
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical design of high performance SPR biosensor based on a dielectric nanoparticle assembly supported onto a gold thin film†
Pier
Berling
a,
Mathias
Dolci
a,
Spyridon
Zafeiratos
 b,
Thomas
Gehin
c,
Cédric
Leuvrey
a,
Céline
Kiefer
a,
Déborah
Wagner
d,
Fouzia
Boulmedais
d and
Benoit P.
Pichon
*ae
b,
Thomas
Gehin
c,
Cédric
Leuvrey
a,
Céline
Kiefer
a,
Déborah
Wagner
d,
Fouzia
Boulmedais
d and
Benoit P.
Pichon
*ae
aUniversité de Strasbourg, CNRS, Institut de Physique et Chimie des Matériaux de Strasbourg, UMR 7504, F-67000 Strasbourg, France. E-mail: benoit.pichon@unistra.fr; Fax: +33 (0)388 10 72 47; Tel: +33 (0)3 88 10 71 33
bUniversité de Strasbourg, CNRS, Institut de Chimie et Procédés pour l'Energie, l'Environnement et la Santé, UMR 7515, 25 Rue Becquerel, 67087 Strasbourg, France
cUniversité de Lyon, Institut des Nanotechnologies de Lyon (INL) – UMR CNRS 5270, Ecole Centrale de Lyon, 36 Avenue Guy de Collongue, 69134 Ecully cedex, France
dUniversité de Strasbourg, CNRS, Institut Charles Sadron, UPR 22, F-67034 Strasbourg Cedex 2, France
eInstitut Universitaire de France, 1 rue Descartes, 75231 Paris Cedex 05, France
First published on 19th July 2022
Abstract
The SPR system is a very efficient tool to investigate original and highly efficient biosensors and to study molecular recognition mechanisms. We report on the design of an original nano-architecture which consists of iron oxide nanoparticle assemblies supported on gold thin films and which acts as a very efficient SPR sensor. The build-up of the sensor consists of a three-step CuAAC “click” reaction process which allows the selective and easy preparation of a robust platform. Iron oxide nanoparticles allow the label-free detection of targeted proteins thanks to their high sensitivity to slight variations in the refractive index in the vicinity of the SPR sensor surface. We investigated the very well-known biotin/streptavidin couple in order to study the selectivity of biomolecular binding of our sensor platform. The nonspecific adsorption of proteins was suppressed by using oligo ethylene oxide (EO) chains. A study of the kinetics revealed the remarkably low limit of detection (down to 1.1 nM) which is ascribed to the enhanced accessibility of biotin groups and to the biotin/SA affinity (2.45 × 107 M). Indeed, the high radius of curvature of the nanoparticles avoids steric hindrance of the biotin groups and the flexibility of the EO linkers favors their mobility in an aqueous medium. An optimized sensing platform was built by taking advantage of the adequate surface functionalization of iron oxide nanoparticle assemblies supported on a gold thin film.
1. Introduction
SPR biosensors have become increasingly popular in research fields related to medicine, biology and ecology.1,2 Their success is ascribed to quantitative analysis for dosage control and toxicity risk management as well as excellent reutilization performance and outstanding reproducibility. Furthermore, SPR sensors allow kinetics and the affinity of biomolecular binding to be measured in real time. SPR sensors usually consist of a metal thin film whose plasmon resonance characteristics are highly sensitive to the variation in refractive index in the vicinity of its surface. This is ascribed to the strong evanescent field which is generated at the metal/dielectric interface by the surface plasmon wave. Therefore, the adsorption of analytes onto the metal surface induces a variation in the plasmonic signal. Since the signal variation is directly dependent on the variation in the refractive index, it can be limited by low molar weight and low concentration of analytes, which are critical parameters for the development of efficient SPR sensors.Dielectric over-layers (n = 1.48 to 2.6) supported on metal thin films were recently reported to increase the intensity of the electromagnetic field at the metal/dielectric interface.3 Such lamellar structures opened up new perspectives for SPR biochips.4 The sensitivity of plasmon thin films can be significantly increased for small changes in the refractive index in the vicinity of the film surface. Relatively thin layers (5–40 nm) of dielectric material are particularly efficient when taking into account the exponential decay of the intensity field within about 200 nm of the metal surface. Nevertheless, for materials with high refractive index, the broadening of the SPR signal reduces the accuracy of the measurement above a critical layer thickness. The use of top-dielectric layers also stabilizes the chemical composition by protecting plasmonic thin films subject to oxidation, such as silver.5 However, the deposition of thin films requires plasma-related deposition techniques which use expensive equipment and difficult processing.
The sensitivity can also be markedly increased by exploiting labelling with non-plasmonic nanoparticles, which amplify the variation in refractive index after adsorption of analytes. Latex microbeads with sizes up to 120 nm led to a significant enhancement in sensitivity.6 Iron oxide particles with sizes from 10 to 200 nm led to the detection of various analytes with concentrations from a few ng to hundredths of pg mL−1.7–11 However, labelling requires nanoparticles to be conjugated to a recognition element which interacts with the analyte in order to adsorb onto the metal thin film. This approach usually leads to potential aggregation, thus making it difficult to quantify the analytes to be detected.
Assemblies of non-plasmonic nanoparticles emerged very recently as an original approach to enhance the sensitivity. Nanoparticle assemblies can be easily prepared by robust chemical reactions belonging to the area of “click” chemistry.12,13 In contrast to continuous thin films, the structure of nanoparticle assemblies can be modulated according to different structural parameters, such as nanoparticle size and density, which allow adjustment of the optical properties, as we have shown by using high refractive index Fe3–δO4 nanoparticles (n = 2.42).14 A high loading of nanoparticles significantly enhances the intensity of the electromagnetic field, i.e. the sensitivity of plasmonic thin films. Furthermore, the topography resulting from nanoparticle assemblies induces a larger specific surface area than flat surfaces, which enhances the number of binding sites, thus allowing the detection of formaldehyde15 or arsenic16 down to a few ppm. The main interest of nanoparticle assemblies supported on plasmonic metal thin films is certainly the ability to finely tune their structure in order to address the interplay between the highest loading of nanoparticles and the highest accessibility of binding sites.14
Besides the optimization of the optical properties, the surface functionalization of nanoparticle assemblies is also a critical parameter. Considering that analytes to be detected are present at a low concentration in bodily fluids which are very complex mixtures, nonspecific interaction is one of the main limitations of sensor efficiency.17 Thus, the specific detection of biomolecules of interest represents a very important challenge. With this aim, the surface chemistry of sensors has been carefully addressed and has received considerable attention during the last few decades. Ethylene oxide (EO) chains are certainly the most efficient chemical material to avoid protein adsorption onto surfaces.18,19 Therefore, they are very well known to functionalize surfaces while preserving the native form and bioactivity of various biomolecular receptors. Steric hindrance also limits the accessibility of bio-receptors in tightly packed assemblies on flat surfaces, which usually require dilution by inactive groups.20–22
In this context, we report the design of a highly specific SPR sensor which consists of an iron oxide nanoparticle assembly supported on a gold thin film. The nano-architecture was built-up by performing a three-step copper-catalyzed alkyne–azido (CuAAC) cycloaddition reaction, a highly specific and reliable reaction to perform, which belongs to the field of “click” chemistry. Thanks to its high theoretical affinity constant (Ka ≃ 1013 M−1),21,23 the biotin–streptavidin couple was selected to investigate the efficiency of such an original biosensor. We focused on the surface functionalization of the nanoparticle assembly in order to avoid nonspecific interactions and steric hindrance by taking advantage of EO chains and the high radius of curvature of spherical nanoparticles, respectively. We show that such a platform is highly efficient for designing sensors with enhanced bioaffinity with specific detection and a low limit of detection.
