
Hydroxyl-anchored covalent organic crown-based polymers for CO2 fixation into cyclic carbonates under mild conditions†
Yongjing
Hao
a,
Xiuli
Yan
a,
Tao
Chang
 *ab,
Xiaohuan
Liu
a,
Lianwei
Kang
a,
Zheng
Zhu
*a,
Balaji
Panchal
a and
Shenjun
Qin
*a
*ab,
Xiaohuan
Liu
a,
Lianwei
Kang
a,
Zheng
Zhu
*a,
Balaji
Panchal
a and
Shenjun
Qin
*a
aKey Laboratory of CO2 Utilization of Handan City, College of Material Science and Engineering, Hebei University of Engineering, Handan 056038, Hebei, China. E-mail: changt03@sina.com; zhuzheng@hebeu.edu.cn; qinsi528@hebeu.edu.cn; Tel: +86-310-3968760 Tel: +86-310-3967962
bKey Laboratory of Heterocyclic Compounds of Hebei Province, Handan College, Handan 056005, Hebei, China
First published on 22nd November 2021
Abstract
Covalent organic polymers (COPs) are a kind of promising material for carbon dioxide (CO2) reutilization. Herein, a series of covalent organic crown-based polymers (COCP–H, COCP–OH, COCP–CH3, COCP–Bu, COCP–NH2) were designed and synthesized. These synthesized COCPs were characterized by FT-IR, SEM, BET and 13C CP MAS NMR. Notably, the novel COCP–OH catalyst with hydroxyl and crown ether groups on the framework simultaneously was particularly effective in cycloaddition reactions, giving a cyclic carbonate yield of 94.8% after 24 h at 70 °C with 1 bar of CO2, using 0.0735 mmol of KI and 71 mg of COCP–OH. It is worth considering that there is a synergetic effect between the hydroxyl group and KI activated by the crown ether fragment, which plays a crucial role in accelerating the reaction rate. The coordinated interaction of crown ether with KI was also confirmed by X-ray photoelectron spectroscopy (XPS). Thus, this study provides a new approach to fabricate multifunctional COP materials for the coupling reaction of CO2 under mild conditions.
Introduction
The rapid increase in atmospheric CO2 levels is the main reason for climate change. To counter this problem, there are two common solution strategies as below: carbon capture and storage (CCS) and carbon capture and utilization (CCU). In the CCU, CO2 is considered to be a valuable resource for the biological or chemical synthesis, not as waste.1–3 Among the products, five-membered cyclic carbonates (5CCs) which were obtained by utilization of CO2 have received considerable attention due to 100% atom-economic and environment-friendly processes of this reaction. Moreover, 5CCs have wide applications, including as alternative polar aprotic solvents, electrolytes in lithium-ion batteries, polymer precursors and fuel additives.4–6 However, the reaction between CO2 and epoxides is induced only in the presence of a catalyst due to the high bond energy of 750 kJ mol−1 of the CO2 molecule.1,7 Thus, the development of new, efficient and practical catalysts has become an important topic for 5CC synthesis.In the past decade, some catalysts including homogeneous and heterogeneous catalytic systems have been developed for the cycloaddition reaction.4,8–10 Heterogeneous catalytic systems are of more interest due to the properties of easy recovery. Several heterogeneous catalysts including covalent organic polymers (COPs),8,11–15 metal organic frameworks (MOFs),16–19 silica based catalysts,20,21 zeolites,21–23 metal oxides24–26 and carbon-based catalysts27,28 have also been reported. COPs are one of the promising heterogeneous catalysts which are composed of well-defined periodic frameworks, skillfully constructed via strong covalently bonded organic groups.29,30 To date, increasing attention has been paid to this kind of catalyst due to the advantages of chemical and thermal stability, ready availability, affordability, non-toxicity and ease of functionalization.3,29 For instance, Liu and co-authors developed an organic polymer containing triazine toward fixation of atmospheric CO2 with epoxides.13 Kureshy et al. employed a nitrogen-rich cyanuric–urea polymer (CUP) for this reaction.14 Recently, Yavuz's group reported a hydrocarbon framework of quaternary ammonium polymer applied in the CO2 cycloaddition reaction.15 Although significant achievements have been made in this area, most of these catalytic systems require harsh conditions such as high temperature, high pressure, multi-step synthesis of catalysts or the use of metals, and thus CO2 fixation under mild conditions is still a challenge. Thus, developing efficient and novel heterogeneous catalysts for the cycloaddition reaction is highly desirable.
In developing COPs, introduction of high-quality functional groups is the critical factor. Early studies have established that crown ethers are good activated agents for KI in the conversion of CO2 with epoxides.31 Meanwhile, hydroxyl groups as hydrogen-bond donors (HBDs) play an important role in the cycloaddition reaction of CO2 as reported in the literature.32 Epoxides could be activated via hydrogen bonding between the hydroxyl groups and O atoms in epoxides. It is envisioned that polymers with multifunctional groups such as crown ether groups and strong hydrogen bond sites will present efficient catalytic activity for the cycloaddition reaction.
