
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective editing of a peptide skeleton via C–N bond formation at N-terminal aliphatic side chains†
Yujie
Han
,
Junjie
Shi
,
Songrong
Li
,
Tingting
Dan
,
Wenwen
Yang
and
Mingyu
Yang
 *
*
Key Laboratory of Applied Surface and Colloid Chemistry of MOE & School of Chemistry and Chemical Engineering Shaanxi Normal University, 620 West Chang'an Ave, Xi'an, 710119, China. E-mail: yangmy05@snnu.edu.cn
First published on 22nd November 2022
Abstract
The applications of peptides and peptidomimetics have been demonstrated in the fields of therapeutics, diagnostics, and chemical biology. Strategies for the direct late-stage modification of peptides and peptidomimetics are highly desirable in modern drug discovery. Transition-metal-catalyzed C–H functionalization is emerging as a powerful strategy for late-stage peptide modification that is able to construct functional groups or increase skeletal diversity. However, the installation of directing groups is necessary to control the site selectivity. In this work, we describe a transition metal-free strategy for late-stage peptide modification. In this strategy, a linear aliphatic side chain at the peptide N-terminus is cyclized to deliver a proline skeleton via site-selective δ-C(sp3)–H functionalization under visible light. Natural and unnatural amino acids are demonstrated as suitable substrates with the transformations proceeding with excellent regio- and stereo-selectivity.
Introduction
Late-stage modification of peptides and peptidomimetics exhibited its potential utility in the field of therapeutic and diagnostic agents, drug candidates, and biological tool compounds (Scheme 1).1–6 Although peptides are abundant in nature, non-natural peptides often demonstrate improved biological and pharmaceutical properties compared with their parent analogs. Therefore, straightforward and efficient methods for the late-stage modification of diverse peptides are highly desirable. To date, various methodologies,7–17 including methylation,12 phosphorylation,13 S-alkylation/arylation,14,15 and glycosylation16 of polar functional groups, have been revealed for the successful chemical modification of natural peptides. Inspired by the development of direct C–H bond functionalization methods, site-specific C–H functionalization has recently emerged as a valuable tool for the late-stage modification of peptides and proteins.18–38 For examples, electrophilic aromatic substitution,18,19 Rh-catalyzed carbenoid insertion,21 Pd-catalyzed arylation/alkenylation,22–26 and photo-induced His-alkylation27 have all been used in this capacity.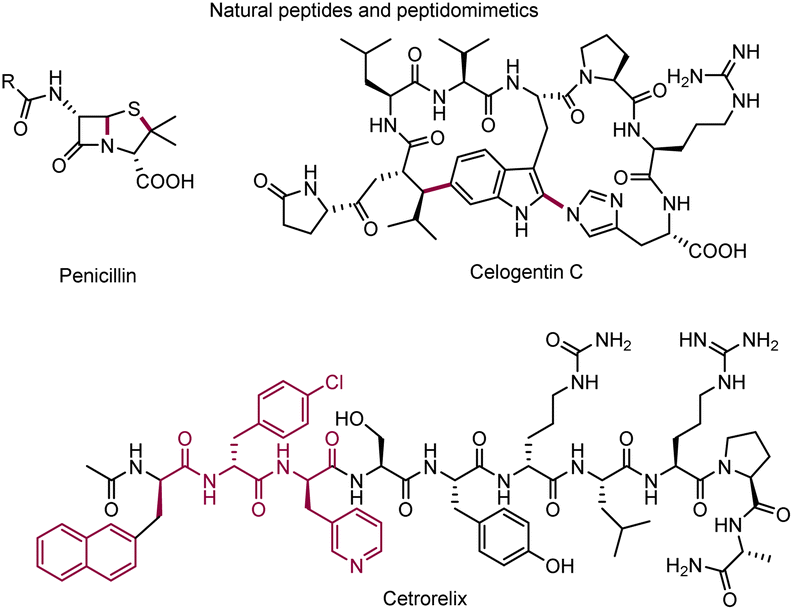 | ||
| Scheme 1 Natural peptides/peptidomimetics. | ||
The modification of aliphatic side chains is a greater challenge than the modification of aromatic residues. In pioneering work by Li and coworkers, copper-catalyzed alkynylation of the α-C–H bond of N-aryl substituted glycine was used to selectively modify short peptides in a racemic fashion.39 Wang and Xu reported the alkylation of N-phenyl glycine residues in peptides via the decarboxylation of N-hydroxyphthalimide esters using a copper photocatalyst.40 Shi and coworkers reported a chemoselective peptide modification strategy involving a photocatalytic tryptophan β-position conjugation;41 oxidation of indole nitrogen initiates a single-electron transfer from the tryptophan β-position to achieve the conjugation. However, these methods are mainly limited to weak C–H bonds, such as benzylic C–H bonds or those adjacent to a heteroatom (Scheme 2a).
