
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical approach for recognition and quantification of p-phenylenediamine: a review
Manorama
Singh
 *a,
Smita R.
Bhardiya
a,
Ankita
Rai
b and
Vijai K.
Rai
a
*a,
Smita R.
Bhardiya
a,
Ankita
Rai
b and
Vijai K.
Rai
a
aDepartment of Chemistry, Guru Ghasidas Vishwavidyalaya, Bilaspur-495 009, C. G, India. E-mail: manoramabhu@gmail.com; Tel: +91 7587401982
bSchool of Physical Sciences, Jawaharlal Nehru University, New Delhi, 110 067, India
First published on 10th March 2022
Abstract
p-Phenylenediamine (p-PDA) is a toxic constituent utilized in textiles, black henna tattoos, hair dyes, pesticides, polymers etc. The metabolites (quinone etc.) produced by p-PDA cause mutagenic and carcinogenic effects in humans as it is quickly absorbed by the blood after oral intake or intake through the skin. Therefore, the literature reports several conventional approaches (such as colorimetry and chromatography) and electrochemical/voltammetric approaches to determine and quantify p-PDA. Amongst them, the electrochemical approach is the most preferable because of the short analysis time, cost-effectiveness, ease of use, ease of miniaturization etc. This is the first review to discuss the sources and toxic effects, and make a comparison between conventional and electrochemical methods for the determination and quantification of p-PDA in analytical samples.
1. Introduction
1.1. P-Phenylenediamine (sources and its applications)
p-Phenylenediamine (p-PDA), also named 1,4-diaminobenzene or 1,4-phenylenediamine, is an azo dye intermediate and has been utilized as a component in the hair dye, polymer and textile industries.1,2 As a chief component, it has been applied since the 19th century in thousands of hair (oxidative) dyes. Nowadays, people use hair dyes to improve their physical appearance and stylishness. Oxidative chemical reactions are involved in coloring the hair color dark.3 Hair dyes may be categorized into three important categories (a) temporary: which have a high molecular mass and color the hair for a short time without penetrating the cortex of the hair; (b) semi-permanent: low molecular mass dyes which persist for a long time compared to temporary dyes due to their ability to penetrate the cortex; and (c) permanent: perpetual coloring is possible by an oxidation method and a coupling reaction.4Permanent hair dyes are generally composed of three constituents: i.e. a precursor agent (p-PDA),5,6 a coupling agent (electron-donating group substituted aromatic compounds, for example resorcin) and an alkaline medium based oxidizer (e.g. H2O2–ammonia mixture).7 The attractive forces in oxidative dyes are possible due to the presence of amino and hydroxyl groups along with their positions. Amino (–NH2) groups present on either o- or p-positions in p-PDA are used as precursors, whereas m-dihydroxybenzenes are used as couplers.1 Furthermore, p-PDA is used for the preparation of conductive or non-conductive polymers through chemical or electrochemical polymerization and has been reported as a redox mediator to transport electrons between electrode and oxidase enzymes.2
1.2. Toxic effects of p-PDA
Since 1863, the dying process has been facilitated by employing p-PDA in oxidizable hair dyes8 and, therefore, many countries have legally limited the use of p-PDA in cosmetic products (<4%).9 In terms of toxicity, p-PDA and o-PDA were found to be more toxic than m-PDA and phenol derivatives.10 A huge number of reports and case studies8,11–16 of using p-PDA in hair dye products have paid attention to their poisoning,8,11 allergic contact dermatitis,12–15 optic atrophy16etc. Convulsion, cyanosis and facial oedema were characteristic symptoms in twenty (20) fatal p-PDA poisoning cases, whereas the swelling of the vocal folds and epiglottis is very common.17 Other symptoms from oral intake are vomiting, pain in the epigastric region, swelling of the neck, shortness of breath, methemoglobinemia, rhabdomyolysis, kidney and liver failure, hemolysis etc.17–19 After oral intake, the blood of the digestive tract absorbs the p-PDA quickly and it is metabolized into quinone-diimine (a cytotoxin). Further, detoxification occurs due to its acetylation into two major metabolites, N-acetyl-p-PDA and N,N-diacetyl-p-PDA, which are expelled through urination.20,21 These metabolites and intermediates (Brondowski's bases) are responsible for ailments in humans such as in vitro mutagenicity and in vivo carcinogenicity.22 Some reports show the involvement of p-PDA in homicidal, suicidal and accidental poisoning cases too.23–25 Hence, the detection of p-PDA levels in hair dye samples is a significant point of concern to ensure the adequate quality of products.4Generally, oxidative hair dye formulations contain p-PDA as a chromophoric constituent. In some parts of Africa, such as Egypt, Sudan, Morocco, and also in the Indian scenario, either p-PDA or a combination of henna and p-PDA is used to color the hair or skin. Al-Suwaidi et al. 2010 reported that black henna contains p-PDA concentrations in the range of ≥0.4% to 29.5%. Therefore, cases of allergic eczema due to black henna use were reported to be high in the UAE.26
Mostly, N-mono- and N,N-9-diacetylated products (MAP-PDA, DAP-PDA) are produced by extreme skin contact with p-PDA. p-PDA and metabolites are largely expelled with urine. Despite less toxicity in use, several cases of toxicity have been reported in some countries per annum, which lead not only to a severe clinical disorder but also rhabdomyolysis, laryngeal oedema, renal problems, and nervous and hepatic toxicity.3
To the best of our knowledge, this is the first review to discuss the sources and toxic effects of p-PDA, and make a comparison between conventional (e.g. colorimetry, spectrophotometric, chromatography etc.) and electrochemical methods (reported from 1935 to 2021) to detect and quantify p-PDA present in analytical samples. The performance of the reported methods has been summarized and compared in terms of the limit of detection, linear range, selectivity etc. (Table 1).
| S N | Method | Electrode | Electrolyte | Linear range | LOD | Interferents | Ref. |
|---|---|---|---|---|---|---|---|
| Abbreviations: HPLC-ESI-MS/MS – HPLC with electrospray ionization tandem mass spectrometry, GC-NICI-MS – gas chromatography with negative ion chemical ionization mass spectrometry, LC-MEKC – liquid chromatography micellar electrokinetic chromatography, ECD – electrochemical detector, SWV – square wave voltammetry, HRP – horseradish peroxidase, PBS – phosphate buffer solution, BRB – BR-buffer, Cat – catechol, 4-AP – 4-aminophenol, CZE-AD – capillary zone electrophoresis coupled with amperometric detection, IL – ionic liquid, HQ – hydroquinone, GO – graphene oxide, rGO – reduced graphene oxide, PANI – polyaniline, GS – graphene sheets, AMP – acetaminophen, DPV – differential pulse voltammetry, CV – cyclic voltammetry, PRM – pyrimethamine, IMP – imipramine, DPM – desipramine, PA – phenylamine, RCN – resorcinol. | |||||||