2. Experimental section
2.1. Materials and products
The materials were: iron II stearate (Strem Chemicals), dioctylether (99%, Aldrich), oleic acid (99%, Alfa Aesar), 10-undecyn-1-ol (96%, Alfa Aesar), triethylamine (99%, Acros Organics), N-[2-[2-[2-(2-azidoethoxy)ethoxy]-ethoxy]ethyl]biotinamide (>97%, Sichem), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hypochloride (>98%, TCI), bromotris(phenylphosphine) copper I (98%, Aldrich). D-(+)-Biotin (>97%, Aldrich), biotin-PEG8-alkyle (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C-EO7-biotin, >97%, Sichem), (11-undec-1-ynyl)thiol were synthesized as we previously reported.12N-Propargylbiotinamide (CC-C3-biotin) was inspired by Nainar et al.24 10 nm-sized Fe3–δO4 nanoparticles were synthesized following the procedure we reported earlier.12
C-EO7-biotin, >97%, Sichem), (11-undec-1-ynyl)thiol were synthesized as we previously reported.12N-Propargylbiotinamide (CC-C3-biotin) was inspired by Nainar et al.24 10 nm-sized Fe3–δO4 nanoparticles were synthesized following the procedure we reported earlier.12
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) C-C3-biotin or CC-EO7-biotin).
A gold film with a thickness of 48 nm was evaporated onto a BK7 glass substrate after evaporation of a 2 nm thick chromium adhesion layer. The assembly of Fe3O4@N3 nanoparticles onto an alkyne-terminated gold substrate (SAM-CC) was performed following the exact procedure we reported earlier.25 Then, nanoparticle assemblies supported on gold thin films were introduced in a 50 mL reactor, which was subsequently filled with a solution of 5 mM of CC-C3-biotin or 2 mM of CC-EO7-biotin, 6.5 mg of CuBr(PPh3)3, 0.5 mL of Et3N and 5 mL of THF. The reaction was performed under reflux for 24 hours. The substrates were then rinsed and ultrasonicated for 1 min in THF before being dried under an air stream.
C-C3-biotin or CC-EO7-biotin).
A gold film with a thickness of 48 nm was evaporated onto a BK7 glass substrate after evaporation of a 2 nm thick chromium adhesion layer. The assembly of Fe3O4@N3 nanoparticles onto an alkyne-terminated gold substrate (SAM-CC) was performed following the exact procedure we reported earlier.25 Then, nanoparticle assemblies supported on gold thin films were introduced in a 50 mL reactor, which was subsequently filled with a solution of 5 mM of CC-C3-biotin or 2 mM of CC-EO7-biotin, 6.5 mg of CuBr(PPh3)3, 0.5 mL of Et3N and 5 mL of THF. The reaction was performed under reflux for 24 hours. The substrates were then rinsed and ultrasonicated for 1 min in THF before being dried under an air stream.
Finally, the sample was introduced into a second 50 mL reactor which was filled with a solution of 2 mM mg of PEO7_N3, 6.5 mg of CuBr(PPh3)3 and 0.5 mL of Et3N. The reaction was performed under reflux for 24 hours. The substrates were then rinsed and ultrasonicated for 1 min in THF before being dried under an air stream.
C-EO7-biotin by CC-EO7-OH.
Then, 2 mM solutions of CC-EO7-biotin and CC-EO7-OH were prepared separately in THF, then mixed in the required proportions. The click reaction follows the same procedure as described above. 6.5 mg of CuBr(PPh3)3 and 0.5 mL of Et3N were stirred in 5 mL of a CC-EO7-biotin/CC-EO7-OH mixture in the chosen proportion in a 50 mL reactor. The reaction was performed under reflux for 24 hours. The substrates were then rinsed and ultrasonicated for 1 min in THF before being dried under an air stream.
2.2. Sample characterization
Transmission electron microscopy (TEM), high resolution TEM (HRTEM), and electron diffraction (ED) were performed using a JEOL ARF200F microscope operating at 200 kV. The size distribution was calculated from the size measurement of more than 100 nanoparticles using Image J software. Granulometry measurements were performed using a Malvern nanosizer (nano ZS) apparatus for each NP suspension. Fourier transform infrared (FTIR) spectroscopy was performed using a PerkinElmer Spectrum Two spectrophotometer in the energy range 4000–400 cm−1. Scanning electron microscopy (SEM) was performed using a Zeiss Gemini SEM 500 microscope equipped with a field emission gun (SEM-FEG) operating at an accelerating voltage of 1 kV. Atomic force microscopy (AFM) was performed in the tapping mode using a Digital Instrument 3100 microscope coupled to a Nanoscope IIIa recorder. The collected data were analyzed using Nanotec WSXM software. Phase modulation infrared reflection absorption spectroscopy (PM-IRRAS) spectra were collected on a Thermo 6700 FTIR spectrometer equipped with an external optical bench. Measurements of IR spectra were carried out with the photoelastic modulator set for half-wave retardation at 2500 cm−1 and the angle of incidence of the infrared beam was set to 82°. The instrument resolution was set to 4 cm−1 and 1000 scans were averaged. X-ray photoelectron spectroscopy (XPS) was performed in a spectrometer described previously.26 An Al Kα X-ray source (1486.6 eV) was used as the incident radiation, while due to the low signal of the photoemission peaks of interest, the XPS spectra were recorded using 100 eV pass energy. The binding energy scale was corrected by the Au 4f7/2 line of the gold substrate at 84 eV as an internal reference. The N 1s spectra were fitted using symmetric Gaussian–Lorentzian functions, while for the S 2p peaks an asymmetric profile was used to account for the unresolved 2p3/2–2p1/2 spin–orbit split doublet. N 1s, S 2p and C 1s core level peaks were properly normalized to the photoemission cross section, assuming a homogeneous distribution of these elements on the surface.An MP-SPR Navi commercial system (Bionavis©, Finland) working in the Kretschmann configuration at two fixed wavelengths (670 nm and 785 nm) was used to monitor the SPR signal by changing the resonance angle position. Substrates were inserted inside the system after building up the detection platform. This system was combined into a microfluidic device which was also used to study the kinetics of the recognition process. Aqueous solutions of protein were injected at a flow rate of 50 μL min−1. Commercial substrates consist of a thin gold layer of 50 nm which is deposited onto a glass substrate (BK7) coated with a chromium thin film (thickness 2 nm) used as an adhesive layer.
Streptavidin was extracted from Streptomyces avidinii bacteria. A buffer solution is commonly used for protein stabilization and to avoid denaturation or conformational change during storage. The streptavidin was used in pure water to avoid unspecific adsorption of salt on the surface, which may hamper the recognition process. 250 μL of streptavidin solution were injected into a microfluidic channel with a flow rate of 50 μL min−1. Once the signal was stabilized, the microfluidic channel was cleaned by injecting pure water.