 | ||
| Scheme 2 The late-stage modification of peptides via C(sp3)–H activation. | ||
Recently, directing group assisted transition-metal catalyzed functionalization of inert C(sp3)–H bonds within aliphatic residues of peptides has shown enormous potential, with applications in biochemistry and drug discovery.44–59 Innovative contributions have been made by the groups of Yu,42,43 Ackermann,44–46 and Shi.47,48 Both radical-induced and directing group assisted transition-metal catalyzed modifications functionalize the peptide at a single site (i.e., point modifications) (Scheme 2b). To improve the skeletal diversity of peptides (Scheme 2c), Noisier/Albericio49 and Wang50 independently reported intramolecular β-C(sp3)–H arylations for peptide macrocyclization. In related work, Chen and coworkers51 reported an elegant protocol for the preparation of constrained cyclic peptides via Pd-catalyzed intramolecular C(sp3)–H arylation. Because these modifications are achieved via transition metal catalysis, residual metal impurities may affect the biocompatibility of the peptide product and may cause toxicity in pharmaceutical applications. In addition, the installation of a directing group is necessary to control the reactivity and selectivity. Furthermore, the reaction scope is restrictive, occurring predominantly at the β- or γ-C(sp3)–H position of a small set of amino acid residues (mainly alanine). Skeletal modifications involving C–N bond formation are rarely reported. Continuing our studies on radical-transfer reactions,52–55 here we describe a transition-metal free strategy for late-stage peptide modification (Scheme 2d). In our strategy, an N-terminal linear aliphatic side chain cyclizes to construct a proline skeleton via site-selective δ-C(sp3)–H functionalization under visible light. The advantages of this protocol include: (1) transition metal-free conditions that cause skeletal modification via C–N bond formation; (2) compatibility with a broad range of amino acid residues; (3) no extra directing groups are required to promote reaction selectivity and activity.
Results and discussion, experimental