| 1 | Laboratory test | — | — | — | — | — | 27 |
| Colorimetric/spectrophotometric/fluorescence | |||||||
| 2 | Colorimetric | — | pH 6 | 0.002–1.5 mM | 0.53 μM | o- and m-PDA, o-toluidine, 4-chloroaniline, aminobiphenyl derivatives, o-dianisidine | 28 |
| 3 | Colorimetric/spectrophotometry | — | pH 3.5–6 | — | — | — | 29 |
| 4 | Spectrophotometry | — | — | 1–5 mcg mL−1 | — | — | 30 |
| 5 | Fluorescence | — | 50 mM PBS pH 7.0 | 0.03–0.40 mM | 3 × 10−5 M | — | 31 |
| 6 | Colorimetric | — | Conc. H2SO4 | 1–100 mg L−1 | 0.001 ppm | 33 | |
| 7 | Colorimetric | — | — | — | 2 mg L−1 in blood, urine, and gastric contents and 2 mg kg−1 in liver | AMP, p-AP, aniline, PRM, IMP, DPM, amantadine, amitriptyline, citalopram, fluvoxamine, ephedrine, pseudoephedrine, amphetamine, methamphetamine etc. | 34 |
| 8 | Fluorescence | — | 20 mM borate (pH 8.0) containing 10 mM Brij 35 and 35% (v/v) methanol | — | 50 nM | — | 36 |
| 9 | Fluorescence | — | — | 10–100 and 100–1000 μg L−1 | 5.35 μg L−1 | o-PDA, p-PDA, PA | 37 |
| Chromatographic/spectroscopic/hyphenated | |||||||
| 10 | Fused-silica capillary gas chromatographic | — | — | 0.03–0.1% | — | — | 38 |
| 11 | HPLC | — | — | 0.93–3700 μM | 9.25 μM | — | 39 |
| 12 | HPLC method with spectrophotometry | — | — | 0.05–0.16 mg mL−1 | — | — | 10 |
| 13 | LC-ECD | — | CH3OH-octylammonium orthophosphate | 0.213–0.018 l g mL−1 | — | — | 40 |
| pH 6.3 | |||||||
| 14 | HPLC-ESI-MS/MS and GC-NICI-MS | — | — | — | — | — | 43 |
| 15 | GC-MS ion trap analysis | — | pH 9.1 | — | 0.1 pg | — | 44 |
| NH4OH | |||||||
| 16 | LC-MEKC | — | Borate buffer pH 9.2 | 0.5–70 μg mL−1 | 0.19 μg mL−1 | — | 45 |
| 17 | GC-MS | — | — | — | 0.05 mg mL−1 | — | 46 |
| 18 | HPLC with amperometric detection | — | 0.1 M acetate buffer, pH 4.5 | 0.3–300 ng | 15–40 pg | — | 47 |
| 19 | GC-MS | — | — | 0.02–0.20 ng g−1 | — | — | 48 |
| 20 | HPLC method using a diode array detector (DAD)-ECD | — | — | 0.05–50 M | 0.55 M | — | 49 |
| 21 | GC-MS | — | 1,4-PDA-2,3,5,6-d4 | 1.0–1275 g mL−1 | 0.10 g mL−1 | — | 50 |
| 22 | Micro-column extraction and MEKC | — | — | 5 × 10−5–1 × 10−2 M | 1.97 × 10−7 M | — | 51 |
| 23 | LC-MS/MS | — | — | 10–2000 ng mL−1 | 5 ng mL−1 | — | 25 |
| Electrophoretic | |||||||
| 24 | Hydrodynamic voltammetry | CFE | PBS | 0.5–500 μM | 0.075 μM | — | 54 |
| 10 mM | |||||||
| 25 | CZE-AD | — | PBS pH 8 | 2 × 10−6–2 × 10−4 M | 1.57 × 10−7 M | — | 22 |
| 26 | CZE-separation models | — | PBS pH 7 | — | 0.1 μM | — | 55 |
| Voltammetric/electrochemical | |||||||
| 27 | SWV | Composite based electrode material | PBS–KCl | 2–20 μM | 0.6 μM | — | 4 |
| pH 7 | |||||||
| 28 | Voltammetric | Nitrogen-doped graphene | — | 2–500 μM | 0.67 μM | — | 56 |
| 29 | Chrono-amperometry | β-MnO2 nanowires/GCE | PBS, pH 7 | 0–150 mM | 50 nM | o-PDA, m-PDA, cat, RCN, p-di-hydroxybenzene | 57 |
| 30 | Voltammetric | MWNTs–CHT/GCE | BRB, pH 7 | 0.55–21.2 mM | 7.31 μM | — | 58 |
| 31 | Photoelectrochemical | CdS quantum dots-GS coated on F-doped SnO2 (FTO) | PBS pH 6 | 0.1–3 μM | 43 nM | — | 59 |
| 32 | Electrochemical method | HRP/NPG/GCE | PBS pH 7 | — | — | Cat, 4-AP, o-PDA | 60 |
| 33 | Electrochemical method | SWV | PBS pH 7 | 0.12–3.00 μM | 71 nM | — | 61 |
| 34 | Electrochemical method | TiO2 nanotube (TiO2NTs) electrodes | KH2PO4, pH 10 | 0.5–98.6 μM | 0.055 μM | — | 62 |
| 35 | CV and AC impedance | PB/SAC | PBS pH 7 | 2 × 10−7–1 mM | 6.47 × 10−8 M | — | 63 |
| 36 | CV | PSC82/GCE | PBS pH 7 | 0.5–2900, 2900–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 μM 400 μM |
0.17 μM | 4-AP, RCN, HQ, Cat, AA, NaCl, KBr | 64 |
| 37 | DPV | PANI/ZnO–starch–rGO/GCE | PBS pH 7 | 1–180 μM | 0.24 μM | H2O2, NH3, RCN, Mg2+, SO42− | 65 |
| 38 | DPV | IL-GO@Cu Ag/GCE | PBS pH 7 | 0.018–22 μM | 3.96 nM | m-PDA, o-PDA, RCN, HQ, H2O2, NH3 | 66 |
2. Different methods for p-PDA detection
Numerous analytical approaches have been established to separate and/or determine p-PDA, including laboratory tests;27 spectrophotometry/colorimetric/fluorescence;28–31,33–37 chromatography, such as high performance liquid chromatography (HPLC), gas chromatography-mass spectrometry (GC-MS), and micellar electro-kinetic chromatography (MEKC),10,25,38–40,43–51 capillary zone electrophoresis;22,54,55 electrochemical (i.e. cyclic voltammetry, square wave voltammetry etc.);4,56–66 and nuclear resonance spectrometry (NMR)67etc. But some are still in their primary stage.2.1. Laboratory tests
Heim et al., 193527 reported a preliminary test based on a laboratory test for the detection of p-PDA. In this, the test specimen is treated with a small amount of a 3% solution of CH3COOH. A small piece of fur is treated with 1 to 2 cc of acid at about 45 °C and the liquid squeezed out into a ceramic crucible. Finally, 1 drop of an aniline solution in water (1 drop/50 cc) is mixed and a small amount of crystalline solid potassium persulfate added. The presence of p-PDA or its derivatives is confirmed by the appearance of a blue-green color within 5 s.2.2. Colorimetric/spectrophotometric/fluorescence method
Colorimetry is a very popular method for detecting p-PDA because of its convenient ‘naked eye observation’ and simple preparation methods.28 Other methods such as fluorescence are extensively used to detect derivatives of p-PDA (used as antiozonants in rubber products). The newest and perhaps the most influential chemical is flexone 3C (N-iso-propyl-N-phenyl-p-PDA) manufactured by the Naugatuck Chemical Division, U.S. Rubber Co. Hilton et al., 1960 reported work to identify and estimate these compounds by oxidation into corresponding Wurster salts using a colorimetric method.29In 2010, Jadhav et al. reported a validated visible spectrophotometric method for the sensitive quantification of p-PDA in henna hair dye samples. In this method, diazotization of p-PDA was performed with NaNO2, HCl and ammonium sulfamate in chilled conditions followed by coupling between N-(1-naphthyl) ethylenediamine to form a diazotized salt. This resulted in the production of a chromogen (magenta-colored) which was absorbed at λmax = 535 nm. The linear concentration range was obtained from 1 to 5 mcg mL−1.30
In 2012, Ngamdee et al. utilized the fluorescence method using micelles with an alizarin–boronic acid adduct for the selective detection of p-PDA. Boronate ester adduct (ARS/4-FPBA) was yielded when the reaction occurred between alizarin red S (ARS) and 4-formylphenylboronic acid (4-FPBA), resulting in the tuning of fluorescence emission. However, the fluorescence intensity of the ARS/4-FPBA adduct started decreasing with an increasing concentration of p-PDA in a linear function in cetyltrimethylammonium bromide (CTAB) (2.0 mM) in PBS pH 7.0 (50 mM). The effect of several optimization parameters, such as 4-FPBA concentration, solution pH, and types and concentrations of surfactants, on fluorescence quenching was investigated. The fluorescence intensity of the ARS/4-FPBA adduct was significantly 11 times higher in CTAB than in a buffer. The calibration curve was recorded in the concentration range 0.03–0.40 mM with a sensitivity 48 times higher than in buffer solution alone with good selectivity. Satisfactory results were found in determining p-PDA in spiked water samples.31