3. Results and discussion
Iron oxide nanoparticles were assembled onto a gold thin film (thickness 48 nm) by performing a click chemistry approach, as we reported earlier (Fig. 1).12,27 According to TEM micrographs, iron oxide nanoparticles exhibiting a narrow size distribution centered on 10.3 ± 1 nm with well-defined spherical shape were functionalized by grafting of 1(12-azidododecyl)phosphonic acid molecules in order to obtain a highly stable colloidal suspension in tetrahydrofuran (see ESI†). These azide-terminated nanoparticles (NP@N3) were assembled onto an alkyne-terminated gold thin film which was functionalized by a self-assembled monolayer of (11-undec-1-ynil)thiol molecules (SAM-CC). The copper-catalyzed alkyne–azide cycloaddition (CuAAC) “click” reaction resulted in the irreversible formation of triazol bonds between nanoparticles and the SAM, i.e. the formation of a robust nanoparticle assembly onto a gold thin film.13
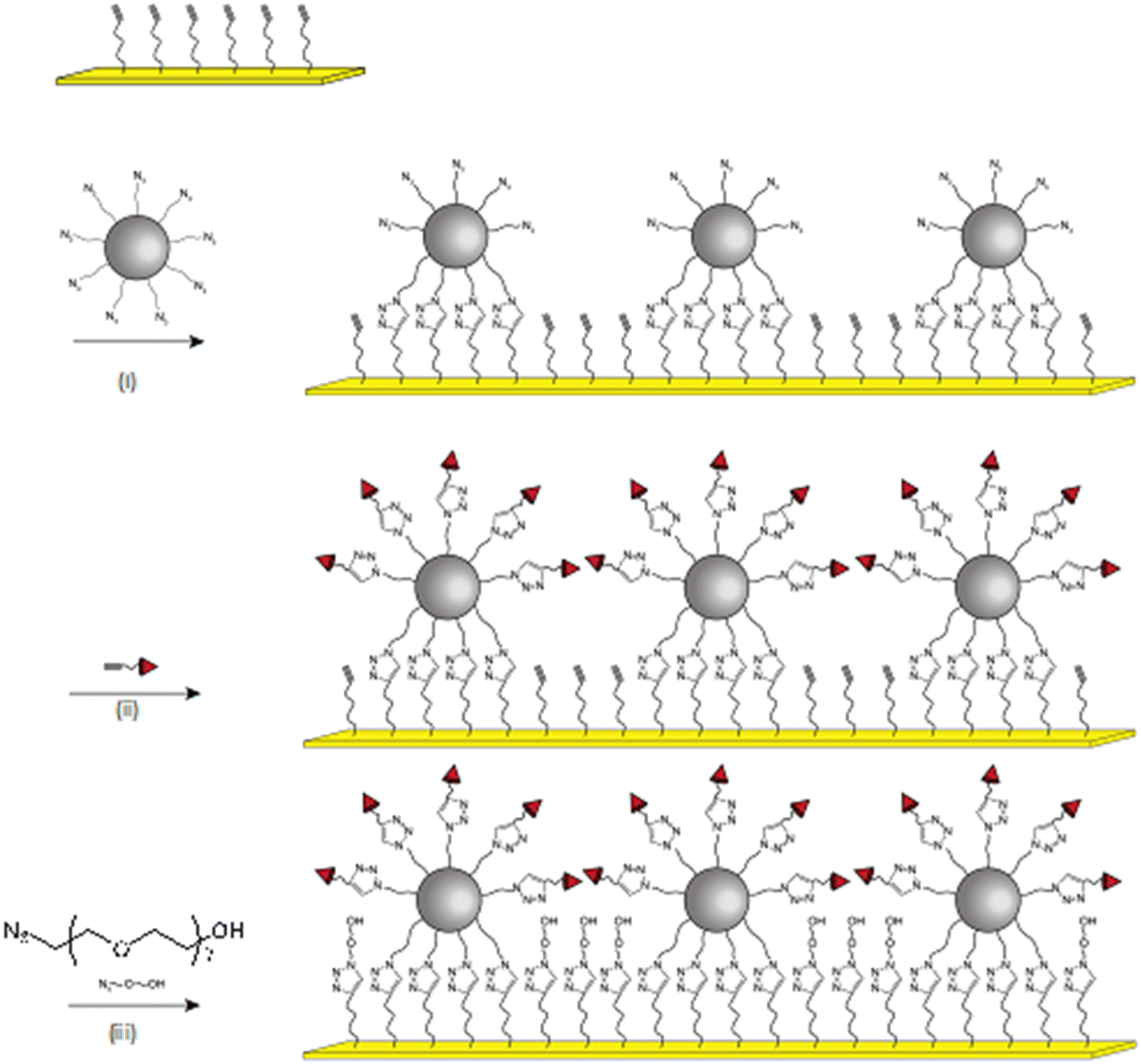 | ||
| Fig. 1 Schematic illustration of the sensor build-up by successively performing the CuAAC “click” reaction up to three times. (i) Assembly of NP@N3, (ii) grafting of CC-EO7-biotin onto nanoparticles, (iii) grafting of N3-EO7-OH onto uncovered areas of SAM-CC. | ||
Scanning electronic microscopy (SEM) images show that the assembly structure consists of a monolayer of nanoparticles with high density (3500 NP ± 120 μm−2) (Fig. 2a), corresponding to an average inter-particle distance of 3.8 nm.14 The nanoparticle density is much higher than that expected according to a random sequential adsorption (RSA) process (54% hexagonal compact packing (hcp) structure).28 Indeed, dipolar interactions contribute to the assembly process, which results in closer nanoparticles (up to 80% hcp).29 Atomic force microscopy (AFM) provided complementary information on the structure of the nanoparticle assembly (Fig. 2b and c). Height profiles with an average value of 9.1 nm and a roughness of 2.6 nm confirmed the formation of a monolayer of nanoparticles.
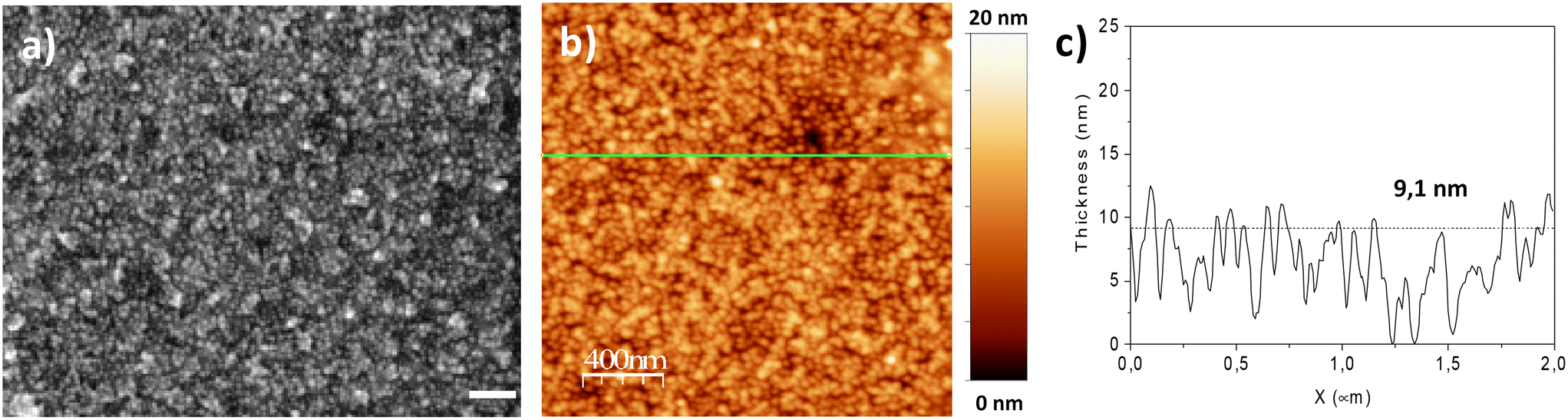 | ||
| Fig. 2 Structure of the nanoparticle assembly obtained by the CuAAC “click” reaction. a) SEM image. b) AFM image and the c) the height profile corresponding to the green line in b). | ||
Polarization-modulation infrared reflection–adsorption spectroscopy (PM-IRRAS) showed typical bands corresponding to the CH2 stretching of hydrocarbon chains (νas = 2928 cm−1 and νs = 2855 cm−1), phosphonic acid groups (νP![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O = 1265 cm−1 and νFe–O–P = 1070–1040 cm−1) and triazol groups (νC–N = 1180 cm−1) (Fig. 3).13 A band centered at 2100 cm−1 is typical of the stretching vibration mode of azide groups (vN3) which remain at the surface of the nanoparticles. This result is in agreement with X-ray photoelectron spectroscopy (XPS) whose spectra showed typical signals of azide groups in the N 1s region at binding energies of 401.8 eV (NNN) and 405.3 eV (NNN), as we reported previously.30
O = 1265 cm−1 and νFe–O–P = 1070–1040 cm−1) and triazol groups (νC–N = 1180 cm−1) (Fig. 3).13 A band centered at 2100 cm−1 is typical of the stretching vibration mode of azide groups (vN3) which remain at the surface of the nanoparticles. This result is in agreement with X-ray photoelectron spectroscopy (XPS) whose spectra showed typical signals of azide groups in the N 1s region at binding energies of 401.8 eV (NNN) and 405.3 eV (NNN), as we reported previously.30
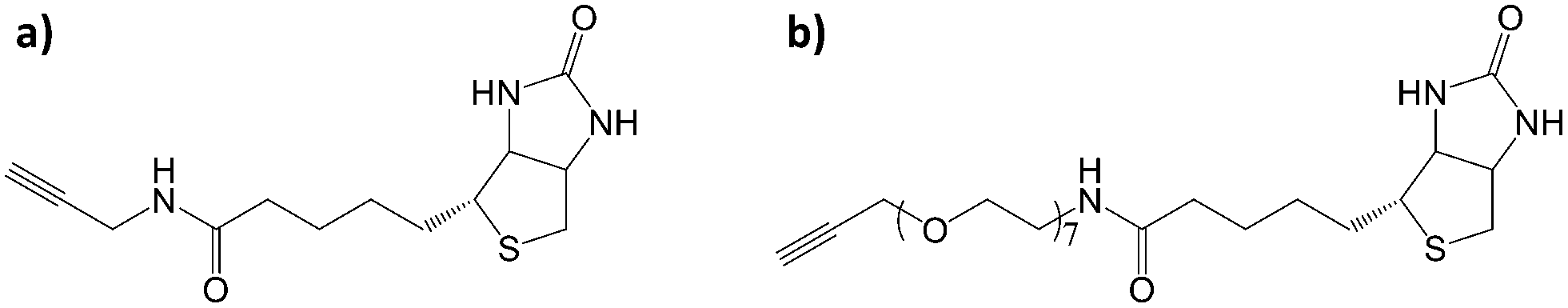 | ||
| Fig. 3 Schematic representation of a) CC-C3-biotin and b) CC-EO7-biotin. | ||
Considering the presence of azide groups at the surface of the nanoparticles, we used them to graft biotin derivatives in order to detect streptavidin (SA). We synthesized two different biotin derivatives with an alkyne group which can react with the azide-terminated nanoparticles. An oligoethylene glycol (EO7) chain was used as a linker between the alkyne and the amide group of N-propargylbiotinamide in order to consider N-[2-[2-[2-(2-azidoethoxy)ethoxy]-ethoxy]ethyl]biotinamide, respectively named CC-C3-biotin and CC-EO7-biotin, (Fig. 3; see ESI† for the synthesis procedure and characterization). Both molecules were separately grafted at the nanoparticle surface by repeating the CuAAC “click” reaction. The presence of biotin groups was confirmed by PM-IRRAS spectra, which showed the disappearance of the azide band (Fig. 4). Furthermore, the broadening and increase in intensity of the band centered at 1591 cm−1 after grafting the biotin derivatives agree with the presence of amide I (νCO) and amide II (δNH) contributions of biotin.31 The broadening of the νP–O–Fe band (1070–1040 cm−1) also results from an additional contribution centered at 1180 cm−1 (δC–N) corresponding to biotin. The higher intensity of the νC–H bands at 2922 cm−1 and 2850 cm−1 agree with extra propyl and ethylene oxide chains.