To begin our investigation, we used intermediate 1a as a model substrate (Table 1). After examining a range of reaction parameters, we were pleased to find that the intermediate 1a can be easily transferred to the desired product 2a in 85% isolated yield using only visible light irradiation and basic conditions. Comparative experiments were conducted to test the efficiency under different conditions. Compared with other solvents (Table 1, entries 2–7), DCE proved to be the best choice for this transformation. Next, the effect of the photocatalyst was investigated. Ru-complexes, Ir-complexes, and the eosin Y organophotocatalyst gave similar results under the standard conditions (entries 8–11). Finally, we found that the transformation was inhibited in the dark; the starting material was recovered in almost quantitative yield (entry 12, 99%).|
|
||
|---|---|---|
| Entry | Variations from “standard” conditions | Yieldb (%) |
| a Reaction was conducted on a 0.1 mmol scale. b Yield was determined by 1H NMR. c Isolated yield in parentheses. | ||
| 1 | None | 89c(85) |
| 2 | CHCl3 instead of DCE | 32 |
| 3 | DCM instead of DCE | 85 |
| 4 | THF instead of DCE | 43 |
| 5 | CH3CN instead of DCE | 83 |
| 6 | PhCF3 instead of DCE | 73 |
| 7 | DMF instead of DCE | 56 |
| 8 | Added Ru(bpy)2Cl2 | 89 |
| 9 | Added Ir(ppy)3 | 88 |
| 10 | Added [Ir(ppy)2dtbpy]PF6 | 88 |
| 11 | Added eosin Y | 89 |
| 12 | Under dark | 0 |

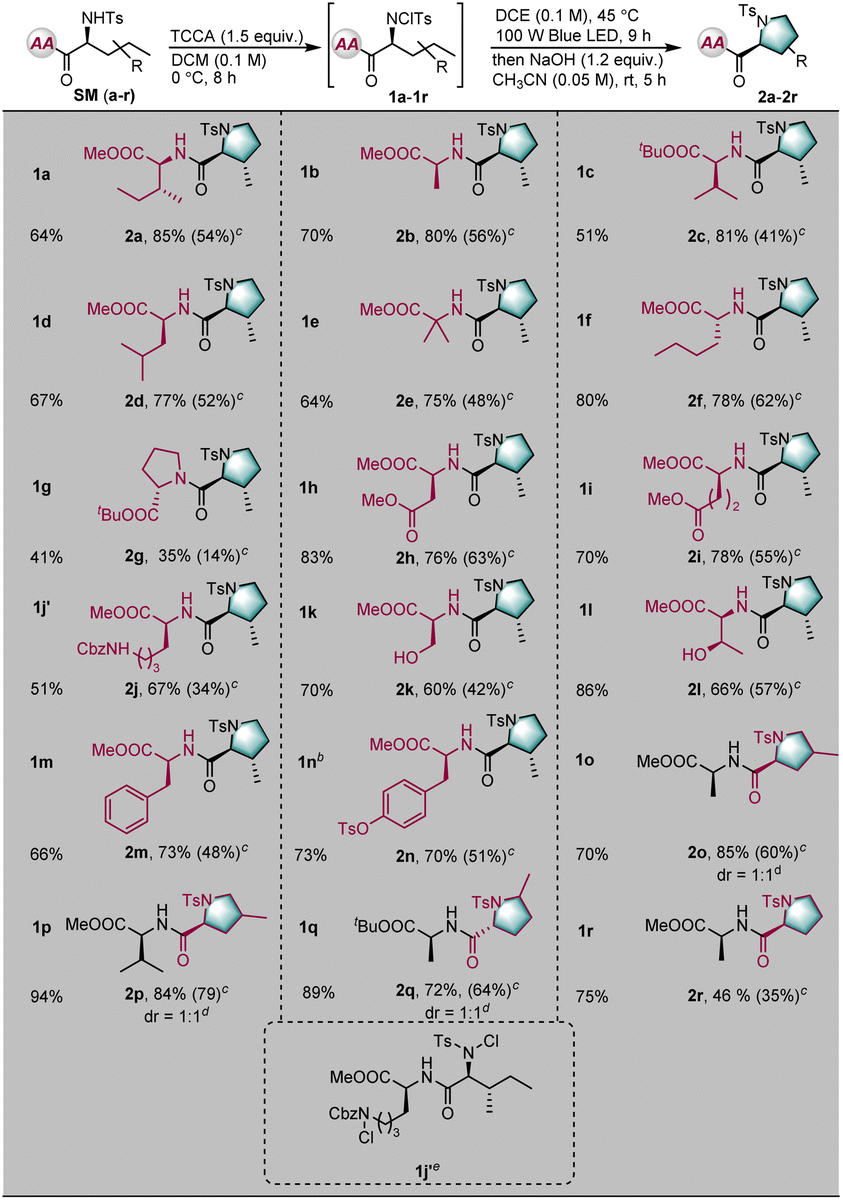
With optimized reaction conditions in hand, the tolerance of the reaction to various peptide substrates was examined. The peptide substrates were generally prepared through the condensation of amino acids and/or short peptides under the solvent conditions. First, to test the selectivity and efficiency of the transformation, various amino acid derivatives were installed in the C-terminal position of a dipeptide substrate (Table 2). Amino acid derivatives of isoleucine, alanine, valine, leucine, 2-aminoisobutyric acid, norleucine, proline, aspartate, glutamic acid, lysine, serine, threonine, phenylalanine, and tyrosine were well-tolerated, giving the desired modified peptides in excellent diastereoselectivities and good yields (SMa–SMn). Notably, when dipeptide SMj was used as a substrate, the cyclization reaction occurred exclusively at the N-terminal position; the lysine side chain was untouched by the reaction, thereby demonstrating the unique selectivity of this transformation (2j). Furthermore, the aliphatic residues at the N–terminal positions were also examined (SMo–SMq). Cyclization occurred at both primary and secondary C–H bonds, selectively yielding the corresponding proline skeleton derivatives. Significantly, the linear unnatural amino acid 2-aminopentanoic acid was converted into a natural proline derivative, albeit in moderate yield, indicating the power of this method (2r).