Two simple, fast, robust, sensitive, and reproducible spectrophotometric methods were developed to determine p-PDA in hair dyes. p-PDA was analyzed using alkaline Folin's reagent solution and ninhydrin reagent in CH3OH at 453 nm and 431 nm, respectively. Linear calibration curves were recorded from 2 to 12 μg mL−1 and from 0.1 to 0.6 μg mL−1 for alkaline Folin's reagent and ninhydrin reagent, respectively. These methods can be applied as routine analysis methods to determine p-PDA in marketed products. The precision of results in terms of RSD values (intra-day and inter-day) were found to be satisfactory (RSD < 2%).32
A simple colorimetric method was developed by Nhokaew et al., 2016 using acidic NaNO3 to determine five aromatic amines: 2-aminodiphenyl (2-ADP), diphenylamine (DPA), 4-aminodiphenyl (4-ADP), p-PDA and o-PDA. Under optimized conditions, the reaction colors of DPA, 2-ADP, 4-ADP, p-PDA and o-PDA were obtained as blue, dark pink, red, light pink and brown, respectively. The linear calibration curves were recorded for DPA, 2-ADP, 4-ADP, p-PDA and o-PDA in the ranges 1–50, 1–170, 1–20, 1–100 and 1–150 mg L−1 while the limits of detection were calculated to be 0.052, 0.081, 0.054, 0.001 and 0.009 mg L−1, respectively.33
A fast colorimetric method for p-PDA determination in several biological samples was established. The test for acetaminophen detection using o-cresol has been adapted to detect p-PDA in urine, blood, gastric contents, and the liver. After precipitating protein with CCl3COOH (2 mL, 10% w/v), biological specimens hydrolyzed p-PDA metabolites to p-PDA in an acidic medium. Finally, o-cresol solution (1 mL, 1% w/v), H2O2 (200 mL, 3% v/v), and conc. NH4OH (0.5 mL) were added to the biological samples. The presence of p-PDA was studied by the production of a violet color turning to a bluish-green color within 10–15 min. The limit of detection was found to be 2 mg kg−1 in the liver and 2 mg L−1 in blood, urine, and gastric contents, respectively. Under optimized conditions, the developed method was selective in the presence of acetaminophen, p-aminophenol etc. and was effectively applied to 13 severe cases of p-PDA poisoning.34
Due to their unique optical and electrical properties, silver nanoparticles (AgNPs) enable strong localized surface plasmon resonance (LSPR) absorption with extremely sharp extinction bands and high extinction coefficients.35 Hence, they behave as an ideal indicator for colorimetric detection. Lee et al., 2017 reported poly (ethylene glycol) methyl ether thiol (PEG·MET) functionalized AgNPs for the efficient colorimetric detection of p-PDA in an aqueous medium. Here, hydrogen bonding was established between –NH2 groups of p-PDAs and oxygen atoms of PEG·MET–AgNPs, which induced aggregation in PEG·MET–AgNPs by lessening the interparticle distance. The resulting color of the aggregate changes dramatically from yellow to reddish-brown. The linear calibration curve was recorded with different concentrations of p-PDA from 0.002 to 1.5 mM with a limit of detection of 0.53 μM.28
Ko et al., 2019 analyzed the four different PDAs [o-, m- & p-PDA, toluene-2,5-diamine (PTDA)] using the fluorescent derivatization strategy to which micellar electrokinetic chromatography (MEKC) was also coupled. In addition, a fluorescent reagent [5-(4,6-dichlorotriazinyl) amino fluorescein] (DTAF) was used and underwent a nucleophilic substitution reaction to improve the detection sensitivity. The optimized separation conditions were 20 mM borate (pH 8.0) containing 10 mM Brij 35 and 35% (v/v) methanol. The limits of detection were found to be 25, 25, 50 and 100 nM for m-PDA, PTDA, p-PDA and o-PDA, respectively.36
Zhang et al., 2020 reported the immobilization of p-PDA on a dialdehyde cellulose membrane reacted with salicylaldehyde to develop a fluorescent sensor for the detection of p-PDA by the emission of yellow fluorescence under 36 nm UV light. Under optimized conditions, the limit of detection was obtained as 5.35 μg L−1 with two linear ranges of 10–100 and 100–1000 μg L−1. Good selectivity was recorded because the detection of p-PDA was barely affected by the presence of 1 ppm of o-PDA, m-PDA or phenylamine. In this method, a two-step Schiff base reaction was involved and fluorescence emission was boosted by hindering nonradiative intramolecular rotation decay of the excited molecules. Satisfactory results were found in spiked hair dye samples (Fig. 1).37
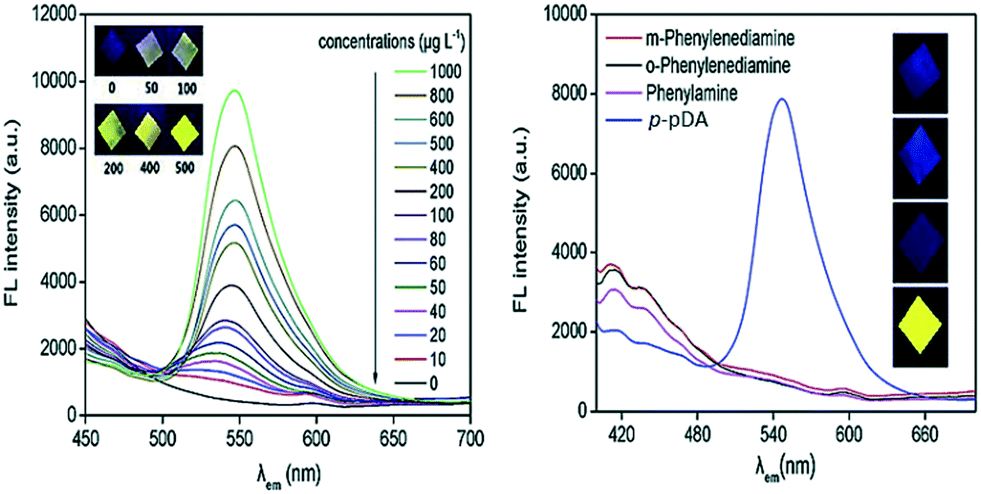 | ||
| Fig. 1 Fluorescence emission spectra with increasing concentrations of p-PDA on S-p-PDA-DCM (inset: colors of part of the prepared S-p-PDA-DCM under 365 nm UV light) [left figure]; fluorescence emission spectra of the samples prepared in 1 mg L−1 of phenylamine, o-PDA, m-PDA and p-PDA solutions, respectively [right figure]. Reproduced from ref. 37 with permission from Elsevier, copyright 2020. | ||
2.3. Chromatography/spectroscopic/hyphenated method
Numerous chromatographic methods have been employed for the separation and/or detection of p-PDA. In 1986, the determination of dye intermediates in oxidative hair dye was reported by Tokuda et al. using the fused-silica capillary gas chromatographic method. A suitable amount of hair dye sample was dissolved in CH3OH (10 mL) with HSCH2CO2NH4 (0.25 g) and 2-amino-4-methylphenol as an internal standard. A gas chromatograph was injected with this solution. Excellent resolution of dye intermediates was obtained using a fused-silica capillary column crosslinked with methyl silicone OV-1 or SE-54 as a liquid phase. Optimization conditions were discussed and good recovery and good reproducibility were obtained. Selective detection of N-containing commercial hair dye intermediates was undertaken by using the nitrogen-phosphorus detector.38Although p-PDA and related compounds are used as antioxidants in rubber products, they commonly display sensitizing properties and are associated with contact dermatitis. It is very important to investigate the p-PDA antioxidant present in commercially available rubber, preventing the risk of contact dermatitis, because the sensitization potential is influenced by the N-substitution of p-PDA. Ikarashi et al., 2000 established a process to determine the derivatives of p-PDA, such as N-1,3-dimethylbutyl-N′-phenyl-p-phenylenediamine (DMB-p-PDA), N-isopropyl-N′-phenyl-p-phenylenediamine (IP-PDA) and N-(1-methylheptyl)-N′-phenyl-p-phenylenediamine (MHP-PDA) using HPLC and also to examine the p-PDA antioxidant present in rubber boots worn by farmers. A combined solution of acetone:chloroform (1:1) was utilized for the extraction of p-PDA derivatives. The silica-gel column was loaded with the extract followed by elution performed with 50 mL of di-ethyl ether:hexane mixtures in the order of 5:95, 10:90, 20:80 and 50:50. The elution for MHP-PDA and DMBP-PDA was performed in the di-ethyl ether:hexane 10:90 fraction. Good recovery of MHP-PDA was obtained by fractionation: 95 ± 8% (n = 5). The detection of P-PDA was done in the di-ethyl ether:hexane 20:80 fraction. The residue was dissolved in dichloromethane after evaporation and HPLC was shown with an ODS column and a UV detector (λmax = 290 nm). The mobile phase was taken, CH3OH:H2O (85:15). No overlap for retention times of this p-PDA was observed after fractionation. The linear concentration ranges of MHP-PDA, DMBP-PDA and IP-PDA were observed from 0.1 to 400 μg mL−1. A total of eight (8) types of rubber boot were investigated in which no MHP-PDA was found while DMBP-PDA and IP-PDA were detected in rubber boots.39
