10.1021/acs.chemrev.7b00072.
- K. Malik, S. Singh, S. Basu and A. Verma, Electrochemical Reduction of CO2 for Synthesis of Green Fuel, Wiley Interdiscip. Rev.: Energy Environ., 2017, 6, e244, DOI:10.1002/wene.244.
- Y. Hori, K. Kikuchi and S. Suzuki, Production of CO and CH4 in Electrochemical Reduction of CO2 at Metal Electrodes in Aqueous Hydrogencarbonate Solution, Chem. Lett., 1985, 14(11), 1695–1698, DOI:10.1246/cl.1985.1695.
- Y. Song, R. Peng, D. K. Hensley, P. V. Bonnesen, L. Liang, Z. Wu, H. M. Meyer, M. Chi, C. Ma, B. G. Sumpter and A. J. Rondinone, High-Selectivity Electrochemical Conversion of CO2 to Ethanol Using a Copper Nanoparticle/N-Doped Graphene Electrode, ChemistrySelect, 2016, 1(19), 6055–6061, DOI:10.1002/slct.201601169.
- C. W. Lee, K. D. Yang, D.-H. Nam, J. H. Jang, N. H. Cho, S. W. Im and K. T. Nam, Defining a Materials Database for the Design of Copper Binary Alloy Catalysts for Electrochemical CO2 Conversion, Adv. Mater., 2018, 1704717, DOI:10.1002/adma.201704717.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Tailoring Copper Nanocrystals towards C2 Products in Electrochemical CO2 Reduction, Angew. Chem., Int. Ed., 2016, 55(19), 5789–5792, DOI:10.1002/anie.201601582.
- N.-T. Suen, Z.-R. Kong, C.-S. Hsu, H.-C. Chen, C.-W. Tung, Y.-R. Lu, C.-L. Dong, C.-C. Shen, J.-C. Chung and H. M. Chen, Morphology Manipulation of Copper Nanocrystals and Product Selectivity in the Electrocatalytic Reduction of Carbon Dioxide, ACS Catal., 2019, 9(6), 5217–5222, DOI:10.1021/acscatal.9b00790.
- H. Wang, E. Matios, C. Wang, J. Luo, X. Lu, X. Hu and W. Li, Rapid and Scalable Synthesis of Cuprous Halide-Derived Copper Nano-Architectures for Selective Electrochemical Reduction of Carbon Dioxide, Nano Lett., 2019, 19, 3932, DOI:10.1021/acs.nanolett.9b01197.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, Particle Size Effects in the Catalytic Electroreduction of CO2 on Cu Nanoparticles, J. Am. Chem. Soc., 2014, 136(19), 6978–6986, DOI:10.1021/ja500328k.
- M. König, J. Vaes, E. Klemm and D. Pant, Solvents and Supporting Electrolytes in the Electrocatalytic Reduction of CO2, iScience, 2019, 19, 135–160, DOI:10.1016/j.isci.2019.07.014.
- Y. Oh, H. Vrubel, S. Guidoux and X. Hu, Electrochemical Reduction of CO2 in Organic Solvents Catalyzed by MoO2, Chem. Commun., 2014, 50(29), 3878–3881, 10.1039/c3cc49262a.
- N. Rashid, M. A. Bhat, A. Das and P. P. Ingole, Unprecedented Lower Over-Potential for CO2 Electro-Reduction on Copper Oxide Anchored to Graphene Oxide Microstructures, J. CO2 Util., 2020, 39, 101178, DOI:10.1016/j.jcou.2020.101178.
- S. Subramanian, K. R. Athira, M. Anbu Kulandainathan, S. Senthil Kumar and R. C. Barik, New Insights into the Electrochemical Conversion of CO2 to Oxalate at Stainless Steel 304L Cathode, J. CO2 Util., 2020, 36, 105–115, DOI:10.1016/j.jcou.2019.10.011.
- M. C. Figueiredo, V. Trieu and M. T. M. Koper, Electrochemical Conversion of CO2 into Organic Carbonates - Products and Intermediates, ACS Sustainable Chem. Eng., 2019, 7(12), 10716–10723, DOI:10.1021/acssuschemeng.9b01390.