| a Unless otherwise noted, the reaction conditions were as follows: SM (0.2 mmol), trichloroisocyanuric acid (TCCA, 1.5 equiv.), DCM 2.0 mL (c = 0.1 M), 0 °C, and 8 h; 1 (0.1 mmol), DCE 1.0 mL, blue LED (100 W), 45 °C, and 9 h; then NaOH (1.2 equiv.), CH3CN (2.0 mL), rt, and 5 h. b 2.5 mmol of SM (n) was used, c = 0.1 M. c In parentheses was the calculated total yield. d dr was determined by crude 1H NMR. e Dichloro-product 1j′ was obtained. |
|---|
|
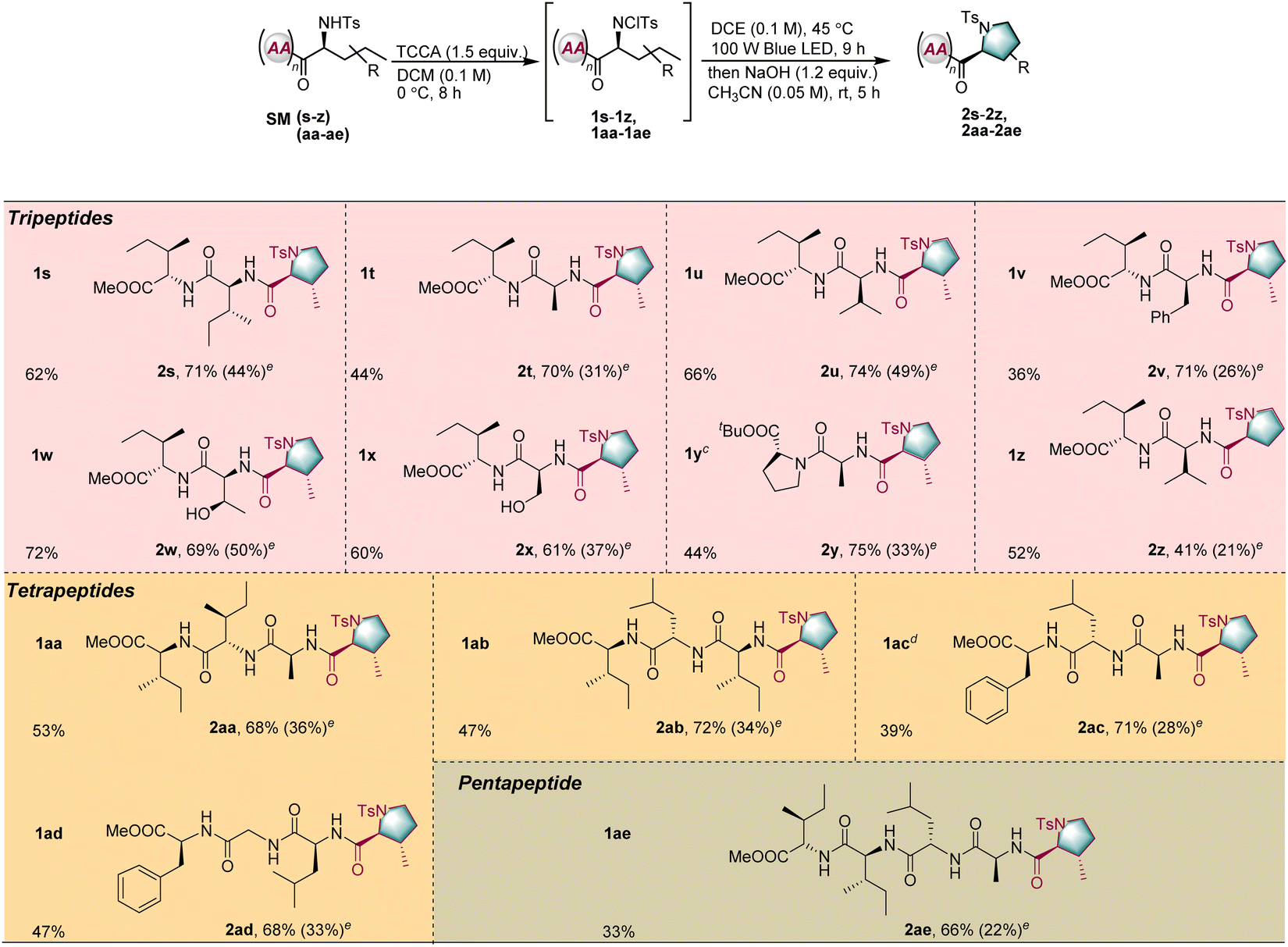
Encouraged by the successful results obtained for dipeptides, the application of the reaction to oligopeptides consisting of three to five amino acid units was explored (Table 3, SMs–z, and SMaa–ae). Because of the larger number of peptide bonds, selective modification of longer peptides is more challenging. However, for all peptides, the transformation proceeded in moderate to good yield with excellent diastereoselectivity under the standard conditions (2s–2z and 2aa–2ae). Unprotected serine or threonine moieties adjacent to the N-terminal position were not detrimental to the reaction (2w and 2x). Furthermore, the phenylalanine moiety, with its highly active benzylic C–H bonds, was untouched during the transformation (2v).