We were interested in the conversion of CO2 with epoxides and a series of catalysts have been developed and studied.8,11,12,33–36 Herein, a series of covalent organic crown-based polymers (COCP–H, COCP–OH, COCP–CH3, COCP–Bu and COCP–NH2) all decorated with crown ether functional groups were designed and synthesized for the cycloaddition reaction of CO2 and epoxide, as shown in Scheme 1. Among the COCPs, the novel COCP–OH catalysts decorated with hydroxyl and crown ether groups simultaneously were particularly effective in cycloaddition reactions. The properties of COCP–OH catalysts could establish them as perfect multifunctional catalysts for the CO2 fixation reaction with epoxides under mild conditions.
 | ||
| Scheme 1 Cycloaddition reaction catalyzed by COCPs and alkali-metal salts. | ||
Results and discussion
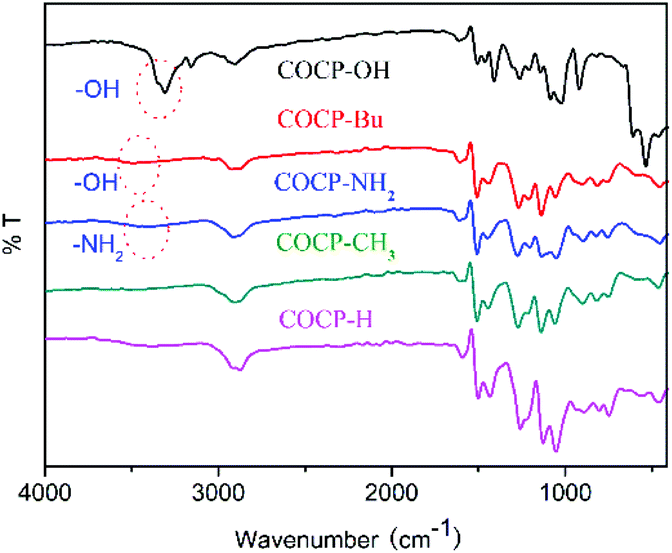
The COCPs as catalysts were synthesized through the Friedel–Crafts alkylation reaction from 1, 4-bis(bromomethyl)benzene, dibenzo-18-crown and different substituted aromatics as active sites, respectively, as shown in Scheme 1. In the five COCPs, crown ether functional groups as the excellent activated sites of KI were all introduced. Notably, the two novel COCP–OH and COCP–Bu contained crown ether groups and the active sites of –OH were considered as strong HBDs. The COCPs are not soluble in common reagents.The FT-IR spectra of COCPs are demonstrated in Fig. 1. As seen from Fig. 1, the spectra of COCP–H and COCP–CH3 are similar, while in the spectra of COCP–OH and COCP–Bu, the peaks appeared at about 3350 cm−1 and 3490 cm−1 which were ascribed to the stretching of the –OH group in the skeleton.37 In the spectrum of COCP–NH2, the signal at 3350 cm−1 attributed to –NH2 was detected. The morphologies of COCPs were further identified from scanning electron microscopy (SEM) images shown in Fig. 2. The results confirmed that all COCPs are composed of small particles. In the TEM images of COCP–OH, sponge-like micropores and mesopores with disordered arrangements can be observed, as shown in Fig. S1.†
 | ||
| Fig. 1 The FT-IR spectra of the COCPs. | ||
 | ||
| Fig. 2 SEM images of COCP–H (a and b), COCP–OH(c and d), COCP–CH3 (e and f), COCP–Bu (g and h) and COCP–NH2 (i and j). | ||
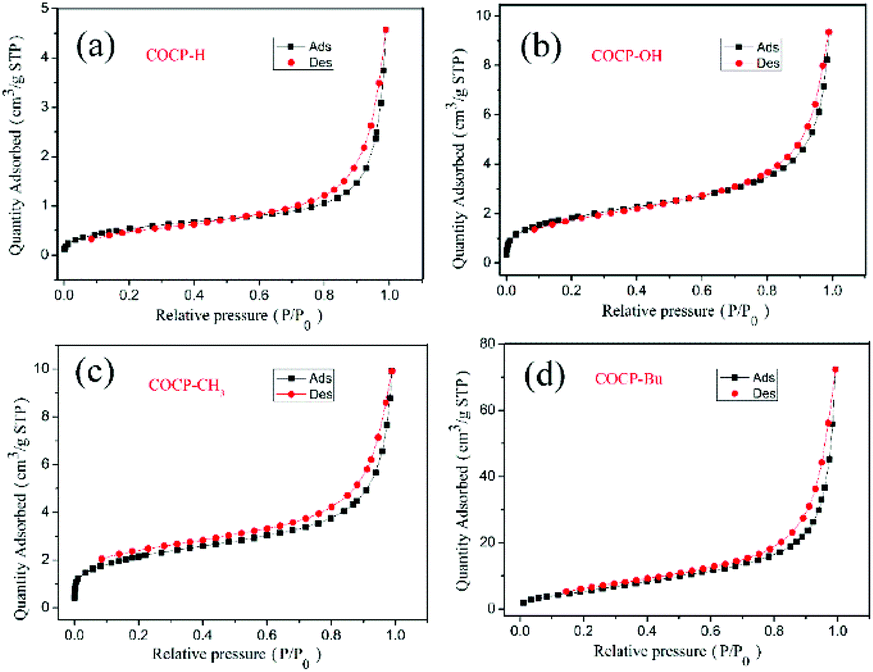
The BET surface area and the pore structure of COCPs are very important factors in the study of heterogeneous catalysts, and thus the N2 adsorption–desorption properties were tested. As demonstrated in Fig. 3 and S2,† the N2 adsorption–desorption isotherms all belong to type III as reported.14 The BET surface areas of COCP–H, COCP–OH, COCP–CH3, COCP–Bu and COCP–NH2 are 2.0, 6.2, 7.2, 20.7 and 6.7 m2 g−1, respectively. The pore volumes are 0.0071, 0.011, 0.015, 0.032 and 0.0084 cm3 g−1, respectively. The average pore diameters are 14.1, 6.7, 7.9, 4.9 and 4.9 nm, respectively. Although the surface areas of the COCPs are relatively low, they can activate the cycloaddition reaction of CO2 with epoxides through crown ethers and hydroxyl functional groups. The porous structure is consistent with the results of SEM and TEM.