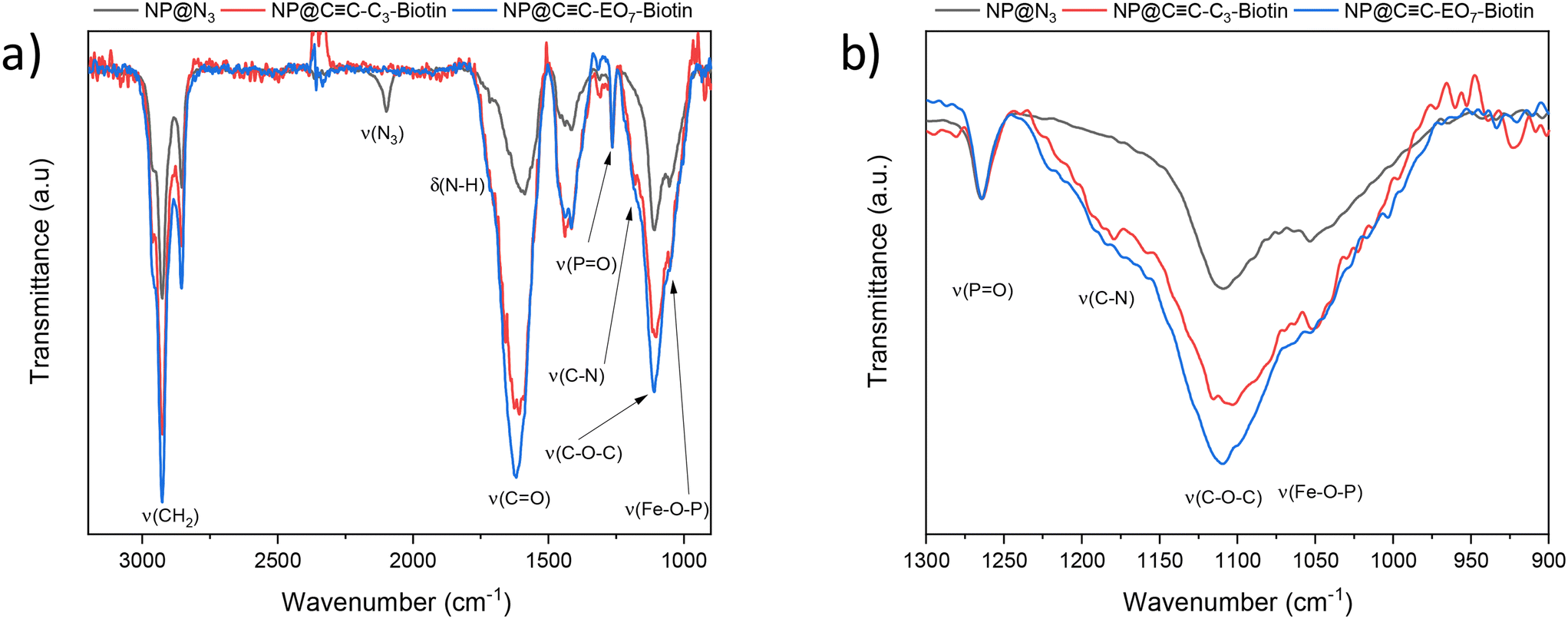 | ||
| Fig. 4 PM-IRRAS spectra of nanoparticle assemblies before and after functionalization by CC-C3-biotin and CC-EO7-biotin and N3-EO7-OH. Spectra were normalized to the PO band. a) Full spectra. b) Enlargement of the low-wavenumber region. | ||
In addition to the functionalization of the nanoparticle surface by CC-EO7-Biotin, areas not covered by nanoparticles were functionalized by ethylene oxide chains (CC-EO7-OH) in order to avoid nonspecific interactions upon detection of protein thanks to repellent properties.18,19 Therefore, O-(2-azidoethyl)heptaethylene glycol (N3-EO7-OH) molecules were grafted at the surface of alkyne-terminated SAM by performing the CuAAC reaction for the third time. The PM-IRRAS spectrum of the sample named CC-EO7-biotin/N3-EO7-OH showed an increase in the δC–O band (1120 cm−1) which agrees with the grafting of N3-EO7-OH molecules (Fig. 4).
Because the SPR signal of the gold thin film is highly sensitive to a variation in the refractive index in its vicinity, it was monitored to study the sensor build-up by using the Kretschmann configuration (Fig. 5a). Measurements were performed at the highest wavelength (785 nm) available in order to take advantage of the better resolution. The incident angle corresponding to the minimum intensity of the SPR signal (SPR dip) varied after each building step. The resonance angle of the gold thin film (65.594°) was successively shifted to higher values after the SAM preparation (66.013°), the nanoparticle assembly (68.134°), the grafting of CC-EO7-biotin at the nanoparticle surface (68.451°) and grafting of additional N3-EO7-OH onto uncovered areas by nanoparticles (68.472°). The gradual shift of the SPR signal to higher angles after each “click” chemistry step agrees with the build-up of a robust nano-architecture through irreversible triazol bonds. The structure of the nanoparticle assembly and organic functionalities are preserved due to the mild operating conditions (60 °C) and specific chemical reactions. Heterogeneous nanostructures such as porous metallic films have been reported to influence the SPR signal.3 Assemblies of nanoparticles also usually lead to a broadening of the SPR-dip as well as a lower decrease in intensity because they absorb and diffuse light.32,33 In our study, the assembly of iron oxide nanoparticles has a very limited influence on the intensity and full width at half maximum of the SPR-dip.
