
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuper-resolved dynamics of isolated zinc formation during extremely fast electrochemical deposition/dissolution processes†
Jiaxin
Mao‡
a,
Guopeng
Li‡
a,
Muhammad
Saqib‡
ac,
Jiantie
Xu
 b and
Rui
Hao
*a
b and
Rui
Hao
*a
aDepartment of Chemistry, Research Center for Chemical Biology and Omics Analysis, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: haor@sustech.edu.cn
bSchool of Environment and Energy, South China University of Technology, Guangzhou 510640, China
cInstitute of Chemistry, Khwaja Fareed University of Engineering & Information Technology, Rahim Yar Khan 64200, Pakistan
First published on 12th October 2022
Abstract
The development of zinc–air batteries with high-rate capability and long lifespan is critically important for their practical use, especially in smart grid and electric vehicle application. The formation of isolated zinc (i-Zn) on the zinc anode surface, however, could easily lead to deteriorated performance, such as rapid capacity decay. In particular, under the fast charging/discharging conditions, the electrochemical activities on the anode surface are complicated and severely suppressed. Thus, it is highly desirable to deeply understand the formation mechanism of i-Zn and its relationship with the electrochemical performance during extremely high-rate cycling. Herein, we employed a super-resolution dark-field microscope to in situ analyze the evolution dynamics of the electrolyte–Zn interface during the extremely fast electrochemical deposition/dissolution processes. The unique phenomenon of nanoscopic i-Zn generation under the condition is unveiled. We discovered that the rapid conversion of nanoscopic i-Zn fragments into passivated products could greatly exacerbate the concentration polarization process and increase the overpotential. In addition, the role of large-sized i-Zn fragments in reducing the coulombic efficiency is further elucidated. This information could aid the rational design of highly effective anodes for extremely high-rate zinc-based batteries and other battery systems.
Introduction
The development of energy-storage systems with excellent rate capability, long lifespan, and low cost for sustainable energy storage grid and electrical vehicles and portable electronics has become critically urgent.1–3 Zinc–air batteries have been widely considered as a promising candidate of energy-storage systems,4,5 due to zinc being the fourth most abundant element in the Earth's crust6 as well as 3/2.2 times higher theoretical volumetric/specific energy densities than those of conventional lithium-ion batteries.7–10However, owing to the easy formation of dendrites on the surface of the zinc anode, the zinc air batteries often suffer from poor electrochemical performance (e.g., rapid capacity decay and poor rate capability11–13) and the related safety issues.14–16 In particular, during the dissolution of zinc dendrites, the isolated zinc (i-Zn) with the fragment patterns, losing their electric connection with the current collector, was generally identified as electrochemically inactive or dead during further deposition and dissolution processes.17,18 The accumulation of i-Zn or dead Zn causes loss of active material and reduction of mass transportation,18 and thus leads to deteriorated electrochemical performance.19 Approaches have been proposed to suppress the generation of i-Zn by optimizing the electrode architecture, interfacial properties, and electrolyte chemistry.5,14,20–22 On the other side, however, mechanistic studies have been focused on characterizing23,24 and quantifying25–27 isolated lithium. In-depth analysis of the i-Zn formation is still rare and highly needed.
Moreover, the formation of i-Zn and its detrimental effects on the electrochemical performance of zinc ion batteries (including zinc air batteries) are relatively severe during the overcharging and fast-charging processes.28 High current densities are crucial for smart grid and mobile energy-storage devices and related research studies have received lots of attention.1,29,30 Recently, Pang et al. proposed an aluminum battery with cycling ability up to 200C.31 Grey and co-workers studied single particle anode materials in a lithium ion battery at 30C.32 The evolution of zinc anodes, especially i-Zn formation, has rarely been examined during extremely fast cycling. In this case, zinc dendrites dissolving from multiple locations simultaneously could result in a large number of i-Zn fragments in a very short period, which is far different from normal low-rate situation. Therefore, it is highly desirable to monitor the morphological evolution of zinc in real-time during the extremely fast charging/discharging process under realistic battery operation.
The use of in situ or operando optical imaging to analyze nanoscale electrochemical interfaces has several advantages over other imaging techniques, including cost effectiveness, noninvasiveness, ease of operation and access, nonvacuum ambient conditions, and simplicity of design.33,34 With optical imaging techniques, the microscopic zinc plating behavior35–37 and the SEI formation process38 can be studied. We had previously developed a total internal reflection dark-field microscopy (TIRDFM) system,39 which allows the direct imaging of the dendrite nucleation and growth processes with millisecond and nanometer resolutions.
In this work, we employed in situ TIRDFM to monitor the electrochemical deposition/dissolution processes in a model zinc air battery system at extremely high current densities and were able to unveil, for the first time, a unique nanoscopic i-Zn phenomenon. The formation of i-Zn was investigated systematically at different charging/discharging rates using representative electrolytes. Super-resolution localization and semiquantitative analyses were performed to systematically study the i-Zn behavior. We found that the formation of the nanoscopic i-Zn and the subsequent excessive generation of passivated products under certain conditions could dramatically increase the overpotential of the battery, with the decrease in the coulombic efficiency being attributable primarily to the microscopic and larger i-Zn.