 | ||
| Fig. 3 The N2 adsorption and desorption isotherms of COCPs: (a) COCP–H, (b) COCP–OH, (c) COCP–CH3, and (d) COCP–Bu. | ||
In order to screen out the optimum catalyst among the COCPs, the cycloaddition reaction of CO2 with epichlorohydrin (ECH) was selected as a representative reaction, and every run was performed under the same conditions. The results are illustrated in Table 1. When only KI was introduced but no COCP was added into the system, the yield of cyclic carbonate was only 5% due to the low catalytic activity and solubility of KI, which is similar to that reported in the literature (entry 5).11,32,38 However, when COCPs and KI were added into the catalytic system, a significant increment in the yields of products was observed (entries 1–5), which is attributed to the synergistic interaction of crown ether with KI, along with activation of KI and improvement of the nucleophilicity of iodine anions.31,39–41 To explore the effect of substituted modifiers connected with the phenyl group on the catalytic performance in CO2 fixation reaction, initially, we compared the activity of COCP–H and COCP–CH3 with the modifier methyl group and unsubstituted phenyl group, respectively. These two catalysts exhibit commensurate conversions of 55.7% and 57.9%, because of a similar secondary structure (entries 1, 3). Comparison of the influence of HBDs including hydroxyl and amino groups (COCP–OH and COCP–NH2) was performed (entries 2 and 5).
Apparently, increased yields of 94.8% and 67.1% were detected, which are better than those of non-HBD catalysts, because HBD groups could activate epoxides by H-bonding resulting in nucleophilic attack of the iodine anion easily, that is the speed-determining step of CO2 fixation reaction. Essentially, oxygen possesses more electronegativity than nitrogen, and therefore the hydroxyl group exhibits stronger hydrogen bond activation ability than the amino group and an outstanding yield was observed. A yield of 89.4% was also detected when ortho t-butyl was linked on the phenyl group, which is slightly lower than that of the hydroxyl group only (entry 4), due to the steric hindrance and electron-donating property of t-butyl in COCP–Bu, which are other critical factors in catalytic performance.42
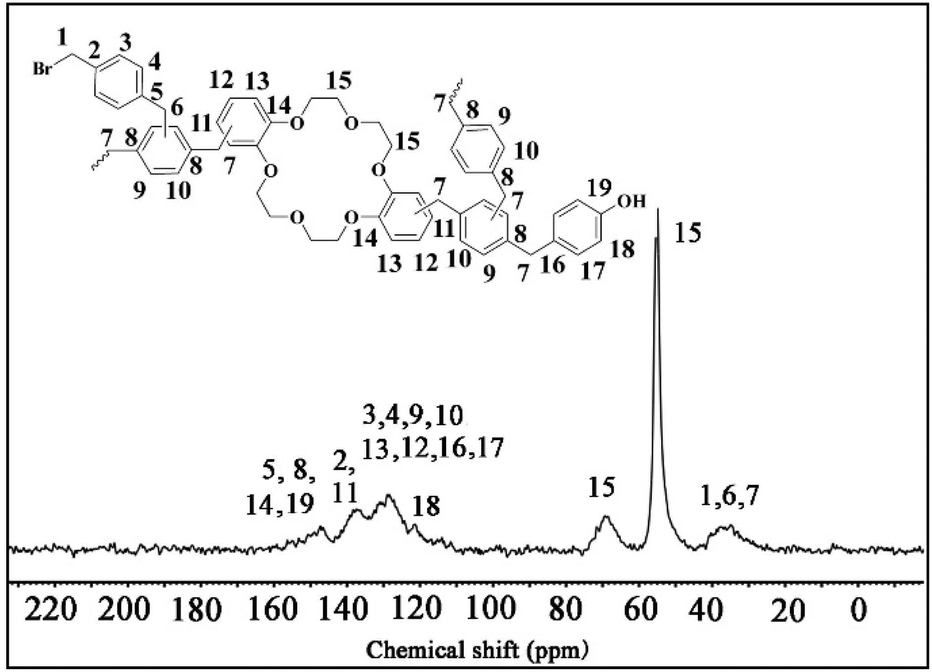
In order to obtain the skeleton information of COCP–OH, the 13C CP MAS NMR spectrum was recorded. As demonstrated in Fig. 4, a band at 147.3 ppm is attributed to C5, C8, C14 and C19 in the benzene rings, while a broad peak with four signals at 137.3 ppm (C2 and C11 on the aromatic rings), 131.0 ppm (C3, C4, C9 and C10 on the benzene rings), 128.4 ppm (C12, C13, C16 and C17 on the aromatic rings) and 121.5 ppm (C18 on the benzene rings) could be seen. The peaks at 68.8 ppm and 55.0 ppm are ascribed to C15 in the methylene of the crown ether skeleton. The signal at 36.4 ppm belongs to C1, C6 and C7 in methylene, respectively.