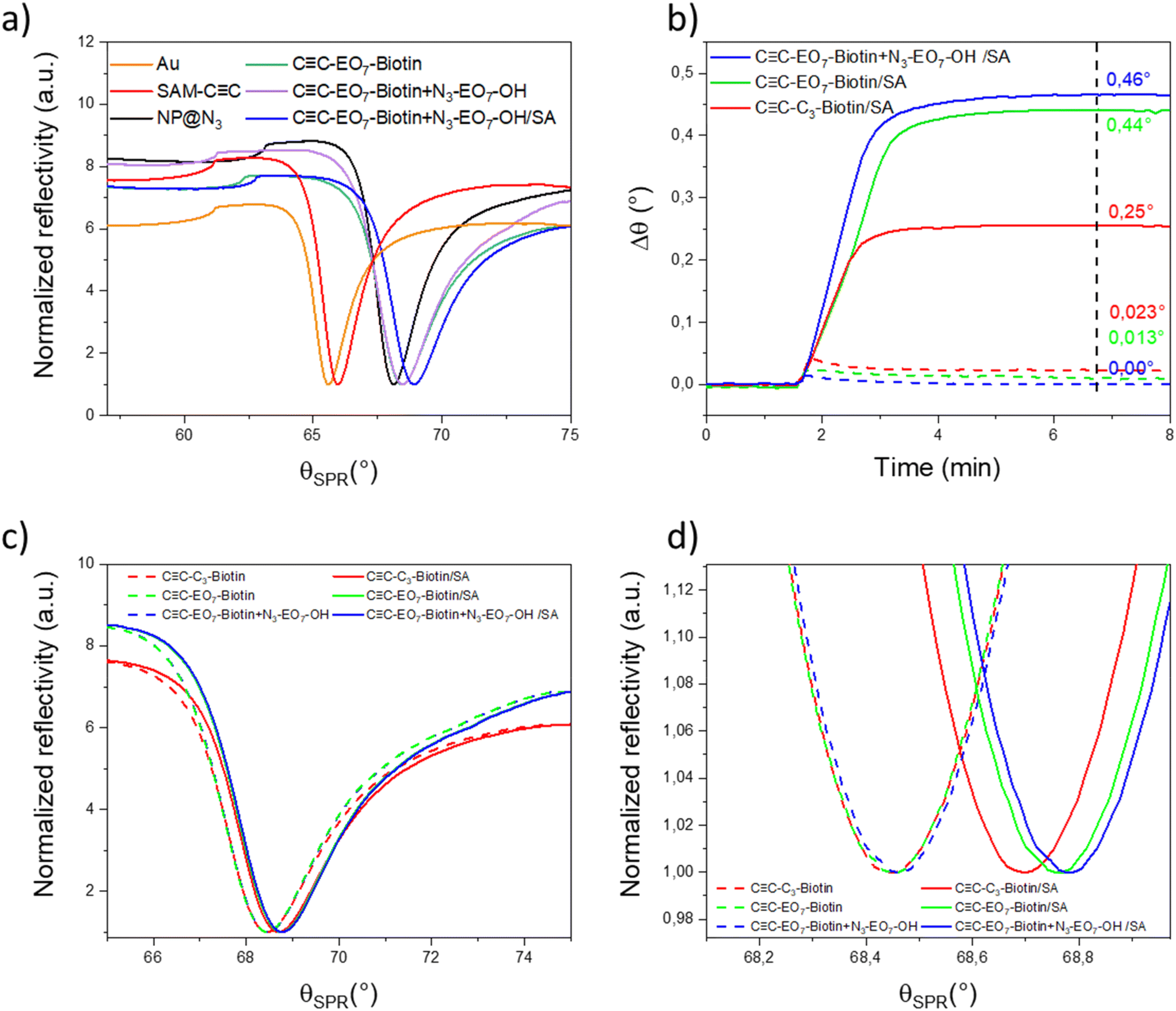 | ||
| Fig. 5 SPR measurements. a) Reflectivity versus angular response (θSPR) measured after each build-up step at a fixed wavelength of 785 nm: gold thin film (Au), self-assembled monolayer of (SAM-CC), nanoparticle assembly (NP@N3), grafting of CC-EO7-biotin (CC-EO7-biotin), grafting of N3-EO7-OH (CC-EO7-biotin + N3-EO7-OH). b) Variation of the incident angle as a function of time after injection of SA (solid lines) and BSA (dashed lines). Dashed curves correspond to BSA injection. The vertical dashed line corresponds to the injection of pure water (washing step). c) Reflectivity versus resonance angle measured before and after injection of SA. d) A closer view of c). | ||
The SPR signal was also monitored to study the absorption of SA as a function of the surface functionalization. The injection of SA (10 μg mL−1) resulted in a significant angular shift for each sample (Fig. 5b–d). Such behavior agrees with the increase in the refractive index in the vicinity of the biotin-terminated nanoparticle assembly supported on a gold thin film. A couple of minutes after stabilization of the SPR signal, pure water was injected to remove proteins from the solution. The position of the resonance peak did not vary, in agreement with strong biomolecular interactions between SA and biotin groups. The angular shift is significantly larger in the presence of CC-EO7-biotin (0.44 ± 0.002°) than CC-C3-biotin (0.25 ± 0.002°). This is ascribed to the high solubility of the PEO7 chain which is extended up to 2 nm in aqueous media, which enhances the mobility of the biotin groups. The SPR angle also shifted to 0.460 ± 0.002° after grafting of N3-EO7-OH onto uncovered areas between nanoparticles (CC-EO7-biotin + N3-EO7-OH).
In order to study the specificity of molecular interactions between biotin and SA, similar experiments were performed by injecting bovine serum albumin (BSA), a well-known protein which adsorbs onto a surface through nonspecific interactions (Fig. 5b). BSA was injected as a 10-fold mass concentration of SA. A shift in the angle (0.0230 ± 0.002°) corresponding to 10% of the shift in the presence of SA was observed for CC-C3-biotin. This shift is significantly reduced for CC-EO7-biotin (0.013 ± 0.002°). The EO7 linker in the biotin derivative significantly reduces nonspecific interactions. For CC-EO7-biotin + N3-EO7-OH, no angular shift was observed at the limit of the resolution of the experiment (0.001°). This shows that the combination of the EO7 linker with biotin and additional N3-EO7-OH chains grafted onto areas not covered by nanoparticles is a very efficient combination to suppress nonspecific interactions.
A high density of functional groups on surfaces is well known to alter their ability to interact with targeted biomolecules because it favors steric hindrance.20–22,34 Dilution of biotin by inactive groups is a well-known approach to enhance interactions with SA. CC-EO7-biotin was mixed with O-(3-butyne)heptaethylene glycol (CC-EO7-OH) in order to graft both molecules at the nanoparticle surface by performing the CuAAC “click” reaction (see Experimental section). CC-EO7-OH was selected in order to favor the homogeneous distribution with CC-EO7-biotin (no segregation phase) thanks to the same chemical composition and chain length. CC-EO7-OH should also avoid nonspecific interactions while preserving the strong affinity of the biotin groups with SA.