| a Unless otherwise noted, the reaction conditions were as follows: for tripeptides SM(s–z) 0.4 mmol, trichloroisocyanuric acid (TCCA, 1.5 equiv.), DCM 4.0 mL (c = 0.1 M), 0 °C, and 8 h; 1 (0.1 mmol), DCE 1.0 mL, blue LED (100 W), 45 °C, and 9 h; then NaOH (1.2 equiv.), CH3CN (2.0 mL), rt, and 5 h. For tetrapeptides SM(aa–ad) or pentapeptide SM(ae) 0.1 mmol, trichloroisocyanuric acid (TCCA, 1.5 equiv.), DCM 20.0 mL (c = 0.005 M), 0 °C, and 8 h; 1 (0.1 mmol), DCE 1.0 mL, blue LED (100 W), 45 °C, and 9 h; then NaOH (1.2 equiv.), CH3CN (2.0 mL), rt, and 5 h. b Isolated yield. c 0.4 mmol of tripeptide SM(y) was used. d 1 mmol of tetrapeptide SM(ac) was used. e In parentheses was the calculated total yield. |
|---|
|
To further understand the reactivity of this catalytic system, the reactivity of an isoleucine derivative Ile-NCl and the isoleucine-derived dipeptide 1a was compared. After irradiation with visible light, dipeptide 1a was successfully converted to the corresponding product 2a′ in quantitative yield, whereas the amino acid only yielded the corresponding product Ile-Cl in 56% yield; this highlights the superior reactivity of peptide substrates over single amino acids (Scheme 3a, details in the ESI†).58 Such a superior reactivity of peptides may have resulted from the hydrogen bonding between amide and chloride in dipeptides, which could assist the C–Cl bond formation.59 Finally, gram-scale experiments were conducted to evaluate the scalability of the transformation. Within two tripeptide substrates, the N-terminal amino acid moiety was transformed into the corresponding cyclic proline derivatives in acceptable yields (Scheme 3b).
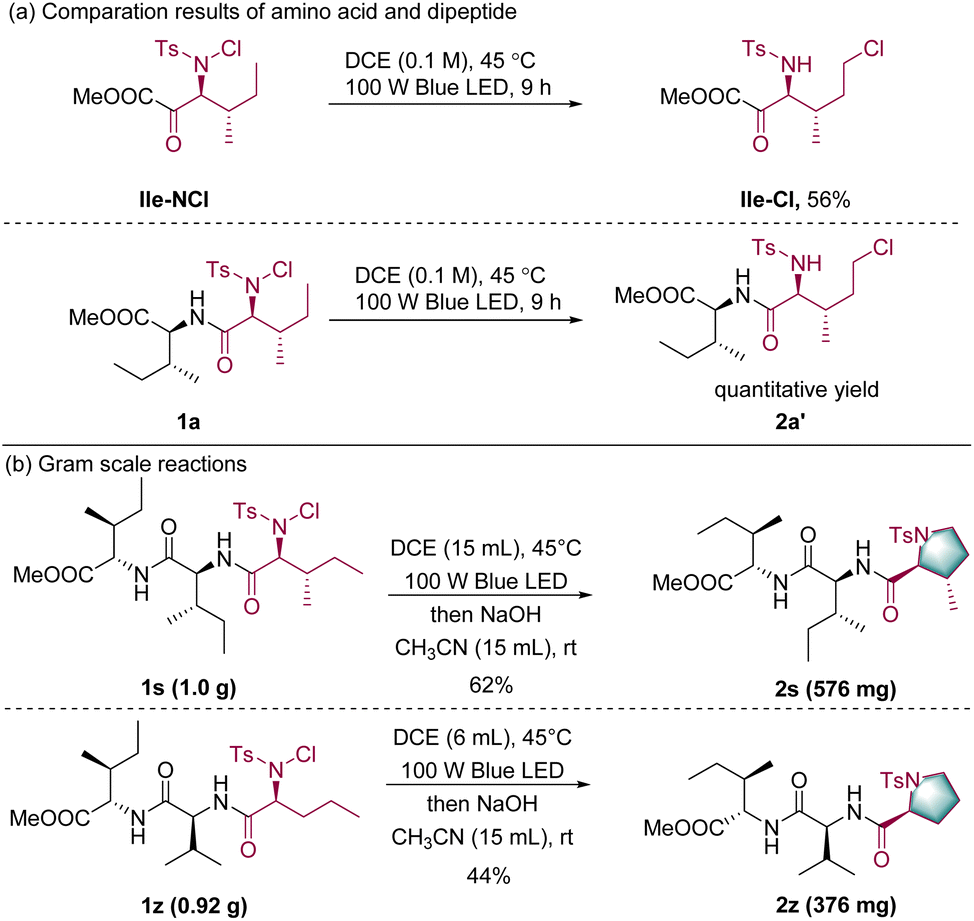 | ||
| Scheme 3 (a) Comparation results of amino acids and dipeptides; (b) gram scale reactions. | ||