Penner et al., 2000 studied the retention of polar organic molecules such as amino phenols, di-phenols and PDAs on a column (250 × 4.6 mm) packed with hyper cross-linked polystyrene chromalite 5HGN (Purolite, 5 mm). The effect of several separation parameters on their retention was studied, such as acetonitrile concentration, phosphate buffer, citrate buffer, pH and ionic strength of the eluent. Eight dye intermediates could be separated in 20 min. An HPLC-spectrophotometric method was reported for the simultaneous determination of resorcinol, pyrocatechol, hydroquinone, isomers of amino phenols and p-PDA in commercial hair dyes. The limits of detection of these compounds were in the range of 0.05–0.16 mg mL−1.10
Wang et al., 2003 established a liquid chromatography-electrochemical detector method to determine the concentration of three diamine derivatives (4-aminoacetanilide, N,N-p-phenylene bis-acetamide, and p-PDA) not only in rabbit blood, urine etc. but also in human urine. The electrochemical signals were recorded in a detection cell containing a glassy carbon electrode dipped in a supporting electrolyte CH3OH—octyl ammonium orthophosphate (pH 6.3). This method was validated with HPLC-UV.40
The risk of bladder cancer from aromatic amines present in hair dyes was reported by a recent epidemiologic study.41 Moreover, an initial study was done with a combination of hair dye with an enhanced concentration of DNA adducts (4-aminobiphenyl in human epithelial breast cells).42 Therefore, an investigation was undertaken to determine the concentration of 4-ABP (a recognized human urinary bladder carcinogen) present in commercial hair dyes. The presence of 4-ABP was recognized by both HPLC-MS and GC-MS. The concentration range for 4-ABP was recorded from a negligible amount <0.29 ppb to 12.8 ppb. p-PDA is a key constituent for the color development of many permanent hair dyes. Sometimes, due to contamination of 4-ABP (up to 500 ppb) and 2-ABP (up to 70 ppm) in chemical research-grade p-PDA, 4-ABP may be the reason for hair dye toxicity.43
A new method was developed by Stambouli et al. 2004 for the determination of p-PDA in biological fluids (blood, urine and gastric content). This analytical procedure involves deproteinization or hydrolysis, liquid–liquid extraction, derivatization with TFA followed by GC-MS ion trap analysis. Benzidine was the internal standard used for quantification and the extraction recovery test was about 85%. The detection limit of p-PDA was determined to be 0.1 pg (S/N = 10).44
In 2005, both liquid chromatography (LC) and micellar electrokinetic chromatography (MEKC) were utilized by Wang et al. to separate and determine the PDAs and amino-phenols as these compounds are common components in commercial hair dyes. But, only MEKC could be utilized for the quantification. CTAB (55 mM), a borate buffer (pH 9.2), was utilized for the optimum separation condition of MEKC. Finally, a real commercial sample was analyzed by LC and MEKC and no significant difference was found at the confidence level of 99.5%.45
In the same year, Gioia et al. reported an analytical procedure to determine the p-PDA in hair dyes. The methodology involves the transformation of analyte into the corresponding imine derivative treated with C6H5CHO and finally analyzed by GC-MS. Commercial coloring creams were investigated by using an internal standard, N-benzylidene-4-methylbenzene-amine. It was simply prepared by condensation of 4-methylbenzene-amine with C6H5CHO. A linear calibration curve for p-PDA concentrations was observed in the range of 0.1–25 mg mL−1.46
The real-time determination of p-PDA, resorcinol and another seven amino-phenols in hair coloring products was performed by Narita et al., 2007 using an HPLC (with reversed-phase column)–amperometric detector using a mobile phase of 0.1 M acetate buffer (pH 4.5)–CH3OH (90:10, % v/v). A linear calibration curve was recorded in the range of 15–40 pg. Peak heights for the amino-phenols and the two other compounds were found to be linearly related to the amount injected, from 0.3 to 300 ng (R2 > 0.994–0.999). This method reduced the time consumed and non-essential pre-treatment procedures.47
In the next year of 2008, Akyuz et al. reported a GC-MS method to determine the carcinogenic and toxic aromatic amines in hair dye, henna and dyed hair samples. The method includes ion-pair extraction, sonication, conversion into derivatives, and finally GC-MS analysis. Good recoveries were found from 92.2 to 98.4% with RSD 0.7–4.2%. The linear calibration curve was found to range from 0.02 to 0.20 ng g−1. GC-MS analysis was applied for the extraction, identification and quantification of aromatic amines in 54 marketed permanent hair dyes, 35 modified or natural henna and 15 dyed hair samples.48
A series of selective and sensitive HPLC methods with an electrochemical detector has been constructed by Meyer et al., 2009 for the quantification of p-PDA and its acetylated metabolites in biotic samples. The separation was performed using an AQUA C18 column (hydrophilic) and a 25 mM mixture of CH3CN and CH3COONH4 in a ratio of 5:95 (v/v) as the mobile phase. Spectrophotometric analysis was carried out at λmax of either 240 or 255 nm and amperometric detection was done at +400 mV. The calibration curve was plotted for three analytes ranging between 0.05 and 50 M with calculated limits of detection of 0.5 M (p-PDA and N-acetyl-p-PDA) and 1 M (N,N-diacetyl-p-PDA). The oxidation of p-PDA was prevented by using ascorbic acid and sample pre-treatment was not required.49
p-PDA in cosmetic henna tattoos has been determined by Wang et al. 2011. The analytical method involved the extraction of 10 mg of cosmetic product sample in CH3COOC2H5 (10 mL) and further determination was performed by GC-MS in the selected ion-monitoring mode. The calibration curve was obtained within the range of 1.0–1275 g mL−1 with a limit of detection and a limit of quantification of 0.10 and 1.0 g mL−1, respectively. Good recovery was observed for tattoo products with p-PDA and without p-PDA. Extraction efficiency was found to be 98%. Real sample analysis was performed successfully with henna-based products and temporary tattoos.50
In 2011, Hoof et al. established a sensitive and validated wet chemistry assay i.e. MALDI-MS/MS and HPLC-UV, to detect p-PDA and the intermediate products of its metabolic reactions in human urine. A calibration curve was obtained in the range of 50–1000 mM p-PDA. Lastly, the assaying was done for critically spiked human urine samples.3