- N. Rashid, M. A. Bhat and P. P. Ingole, Dendritic Copper Microstructured Electrodeposits for Efficient and Selective Electrochemical Reduction of Carbon Dioxide into C1 and C2 Hydrocarbons, J. CO2 Util., 2020, 38(December 2019), 385–397, DOI:10.1016/j.jcou.2020.02.017.
- N. Uemoto, M. Furukawa, I. Tateishi, H. Katsumata and S. Kaneco, Electrochemical Carbon Dioxide Reduction in Methanol at Cu and Cu2O-Deposited Carbon Black Electrodes, Chem. Eng., 2019, 3(1), 15, DOI:10.3390/chemengineering3010015.
- S. Kaneco, K. Iiba, S. K. Suzuki, K. Ohta and T. Mizuno, Electrochemical Reduction of Carbon Dioxide to Hydrocarbons with High Faradaic Efficiency in LiOH/Methanol, J. Phys. Chem. B, 1999, 103(35), 7456–7460, DOI:10.1021/jp990021j.
- M. Moura de Salles Pupo and R. Kortlever, Electrolyte Effects on the Electrochemical Reduction of CO2, ChemPhysChem, 2019, 20(22), 2926–2935, DOI:10.1002/cphc.201900680.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, Promoter Effects of Alkali Metal Cations on the Electrochemical Reduction of Carbon Dioxide, J. Am. Chem. Soc., 2017, 139(32), 11277–11287, DOI:10.1021/jacs.7b06765.
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager and A. T. Bell, Hydrolysis of Electrolyte Cations Enhances the Electrochemical Reduction of CO2 over Ag and Cu, J. Am. Chem. Soc., 2016, 138, 13006–13012, DOI:10.1021/jacs.6b07612.
- R. B. Song, W. Zhu, J. Fu, Y. Chen, L. Liu, J. R. Zhang, Y. Lin and J. J. Zhu, Electrode Materials Engineering in Electrocatalytic CO2 Reduction: Energy Input and Conversion Efficiency, Adv. Mater., 2019, 32(27), 1903796, DOI:10.1002/adma.201903796.
- M. T. Mota-Martinez, J. P. Hallett and N. Mac Dowell, Solvent Selection and Design for CO2 Capture-How We Might Have Been Missing the Point, Sustainable Energy Fuels, 2017, 1, 2078–2090, 10.1039/c7se00404d.
- B. Jiang, L. Yu, L. Wu, D. Mu, L. Liu, J. Xi and X. Qiu, Insights into the Impact of the Nafion Membrane Pretreatment Process on Vanadium Flow Battery Performance, ACS Appl. Mater. Interfaces, 2016, 8(19), 12228–12238, DOI:10.1021/acsami.6b03529.
- C. S. Chen, A. D. Handoko, J. H. Wan, L. Ma, D. Ren and B. S. Yeo, Stable and Selective Electrochemical Reduction of Carbon Dioxide to Ethylene on Copper Mesocrystals, Catal. Sci. Technol., 2015, 5(1), 161–168, 10.1039/c4cy00906a.
- N. Rashid, M. A. Bhat, U. K. Goutam and P. P. Ingole, Electrochemical Reduction of CO2 to Ethylene on Cu/CuXO-GO Composites in Aqueous Solution, RSC Adv., 2020, 10(30), 17572–17581, 10.1039/d0ra02754e.
- L. Ou and Z. He, Potential-Dependent Competitive Electroreduction of CO2 into CO and Formate on Cu(111) from an Improved H Coverage-Dependent Electrochemical Model with Explicit Solvent Effect, ACS Omega, 2020, 5, 12735–12744, DOI:10.1021/acsomega.0c00227.
- Y. Hori, A. Murata and R. Takahashi, Formation of Hydrocarbons in the Electrochemical Reduction of Carbon Dioxide at a Copper Electrode in Aqueous Solution, J. Chem. Soc., Faraday Trans. 1, 1989, 85(8), 2309–2326, 10.1039/F19898502309.
- D. W. DeWulf, T. Jin and A. J. Bard, Electrochemical and Surface Studies of Carbon Dioxide Reduction to Methane and Ethylene at Copper Electrodes in Aqueous Solutions, J. Electrochem. Soc., 1989, 136(6), 1686–1691, DOI:10.1149/1.2096993.