Based on our previous studies,52–55 and other reported research,60 we considered that such transformation proceeded through a 1,5-hydrogen atom transfer (1,5-HAT) process as the Hofmann–Löffler reaction reported.61,62 Under the irradiation of visible light, the homolytic cleavage of the N–Cl bond occurred to generate a nitrogen radial and chloride radical (trapped by TEMPO and detected by ESI-MS, see the ESI†). Subsequently, 1,5-HAT takes place yielding the carbon-centered radical, which could be trapped by the chloride radical to form a C–Cl bond. Then, an intramolecular nucleophilic substitution occurred under the base conditions to form the target product.
Conclusions
In summary, we have developed a visible light-promoted selective editing of peptide skeletons via C–N bond formation at N-terminal aliphatic side chains. A proline skeleton was constructed in peptides under such transition metal free conditions. The excellent diastereoselectivity and broad substrate scope of the reaction highlight its potential utility in biochemistry, medicinal chemistry, and/or drug discovery.Author contributions
Y. H. and S. L. performed the experiments and analyzed the data. J. S., T. D. and W. Y. assisted the purification of compounds and analysis of data. M. Y. designed and directed the whole project and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support by the National Natural Science Foundation of China (22171175), Fundamental Research Funds for the central Universities (GK202103049), and start-up funds from Shaanxi Normal University, and Shaanxi Provincial Natural Science Foundation (2021JM-195).Notes and references
- K. L. Dunbar, D. H. Scharf, A. Litomska and C. Hertweck, Chem. Rev., 2017, 117, 5521–5577 CrossRef CAS PubMed
.