Wu et al., 2011 reported a novel micellar electrokinetic chromatography (MEKC) method for the determination of PDA isomers in hair dyes. 12 mM ionic liquid (1-butyl-3-methylimidazolium hexafluorophosphates) and 8 M β-cyclodextrins are used as additives. Under optimization conditions, the limits of detection were found to be 1.97 × 10−7 M (p-PDA), 0.99 × 10−7 M (m-PDA) and 0.61 × 10−7 M (o-PDA) with an enrichment factor (EF) of 33.3. Satisfactory results were found for the determination of PDA isomers in hair dyes.51
Mohamed et al., 2015 established a validated liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS) method for the instantaneous detection of p-PDA and its metabolites N-acetyl-p-PDA and N,N-diacetyl-p-PDA in human plasma in the presence of an internal standard ‘Acetanilide’. The operation of LC-MS/MS involved multiple reaction monitoring modes. The linearity was found to be 10–2000 ng mL−1 for p-PDA and other metabolites. Absolute recoveries were found for p-PDA and other metabolites. This new procedure offered availability for the determination of p-PDA, N-acetyl-p-PDA and N,N-diacetyl-p-PDA in clinical plasma samples.25
Recently, one real work-study was reported for the HPLC analysis of p-PDA in marketed hair dye samples (ten) which had been purchased from the local market of El-Bieda-Libya. Here, 50% methanol solution was utilized as a solvent for the determination of p-PDA. A wide linear range was found, ranging from 5 to 25 μg mL−1 with R2 > 0.997. The LOQ and LOD were 3.67 μg mL−1 and 1.21 μg mL−1, respectively. It was found that a Beauty Touch (Blonde) sample contains 0.0855% w/w and a Jourin sense cosmetics (black blue) sample contains 2.2526% w/w. Black color samples showed the highest level of p-PDA. The method analyzed a low-level of p-PDA in the samples as given by the US Food and Drugs Administration.52
Sample preparation and chromatographic separation are common bottlenecks in analyses since these steps are often tedious, time-consuming and expensive.53
2.4. Electrophoretic method
Capillary electrophoresis (CE) of positively charged aromatic amines undertaken at a carbon fiber-microdisk electrode followed by their electrochemical detection has been reported by Huang et al., 1999. Under optimization conditions, the limits of detection were found to be 0.9 (3,4-dihydroxybenzylamine), 0.03 (N,N-dimethylaniline), 0.075 (p-PDA), 1.2 (p-aminophenol) and 0.15 mM (aniline sulfate). Linearity in responses was observed ranging from 5 ± 1000 (3,4-dihydroxybenzylamine), to 0.5 ± 500 (p-PDA & p-aminophenol), 0.1 ± 500 (N,N-dimethylaniline), and 1 ± 200 mM (aniline sulfate). The influence of pH (buffer) and electrode position was also investigated.54In 2008, capillary zone electrophoresis coupled with amperometric detection (CZE-AD) was utilized by Dong et al., 2008 for the instantaneous detection of o-, m- and p-PDA, along with resorcinol and catechol in hair dye samples. Optimization conditions for CZE-AD were investigated in terms of applied working electrode potential, buffer pH and concentration, injection time and separation voltage. Linearity in the concentration of five analytes was found to be in the range of 1–100 μM in borate (0.3 M)–phosphate buffer mixture (0.4 M) (pH 5.8) and the limits of detection were as low as 0.1 μM. Good recovery results were found for 8 types of hair dye samples ranging from 91 to 108% (RSDs < 5.0%). A platinum electrode was used as a detector in CZE-AD, which was fast, easy, reproducible and highly sensitive for real samples.22
Dong et al., 2009 reported capillary zone electrophoresis (CZE) with amperometric detection to determine three positional isomers of PDA (o-, m-, and p-) and di-hydroxybenzene in a weak acidic buffer and weak basic buffer, respectively. Applying two different running buffers in suitable time and order, approx. 20 min was required to separate six analytes and the limit of detection was found to be 0.1 μM. Furthermore, injection time, the influence of working potential, buffer concentration and pH, and applied voltage for separation on detection were also studied. Two groups of isomers with unlike pKa were determined successfully by using this method and it showed some merits in terms of the requirement for less sample, high sensitivity and good reproducibility.55
CE showed good advantages such as low reagent consumption, small sample volume requirement, and high separation efficiency with high-speed analysis.51 However, its short optical path length and sample injection at low volume give poor detection sensitivity.36
However, most of the above-mentioned techniques suffer various disadvantages: for example, costly instrumentation, poor selectivity and long sample treatment process.10 Combinations of CZE and several other detection techniques have also been developed to determine different analytes: for example, mass spectrometry, UV-visible absorbance, fluorescence detection and amperometry.12,13 The use of the electroanalytical technique is an appropriate method to determine p-PDA compounds and their derivatives, because of their electroactive –NH2 groups. Several advantages of this method, such as speed, simplicity, cost-effectiveness, ease of miniaturization, high sensitivity and selectivity, freedom from derivatization etc. make it an excellent method for p-PDA detection.
2.5. Voltammetric/electrochemical methods
p-PDA is an electrochemically active chemical compound and gives well-defined redox peaks. This is the basis of electroanalytical detection and its quantification. Generally, the electro-oxidation of p-PDA involves a two-electron and two-proton mechanism. In 2015, Dai et al., reported that two strong peaks were found at peak potentials 0.245 V and 0.575 V at NG@GCE which also proves the involvement of a two-step mechanism in p-PDA oxidation (Scheme 1).56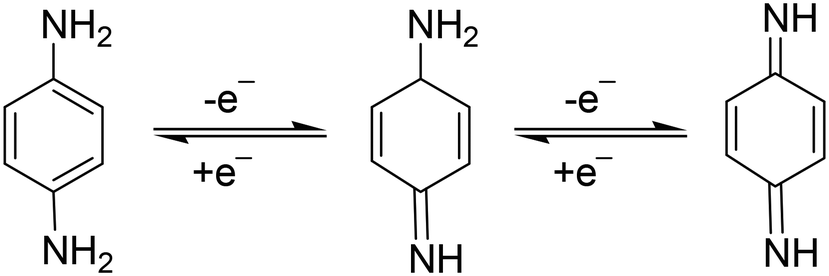 | ||
| Scheme 1 The reaction showing involvement of a single-electron-based reversible mechanism towards p-PDA oxidation at N-doped graphene@GCE. | ||
This mechanism (Scheme 2) showed the stepwise oxidation of two amino groups of p-PDA to quinone diamine. In the electrochemical mechanism, two small reduction peaks also appeared, which showed that the p-PDA is reversible.58 The involvement of a two-electron and two-proton system can also be confirmed by plotting a graph of Epavs. pH.
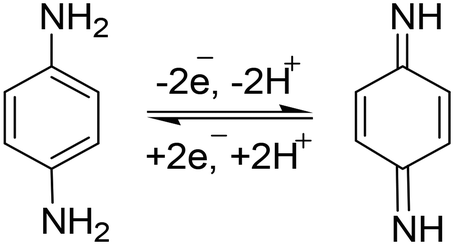 | ||
| Scheme 2 Reversible two-electron and two-proton catalytic mechanism involved in p-PDA oxidation at PANI/ZnO–starch–rGO/GCE. | ||
Detection of p-PDA has been carried out with voltammetric methods using a three-electrode system in which the use of a modified electrode with a composite-based electrode material is an attractive approach. To date, the development of some electrochemical sensors has been reported in the literature for the fabrication of electrochemical sensors for the detection of p-PDA.