XPS analysis was performed for samples with 100%, 50%, 5% and 0% biotin content (Fig. 6). The survey spectra indicate the presence of elements due to the substrate (Au 4f) as well as the over-layer (N 1s, C 1s, S 2p, O 1s, Fe 2p) (see ESI†). In the N 1s region of the 0% sample, two peaks can be resolved around 404 eV and 401 eV, which can be attributed to the azide and triazol groups, respectively.30,35 The peak related to the azide groups (404 eV) decreases for 5% of CC-EO7-biotin and disappears completely for a higher content of CC-EO7-Biotin, which agrees with the formation of triazol bonds.13 This is followed by a broadening and shift of the N 1s peak at lower binding energies. These changes are consistent with the appearance of the amide groups of biotin, which typically appear at 400.7 eV.36 Unfortunately, due to the close proximity of the binding energies of biotin triazol groups, it is not possible to resolve the two components in the N 1s spectra. Nevertheless, the peak broadening (the full width at half maximum increases from 3.4 to 3.6 eV) is an indirect confirmation of the overlapping of the two components in the N 1s peak. The S 2p signal shows the contributions of two peaks. The first is centered on 162.5 eV which is typical of S atoms linked to Au atoms.30 An additional contribution around 168.8 eV is ascribed to sulfonate or sulfate groups, i.e. oxidized S atoms.37 Although the signal to noise ratio of the S 2p peak is relatively low, a third contribution centered at 165 eV can be resolved, which increases with the% biotin content. This component depicts the increase in biotin groups at the surface of the nanoparticles.38 In the C 1s region, the main contribution (285 eV) is typical of C–C bonds (see ESI†). An additional contribution was observed around 288 eV, which can be ascribed to CO bonds of the biotin groups (289 eV)39 and C–O of the ethylene oxide chains (287.5 eV).38
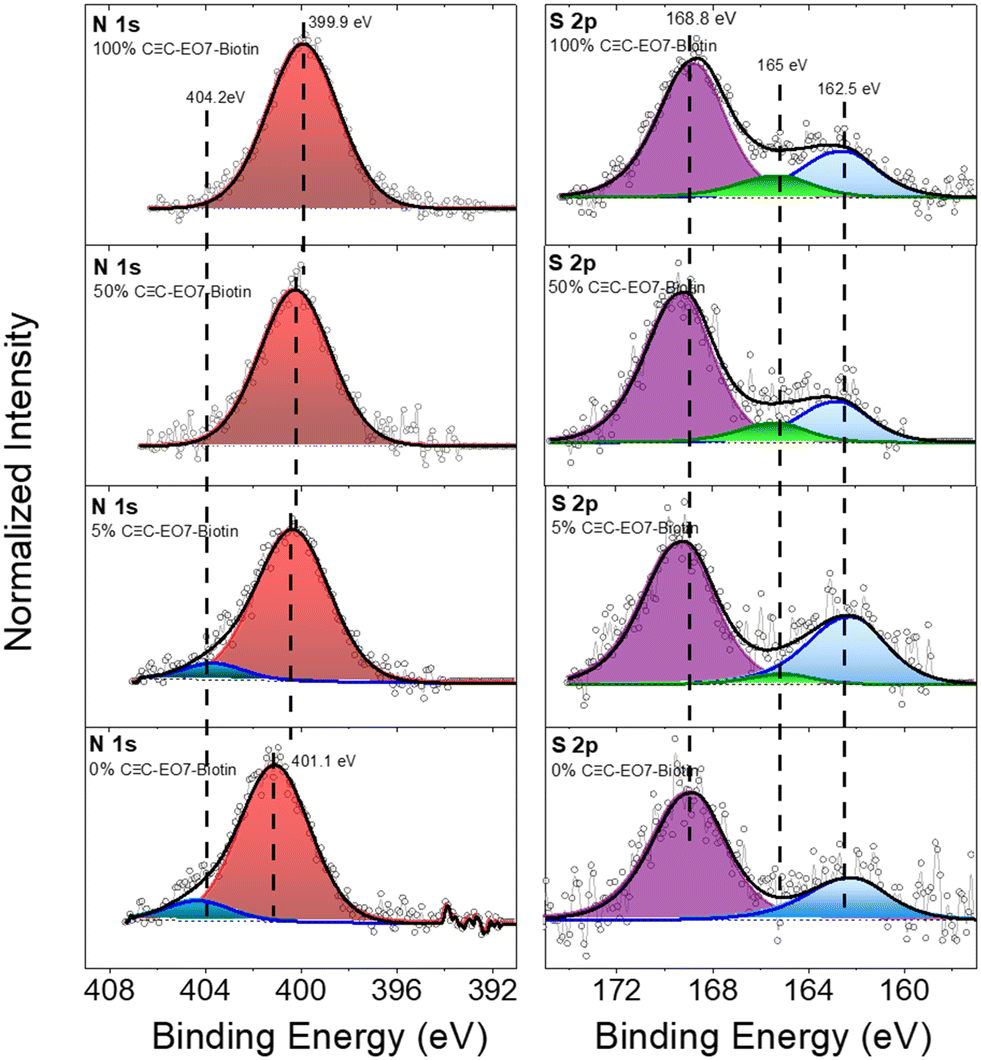 | ||
| Fig. 6 XPS spectra recorded in N 1s and S 2p regions for samples after grafting different mol% of CC-EO7-biotin at the surface of the nanoparticle assemblies. | ||
The SPR angle was monitored to study the absorption of SA as a function of the gradual replacement of CC-EO7-biotin by CC-EO7-OH at the surface of the nanoparticles (Fig. 7). Both molecules were mixed in solution with different molar ratios before performing the CuAAC reaction. A significant decrease in the angular shift was observed when CC-EO7-biotin decreased to 50%. This correlates with lower amounts of SA adsorbed at the surface of the nanoparticles. In contrast, similar angular shifts were observed for amounts down to 5%. No variation in the angular shift was observed when CC-EO7-biotin was totally replaced by CC-EO7-OH, in agreement with the repellent properties of EO chains. Such a variation in the angular shift is unexpected. It is well known that the high loading of molecules onto a surface undergoes tight packing of functional groups, which hampers their accessibility. Therefore, the highest amount of adsorbed SA was expected for an intermediate biotin content, which corresponds to the interplay with the lowest steric hindrance. The observed results are ascribed to the high radius of curvature of spherical nanoparticles which avoids the steric hindrance of biotin groups. The flexibility of the EO7 linker, which enhances the mobility of biotin groups, also contributes to the absorption of higher amounts of SA. It may also compensate for low biotin content (below 50%), resulting in a similar amount of adsorbed SA. It is worth noting that only 5% of CC-EO7-biotin results in a significant angular shift of 0.26, in agreement with the exceptional biomolecular affinity of the biotin/streptavidin couple.
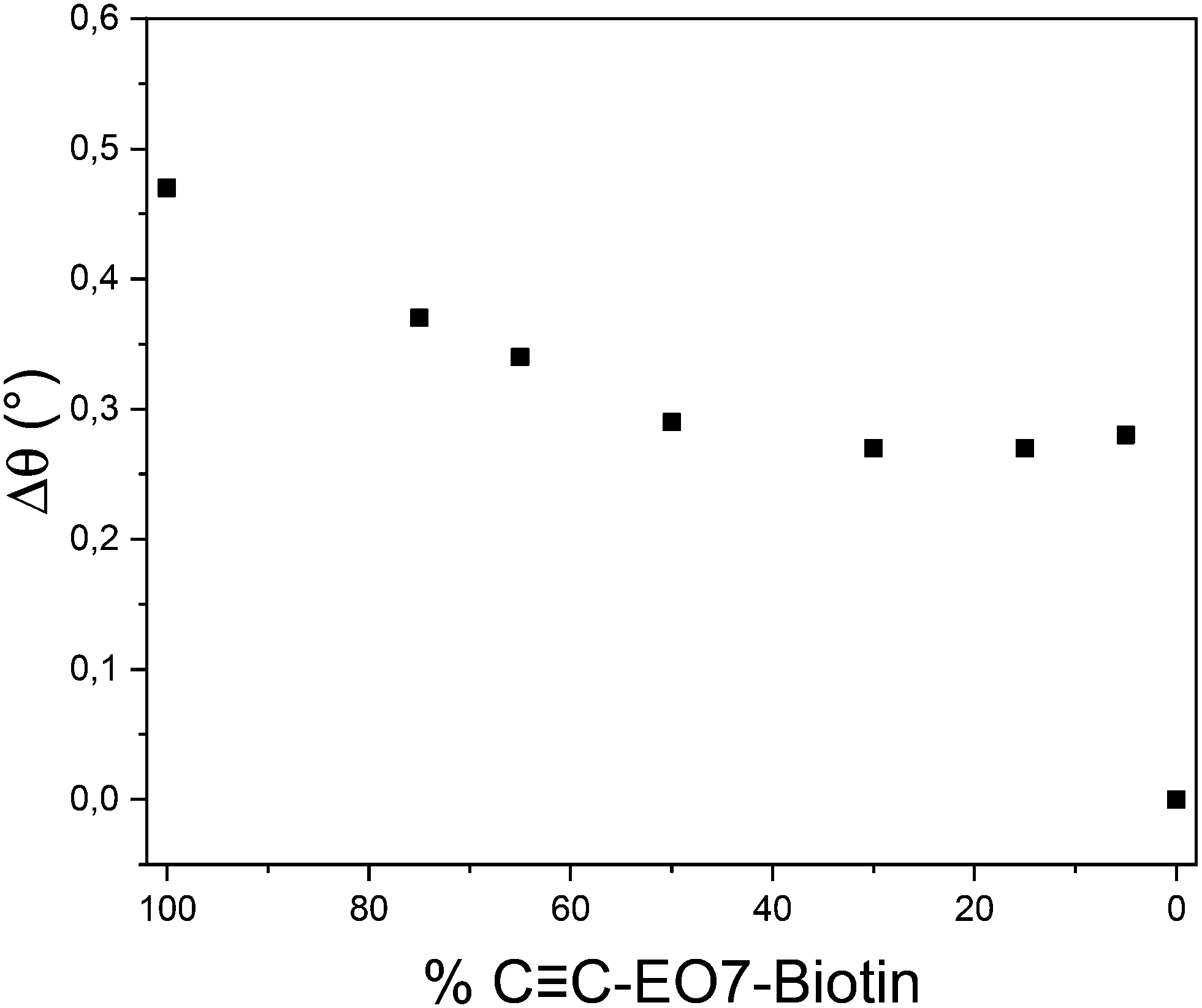 | ||
| Fig. 7 Angular shift measured from sensorgrams as a function of the CC-EO7-biotin mol% after injection of an aqueous solution of SA (10 μg mL−1). | ||
The shift in the SPR signal to higher angles (Δθ) corresponds to the increase in the refractive index at the sensor surface resulting from the adsorption of material which can be quantified according to the Campbell model:40

Considering the sensor build-up, the largest shift in the resonance angle is ascribed to the assembly of nanoparticles because it corresponds to the highest variation in the refractive index. According to the nanoparticle density and organic molecules grafted onto the nanoparticle surface, we have calculated an average refractive index nadsorbate of 1.64 (see ESI†) which is higher than that of organic molecules (n = 1.42). Considering a 48 nm-thick gold thin film covered by a nanoparticle assembly and a wavelength of 785 nm, we calculated a decay length (ld) of 204 nm and a sensitivity factor m of 114 ± 2°/RIU (see ESI†). Therefore, the angular shift (2.121°) corresponds to an effective layer thickness of 12.2 nm, which agrees with the size of the nanoparticles surrounded by organic moieties. The angular shifts after grafting of CC-EO7-biotin (0.317°) and N3-EO7-OH (0.021°) correspond to much thinner adsorbate layers of streptavidine (2.1 nm and 0.1 nm, respectively). The amount of adsorbed SA (nstreptavidin = 1.47 (ref. 41 and 42)) was also calculated from the angular shift measured for each CC-EO7-biotin content (see ESI†). The effective adsorbate layer of SA decreases from 3.1 nm to 1.6 nm when diluting biotin groups with CC-EO7-OH chains at the surface of the nanoparticles. The effective adsorbate layer thickness is also correlated with the mass of SA per area unit (Γ) according to the Freiter equation:43