Results and discussion
Imaging of i-Zn
To monitor both the real-time (dynamic) nanoscopic i-Zn formation process and the dynamic changes in the dendrite morphology, a model zinc air battery system (Fig. 1a) was constructed using a nanoscale specular ring working electrode. The thickness of the electrode was approximately 400 nm to ensure that representative information related to the i-Zn formation process could be obtained with ease. We had previously confirmed the feasibility of using a TIRDFM system (Fig. 1b) to in situ visualize the location of sparse zinc nuclei on the electrode surface with a precision of ∼10 nm.39 In this study, we applied a similar super-resolution localization technique to observe the zinc-stripping process in extremely high-rate batteries. Total internal reflection (TIR) produces an evanescent wave with an exponentially decaying amplitude. Thus, only objects in the evanescent field near the interface can be illuminated, which can effectively reduce the background signal, improve the signal-to-noise ratio, and increase the axial resolution. Fig. 1b shows that only scattered light from the zinc plating/stripping process was recorded by the camera, whereas both the TIR light (from the glass) and specular reflection (from the electrode) were rejected. This unique setup allowed us to successfully distinguish the deposited zinc structures from the bulk electrode and detect extremely small zinc particles (approximately down to 10 nm in diameter). By equipping the TIRDFM system with a high-speed scientific complementary metal oxide semiconductor (sCMOS) camera, the zinc plating and stripping process could be monitored with an exposure time of 2 ms at a current of 1, 2, and 5 μA, which corresponded to current densities of 38, 76, and 189 mA cm−2 respectively. In a zinc-ion coin cell with an areal capacity of 1 mA h cm−2, these current densities corresponded to rates of 38, 76, and 189C, respectively. | ||
| Fig. 1 Schematic illustration of the experimental setup. (a) A model zinc air battery with the cross-section view of the cell in the upper-left corner (electrochemical setup) and (b) in situ TIRDFM (reflected light is rejected)-based set-up used to collect scattered light during the zinc-plating process. | ||
Fig. 2a–c show a montage of a 150 s-long zinc electrochemical dissolution process with 1 M ZnSO4 electrolyte at a current density of 76 mA cm−2. The bright-field (BF) recording (Fig. 2a) shows the gradual dissolution of zinc dendrites and the formation of a passivation layer (loose) as well as i-Zn (large and microscopic). The intensity of the BF images was normalized using the ratio of the intensity of the dendrite area to that of the electrolyte area (taken as the transmittance) to highlight the degree of dendrite dissolution over time. Clearly, the transmittance will increase with the zinc dissolution in BF mode. The black curve in Fig. 2f shows the changes in the transmittance of the deposited zinc during the BF recording. The transmittance increases slowly in the beginning and then rapidly in the later stage, indicating a gradual decrease in the amount of zinc deposited.
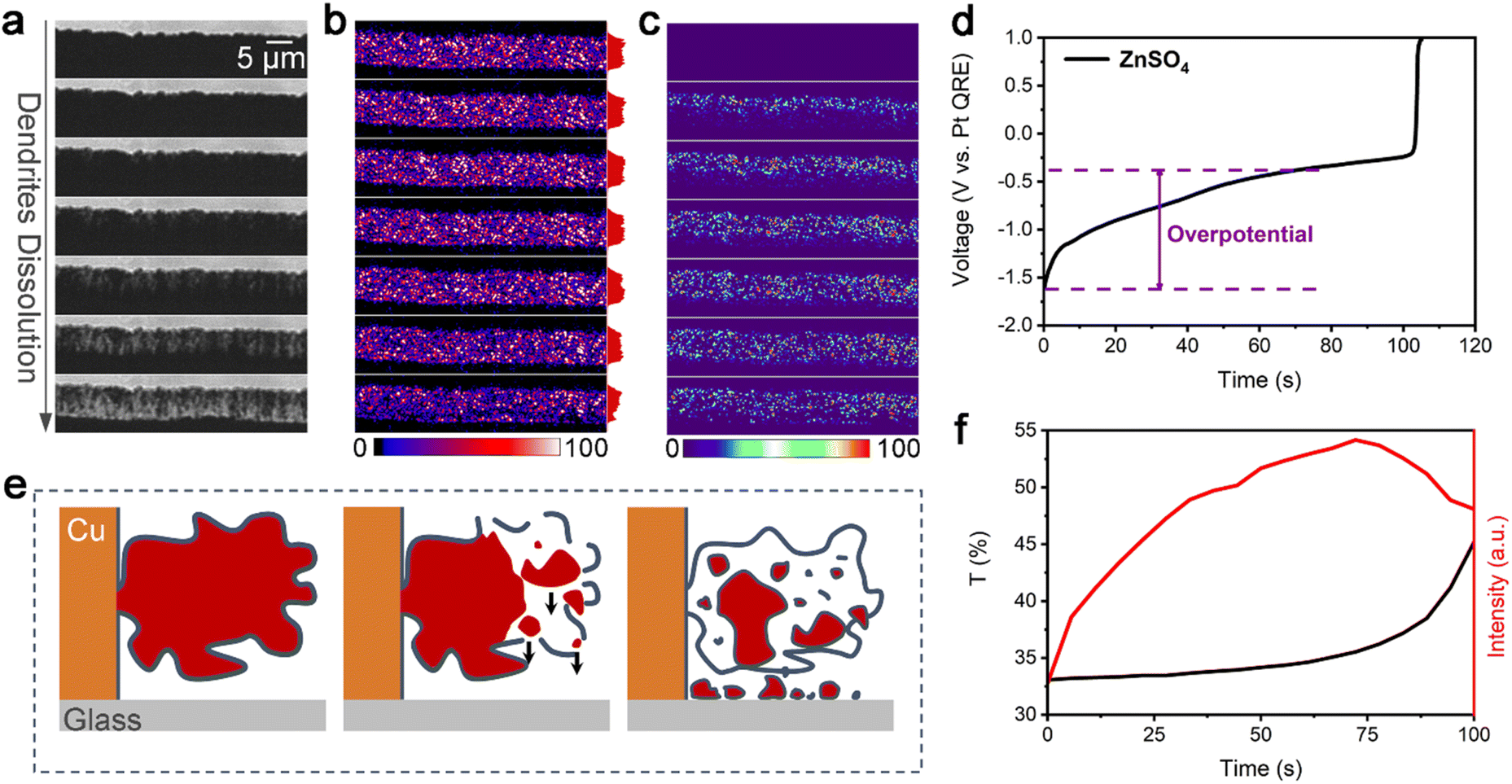 | ||
| Fig. 2 Recording and schematic illustration of isolated zinc formation. Montages of images (15 s interval) from (a) BF recording, (b) TIRDF recording, and (c) processed TIRDF recording obtained by subtracting the first image of dissolution of zinc dendrites at a discharging current density of 76 mA cm−2. Zinc dendrites were deposited at 76 mA cm−2 and 300 μC (deposition time of 150 s). The red plots on the right in (b) show the changes in relative scattering intensity during the dissolution of dendrites. (d) Galvanostatic dissolution curve corresponding to the dissolution of zinc dendrites (1 M ZnSO4 electrolyte at 76 mA cm−2 in the model zinc air battery). (e) Schematic illustration of the dissolution of zinc dendrites. (f) Transmittance (T)-time curve (black) of dendrites during BF recording and mean intensity-time curve (red) for nanoscopic i-Zn formation based on the processed TIRDF recording. | ||
The montage of the TIRDF recording in Fig. 2b consists of images with a much higher contrast and greater detail. On analysing the scattering intensity profiles (from the electrode to the solution, determined by the size and amount of metal particles) of the TIRDF images, a counterintuitive phenomenon was found: the scattering intensity of dendrites and i-Zn remains almost constant and even increases slightly during most of the zinc dissolution process (Fig. S1, ESI†). The TIRDF recordings were processed by subtracting the first image to highlight the intensity change. The obtained images (Fig. 2c) indicate the formation of new materials with a high degree of scattering within the illumination range corresponding to TIR (∼200 nm). Clearly, this unusual “reverse” growth phenomenon was not observed in the BF images.