To the best of our knowledge, Lawrence et al., 2001 were the first to report an electrochemical square wave voltammogram (SWV) showing the detection of p-PDA in hair dyes. The SWVs showed the appearance of a distinct anodic peak at a positive potential of 0.13 V with an increase in anodic current in the calibration range of 2–20 mM. The detection limit was obtained to be 0.6 mM.4
Bai et al., 2010 employed β-MnO2 nanowires to modify a glassy carbon electrode to determine p-PDA in hair dyes via an electrochemical co-deposition process. Cyclic voltammetric and UV-visible studies showed the involvement of a distinct chemical reaction–electron transfer (CE) process (Fig. 2). The reaction between p-PDA and MnO2 nanowires produced di-imine involving 2e− and 2H+. The oxidation of p-PDA was uninfluenced by the effect of different interferents at a lower overpotential of 0 V vs. SCE, such as other positional isomers of PDA (i.e. o-PDA and m-PDA) and hydroxybenzene due to low chemical reaction activity with manganese dioxide. The calibration range was recorded in the range of 0.2–150 mM with a limit of detection of 50 nM. Real sample analysis was performed with the modified electrode, successfully excluding proper sample preparation.57
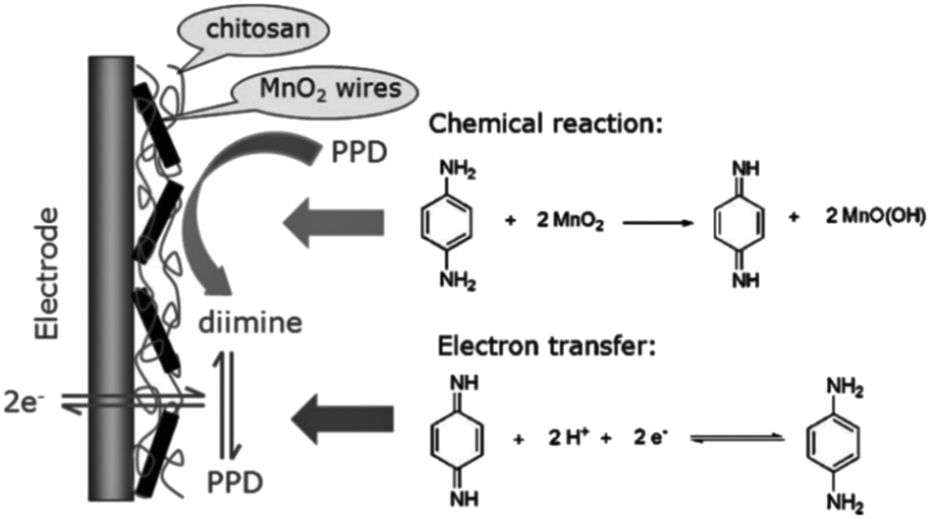 | ||
| Fig. 2 The chemical–electrochemical process of p-PDA on β-MnO2 nanowire/GCE. Reproduced from ref. 57 with permission from John Wiley and Sons, copyright 2010. | ||
Hudari et al., 2014 reported an easy and cost-effective method to functionalize multiwall carbon nanotubes with chitosan for the instantaneous low-level detection of p-PDA and resorcinol in marketed hair dye formulation and tap water. The oxidation of p-PDA occurred at +0.17 V while resorcinol was oxidized at +0.61 V (vs. Ag/AgCl) with normalized current densities 10% and 70% greater than that of a plain GCE, respectively. The linear range was recorded for both p-PDA and resorcinol from 0.55 to 21.2 ppm with limits of detection 0.79 and 0.58 ppm, respectively. This method was applied to determine p-PDA and resorcinol in marketed dye products and tap water with a satisfactory average recovery ∼97%. The reaction products (p-quinone di-imine and Bandrowski's bases) of p-PDA and resorcinol were determined by UV-vis spectrophotometry and LC-MS/MS.58
Zhu et al., 2015 utilized graphene and CdS quantum dot-based photo-electroactive film modified on F-doped SnO2 to investigate the response to photo-irradiation of p-PDA. Based on the photo-electrocatalytic activity of p-PDA in natural light, the recorded cyclic voltammogram was found to be sigmoidal. Optimization of different ratios of CDS and graphene influenced the photo-electrocatalytic activity of p-PDA responses. The effect of pH and scan rates on the photo-electrocatalytic/voltammetric response involved in p-PDA detection was also studied. In addition, the prepared film was explored to determine the p-PDA concentration. Under optimized conditions, a linear calibration curve was obtained in the range of 0.1–3 μM with a limit of detection of 43 nM.59 Dai et al., 2015 synthesized N-doped graphene by thermal annealing of NH2CONH2 and graphene oxide together. Incorporation of nitrogen in the graphene skeleton offered exclusive properties generating high electrocatalytic activity for p-PDA oxidation. In addition, a linear concentration range was plotted from 2 to 500 μM and a low detection limit was calculated to be 0.67 μM. This new approach was employed successfully for the fast sensing of p-PDA in environmental samples.56
Chao et al., 2016 reported that nano-porous gold can carry the enzyme horseradish peroxidase (HRP) in developing an electrochemical biosensor. Nano-porous gold was found to be efficient for oxidizing phenols and aromatic amines selectively. Therefore, it was assumed that HRP and nano-porous gold might have been co-catalyzed on HRP immobilized with a nano-porous gold modified glassy carbon bioelectrode. Linear calibration curves were recorded for the ortho- and para-isomers of PDA, 4-aminophenol and catechol with a low limit of detection, high selectivity, high reproducibility and a negligible effect from interference. The presence of phenols and amines was detected by real sample analysis in a seawater sample, and it offered an efficient and reliable bioelectrode for the simultaneous sensing of these compounds.60 Mou et al., 2017 reported a method to determine p-PDA electrochemically on a carbon paste electrode with a high electrocatalytic effect in neutral phosphate buffer solution. Under optimized conditions, a square wave voltammogram recorded responses linearly ranging from 0.12 to 3.00 μM with a low limit of detection of 71 nM. This procedure was also successfully applied for an extracted sample.61
In 2017, semiconductor titanium dioxide nanotubes were employed for electrochemical sensors. In the anodic region, the electrochemical response was recorded by its prior activation using cathodic polarization. First, a ferricyanide/ferrocyanide redox reaction was studied with a polarized modified electrode and, later, oxidation of p-PDA. Linear sweep adsorptive stripping voltammetry (LSAdSV) was employed to record the responses of increasing the concentration of p-PDA linearly from 0.5 to 98.6 μM with a limit of detection of 0.055 μM.62
In 2018, Hu et al. reported a novel composite of Prussian blue/super-activated carbon to develop an electrochemical sensor for the efficient electrocatalytic sensing of p-PDA in a watery medium. Cyclic voltammetry and AC impedance explained a good electron transfer process in a special chemical reaction involved in p-PDA oxidation. Differential pulse voltammetry was applied for recording the increasing current values with the addition of increasing p-PDA concentration from 0.2 μM to 1 × 10−3 M with a calculated limit of detection of 6.47 × 10−8 M. Good recovery results were found to be in the range of 97–104%, which established the sensor had good operational stability and good repeatability.63
To develop a simple amperometric sensor, He et al., 2019 utilized a perovskite oxide-based electrode because of its cost-effectiveness, ease of carrying and fast response properties. A series of PrCoO3-δ perovskite oxides with the composition Pr1−xSrxCoO3-δ (x = 0, 0.2, 0.4, 0.6, 0.8 and 1) and doped with Sr were synthesized. The prepared PSC82 was further utilized to modify a GC electrode for the sensing of p-PDA in a basic medium. High sensitivities were found to be 655 and 308 μA mM−1 cm−2 in linear calibration ranges 0.5–2900 and 2900–10400 μM, respectively. The limit of detection was calculated to be 0.17 μM and there was good storage stability for 30 days. The fabricated electrochemical sensor confirmed that the p-PDA concentration level was <0.5% in the marketed hair dye sample (Fig. 3).64