 | ||
| Fig. 4 The 13C CP MAS NMR spectrum of the catalyst of COCP–OH. | ||
To evaluate the effects of co-catalyst on the conversion of CO2, alkali metal salts were varied while other conditions were kept unchanged. As exhibited in Fig. 5, potassium iodide (KI), sodium iodide (NaI) or lithium iodide (LiI) with COCP–OH gave the yields of 51.4%, 39.1% and 29.9% respectively. Compared to the co-catalyst of KI, potassium bromide (KBr) and potassium chloride (KCl) with COCP–OH demonstrated lower catalytic activity, and the yields were 43.1% and 34.4 respectively. The co-catalyst of KI showed the best catalytic activity. These results are similar to previous reports.8,32,43
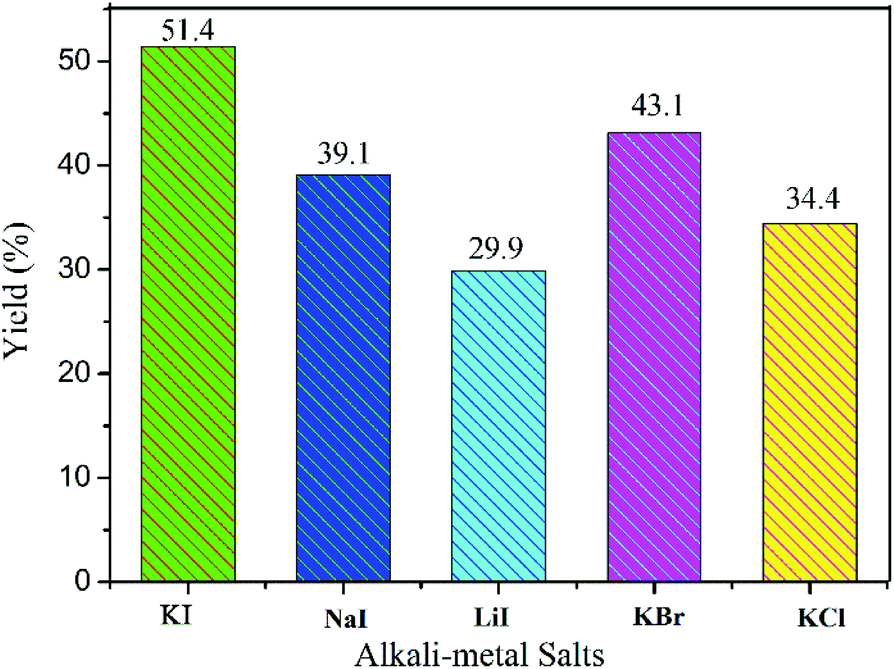 | ||
| Fig. 5 Effects of co-catalyst on the conversion of CO2 with ECH. Reaction conditions: ECH (10 mmol), CO2 (1 atm), COCP–OH (71 mg), co-catalyst (0.0735 mmol), 70 °C, 6 h. The yields were determined by 1H-NMR. | ||
The catalyst amount of COCP–OH–KI has a great influence on the catalytic activity of the reaction. Fig. 6 shows the effect of catalyst amount on the yield. When KI was kept as 12.2 mg and the COCP–OH amount was increased from 45 to 97 mg, the yields of cyclic carbonate increased (Fig. 6(a)). When the COCP–OH amount was 71 mg, the product yield reached 94.8%, and on further increasing the COCP–OH amount to 97 mg, a slight increase of the yield (95.2%) was obtained, while when the COCP–OH amount (71 mg) was kept unchanged and the KI amount was varied from 7.8 to 21 mg, a similar result was observed (Fig. 6(b)). Thus, the optimum catalyst amounts are 71 mg of COCP–OH and 12.2 mg of KI.
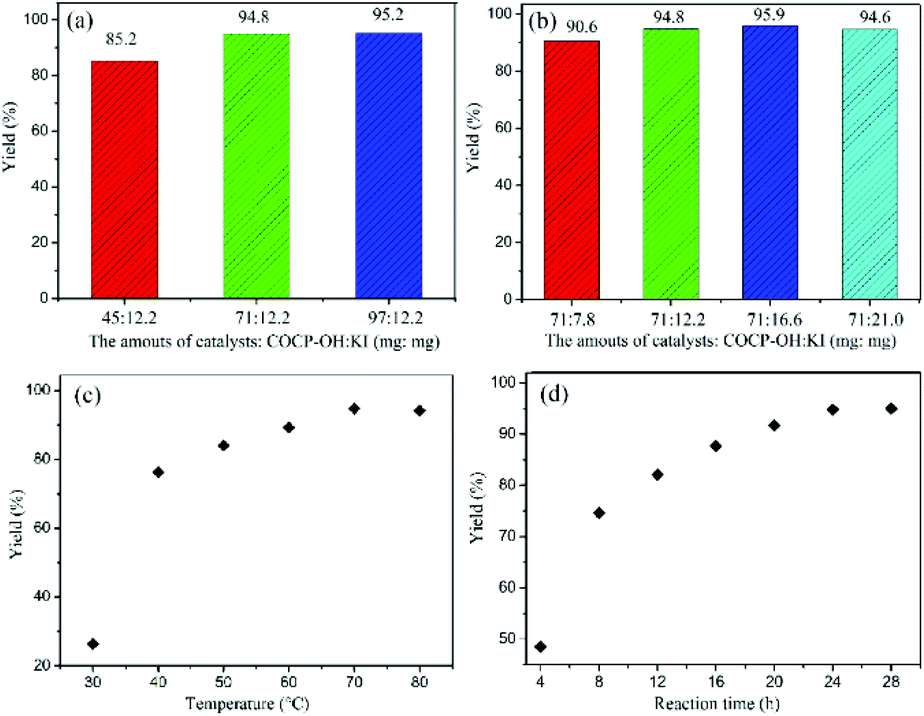 | ||
Fig. 6 Effects on the conversion of CO2 with ECH (a) the effect of COCP–OH content on the yield (AM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :KI = mg:mg); (b) the effect of KI content on the yield (AM:KI = mg:mg); (c) the effect of reaction temperature on the yield (reaction conditions: ECH (10 mmol), CO2 (1 atm), COCP–OH (71 mg), KI (0.0735 mmol, 12.2 mg), 24 h); (d) the effect of reaction time on the yield (reaction conditions: ECH (10 mmol), CO2 (1 atm), COCP–OH (71 mg), KI (0.0735 mmol, 12.2 mg), 70 °C). The yields were determined by 1H-NMR. :KI = mg:mg); (b) the effect of KI content on the yield (AM:KI = mg:mg); (c) the effect of reaction temperature on the yield (reaction conditions: ECH (10 mmol), CO2 (1 atm), COCP–OH (71 mg), KI (0.0735 mmol, 12.2 mg), 24 h); (d) the effect of reaction time on the yield (reaction conditions: ECH (10 mmol), CO2 (1 atm), COCP–OH (71 mg), KI (0.0735 mmol, 12.2 mg), 70 °C). The yields were determined by 1H-NMR. | ||
Reaction temperature is an important factor for the cycloaddition reaction. The influence of temperature was investigated in the range of 30 to 80 °C. As demonstrated in Fig. 6(c), the yields of cyclic carbonate increased from 26.3 to 94.8% when the reaction temperature was varied from 30 to 70 °C. However, the yield was no longer changed with further improvement of temperature to 80 °C, and therefore, the optimal reaction temperature was identified to be 70 °C.