The change in scattering intensity of zinc dendrites is plotted against time as a red curve in Fig. 2f. A rapid increase occurs at the beginning, and the rate of change decreases in a later stage, followed by a decline towards the end of the recording. The galvanostatic dissolution curve (Fig. 2d) shows that zinc has a high overpotential of 1.21 V at a current density of 76 mA cm−2. The efficiency of the battery is approximately 70%. Interestingly, a rapid increase in the TIRDF scattering intensity coincides with a rapid increase in overpotential, suggesting a possible correlation between them.
We believe that the “reverse” growth process highlights the formation of i-Zn. The changes in the scattering intensity in the processed TIRDF recording suggest the formation of microscopic and nanoscopic i-Zn. We propose the following mechanism to explain this phenomenon: during the dissolution of the dendrites, i-Zn of various sizes, including large, microscopic, and nanoscopic ones, forms, while the passivation layer (usually zinc hydroxide sulfate when ZnSO4 is used as the electrolyte) breaks down owing to the loss of support.25 The larger i-Zn fragments are more likely to be stationary, but the microscopic and nanoscopic i-Zn fragments can fall through the cracks in the passivation layer (Fig. 2e). There is no exact criterion for defining the various types of i-Zn. Based on the resolution and sensitivity of the method used, we classified i-Zn fragments with a size of 1 μm to 200 nm as microscopic and those smaller than 200 nm as nanoscopic.
Effects of charging/discharging current density
To elucidate the formation mechanism of the microscopic and nanoscopic i-Zn during the zinc dissolution process in a spatiotemporally resolved manner, a super-resolution localization technique was used. All the microscopic and nanoscopic i-Zn fragments that fell in the illuminating areas of the evanescent wave could be located using this technique. Although the scattering intensities of i-Zn fragments of different sizes showed large differences, the number of i-Zn fragments could be determined using this technique. The spatiotemporal mapping of the nanoscopic i-Zn under different conditions yielded temporal color-coded super-resolution images. In the case of 1 M ZnSO4 as the electrolyte, two charging/discharging current densities (38 and 189 mA cm−2, respectively; denoted as S (slow) and F (fast), respectively), were used. To enable a better comparison of the zinc dissolution process at the different current densities, the electric charge for the entire deposition process was kept at 300 μC.Fig. 3a–c show color-merged initial-final (green-red) TIRDF images, final BF images, and temporal color-coded super-resolution images of the zinc dissolution process under different electrochemical deposition/dissolution conditions. The images in Fig. 3a exhibit a clear contrast between the start and end stages of the zinc dissolution process. The presence of particles with a higher scattering intensity (green region) at a low charging/discharging rate indicates that the number density of the dendrites was higher in this case and that they had a larger particle size. In contrast, a high charging rate led to loosely grown dendrites with a smaller particle size. The red region shows the remaining zinc (microscopic and nanoscopic i-Zn) after the dendrite dissolution process. The final BF images of the dendrite deposition process (Fig. S2†) provide morphological information about the dendrites at the two current densities with limited details. The galvanostatic deposition curve (Fig. S3†) corresponding to a current density of 38 mA cm−2 has a lower overpotential (0.76 V) than that corresponding to a current density of 189 mA cm−2; this can be ascribed to concentration polarization because of faster ion consumption and the resulting ion concentration gradient.40
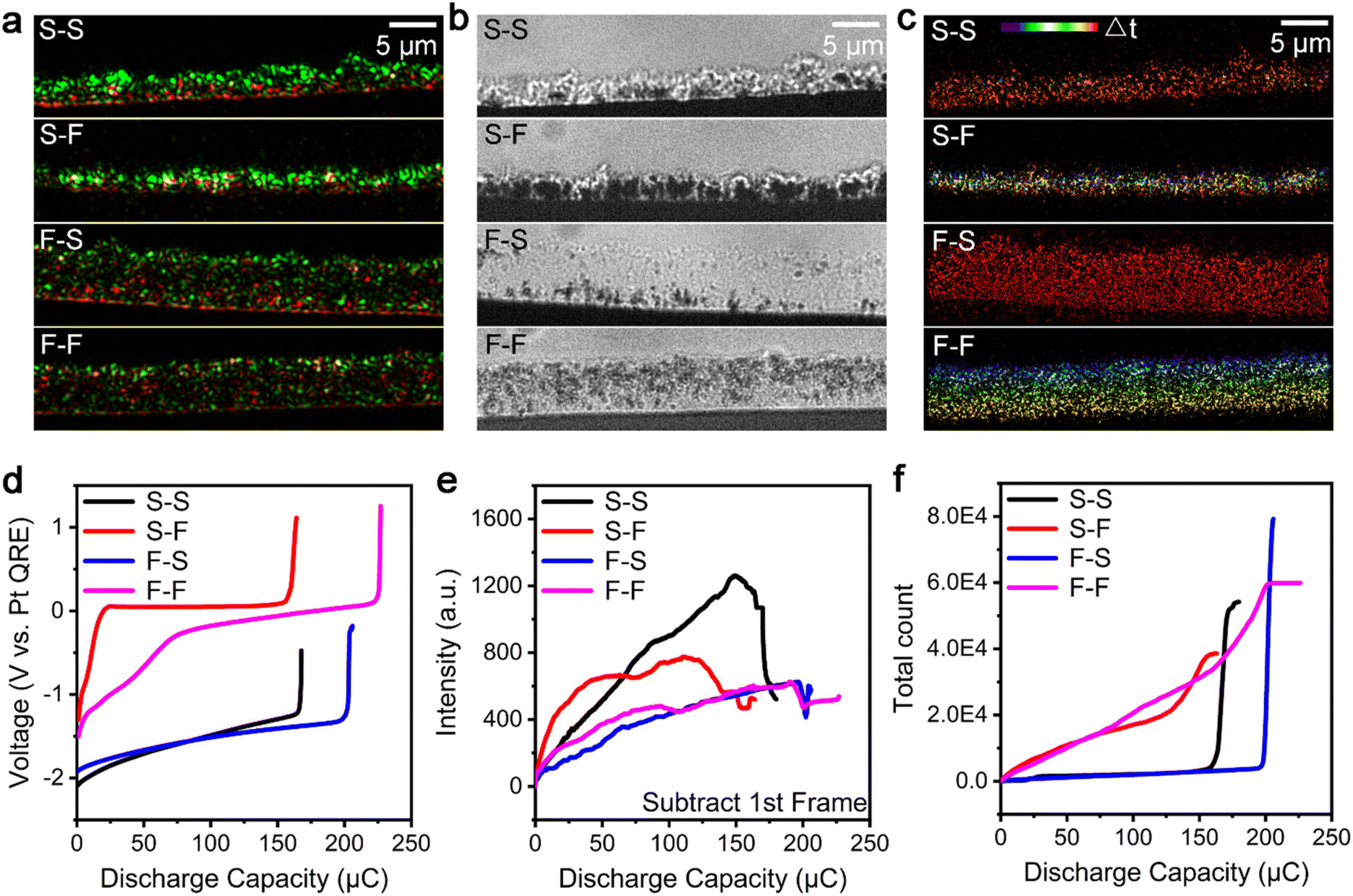 | ||