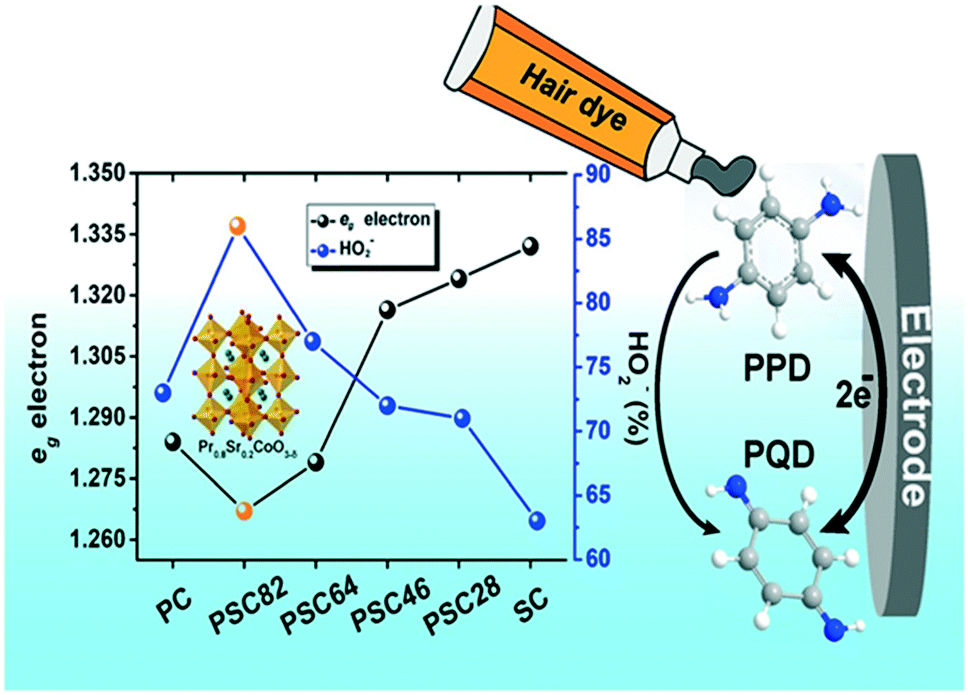 | ||
| Fig. 3 Conversion of p-PDA into p-PQD at PSC82/GCE. Reproduced from ref. 64 with permission from Elsevier, copyright 2019. | ||
In a continuation of these efforts, our group has also prepared different nanocomposite material modified electrodes for the sensitive and selective detection of p-PDA.65,66 In 2019, the construction of a new nanocomposite ‘polyaniline/ZnO-anchored bio-reduced graphene oxide (PANI/ZnO–starch–rGO/GCE)’ was undertaken through π–π interaction and electrostatic interaction. Firstly, bio-reduction of GO was performed by starch and it was further doped along with ZnO in in situ chemical polymerization of aniline for the preparation of a nanocomposite. The structure of the prepared nanocomposite was elucidated by using FTIR spectroscopy, XRD, Raman and XPS spectroscopy while its morphology was studied by using HR-TEM and SEM. Cyclic voltammetry was utilized for electrochemical studies of the nanocomposite. Further, a prepared nanocomposite immobilized in Nafion-modified GCE was utilized for the electrochemical sensing of ‘p-PDA’ in 0.1 M phosphate buffer saline (pH 7) using differential pulse voltammetry in the linear range 1–180 μM with a limit of detection of 0.1 μM. A good selectivity test was obtained with a 50-fold concentration of NH3, resorcinol, magnesium, sulfate and hydrogen peroxide. The prepared electrode was also utilized for real sample analysis with good recovery results (Fig. 4).65
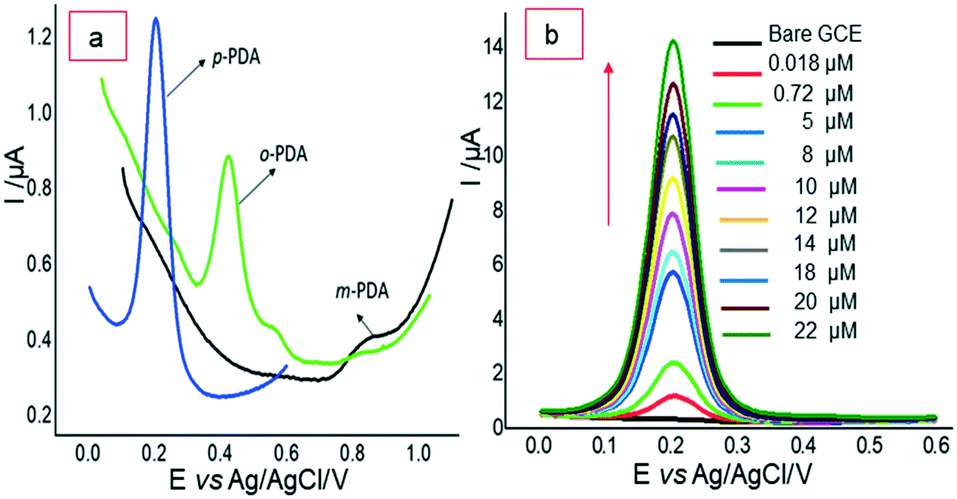 | ||
| Fig. 4 (a) DPVs for electrocatalytic oxidation of p-PDA, o-PDA and m-PDA on IL-GO@Cu–Ag/GCE at different potentials. (b) DPVs of p-PDA oxidation with different concentrations. Reproduced from ref. 65 with permission from Elsevier, copyright 2021. | ||
Recently, in 2021, our group reported a selective electrochemical sensor based on a newly synthesized bimetallic nanocomposite ‘ionic liquid functionalized graphene oxide wrapped Cu–Ag nanoalloy particles’. Here, the synergism between Cu–Ag nanoalloy and ionic liquid–GO offers an improved active surface area and boosted electron transportation, which helped in the excellent electrocatalytic selective detection of p-PDA at 0.21 V (vs. Ag/AgCl) even in the presence of other isomers o-PDA and m-PDA. The electrochemical sensing of p-PDA was recorded in a linear calibration range of 0.018–22 μM with a detection limit of 3.96 nM [Fig. 4]. The effect of the optimization of different experimental conditions, such as the amount of GO, amount of IL, amount of ionic liquid–GO@Cu–Ag, the ratio of ionic liquid–GO@Cu–Ag, and the effect of pH, on the performance of the electrode was also studied. Excellent selectivity was obtained in the presence of o-PDA, m-PDA, hydroquinone, resorcinol, ammonia and hydrogen peroxide. Good recovery results were obtained in water and spiked dye samples, which recognized the nanocomposite material as a good electrochemical scaffold for developing a tool with high simplicity and high selectivity.66
3. Concluding remarks and future prospects
In this review article, the authors have tried to summarize the literature reports (from 1935 to 2021) on conventional and electrochemical methods applied for the determination of p-PDA and compared their performance. It was found that the use of the electrochemical method is preferable because of its simplicity, speed, easy sample preparation, high sensitivity, selectivity etc. To date, a smaller number of modified electrode-based electrochemical sensors have been reported for the detection and quantification of p-PDA in environmental samples. Therefore, great attention is still required to meet the challenges in terms of the fabrication of new sensitive materials, selectivity, stability, lifespan etc.Conflicts of interest
None declared.Acknowledgements
Authors are grateful to CSIR, New Delhi [No. F. 01(3017)/21/EMR-II] for financial assistance. SRB is thankful to CSIR, New Delhi for Senior Research Fellowship.References
- Y. H. Chen, Y. Y. Liu, R. H. Lin and F. S. Yen, J. Hazard. Mater., 2009, 163, 973–981 CrossRef CAS PubMed.
- M. Hebert and D. Rochefort, Electroanalysis, 2008, 53, 5272–5279 CAS.
- G. P. Hooff, N. A. Van Huizen, R. J. W. Meesters, E. E. Zijlstra, M. Abdelraheem, W. Abdelraheem, M. Hamdouk, J. Lindemans and T. M. Luider, PLoS One, 2011, 6, e2219 CrossRef PubMed.
- S. Lawrence, E. L. Beckett, J. Davis and R. G. Compton, Analyst, 2001, 126, 1897–1900 RSC.
- J. Araldi and S. S. Guterres, Tinturas Capilares: Existe Risco De Câncer Relacionado À Utilização Desses Produtos?, Infarma, 2005, 17, 78 Search PubMed.
- G. J. Nohynek, R. Fautz, F. Benech-Keffer and H. Tountain, Food Chem. Toxicol., 2004, 42, 517–543 CrossRef CAS PubMed.
- T. Kojima, H. Yamada, T. Yamamoto, Y. Matsushita and K. Fukushima, Colloids Surf., B, 2013, 106, 140–144 CrossRef CAS PubMed.
- W. Ashraf, S. Dawling and L. J. Farrow, Hum. Exp. Toxicol., 1994, 13, 167–170 CrossRef CAS PubMed.
- (a) P. W. Wu, M. I. Liaw, C. C. Cheng and S. S. Chou, J. Food Drug Anal., 1997, 5, 99 CAS; (b) H. P. Chong, K. Reena, K. Y. Ng, R. Y. Koh, C. H. Ng and S. M. Chye, J. Environ. Anal. Toxicol., 2016, 6(5), 100403–100410 Search PubMed.
- N. A. Penner and P. N. Nesterenko, Analyst, 2000, 125, 1249–1254 RSC.
- H. Yagi, A. M. Elhind and S. I. Khalil, East Afr. Med. J., 1991, 68, 404–411 CAS.
- J. P. McFadden, S. H. Wakelin, D. B. Holloway and D. A. Basketter, Contact Dermatitis, 1998, 38, 79–81 CrossRef PubMed.
- Z. Xie, R. Hayakawa, M. Sugiura, H. Kogma, H. Konishi, G. Ichihara and Y. Takeuchi, Contact Dermatitis, 2000, 42, 270–275 CrossRef CAS PubMed.
- C. J. Le Coz, C. Lefebvre, F. Keller and E. Grosshans, Arch. Dermatol., 2000, 136, 1515–1522 CAS.
- A. Deo Klein and O. G. Rodman, Mil. Med., 1981, 146, 46 CrossRef.
- H. Yagi, A. M. El Hendi, A. Diab and A. A. Elshikh, Hum. Exp. Toxicol., 1996, 15, 617–638 CrossRef CAS PubMed.
- N. Takahashi, J. Ishizawa and M. Yamashita, Jpn. J. Acute Med., 1988, 12, 1509 Search PubMed.
- J. Ishizawa and N. Takahashi, Jpn. J. Acute Med., 1996, 20, 1584 Search PubMed.
- H. Shima, Med. J. Nihon Univ., 1960, 19, 2633 Search PubMed.