The effect of reaction time on the coupling reaction has also been examined. As illustrated in Fig. 6(d), with the reaction time prolonged from 4 to 24 h, the yield of cyclic carbonate was increased from 48.5 to 94.8%. However, when the reaction time was further prolonged to 28 h, there was only a little change of the yield. Thus, 24 h was identified to be the optimal reaction time.
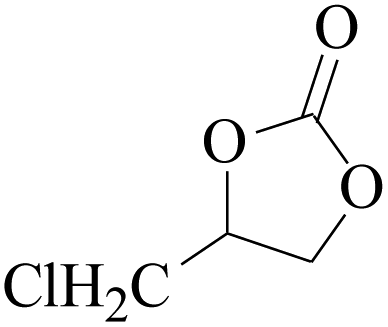
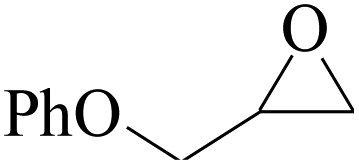
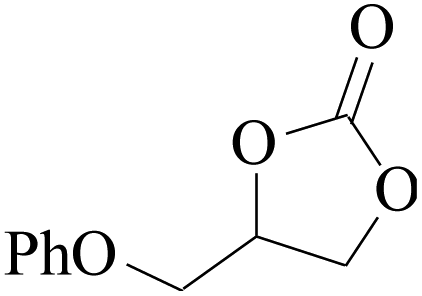
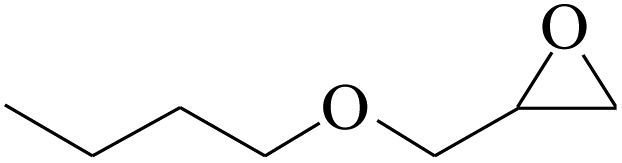
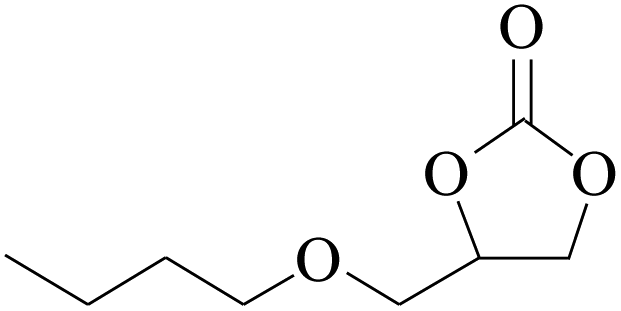
Furthermore, the cycloaddition reaction scope of substrates has been studied under the optimal reaction conditions, such as: epoxide concentration (10 mmol), COCP–OH (71 mg), KI (12.2 mg), CO2 (1 atm), and reaction time (24 h) at 70 °C. The experimental results are summarized in Table 2. As shown in Table 2, most of the corresponding cyclic carbonates were obtained with good yields. Aliphatic terminal epoxides including epichlorohydrin and epibromohydrin as substrates gave high yields of 94.8% and 97.1%, respectively (Table 2, entries 1 and 2). The reaction of aromatic or terminal aryloxy epoxides with CO2 also gave excellent yields of cyclic carbonates (Table 2, entries 3 and 6, yield = 97.9% and 94.3%). Substrates with aliphatic terminal ether were also examined using COCP–OH–KI as the catalyst, and all three cyclic carbonates were obtained in high yields (Table 2, entries 4, 5, 7 and 9, yield = 95.2%, 98.0%, 95.7% and 90.9%). When 1,2-epoxyhexane was used as the substrate, 97.1% yield of cyclic carbonate was obtained (Table 2, entry 8). Considering the low reactivity of 2-decyloxirane and cyclohexane oxide, the reaction time was prolonged to 72 h, and the conversion yields of cyclic carbonates were 44.1% and 31.4% respectively (Table 2, entries 10 and 11).