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, Electroreduction of Carbon Monoxide to Methane and Ethylene at a Copper Electrode in Aqueous Solutions at Ambient Temperature and Pressure, J. Am. Chem. Soc., 1987, 109(2), 5022–5023 CrossRef CAS.
- Y. Huang, A. D. Handoko, P. Hirunsit and B. S. Yeo, Electrochemical Reduction of CO2 Using Copper Single-Crystal Surfaces: Effects of CO* Coverage on the Selective Formation of Ethylene, ACS Catal., 2017, 7(3), 1749–1756, DOI:10.1021/acscatal.6b03147.
- J. Kim, W. Choi, J. Woo Park, C. Kim, M. Kim and H. Song, Branched Copper Oxide Nanoparticles Induce Highly Selective Ethylene Production by Electrochemical Carbon Dioxide Reduction, J. Am. Chem. Soc., 2019, 141(17), 6986–6994, DOI:10.1021/jacs.9b00911.
- C. Reller, R. Krause, E. Volkova, B. Schmid, S. Neubauer, A. Rucki, M. Schuster and G. Schmid, Selective Electroreduction of CO2 toward Ethylene on Nano Dendritic Copper Catalysts at High Current Density, Adv. Energy Mater., 2017, 7(12), 1602114, DOI:10.1002/aenm.201602114.
- K. Klingan, T. Kottakkat, Z. P. Jovanov, S. Jiang, C. Pasquini, F. Scholten, P. Kubella, A. Bergmann, B. Roldan Cuenya, C. Roth and H. Dau, Reactivity Determinants in Electrodeposited Cu Foams for Electrochemical CO2 Reduction, ChemSusChem, 2018, 11(19), 3449–3459, DOI:10.1002/cssc.201801582.
- D. Kim, S. Lee, J. D. Ocon, B. Jeong, J. K. Lee and J. K. Lee, Insights into an Autonomously Formed Oxygen-Evacuated Cu2O Electrode for the Selective Production of C2H4 from CO2, Phys. Chem. Chem. Phys., 2015, 17(2), 824–830, 10.1039/c4cp03172e.
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, Enhanced Formation of Ethylene and Alcohols at Ambient Temperature and Pressure in Electrochemical Reduction of Carbon Dioxide at a Copper Electrode, J. Chem. Soc., Chem. Commun., 1988, 17–19, 10.1039/C39880000017.
- M. Ma, K. Djanashvili and W. A. Smith, Controllable Hydrocarbon Formation from the Electrochemical Reduction of CO2 over Cu Nanowire Arrays, Angew. Chem., Int. Ed., 2016, 55(23), 6680–6684, DOI:10.1002/anie.201601282.
- Y. Huang, A. D. Handoko, P. Hirunsit and B. S. Yeo, Electrochemical Reduction of CO2 Using Copper Single-Crystal Surfaces: Effects of *CO Coverage on the Selective Formation of Ethylene, 2017, DOI: DOI:10.1021/acscatal.6b03147.
- S. Liu, H. Tao, L. Zeng, Q. Liu, Z. Xu, Q. Liu and J.-L. Luo, Shape-Dependent Electrocatalytic Reduction of CO2 to CO on Triangular Silver Nanoplates, J. Am. Chem. Soc., 2017, 139, 22, DOI:10.1021/jacs.6b12103.
- K. D. Yang, C. W. Lee, J. H. Jang, T. R. Ha and K. T. Nam, Rise of Nano Effects in Electrode during Electrocatalytic CO2 Conversion, Nanotechnology, 2017, 28(35), 2001, DOI:10.1088/1361-6528/aa7b0b.
- F. Calle-Vallejo and M. T. M. Koper, Theoretical Considerations on the Electroreduction of CO to C2 Species on Cu(100) Electrodes, Angew. Chem., Int. Ed., 2013, 52(28), 7282–7285, DOI:10.1002/anie.201301470.
- Y. Hori, R. Takahashi, Y. Yoshinami and A. Murata, Electrochemical Reduction of CO at a Copper Electrode, J. Phys. Chem. B, 1997, 101(36), 7075–7081, DOI:10.1021/jp970284i.