| Fig. 3 Super-resolution analysis and statistics of isolated zinc formation at different charging/discharging current densities. (a) Color-merged initial-final (green-red) TIRDF images, (b) final BF images, and (c) temporal color-coded super-resolution images of the dendrite dissolution process with 1 M ZnSO4 as the electrolyte at different current densities. The charge capacity during the dendrite growth process was 300 μC. Charging and discharging current densities from top to bottom: 38 and 38 mA cm−2, 38 and 189 mA cm−2, 189 and 38 mA cm−2, and 189 and 189 mA cm−2, respectively. The current densities of 38 and 189 mA cm−2 are denoted as S (slow) and F (fast), respectively. Discharge capacity during the dissolution process from top to bottom: 180, 210, 206, and 200 μC, respectively. (d) Galvanostatic dissolution and (e) scattering intensity-discharge capacity curves obtained from the processed TIRDF recording by subtracting the first image and (f) total count-discharge capacity curves of nanoscopic i-Zn fragments formed during the zinc dendrite dissolution process in a model zinc air battery with 1 M ZnSO4 as the electrolyte. The different colors represent different charging/discharging current densities: 38 and 38 mA cm−2 (black), 38 and 189 mA cm−2 (red), 189 and 38 mA cm−2 (blue), and 189 and 189 mA cm−2 (magenta). We employed the ThunderSTORM plugin of the ImageJ software to count the number of microscopic and nanoscopic i-Zn fragments formed under different conditions. | ||
The BF image of the dissolved dendrites (Fig. 3b) shows many black areas with translucent surroundings; these were large and microscopic i-Zn fragments embedded within the passivated products.41 At a relatively low charging rate (current density of 38 mA cm−2), regardless of the discharging rate, the dendrites dissolved unevenly and formed larger i-Zn fragments, resulting in a passivation layer with many folds and bumps. At a higher charging rate (current density of 189 mA cm−2), the presence of more nucleation sites leads to more ion consumption and severe concentration polarization, forming looser and more enormous dendrites.39,42 At a higher charging rate, the dendrites dissolved more uniformly. As a result, fewer large i-Zn fragments were generated, and the passivation layer was relatively flat and smooth. These results indicate that, at the microscopic level, the i-Zn and passivated products are heavily influenced by the deposition morphology of the zinc dendrites. At a lower charging rate,30 the dendrites were formed at a lower rate and thus had more time to react with the electrolyte, leading to a thicker passivation layer during the plating process.43
Spatiotemporal mapping was performed to study the formation of nanoscopic i-Zn (Fig. 3c). The results indicated that the discharging rate plays an important role. At a relatively low discharging rate (current density of 38 mA cm−2), nanoscopic i-Zn fragments appeared simultaneously over the entire deposition area in the later stage (red region) of the dendrite dissolution process (Fig. 3c, S–S and F–S). When the charging rate is further increased (current density of 189 mA cm−2), nanoscopic i-Zn fragments began forming uniformly in nearly every stage of the dissolution process (Fig. 3c, S–F and F–F). There was no obvious correlation between the spatial and temporal distributions of i-Zn for the S–F case. However, in the F–F case, the spatiotemporal mapping showed that the i-Zn formation began at the electrolyte side in the beginning and gradually shifted to the electrode side over time.
The galvanostatic dissolution curves of the corresponding zinc dendrite dissolution processes are shown in Fig. 3d. The charging/discharging rates have a determining effect on the battery performance in terms of the coulombic efficiency and overpotential. The overpotential of the dissolution process is higher than that of the deposition process; this may be the reason why more passivated products are generated owing to polarization during the dissolution process (Fig. S3†). Generally, the coulombic efficiency of a model battery is higher for higher charging current densities. The overpotential at the discharging current density of 189 mA cm−2 was much larger than that at 38 mA cm−2. The battery subjected to relatively slow charging and extremely fast discharging exhibited the worst performance while the one subjected to the opposite combination showed the best performance.