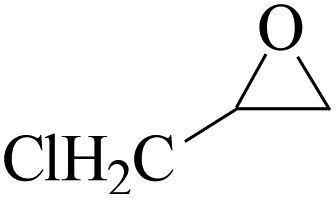
In order to figure out the interaction of COCP–OH with KI, XPS spectra were studied (Fig. 7). Fig. 7(a) shows the O 1s XPS spectra of COCP–OH; in the spectra two distinct signals at 530.0 eV and 531.9 eV can be seen which demonstrated that there are two kinds of O species in COCP–OH. The peak at 531.9 eV was attributed to the oxygen atoms of crown ether and the other one was due to O 1s of the hydroxyl group. Fig. 7(c) shows the O 1s XPS spectrum obtained from the sample COCP–OH reacted with KI. Compared to Fig. 7(a), it can be seen that the peaks of O 1s in crown ether groups showed a significant shift (from 531.9 eV to 532.7 eV). Thus it was proved that the interaction of K+ and I− could be weakened by coordinating the oxygen atoms of crown ether groups with KI, and then the nucleophilicity of I− could be increased. Fig. 7(b) and (d) show the I 3d XPS spectra of KI and COCP–OH–KI, respectively. As illustrated in the spectra, a change in the signals was also observed (from 619.0 eV to 619.4 eV) which further proved the interaction of COCP–OH with KI.
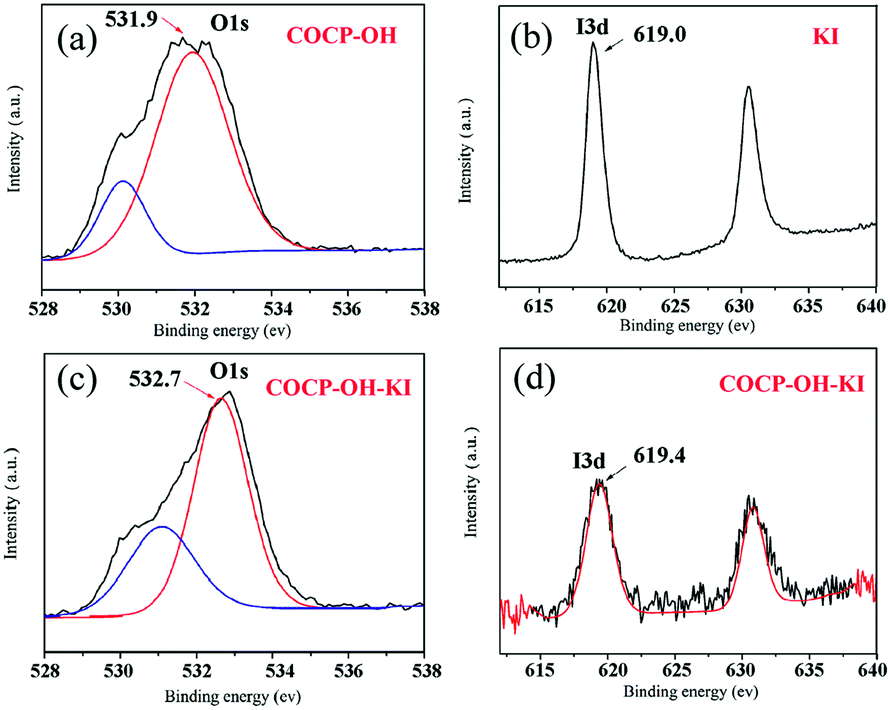 | ||
| Fig. 7 Interaction of COCP–OH with KI identified by XPS. (a) O 1s XPS spectrum of COCP–OH; (b) I 3d XPS spectrum of KI; (c) O 1s XPS spectrum of COCP–OH–KI; (d) I 3d XPS spectrum of COCP–OH–KI. | ||
The coordination of crown ether groups with K+ can significantly improve the nucleophilicity of I− which has been proved by experimental results. The reactant of epoxide could be activated via hydrogen bonding between the hydroxyl groups and O atoms in epoxide as reported in the literature.32 Thus, the possible mechanism of the cycloaddition reaction catalyzed by COCP–OH and KI is demonstrated in Scheme 2. COCP–OH–KI (I) was formed by the interaction of COCP–OH with KI. The nucleophilicity of I− was improved by this step. Then, the reactant epoxide was activated by forming hydrogen bonding between the hydroxyl groups of COCP–OH and O atoms in epoxide, and thus intermediate II was generated. After that, the epoxide undergoes ring-opening reaction under the attack of I− to form intermediate III. Subsequently, CO2 attaches to active alkoxide and intermediate IV was formed. Finally, cyclic carbonate was obtained by ring-closure of intermediate IV with the regeneration of the catalyst.