- S. Sato and H. Nakamura, Molecules, 2019, 24, 3980–3996 CrossRef CAS PubMed
- J. C. Lewis, P. S. Coelho and F. H. Arnold, Chem. Soc. Rev., 2011, 40, 2003–2021 RSC
- A. Henninot, J. C. Collins and J. M. Nuss, Med. Chem., 2018, 61, 1382–1414 CrossRef CAS PubMed
- J. Lu, Y. Li, Z. Bai, H. Lv and H. Wang, Nat. Prod. Rep., 2021, 38, 981–992 RSC
- S. Xu, Z. Zhao and J. Zhao, Chin. Chem. Lett., 2018, 29, 1009–1016 CrossRef CAS
- T. Morioka, A. Nishizawa, K. Nakamura, M. Tobisu and N. Chatani, Chem. Lett., 2015, 44, 1729–1731 CrossRef CAS
- J. N. deGruyter, L. R. Malins and P. S. Baran, Biochemistry, 2017, 56, 3863–3873 CrossRef CAS PubMed
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed
- L. R. Malins, Curr. Opin. Chem. Biol., 2018, 46, 25–32 CrossRef CAS PubMed
- O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed
- K. K. Biggar and S. S. Li, Nat. Rev. Mol. Cell Biol., 2015, 16, 5–17 CrossRef CAS PubMed
- R. Klingberg, J. O. Jost, M. Schümann, K. A. Gelato, W. Fischle, E. Krause and D. Schwarzer, ACS Chem. Biol., 2015, 10, 138–145 CrossRef CAS PubMed
- D. Wang, M. Yu, N. Liu, C. Lian, Z. Hou, R. Wang, R. Zhao, W. Li, Y. Jiang, X. Shi, S. Li, F. Yin and Z. Li, Chem. Sci., 2019, 10, 4966–4972 RSC
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Nature, 2015, 526, 687–691 CrossRef CAS PubMed
- D. P. Gamblin, E. M. Scanlan and B. G. Davis, Chem. Rev., 2009, 109, 131–163 CrossRef CAS PubMed
- M. Yang, X. Wang and J. Zhao, ACS Catal., 2020, 10, 5230–5235 CrossRef CAS
- A. K. Mishra, R. Tessier, D. P. Hari and J. Waser, Angew. Chem., Int. Ed., 2021, 60, 17963–17968 CrossRef CAS PubMed
- M. W. Jones, G. Mantovani, C. A. Blindauer, S. M. Ryan, X. Wang, D. J. Brayden and D. M. Haddleton, J. Am. Chem. Soc., 2012, 134, 7406–7413 CrossRef CAS PubMed
- A. E. Mangubat-Medina, S. C. Martin, K. Hanaya and Z. T. Ball, J. Am. Chem. Soc., 2018, 140, 8401–8404 CrossRef CAS PubMed
- J. M. Antos and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 10256–10257 CrossRef CAS PubMed
- Q. Bai, Z. Bai and H. Wang, Org. Lett., 2019, 21, 8225–8228 CrossRef CAS PubMed
- X. Li, L. Qi, B. Li, Z. Zhao, G. He and G. Chen, Org. Lett., 2020, 22, 6209–6213 CrossRef CAS PubMed
- J. Tan, J. Wu, S. Liu, H. Yao and H. Wang, Sci. Adv., 2019, 5, eaaw0323 CrossRef CAS PubMed
- J. Tang, H. Chen, Y. He, W. Sheng, Q. Bai and H. Wang, Nat. Commun., 2018, 9, 3383 CrossRef PubMed
- Z. Bai and H. Wang, Synlett, 2020, 31, 199–204 CrossRef CAS
- X. Chen, F. Ye, X. Luo, X. Liu, J. Zhao, S. Wang, Q. Zhou, G. Chen and P. Wang, J. Am. Chem. Soc., 2019, 141, 18230–18237 CrossRef CAS PubMed
- J. Ruiz-Rodríguez, F. Albericio and R. Lavilla, Chem.–Eur. J., 2010, 16, 1124–1127 CrossRef PubMed
- L. Mendive-Tapia, S. Preciado, J. García, R. Ramón, N. Kielland, F. Albericio and R. Lavilla, Nat. Commun., 2015, 6, 7160 CrossRef PubMed
- W. Gong, G. Zhang, T. Liu, R. Giri and J. Q. Yu, J. Am. Chem. Soc., 2014, 136, 16940–16946 CrossRef CAS PubMed
- B.-B. Zhan, Y. Li, J.-W. Xu, X.-L. Nie, J. Fan, L. Jin and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 5858–5862 CrossRef CAS PubMed
- Y. Zhu, M. Bauer, J. Ploog and L. Ackermann, Chem.–Eur. J., 2014, 20, 13099–13102 CrossRef CAS PubMed
- J. Tang, Y. He, H. Chen, W. Sheng and H. Wang, Chem. Sci., 2017, 78, 4565–4570 RSC