- J. H. Montoya, C. Shi, K. Chan and J. K. Nørskov, Theoretical Insights into a CO Dimerization Mechanism in CO2 Electroreduction, J. Phys. Chem. Lett., 2015, 6(11), 2032–2037, DOI:10.1021/acs.jpclett.5b00722.
- F. Scholten, I. Sinev, M. Bernal and B. R. Cuenya, Plasma-Modified Dendritic Cu Catalyst for CO2 Electroreduction, ACS Catal., 2019, 9(I), 5496–5502, DOI:10.1021/acscatal.9b00483.
- C. Amatore, J. M. Saveant and J. M. Savéant, Mechanism and Kinetic Characteristics of the Electrochemical Reduction of Carbon Dioxide in Media of Low Proton Availability, J. Am. Chem. Soc., 1981, 103(17), 5021–5023, DOI:10.1021/ja00407a008.
- A. D. Handoko, K. W. Chan and B. S. Yeo, -CH3 Mediated Pathway for the Electroreduction of CO2 to Ethane and Ethanol on Thick Oxide-Derived Copper Catalysts at Low Overpotentials, ACS Energy Lett., 2017, 2(9), 2103–2109, DOI:10.1021/acsenergylett.7b00514.
- T. Qin, Y. Qian, F. Zhang, A. Chen and B. Lin, Enhanced Electrochemical Reduction of CO2 to Ethylene on Electrodeposited Copper in 0.1 M KHCO3, Int. J. Electrochem. Sci., 2018, 13(11), 10101–10112, DOI:10.20964/2018.11.42.
- L. Hou, J. Han, C. Wang, Y. Zhang, Y. Wang, Z. Bai, Y. Gu, Y. Gao and X. Yan, Ag Nanoparticle Embedded Cu Nanoporous Hybrid Arrays for the Selective Electrocatalytic Reduction of CO2 towards Ethylene, Inorg. Chem. Front., 2020, 7(10), 2097–2106, 10.1039/d0qi00025f.
- M. Wu, C. Zhu, K. Wang, G. Li, X. Dong, Y. Song, J. Xue, W. Chen, W. Wei and Y. Sun, Promotion of CO2 Electrochemical Reduction via Cu Nanodendrites, ACS Appl. Mater. Interfaces, 2020, 12, 11562–11569, DOI:10.1021/acsami.9b21153.
- Y. Chen, Z. Fan, J. Wang, C. Ling, W. Niu, Z. Huang, G. Liu, B. Chen, Z. Lai, X. Liu, B. Li, Y. Zong, L. Gu, J. Wang, X. Wang, H. Zhang, J. Wang, X. Wang and H. Zhang, Ethylene Selectivity in Electrocatalytic CO2 Reduction on Cu Nanomaterials: A Crystal Phase-Dependent Study, J. Am. Chem. Soc., 2020, 142(29), 12760–12766, DOI:10.1021/jacs.0c04981.
- Z. Q. Liang, T. T. Zhuang, A. Seifitokaldani, J. Li, C. W. Huang, C. S. Tan, Y. Li, P. De Luna, C. T. Dinh, Y. Hu, Q. Xiao, P. L. Hsieh, Y. Wang, F. Li, R. Quintero-Bermudez, Y. Zhou, P. Chen, Y. Pang, S. C. Lo, L. J. Chen, H. Tan, Z. Xu, S. Zhao, D. Sinton and E. H. Sargent, Copper-on-Nitride Enhances the Stable Electrosynthesis of Multi-Carbon Products from CO2, Nat. Commun., 2018, 9(1), 1–8, DOI:10.1038/s41467-018-06311-0.
- D. Kim, C. S. Kley, Y. Li and P. Yang, Copper Nanoparticle Ensembles for Selective Electroreduction of CO2 to C2–C3 Products, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(40), 10560–10565, DOI:10.1073/pnas.1711493114.
- Y. Li, F. Cui, M. B. Ross, D. Kim, Y. Sun and P. Yang, Structure-Sensitive CO2 Electroreduction to Hydrocarbons on Ultrathin 5-Fold Twinned Copper Nanowires, Nano Lett., 2017, 17(2), 1312–1317, DOI:10.1021/acs.nanolett.6b05287.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1se01255j |
| This journal is © The Royal Society of Chemistry 2022 |