Semiquantitative analysis of i-Zn
To better understand the dissolution processes, we investigated the isolation process semiquantitatively by analysing the changes in the scattering intensity (Fig. 3e) and counting the number of microscopic and nanoscopic i-Zn fragments in the super-resolution images (Fig. 3f). Fig. 3e shows that the magnitude of the change in the scattering intensity was highly dependent on the charging rate. For the relatively low-charging-rate cases (S–S and S–F), the intensity increases sharply in the early stage, and this is followed by a rapid decline at the end of the discharging period. A quasilinear increase in the intensity followed by a small dip was observed for both high-charging-rate cases (F–S and F–F). The decrease in the scattering intensity can be attributed to the polarization electrochemical dissolution of i-Zn, which likely affects the total coulombic efficiency of the battery as well.19The spatiotemporal maps in Fig. 3c and the total number of microscopic and nanoscopic i-Zn fragments seen in Fig. 3f are in contrast to each other, because of the discharging rate. In both cases (S–S and F–S) with relatively low discharging rates, the curve remains flat for most of the dissolution process and shows an abrupt increase at the end of the process. Contrastingly, for both high-discharging-rate cases (S–F and F–F), the curve for the number of nanoscopic i-Zn fragments increases smoothly over time and eventually plateaus.
The overall deposition or dissolution behavior of the microscopic and nanoscopic i-Zn fragments is reflected by the scattering intensity of the processed TIRDF recording after the subtraction of the first image. However, in the case of the nanoscopic i-Zn fragments, the scattering intensity is weak, which could possibly be due to masking by that of their larger counterparts or averaging with that of the blank background. Therefore, it is not possible to use the mean intensity to map the number of i-Zn fragments. We used statistical counting to analyse the formation of the microscopic and nanoscopic i-Zn. Smaller i-Zn fragments have a larger specific surface area and thus higher reactivity. Generally, there are two possible reaction pathways available to the nanoscopic i-Zn: dissolution back into the solution and/or conversion into a passivation layer.44,45 Both pathways have an effect on the battery performance.
On comparing the curves in Fig. 3d and f, one can observe an obvious correlation between the galvanostatic dissolution curves and the curves showing the number of i-Zn fragments formed. The red and magenta (fast discharging; S–F and F–F) galvanostatic dissolution curves indicate a huge overpotential, while the curve showing the number of i-Zn fragments formed indicates the continuous generation of microscopic and nanoscopic i-Zn throughout the dissolution process. The highly reactive nanoscopic i-Zn fragments were converted into excessive passivated products. In this situation, it is difficult for the newly formed zinc ions to diffuse deep into the solution over time, resulting in a higher local ion concentration near the electrode surface. This phenomenon of concentration polarization becomes more pronounced over time, leading to a very large overpotential (∼1.2 V).
Contrastingly, for the relatively slower discharging case, the microscopic and nanoscopic i-Zn were primarily generated in the later stage of the dissolution process, and less passivated products were formed. Thus, the overpotentials were small. Important information can also be obtained by analysing the scattering intensity (Fig. 3e). The black and red curves (relatively low charging rates) show a rapid increase, which indicates the generation of more microscopic i-Zn. Given the smaller specific surface area and thus lower reactivity of the microscopic i-Zn, their primary effect was a loss in capacity, as per the electrochemical recordings in Fig. 3d. This was in good agreement with the results of previous studies.
I-Zn behaviours in different electrolyte systems