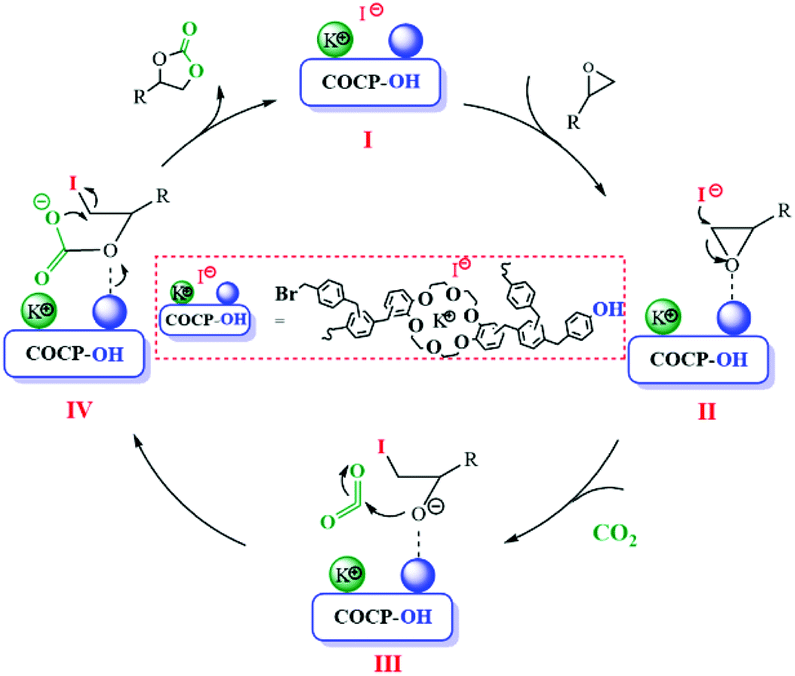 | ||
| Scheme 2 The possible mechanism of the cycloaddition reaction catalyzed by COCP–OH and KI. | ||
The reusability of COCP–OH was also explored under the optimal reaction conditions. The representative reaction of ECH and CO2 was selected. From the results of recyclability investigation, it can be seen that the COCP–OH exhibited good catalytic activity (Fig. S45†). In addition, the yields of each cycle have decreased a bit which was probably attributed to the loss of catalyst. In order to determine the stability of COCP–OH, the fresh and reused COCP–OH were characterized by IR. From the IR spectra, it is found that there is no obvious structural change (Fig. S46†), while a new absorption band at 1795 cm−1 in the IR of the 4th recycled catalyst appeared due to the adsorption of cyclic carbonate on the catalyst surface.12
Experimental section
Materials
All solvents and chemicals were of analytical grade which were commercially available and were used without further purification. In this work, CO2 with a purity of 99.99% was obtained from Handan Anke Factory.Characterization
The IR spectra were obtained using a Bruker IFS 120HR spectrometer (400–4000 cm−1). 1H NMR spectra were recorded on a Mercury plus-400 spectrometer in deuterated chloroform (CDCl3), and tetramethylsilane (TMS) was used as an internal standard. The 13C CP MAS NMR spectrum was recorded with a Bruker AVANCE III 400 WB spectrometer, and the 13C chemical shift was referenced to HMB (hexamethylbenzene). The XPS spectra were collected using a Thermo Fisher Scientific K-Alpha spectrometer using an Al K-Alpha source, and the pass energy was 150.0 eV. The photoelectric peak of C 1s is located at 284.8 eV, and it was used as the criterion to rectify the binding energies of all XPS spectra. SEM was performed on a Hitachi SU-8020 microscope operating at an acceleration voltage of 3.0 kV. The nitrogen adsorption isotherm was obtained from a Micromeritics ASAP 2460 at 77 K, and the sample was degassed at 120 °C for 6 h before adsorption measurements. TEM was carried out on a FEI Tecnai G2 microscope.Synthesis of COCPs and cycloaddition reaction of epoxides with CO2
Conclusions
In summary, five efficient covalent organic crown-based polymers (COCP–H, COCP–OH, COCP–CH3, COCP–Bu and COCP–NH2) with crown ether and/or hydroxyl groups have been designed and synthesized. These COCPs as catalysts demonstrated excellent catalytic activity for the coupling reaction of CO2. Notably, the catalyst of COCP–OH showed excellent activity under the optimum solvent free and mild conditions: 71 mg of COCP–OH, 12.2 mg of KI, 70 °C and 1 atm of CO2 for 24 h, and moderate to excellent yields of cyclic carbonates could be obtained. The study results demonstrate that crown ether and hydroxyl groups play an important role in the cycloaddition reaction. This research provides a new approach to fabricate multifunctional COCP materials for the coupling reaction of CO2 under mild conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) program (21902041), the National Science Foundation of Hebei Province (B2020402002, E2021402017), and the Science and Technology Project of Hebei Education Department (ZD2019002, QN2020165).Notes and references
- L. Guo, K. J. Lamb and M. North, Green Chem., 2021, 23, 77–118 RSC.
- P. Zhang, Z. Wang, P. Cheng, Y. Chen and Z. Zhang, Coord. Chem. Rev., 2021, 438, 213873 CrossRef CAS.
- A. Monfared, R. Mohammadi, A. Hosseinian, S. Sarhandi and P. D. K. Nezhad, RSC Adv., 2019, 9, 3884–3899 RSC.
- A. A. Marciniak, K. J. Lamb, L. P. Ozorio, C. J. A. Mota and M. North, Curr. Opin. Green Sustain. Chem., 2020, 26, 100365 CrossRef.
- X. Cheng, P. Zhao, M. Zhang, S. Wang, M. Liu and F. Liu, Chem. Eng. J., 2021, 418, 129360 CrossRef CAS.
- F. Zhang, S. Bulut, X. Shen, M. Dong, Y. Wang, X. Cheng, H. Liu and B. Han, Green Chem., 2021, 23, 1147–1153 RSC.
- G. Singh, J. Lee, A. Karakoti, R. Bahadur, J. Yi, D. Zhao, K. AlBahily and A. Vinu, Chem. Soc. Rev., 2020, 49, 4360–4404 RSC.
- Y. Hao, T. Tian, Y. Kang, T. Chang, X. Fu, Z. Zhu, X. Meng, B. Panchal and S. Qin, New J. Chem., 2020, 44, 15811–15815 RSC.
- S. Bhunia, R. A. Molla, V. Kumari, S. M. Islam and A. Bhaumik, Chem. Commun., 2015, 51, 15732–15735 RSC.
- P. Bhanja, A. Modak and A. Bhaumik, ChemCatChem, 2019, 11, 244–257 CrossRef CAS.
- Y. Hao, X. Yan, Z. Zhu, T. Chang, X. Meng, X. Fu, B. Panchal, L. Kang and S. Qin, Sustainable Energy Fuels, 2021, 5, 2943–2951 RSC.
- J. Zhang, X. Li, Z. Zhu, T. Chang, X. Fu, Y. Hao, X. Meng, B. Panchal and S. Qin, Adv. Sustainable Syst., 2021, 2000133 CrossRef CAS.