 C-EO7-biotin content, calculated effective adsorbate layer thickness (t), mass of adsorbed SA per area unit (Γ) and number of SA per nanoparticle
C-EO7-biotin content, calculated effective adsorbate layer thickness (t), mass of adsorbed SA per area unit (Γ) and number of SA per nanoparticle
| Mol% CC-EO7-biotin |
Mol% PEO7 | Δθ (°) | t (nm) | Mass (ng cm−2) | SA/NP |
|---|---|---|---|---|---|
| 0 | 100 | 0 | 0 | 0 | 0 |
| 5 | 95 | 0.26 | 1.7 | 113 | 5.1 |
| 15 | 85 | 0.25 | 1.6 | 108 | 4.8 |
| 30 | 70 | 0.25 | 1.6 | 108 | 4.8 |
| 50 | 50 | 0.28 | 1.8 | 121 | 5.4 |
| 65 | 35 | 0.34 | 2.2 | 148 | 6.6 |
| 75 | 25 | 0.37 | 2.4 | 161 | 7.2 |
| 100 | 0 | 0.47 | 3.1 | 205 | 9.2 |
One of the main advantages of SPR sensing is determination of the kinetics of biomolecular interactions. The sensorgram allowed separate observation of the association, equilibrium and dissociation phases.45 Since the CC-EO7-biotin/N3-EO7-OH sample was the most efficient for detecting SA, we studied the adsorption kinetics by injecting SA aqueous solutions with concentrations ranging from 0.1 to 10 μg mL−1 (1.9 to 190 nM). All sensorgrams show an increase in the resonance angle followed by a plateau a few minutes after injection of SA (Fig. 8a). Lowering the SA concentration resulted in lower angular shifts and increased the time to reach the maximum value of the angular shift from 1 to 4 minutes. Sensorgrams could be recorded for concentrations down to an angular shift of 0.04° for an SA mass concentration of 0.1 μg mL−1 (1.9 nM). For lower concentrations, the signal to noise ratio was lower than the equipment resolution (0.001°).
 | ||
| Fig. 8 a) Sensorgrams recorded for CC-EO7-biotin + N3-EO7-OH after the injection of different concentrations of SA with a fixed flow rate of 50 μL min−1. b) Angular shift plotted against SA concentration. The standard deviation is very low (0.001°) in accordance with the equipment. | ||
The association and dissociation constants were calculated by refining Δθ([C)]) curves, namely binding isotherms (Fig. 8b).46 The response at the equilibrium (Δθ) is related to the affinity constant according to non-linear regression:

Finally, the limit of detection (LoD) of the system can be calculated according to:

4. Conclusion
We reported an original approach to designing the surface chemistry of an efficient SPR sensing platform based on an assembly of iron oxide nanoparticles supported on a gold thin film with specific functionalization by organic molecules. The building approach consisting of a three-step CuAAC “click” reaction resulted in a very robust nano-architecture. The mild operating conditions of “click” chemistry preserved the structure of nanoparticle assemblies and the functionalities of organic moieties grafted onto the nanoparticles and uncovered areas. Besides the significant enhancement in sensitivity resulting from the high refractive index of the nanoparticle assembly we reported earlier,13 the topography of the surface and its functionalization by an accurate biotin derivative resulted in a significant enhancement in the amount of absorbed streptavidin (SA). We showed that an ethylene oxide (EO7) a linker between the biotin group and the surface of nanoparticles favored the better mobility of biotin groups thanks to their solubility in an aqueous medium. Such EO7 chains avoided the adsorption of non-target proteins such as BSA thanks to efficient repellent properties. We also showed that the high radius of curvature of the spherical nanoparticles avoids steric hindrance, which is usually correlated with a high loading of bioreceptors on “flat” surfaces. Therefore, the topography of nanoparticle assemblies and their accurate functionalization allowed the combination of the high density and high accessibility of biotin groups. Such an original approach resulted in a high affinity constant (KA = 2.45 × 107 M) of the biotin/SA couple.A remarkable enhancement in the limit of detection (LoD) down to 1.1 nM was calculated compared with state-of-the-art biosensors based on “flat” gold thin films. Such an LoD is also competitive with biosensors involving labelling. Finally, this approach offers interesting perspectives toward the development of new chemically designed SPR sensors based on robust nano-architectures. Thanks to the azide groups localized at the surface of iron oxide nanoparticles, a wide range of receptors can be grafted in order to design sensors for the specific detection of a variety of molecules.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Agence Nationale de la Recherche (ANR) are acknowledged for financial support through the MaChiNaCo project (ANR19-CE09-0018). The Region Grand Est is also acknowledge for finacial support.References
- J. Homola, Chem. Rev., 2008, 108, 462–493 CrossRef CAS PubMed.
- J. Breault-Turcot and J.-F. Masson, Anal. Bioanal. Chem., 2012, 403, 1477–1484 CrossRef CAS PubMed.
- A. Shalabney and I. Abdulhalim, Laser Photonics Rev., 2011, 5, 571–606 CrossRef CAS.
- S. Szunerits, A. Shalabney, R. Boukherroub and I. Abdulhalim, Rev. Anal. Chem., 2012, 31, 15–28 CAS.
- S. Szunerits, X. Castel and R. Boukherroub, J. Phys. Chem. C, 2008, 112, 15813–15817 CrossRef CAS.
- G. A. J. Besselink, R. P. H. Kooyman, P. J. H. J. van Os, G. H. M. Engbers and R. B. M. Schasfoort, Anal. Biochem., 2004, 333, 165–173 CrossRef CAS PubMed.
- J. Wang, Z. Zhu, A. Munir and H. S. Zhou, Talanta, 2011, 84, 783–788 CrossRef CAS PubMed.
- S. D. Soelberg, R. C. Stevens, A. P. Limaye and C. E. Furlong, Anal. Chem., 2009, 81, 2357–2363 CrossRef CAS PubMed.