We also investigated the i-Zn generation (large, microscopic, and nanoscopic i-Zn) and passivated product formation during the zinc dissolution process using different electrolyte systems, such as those consisting of zinc ion electrolytes and additives. Fig. 4a–c show the results of the analysis of the dendrite dissolution process at a current density of 76 mA cm−2. The deposition process was performed for 150 s using three representative electrolytes, namely, 1 M ZnSO4, 1 M ZnSO4 + 100 ppm PEI (PEI for short), and 1 M Zn(OTf)2. The final BF images (Fig. S4†) obtained during the deposition process show the presence of denser dendrites in the ZnSO4 system and looser dendrites with voids in the PEI and Zn(OTf)2 electrolyte systems. Moreover, the ZnSO4 and Zn(OTf)2 electrolyte systems have similar overpotentials, which are lower than that of the PEI system for a charging current density of 76 mA cm−2 (Fig. S5†). The green areas in Fig. 4a show that the zinc dendrites with the highest scattering intensity grew most densely in the ZnSO4 electrolyte system, whereas in the other two systems, dendrites with lower scattering intensities were formed. The Zn(OTf)2 electrolyte system contained more residual zinc after discharging than the other two systems (Fig. 4a, red areas), indicating that a greater number of microscopic and nanoscopic i-Zn fragments formed and remained undissolved in it. The BF images in Fig. 4b show the numbers of larger i-Zn fragments formed in the different systems. At a higher current density of 189 mA cm−2 (Fig. S6†), the PEI system contained smooth, pillar-shaped i-Zn fragments, whereas the Zn(OTf)2 system contained larger clusters with a higher contrast. Generally, the passivation layers in the ZnSO4 and PEI systems were thicker and more uneven than that in the Zn(OTf)2 system. The formation of the passivation layer and its morphology were analyzed for all three electrolytes. The spatiotemporal maps in Fig. 4c and the black line in Fig. 4f indicate that a few microscopic and nanoscopic i-Zn fragments were generated in the ZnSO4 electrolyte throughout the entire process. Contrastingly, in the PEI electrolyte, these two types of i-Zn were highly limited and primarily produced in the later stage (Fig. 4c, yellow and red regions; Fig. 4f, purple line). More i-Zn fragments were generated at a higher current density of 189 mA cm−2 (Fig. S7b, yellow and red regions; Fig. S10, purple line†). The spatiotemporal maps of the Zn(OTf)2 electrolyte in Fig. 4c show a large number of microscopic and nanoscopic i-Zn fragments distributed uniformly from top to bottom. With the increase in the current density, the scattering intensity continued to increase and became higher than that for the ZnSO4 system (Fig. S9, black and magenta lines†). Subsequently, even more microscopic and nanoscopic i-Zn fragments were irregularly generated (Fig. S7c and S10, magenta line†).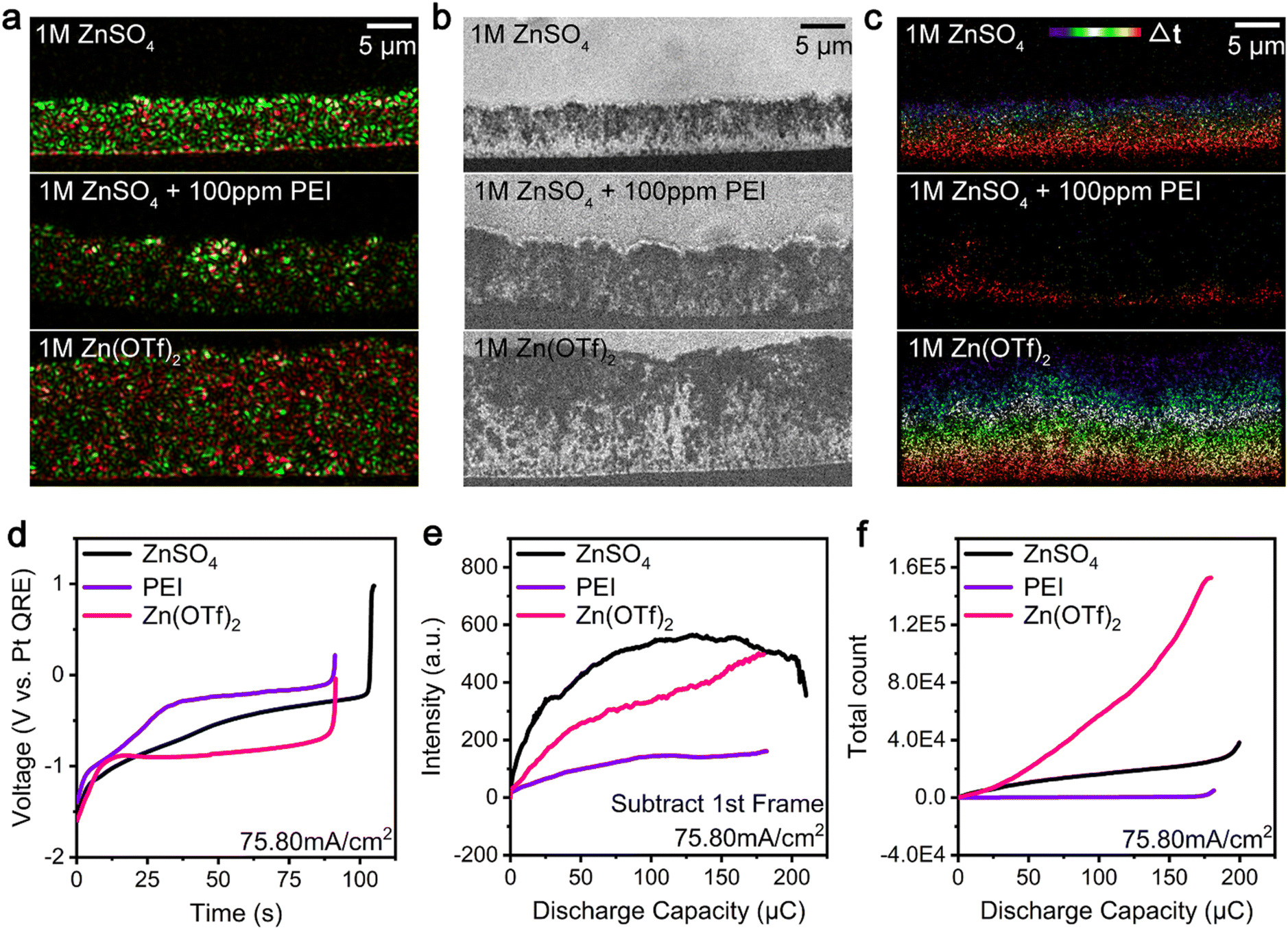 | ||
| Fig. 4 Super-resolution analysis and statistics of isolated zinc formation in different electrolyte systems. (a) Color-merged initial-final (green-red) TIRDF images, (b) final BF images, and (c) temporal color-coded super-resolution images of the dendrite dissolution process using 1 M ZnSO4 (top), 1 M ZnSO4 + 100 ppm PEI (middle), and 1 M Zn(OTf)2 (bottom) as electrolytes at 76 mA cm−2. (d) Galvanostatic dissolution curves of the zinc dendrite dissolution process using 1 M ZnSO4 (black), 1 M ZnSO4 + 100 ppm PEI (purple), and 1 M Zn(OTf)2 (magenta) at 76 mA cm−2 in a model zinc air battery. Zinc dendrites were deposited at 76 mA cm−2 and 300 μC (deposition time of 150 s). Discharge capacity during the dissolution process: 210, 180, and 182 μC, respectively. (e) Scattering intensity-discharge capacity curves for the three electrolytes obtained from the processed TIRDF recording by subtracting the first image. (f) Total number of microscopic and nanoscopic i-Zn-discharge capacity curves for the three electrolytes. | ||
The galvanostatic dissolution curves in Fig. 4d show that, in the case of the PEI and Zn(OTf)2 electrolyte systems, the dissolution process was completed within 91 s, whereas the ZnSO4 electrolyte showed a coulombic efficiency of more than 70%. Compared with the overpotential of the ZnSO4 system, that of the PEI system was higher, while that of the Zn(OTf)2 system was lower. At an even higher discharging rate (Fig. S8†), all the galvanostatic dissolution curves indicated a high overpotential, with the ZnSO4 system exhibiting the highest coulombic efficiency.
Fig. 4e shows the scattering intensity of the microscopic and nanoscopic i-Zn formed in the three systems. The ZnSO4 system exhibited a rapid increase in the intensity followed by a slow decrease, while the Zn(OTf)2 system showed a gradual increase. The change in the scattering intensity for the PEI system was very small compared with those for the other two systems, suggesting that considerably fewer microscopic and nanoscopic i-Zn fragments were generated in the PEI system. The super-resolution images showed a dramatic increase in microscopic and nanoscopic i-Zn formation in the Zn(OTf)2 system (Fig. 4f). Contrastingly, few i-Zn fragments were generated in the PEI system. A similar trend was observed at a higher discharging rate (Fig. S9 and S10†).