- Y. Zheng and W. Song, Org. Lett., 2019, 21, 3257–3260 CrossRef CAS PubMed
- M. Yang, X. Jiang and Z.-J. Shi, Org. Chem. Front., 2015, 2, 51–54 RSC
- E. Hernando, J. Villalva, Á. M. Martínez, I. Alonso, N. Rodriguez, R. G. Arrayás and J. C. Carretero, ACS Catal., 2016, 6, 6868–6882 CrossRef CAS
- B. Li, X. Li, B. Han, Z. Chen, X. Zhang, G. He and G. Chen, J. Am. Chem. Soc., 2019, 141, 9401–9407 CrossRef PubMed
- T. J. Osberger, D. C. Rogness, J. T. Kohrt, A. F. Stepan and M. C. White, Nature, 2016, 537, 214–219 CrossRef CAS PubMed
- L. Zhao, O. Baslé and C. J. Li, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4106–4111 CrossRef CAS PubMed
- C. Wang, R. Qi, H. Xue, Y. Shen, M. Chang, Y. Chen, R. Wang and Z. Xu, Angew. Chem., Int. Ed., 2020, 59, 7461–7466 CrossRef CAS PubMed
- Y. Yu, L.-K. Zhang, A. V. Buevich, G. Li, H. Tang, P. Vachal, S. L. Colletti and Z.-C. Shi, J. Am. Chem. Soc., 2018, 140, 6797–6800 CrossRef CAS PubMed
- W. Gong, G. Zhang, T. Liu, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 16940–16946 CrossRef CAS PubMed
- T. Liu, J.-X. Qiao, M. A. Poss and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 10924–10927 CrossRef CAS PubMed
- M. Bauer, W. Wang, M. M. Lorion, C. Dong and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 203–207 CrossRef CAS PubMed
- W. Wang, M. M. Lorion, O. Martinazzoli and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 10554–10558 CrossRef CAS PubMed
- J. Wu, N. Kaplaneris, S. Ni, F. Kaltenhäuser and L. Ackermann, Chem. Sci., 2020, 11, 6521–6526 RSC
- B.-B. Zhan, J. Fan, L. Jin and B.-F. Shi, ACS Catal., 2019, 9, 3298–3303 CrossRef CAS
- J.-W. Xu, Z.-Z. Zhang, W.-H. Rao and B.-F. Shi, J. Am. Chem. Soc., 2016, 138, 10750–10753 CrossRef CAS PubMed
- A. F. M. Noisier, J. García, I. A. Ionut and F. Albericio, Angew. Chem., Int. Ed., 2017, 56, 314–318 CrossRef CAS PubMed
- J. Tang, Y. He, H. Chen, W. Sheng and H. Wang, Chem. Sci., 2017, 8, 4565–4570 RSC
- X. Zhang, G. Lu, M. Sun, M. Mahankali, Y. Ma, M. Zhang, W. Hua, Y. Hu, Q. Wang, J. Chen, G. He, X. Qi, W. Shen, P. Liu and G. Chen, Nat. Chem., 2018, 10, 540–548 CrossRef CAS PubMed
- M. Yang, B. Su, Y. Wang, K. Chen, X. Jiang, Y.-F. Zhang, X.-S. Zhang, G. Chen, Y. Cheng, Z. Cao, Q.-Y. Guo, L. Wang and Z.-J. Shi, Nat. Commun., 2014, 5, 4707 CrossRef CAS PubMed
- D. Meng, Y. Tang, J. Wei, X. Shi and M. Yang, Chem. Commun., 2017, 53, 5744–5747 RSC
- Y. Tang, Y. Qin, D. Meng, C. Li, J. Wei and M. Yang, Chem. Sci., 2018, 9, 6374–6378 RSC
- Y. Qin, Y. Han, Y. Tang, J. Wei and M. Yang, Chem. Sci., 2020, 11, 1276–1282 RSC
- C. Che, Y.-N. Li, X. Cheng, Y.-N. Lu and C.-J. Wang, Angew. Chem., Int. Ed., 2021, 60, 4698–4704 CrossRef CAS PubMed
- W. Wang, J. Wu, R. Kuniyil, A. Kopp, R. N. Lima and L. Ackermann, Chem, 2020, 6, 3428–3439 CAS
- Q. Qin and S. Yu, Org. Lett., 2015, 17, 1894–1897 CrossRef CAS PubMed
- K. J. Winstanley and D. K. Smith, J. Org. Chem., 2007, 72, 2803–2815 CrossRef CAS PubMed
- A. E. Bosnidou, T. Duhamel and K. Muñiz, Eur. J. Org. Chem., 2020, 40, 6361–6365 CrossRef
- A. W. Hofmann, Chem. Ber., 1883, 16, 558–560 CrossRef
- K. Löffler and C. Freytag, Chem. Ber., 1909, 42, 3427–3431 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc04909k |
| This journal is © The Royal Society of Chemistry 2022 |