- L.-G. Zamfir, I. Geana, S. Bourigua, L. Rotariu, C. Bala, A. Errachid and N. Jaffrezic-Renault, Sens. Actuators, B, 2011, 159, 178–184 CrossRef CAS.
- D. Garibo, K. Campbell, A. Casanova, P. de la Iglesia, M. Fernández-Tejedor, J. Diogène, C. T. Elliott and M. Campàs, Sens. Actuators, B, 2014, 190, 822–828 CrossRef CAS.
- Y. Teramura, Y. Arima and H. Iwata, Anal. Biochem., 2006, 357, 208–215 CrossRef CAS PubMed.
- D. Toulemon, B. P. Pichon, X. Cattoen, M. W. C. Man and S. Begin-Colin, Chem. Commun., 2011, 47, 11954–11956 RSC.
- M. Dolci, J.-F. Bryche, C. Leuvrey, S. Zafeiratos, S. Gree, S. Begin-Colin, G. Barbillon and B. P. Pichon, J. Mater. Chem. C, 2018, 6, 9102–9110 RSC.
- M. Dolci, J.-F. Bryche, J. Moreau, C. Leuvrey, S. Begin-Colin, G. Barbillon and B. P. Pichon, Appl. Surf. Sci., 2020, 527, 146773 CrossRef CAS.
- J. Kim, U. G. Hong, Y. Choi and S. Hong, Colloids Surf., B, 2019, 182, 110303 CrossRef CAS PubMed.
- A. R. Sadrolhosseini, M. Naseri and H. M. Kamari, Opt. Commun., 2017, 383, 132–137 CrossRef CAS.
- A. G. Paulovich, J. R. Whiteaker, A. N. Hoofnagle and P. Wang, Proteomics: Clin. Appl., 2008, 2, 1386–1402 CAS.
- K. L. Prime and G. M. Whitesides, Science, 1991, 252, 1164–1167 CrossRef CAS PubMed.
- S. Herrwerth, W. Eck, S. Reinhardt and M. Grunze, J. Am. Chem. Soc., 2003, 125, 9359–9366 CrossRef CAS PubMed.
- J. Spinke, M. Liley, H. J. Guder, L. Angermaier and W. Knoll, Langmuir, 1993, 9, 1821–1825 CrossRef CAS.
- L. S. Jung, K. E. Nelson, P. S. Stayton and C. T. Campbell, Langmuir, 2000, 16, 9421–9432 CrossRef CAS.
- F. Rios and S. Smirnov, ACS Appl. Mater. Interfaces, 2009, 1, 768–774 CrossRef CAS PubMed.
- N. M. Green, in Advances in Protein Chemistry, ed. C. B. Anfinsen, J. T. Edsall and F. M. Richards, Academic Press, 1975, vol. 29, pp. 85–133 Search PubMed.
- S. Nainar, M. Kubota, C. McNitt, C. Tran, V. V. Popik and R. C. Spitale, J. Am. Chem. Soc., 2017, 139, 8090–8093 CrossRef CAS PubMed.
- D. Toulemon, B. P. Pichon, X. Cattoen, M. W. C. Man and S. Begin-Colin, Chem. Commun., 2010, 47, 11954–11956 RSC.
- W. Luo and S. Zafeiratos, in Metal-free Functionalized Carbons in Catalysis, 2018, pp. 138–176 Search PubMed.
- D. Toulemon, B. P. Pichon, C. Leuvrey, S. Zafeiratos, V. Papaefthimiou, X. Cattoen and S. Begin-Colin, Chem. Mater., 2013, 25, 2849–2854 CrossRef CAS.
- P. Schaaf, J.-C. Voegel and B. Senger, J. Phys. Chem. B, 2000, 104, 2204–2214 CrossRef CAS.
- M. Dolci, Y. Lei, L.-M. Lacroix, C. Kiefer, C. Leuvrey, S. Begin-Colin and B. P. Pichon, J. Phys. Chem. C, 2019, 123, 27927–27936 CrossRef CAS.
- C.-M. Pradier, M. Salmain, L. Zheng and G. Jaouen, Surf. Sci., 2002, 502, 193–202 CrossRef.
- N. A. Lapin and Y. J. Chabal, J. Phys. Chem. B, 2009, 113, 8776–8783 CrossRef CAS PubMed.
- L. A. Lyon, D. J. Peña and M. J. Natan, J. Phys. Chem. B, 1999, 103, 5826–5831 CrossRef CAS.
- B. P. Pichon, G. Barbillon, P. Marie, M. Pauly and S. Begin-Colin, Nanoscale, 2011, 3, 4696–4705 RSC.
- V. H. Pérez-Luna, M. J. O'Brien, K. A. Opperman, P. D. Hampton, G. P. López, L. A. Klumb and P. S. Stayton, J. Am. Chem. Soc., 1999, 121, 6469–6478 CrossRef.
- Y. Liu, N. RamaRao, T. Miller, G. Hadjipanayis and A. V. Teplyakov, J. Phys. Chem. C, 2013, 117, 19974–19983 CrossRef CAS.
- E. H. Williams, A. V. Davydov, A. Motayed, S. G. Sundaresan, P. Bocchini, L. J. Richter, G. Stan, K. Steffens, R. Zangmeister, J. A. Schreifels and M. V. Rao, Appl. Surf. Sci., 2012, 258, 6056–6063 CrossRef CAS.
- Y. Joseph, B. Guse and G. Nelles, Chem. Mater., 2009, 21, 1670–1676 CrossRef CAS.
- F. Taraballi, L. Russo, C. Battocchio, G. Polzonetti, F. Nicotra and L. Cipolla, Org. Biomol. Chem., 2014, 12, 4089 RSC.
- S. Gao, N. Koshizaki, H. Tokuhisa, E. Koyama, T. Sasaki, J.-K. Kim, J. Ryu, D.-S. Kim and Y. Shimizu, Adv. Funct. Mater., 2010, 20, 78–86 CrossRef CAS.
- L. S. Jung, C. T. Campbell, T. M. Chinowsky, M. N. Mar and S. S. Yee, Langmuir, 1998, 14, 5636–5648 CrossRef CAS.
- R. Reiter, H. Motschmann and W. Knoll, Langmuir, 1993, 9, 2430–2435 CrossRef CAS.
- R. D'Agata, P. Palladino and G. Spoto, Beilstein J. Nanotechnol., 2017, 8, 1–11 CrossRef PubMed.
- J. A. De Feijter, J. Benjamins and F. A. Veer, Biopolymers, 1978, 17, 1759–1772 CrossRef CAS.
- W. M. Albers and I. Vikholm-Lundin, in Nano-Bio-Sensing, ed. S. Carrara, Springer, New York, NY, 2011, pp. 83–125 Search PubMed.
- R. B. M. Schasfoort, Handbook of Surface Plasmon Resonance, The Royal Society of Chemistry, 2008 Search PubMed.
- W. M. Albers and I. Vikholm-Lundin, in Nano-Bio-Sensing, Springer, New York, NY, 2011, pp. 83–125 Search PubMed.
- Y. Tang, R. Mernaugh and X. Zeng, Anal. Chem., 2006, 78, 1841–1848 CrossRef CAS PubMed.
- M. Dolci, Design of magnetic iron oxide nanoparticle assemblies supported onto gold thin films for SPR biosensor applications, Université de Strasbourg, 2018 Search PubMed.
- C.-D. Chen, S.-F. Cheng, L.-K. Chau and C. R. C. Wang, Biosens. Bioelectron., 2007, 22, 926–932 CrossRef CAS PubMed.
- G. Sheppard, T. Oseki, A. Baba, D. Patton, F. Kaneko, L. Mao and J. Locklin, Biomicrofluidics, 2011, 5, 026501 CrossRef PubMed.
- N. Nath and A. Chilkoti, Anal. Chem., 2002, 74, 504–509 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TEM micrographs and size distribution of nanoparticles. Additional PM-IRRAS and XPS spectra. Theoretical calculations of decay length, sensitivity, and amount of SA as a function of the nanoparticle size and density. See DOI: https://doi.org/10.1039/d2sd00069e |
| This journal is © The Royal Society of Chemistry 2022 |