We believe that the sharp difference in the overpotentials of the PEI and Zn(OTf)2 systems can be explained by the difference in the reaction paths of the highly active nanoscopic i-Zn: conversion or dissolution. In the PEI system, at a relatively high pH, the nanoscopic i-Zn was rapidly converted into passivated products,46 resulting in a higher overpotential with the adsorption of the additive on the dendrite surfaces36,37 as well as a low efficiency. In the Zn(OTf)2 system, a large number of nanoscopic i-Zn fragments were generated during the dendrite dissolution process; however, only a few of them were converted into passivated products. This difference highlights the role played by the local chemical environment in determining the fate of nanoscopic i-Zn. Interestingly, in the case of the ZnSO4 system, the scattering intensity decreased at the end of the stripping process (Fig. 4e and S9†). As discussed above, a decrease in the intensity correlates with the electrochemical dissolution of the i-Zn. In the cases of the PEI and Zn(OTf)2 systems, the scattering intensity did not decrease in a later stage as in the case of the ZnSO4 system, suggesting the absence of the polarization electrochemical dissolution period;19 thus, lower coulombic efficiencies were obtained.
Ex situ characterization of i-Zn and passivated products
TIRDFM was used to evaluate the changes in the microscopic and nanoscopic i-Zn fragments while an ex situ analysis was performed to observe and evaluate the passivated products. It is known that the structure and morphological evolution of the passivated products are strongly dependent on the electrolyte composition. Helium ion microscopy (HIM) was used to observe the morphology of the passivation layer in the different electrolytes. For the ZnSO4 and PEI systems, at a charging/discharging current density of 76 mA cm−2, the HIM results showed the presence of a dense passivation layer structure containing small, scattered zinc particles (Fig. 5a and b). The passivation layer formed in the Zn(OTf)2 system contained larger voids (Fig. 5c) along with high-contrast flakes of zinc metal. | ||
| Fig. 5 HIM images and XPS spectra. (top) HIM images and (bottom) magnified images of the region outlined in red in the top images of the passivation layer formed in the (a) 1 M ZnSO4, (b) 1 M ZnSO4 + 100 ppm PEI, and (c) 1 M Zn(OTf)2 electrolytes after the first cycle at a (charging/discharging) current density of 76 mA cm−2 in a model zinc air battery. XPS Zn 2p, N 1s, and F 1s spectra of the surface of the passivation layer after the first cycle: (d) 1 M ZnSO4, (e) 1 M ZnSO4 + 100 ppm PEI, and (f) 1 M Zn(OTf)2. | ||
The results of X-ray photoelectron spectroscopy (XPS) (Fig. 5e and f) showed that the passivated products in the PEI and Zn(OTf)2 systems47 contained the elements (nitrogen and fluorine) present in the electrolyte. This confirmed that the constituent elements of the electrolyte were involved in the formation of the passivated products (of the respective systems). The XPS data also showed that zero-valent zinc was present in the Zn(OTf)2 system, in contrast to the case for the ZnSO4 and PEI systems. This is the reason why the Zn(OTf)2 system contained an excessive number of i-Zn fragments, which are less likely to convert into the passivated products. This conclusion is consistent with the HIM imaging results.
On comparing the three electrolyte systems, we concluded that the accumulation of large and microscopic i-Zn during the cycling process is the primary reason for the lower coulombic efficiency, which can be ascribed to a usually irreversible loss of the active material; this is also the commonly accepted view. The unique polarization electrochemical dissolution19 was observed only in ZnSO4, which has a relatively high coulombic efficiency among the three systems. The polarization electrochemical dissolution is a typical bipolar electrochemical coupling process, in which a pair of complimentary redox reactions happen at the two extremities of a metal conductor with a sufficiently high potential difference.48–51 The results indicate that the resistance of the passivation layer formed in the ZnSO4 system is significantly higher than that formed in other systems to provide a sufficiently high potential difference between the conductor extremities and drive the bipolar reaction. Contrastingly, the generation of nanoscopic i-Zn near the electrode–electrolyte interface and its subsequent conversion into passivated products (in the ZnSO4 and PEI systems) increases the resistance to mass transport in the electrolyte. Preventing this conversion process by changing the local chemical environment, as in the case of the Zn(OTf)2 system, could help limit the increase in the overpotential.
Conclusions
In this study, we combined in situ TIRDFM and electrochemical analysis to elucidate the dynamic electrochemical interface evolution process in extremely high-rate zinc–air batteries in detail. The unique phenomenon of the formation of nanoscopic i-Zn was unveiled for the first time. The effects of the charging current density on the generation of microscopic and large i-Zn and those of the discharging current density on the generation of nanoscopic i-Zn during the dissolution of the zinc dendrites in the ZnSO4 electrolyte system were clarified. The formation of nanoscopic i-Zn is accelerated with further current density increase. These results confirm that the formation of large and microscopic i-Zn contributes to the capacity loss of these batteries. The effect of the number of nanoscopic i-Zn fragments formed and their subsequent conversion into the passivated products on the overpotential was also elucidated. Moreover, the performances of three different electrolyte systems were compared to better understand the role of the electrolyte used in nanoscopic and microscopic i-Zn formation.The proposed analytical method offers new capabilities for monitoring the generation of i-Zn at different scales and the conversion of the passivated products under actual battery operation conditions. While the zinc electrochemical dissolution process remains to be understood completely, we believe the ability to perform nondestructive and noninvasive in situ experiments that allow for the real-time visualization of the behavior of i-Zn will guide the design of high-performance metal anodes.
Data availability
Primary data for the galvanostatic deposition/dissolution curves and final BF images under different conditions are provided in the ESI†.Author contributions
The manuscript was written and proofread by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22150410330 and 22074059), the Pearl River Talent Recruitment Program (2019QN01L096), and the Guangdong Innovative and Entrepreneurial Research Team Program (2019ZT08L075). The authors thank Professor Bo Zhang of University of Washington for helpful discussions.References
- Z. Yang, J. Zhang, M. C. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS
.