- N. Goetz, P. Laserre, P. Bore and G. Kalopissis, Int. J. Cosmet. Sci., 1988, 10, 63–73 CrossRef CAS PubMed.
- Y. Kawakubo, H. F. Merk, T. A. Masaoudi, S. Sieben and B. Blomeke, J. Pharmacol. Exp. Ther., 2000, 292, 150–155 CAS.
- S. Dong, L. Chi, S. Zhang, P. He, Q. Wang and Y. Fang, Anal. Bioanal. Chem., 2008, 391, 653–659 CrossRef CAS PubMed.
- S. Kumar, J. Indian Acad. Forensic Med., 2010, 32, 163–164 Search PubMed.
- K. M. Mohamed, M. A. Hilal and N. S. Aly, International Journal of Forensic Science and Pathology, 2014, 2, 19–23 Search PubMed.
- K. M. Mohamed, D. Cromarty and V. Steenkamp, J. Chromatogr. B, 2015, 997, 1–6 CrossRef CAS PubMed.
- A. Al-Suwaidi and H. Ahmed, Int. J. Environ. Res. Public Health, 2010, 7, 1681–1693 CrossRef CAS PubMed.
- O. Heim, Ind. Eng. Chem., 1935, 7, 146 CAS.
- S. Lee, Y. S. Nam, I. W. Nah, Y. Lee and K. B. Lee, J. Nanosci. Nanotechnol., 2017, 7, 3261–3267 CrossRef.
- C. L. Hilton, Anal. Chem., 1960, 32, 1554–1557 CrossRef.
- J. S. Nitin, M. Muthu Lakshmi, S. Meena and S. Duraivel, Int. J. Pharm. Technol., 2010, 2, 900 Search PubMed.
- K. Ngamdee, S. Martwiset, T. Tuntulanid and W. Ngeontae, Sens. Actuators, B, 2012, 173, 682–691 CrossRef CAS.
- C. H. L. Saranya, B. M. Gurupadayya, K. Kinnera and J. C. Thejaswini Turk, J. Pharm. Sci., 2014, 11, 295–306 Search PubMed.
- B. Nhokaew and R. Jitchati, KKU Eng. J., 2016, 43, 137–140 Search PubMed.
- M. Imran, H. F. Usman, H. Shafi, M. Sarwar and M. A. Tahir, J. Forensic Sci., 2016, 62, 483–487 CrossRef PubMed.
- A. D. McFarland and R. P. Van Duyne, Nano Lett., 2003, 3, 1057–1062 CrossRef CAS.
- H. Yu Ko, Y. H. Lin, C. J. Shih and Y. L. Chen, J. Food Drug Anal., 2019, 23, 825–831 CrossRef PubMed.
- S. Zhang, B. Liu, D. Hu, S. Zhang, Y. Pei and Z. Gong, Anal. Chim. Acta, 2020, 1139, 189–197 CrossRef CAS PubMed.
- H. Tokuda, Y. Kimura and S. Takano, J. Chromatogr. A, 1986, 367, 345–356 CrossRef CAS PubMed.
- Y. Ikarashi and M. A. Kaniwa, J. Health Sci., 2000, 46, 467–473 Search PubMed.
- L. H. Wang and S. J. Tsai, Anal. Biochem., 2003, 312, 201–207 CrossRef CAS PubMed.
- M. G. Dominguez, D. A. Bell, M. A. Watson, J. M. Yuan, J. E. Castelao, D. W. Hein, K. K. Chan, G. A. Coetzee, R. K. Ross and M. C. Yu, Carcinogenesis, 2003, 24, 483–489 CrossRef PubMed.
- K. Gorlewska-Roberts, B. Green, M. Fares, C. B. Ambrosone and F. F. Kadlubar, Proc. Am. Assoc. Cancer Res., 2002, 43, 1018–1192 Search PubMed.
- R. J. Turesky, J. P. Freeman, R. D. Holland, D. M. Nestorick, D. W. Miller, D. L. Ratnasinghe and F. F. Kadlubar, Chem. Res. Toxicol., 2003, 16, 1162–1173 Search PubMed.
- A. Stambouli, M. A. Bellimam, N. El Karni, T. Bouayoun and A. El Bouri, Forensic Sci. Int., 2004, 146S, S87–S92 CrossRef PubMed.
- S. P. Wang and T.-H. Huang, Anal. Chim. Acta, 2005, 534, 207–214 CrossRef CAS.
- M. L. D. Gioia, A. Leggio, A. L. Pera, A. Liguori, A. Napoli, F. Perri and C. Siciliano, J. Chromatogr. A, 2005, 1066, 143–148 CrossRef PubMed.
- M. Narita, K. Murakamia and J. M. Kauffmann, Anal. Chim. Acta, 2007, 588, 316–320 CrossRef CAS PubMed.
- M. Akyuz and S. Ata, J. Pharm. Biomed. Anal., 2008, 47, 68–80 CrossRef CAS PubMed.
- A. Meyer, B. Blomeke and K. Fischer, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 1627–1633 CrossRef CAS PubMed.
- P. G. Wang and A. J. Krynitsky, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 1795–1801 CrossRef CAS PubMed.
- Y. Wu, F. Jiang, L. Chen, J. Zheng, Z. Den, Q. Tao, J. Zhang, L. Han, X. Wei, A. Yu and H. Zhang, Anal. Bioanal. Chem., 2011, 400, 2141–2147 CrossRef CAS PubMed.
- G. Almanfe, O. Khreit and O. Abdullaji, Mater. Sci. Forum, 2019, 955, 13–19 Search PubMed.
- W. Chen, T. A. N. Nkosi, S. Combrinck, A. M. Viljoen and C. Cartwright-Jones, J. Pharm. Biomed. Anal., 2016, 128, 119–125 CrossRef CAS PubMed.
- X. Huang, T. You, T. Li, X. Yang and E. Wang, Electroanalysis, 1999, 11, 969–972 CrossRef CAS.
- S. Dong, L. Chi, Z. Yang, P. He, Q. Wang and Y. Fang, J. Sep. Sci., 2009, 32, 3232–3238 CrossRef CAS PubMed.
- S. Dai, Z. Xu, M. Zhu, Y. Qian and C. Wang, Int. J. Electrochem. Sci., 2015, 10, 7063–7072 CAS.
- Y. H. Bai, J. Y. Li, Y. H. Zhu, J. J. Xu and H. Y. Chen, Electroanalysis, 2010, 22, 1239–1247 CrossRef CAS.
- F. F. Hudari, L. C. de Almeida, B. F. de Silva and M. V. B. Zanoni, Microchem. J., 2014, 116, 261–268 CrossRef CAS.
- Y. Zhu, K. Yan, Y. Liu and J. Zhang, Anal. Chim. Acta, 2015, 884, 29–36 CrossRef CAS PubMed.
- W. Chao, L. Zhuang, S. Huihui, W. Xia and X. Ping, Biosens. Bioelectron., 2016, 79, 843–849 CrossRef PubMed.
- A. A. S. Mou, A. Ouarzane and M. E. Rhazi, J. Electrochem. Sci. Eng., 2017, 7, 111–118 CrossRef CAS.
- G. G. Bessegato, F. F. Hudari and M. V. B. Zanoni, Electrochim. Acta, 2017, 235, 527–533 CrossRef CAS.
- L. Hu, J. Q. Li, X. Q. Zhou, T. T. Bai, Q. J. Cui, J. Tang and W. Y. Xu, Indian J. Chem., 2018, 57A, 896–904 CAS.
- J. He, J. Sunarso, J. Miao, H. Sun, J. Dai, C. Zhang, W. Zhou and Z. Shao, J. Hazard. Mater., 2019, 369, 699–706 CrossRef CAS PubMed.
- M. Singh, A. Sahu, S. Mahata, P. Shukla, A. Rai and V. K. Rai, New J. Chem., 2019, 43, 6500–6506 RSC.
- M. Singh, S. R. Bhardiya, A. Asati, H. Sheshma, V. K. Rai and A. Rai, J. Electroanal. Chem., 2021, 894, 115360–115368 CrossRef CAS.
- B. C. Swan, M. M. Tam, C. L. Higgins and R. L. Nixon, Australas. J. Dermatol., 2016, 57, 219–221 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2022 |