- A. Liu, J. Zhang and X. Lv, Chin. J. Catal., 2018, 39, 1320–1328 CrossRef CAS.
- S. Verma, G. Kumar, A. Ansari, R. I. Kureshy and N.-u. H. Khan, Sustainable Energy Fuels, 2017, 1, 1620–1629 RSC.
- D. Kim, S. Subramanian, D. Thirion, Y. Songa, A. Jamal, M. S. Otaibi and C. T. Yavuz, Catal. Today, 2020, 356, 527–534 CrossRef CAS.
- H. He, Q. Sun, W. Gao, J. A. Perman, F. Sun, G. Zhu, B. Aguila, K. Forrest, B. Space and S. Ma, Angew. Chem., Int. Ed., 2018, 57, 4657–4662 CrossRef CAS PubMed.
- J.-H. Chen, C.-H. Deng, S. Fang, J.-G. Ma and P. Cheng, Green Chem., 2018, 20, 989–996 RSC.
- N. Wei, Y. Zhang, L. Liu, Z.-B. Han and D.-Q. Yuan, Appl. Catal., B, 2017, 219, 603–610 CrossRef CAS.
- S. K. Das, S. Chatterjee, S. Bhunia, A. Mondal, P. Mitra, V. Kumari, A. Pradhan and A. Bhaumik, Dalton Trans., 2017, 46, 13783–13792 RSC.
- S. Zhang, X. Liu, M. Li, Y. Wei, G. Zhang, J. Han, X. Zhu, Q. Ge and H. Wang, Catal. Today, 2019, 324, 59–65 CrossRef CAS.
- D. Liu, G. Li, J. Liu and Y. Yi, Fuel, 2019, 244, 196–206 CrossRef CAS.
- D. Liu, G. Li, J. Liu and Y. Yi, Chin. J. Chem., 2018, 36, 187–193 CrossRef.
- R. Babu, S.-H. Kim, J. F. Kurisingal, H.-J. Kim, G.-G. Choi and D.-W. Park, J. CO2 Util., 2018, 25, 6–13 CrossRef CAS.
- P. R. Tambe and G. D. Yadav, Clean Technol. Environ. Policy, 2018, 20, 345–356 CrossRef CAS.
- K. B. Rasal, G. D. Yadav, R. Koskinen and R. Keiski, Mol. Catal., 2018, 451, 200–208 CrossRef CAS.
- L.-Y. Zhao, J.-Y. Chen, W.-C. Li and A.-H. Lu, J. CO2 Util., 2019, 29, 172–178 CrossRef CAS.
- J. L. Vidal, V. P. Andrea, S. L. MacQuarrie and F. M. Kerton, ChemCatChem, 2019, 11, 4089–4095 CrossRef CAS.
- S. Zhang, H. Zhang, F. Cao, Y. Ma and Y. Qu, ACS Sustainable Chem. Eng., 2018, 6, 4204–4211 CrossRef CAS.
- M. Fan, W. D. Wang, X. Wang, Y. Zhu and Z. Dong, Ind. Eng. Chem. Res., 2020, 59, 12677–12685 CrossRef CAS.
- J. Zhao, Z. Si, H. Shan, D. Cai, S. Li, G. Li, H. Lin, J. Baeyens, G. Wang, H. Zhao and P. Qin, Ind. Eng. Chem. Res., 2020, 59, 14569–14577 CrossRef CAS.
- W. Desens, C. Kohrt, M. Frank and T. Werner, ChemSusChem, 2015, 8, 3815–3822 CrossRef CAS PubMed.
- S. Kaneko and S. Shirakawa, ACS Sustainable Chem. Eng., 2017, 5, 2836–2840 CrossRef CAS.
- T. Chang, X. Gao, L. Bian, X. Y. Fu, M. Yuan and H. Jing, Chin. J. Catal., 2015, 36, 408–413 CrossRef CAS.
- X. Fu, P. Xie, Y. Lian, L. He, W. Zhao, T. Chang and S. Qin, Chin. J. Catal., 2018, 39, 1854–1860 CrossRef CAS.
- T. Chang, X. P. Li, Y. J. Hao, L. W. Kang, T. Tian, X. Y. Fu, Z. Zhu, B. Panchal and S. J. Qin, RSC Adv., 2021, 11, 30222–30228 RSC.
- X. Fu, D. Zhou, K. Wang and H. Jing, J. CO2 Util., 2016, 14, 31–36 CrossRef CAS.
- Y. Zhang, K. Zhang, L. Wu, K. Liu, R. Huang, Z. Long, M. Tong and G. Chen, RSC Adv., 2020, 10, 3606–3614 RSC.
- Q. Li, H. Yang, M. Li, K. Zhao, B. Duan, C. Zhang, H. Wang, C. Qiao, H. Chang and T. Lin, Ind. Eng. Chem. Res., 2020, 59, 8136–8144 CrossRef CAS.
- M. Zhang, Y. Yang and L. B. Chen, Chin. J. Catal., 2015, 36, 1304–1311 CrossRef CAS.
- J. Steinbauer and T. Werner, ChemSusChem, 2017, 10, 3025–3029 CrossRef CAS PubMed.
- J. Steinbauer, A. Spannenberg and T. Werner, Green Chem., 2017, 19, 3769–3779 RSC.
- Y. P. Song, W. Sun, P. C. Lan and S. Q. Ma, ACS Appl. Mater. Interfaces, 2020, 12, 32827–32833 CrossRef CAS PubMed.
- J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC.
- X. Y. Fu and H. W. Jing, J. Catal., 2015, 329, 317–324 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1se01120k |
| This journal is © The Royal Society of Chemistry 2022 |