- L. Ma, M. A. Schroeder, O. Borodin, T. P. Pollard, M. S. Ding, C. Wang and K. Xu, Nat. Energy, 2020, 5, 743–749 CrossRef CAS
- P. Ruan, S. Liang, B. Lu, H. J. Fan and J. Zhou, Angew. Chem., Int. Ed. Engl., 2022, 61, e202200598 CrossRef CAS PubMed
- F. Wan, X. Z. Zhou, Y. Lu, Z. Q. Niu and J. Chen, ACS Energy Lett., 2020, 5, 3569–3590 CrossRef CAS
- J. Ming, J. Guo, C. Xia, W. X. Wang and H. N. Alshareef, Mater. Sci. Eng. R Rep., 2019, 135, 58–84 CrossRef
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Nat. Mater., 2018, 17, 543–549 CrossRef CAS PubMed
- T. H. Wu, Y. Zhang, Z. D. Althouse and N. Liu, Mater. Today Nano, 2019, 6, 100032 CrossRef
- K. Y. Kwon, T. H. Jo, J. S. Kim, F. Hasan and H. D. Yoo, ACS Appl. Mater. Interfaces, 2020, 12, 42612–42621 CrossRef CAS
- Y. Jin, K. S. Han, Y. Y. Shao, M. L. Sushko, J. Xiao, H. L. Pan and J. Liu, Adv. Funct. Mater., 2020, 30, 2003932 CrossRef CAS
- A. Bayaguud, X. Luo, Y. P. Fu and C. B. Zhu, ACS Energy Lett., 2020, 5, 3012–3020 CrossRef CAS
- W. Lu, C. Xie, H. Zhang and X. Li, ChemSusChem, 2018, 11, 3996–4006 CrossRef CAS
- L. E. Blanc, D. Kundu and L. F. Nazar, Joule, 2020, 4, 771–799 CrossRef CAS
- B. S. Lee, S. Cui, X. Xing, H. Liu, X. Yue, V. Petrova, H. D. Lim, R. Chen and P. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 38928–38935 CrossRef CAS
- S. Huang, H. Li, P. Pei, K. Wang, Y. Xiao, C. Zhang and C. Chen, iScience, 2020, 23, 101169 CrossRef CAS
- Q. Zhang, J. Luan, L. Fu, S. Wu, Y. Tang, X. Ji and H. Wang, Angew. Chem., Int. Ed. Engl., 2019, 58, 15841–15847 CrossRef CAS
- Y. Jin, Z. Zheng, D. Wei, X. Jiang, H. Lu, L. Sun, F. Tao, D. Guo, Y. Liu, J. Gao and Y. Cui, Joule, 2020, 4, 1714–1729 CrossRef CAS
- I. Yoshimatsu, T. Hirai and J. Yamaki, J. Electrochem. Soc., 1988, 135, 2422–2427 CrossRef CAS
- J. Steiger, D. Kramer and R. Moenig, Electrochim. Acta, 2014, 136, 529–536 CrossRef CAS
- F. Liu, R. Xu, Y. Wu, D. T. Boyle, A. Yang, J. Xu, Y. Zhu, Y. Ye, Z. Yu, Z. Zhang, X. Xiao, W. Huang, H. Wang, H. Chen and Y. Cui, Nature, 2021, 600, 659–663 CrossRef CAS PubMed
- Y. M. Sun, N. A. Liu and Y. Cui, Nat. Energy, 2016, 1, 16071 CrossRef CAS
- S. J. Zhang, J. N. Hao, D. Luo, P. F. Zhang, B. K. Zhang, K. Davey, Z. Lin and S. Z. Qiao, Adv. Energy Mater., 2021, 11, 2102010 CrossRef CAS
- J. L. Yang, J. Li, J. W. Zhao, K. Liu, P. Yang and H. J. Fan, Adv. Mater., 2022, 34, e2202382 CrossRef
- A. J. Sanchez, E. Kazyak, Y. X. Chen, K. H. Chen, E. R. Pattison and N. P. Dasgupta, ACS Energy Lett., 2020, 5, 994–1004 CrossRef CAS
- D. Tewari, S. P. Rangarajan, P. B. Balbuena, Y. Barsukov and P. P. Mukherjee, J. Phys. Chem. C, 2020, 124, 6502–6511 CrossRef CAS
- C. Fang, J. Li, M. Zhang, Y. Zhang, F. Yang, J. Z. Lee, M. H. Lee, J. Alvarado, M. A. Schroeder, Y. Yang, B. Lu, N. Williams, M. Ceja, L. Yang, M. Cai, J. Gu, K. Xu, X. Wang and Y. S. Meng, Nature, 2019, 572, 511–515 CrossRef CAS PubMed
- A. Aryanfar, D. J. Brooks, A. J. Colussi and M. R. Hoffmann, Phys. Chem. Chem. Phys., 2014, 16, 24965–24970 RSC
- A. B. Gunnarsdottir, C. V. Amanchukwu, S. Menkin and C. P. Grey, J. Am. Chem. Soc., 2020, 142, 20814–20827 CrossRef CAS
- P. H. Cao, X. Y. Zhou, A. R. Wei, Q. Meng, H. Ye, W. P. Liu, J. J. Tang and J. Yang, Adv. Funct. Mater., 2021, 31, 2100398 CrossRef CAS
- Y. Y. Liu, Y. Y. Zhu and Y. Cui, Nat. Energy, 2019, 4, 540–550 CrossRef
- Z. Cai, J. Wang, Z. Lu, R. Zhan, Y. Ou, L. Wang, M. Dahbi, J. Alami, J. Lu, K. Amine and Y. Sun, Angew. Chem., Int. Ed. Engl., 2022, 61, e202116560 CAS
- Q. Pang, J. Meng, S. Gupta, X. Hong, C. Y. Kwok, J. Zhao, Y. Jin, L. Xu, O. Karahan, Z. Wang, S. Toll, L. Mai, L. F. Nazar, M. Balasubramanian, B. Narayanan and D. R. Sadoway, Nature, 2022, 608, 704–711 CrossRef CAS PubMed
- A. J. Merryweather, Q. Jacquet, S. P. Emge, C. Schnedermann, A. Rao and C. P. Grey, Nat. Mater., 2022 DOI:10.1038/s41563-022-01324-z
- Y. T. Wu and N. A. Liu, Chem, 2018, 4, 438–465 CAS
- A. M. Tripathi, W. N. Su and B. J. Hwang, Chem. Soc. Rev., 2018, 47, 736–851 RSC
- S. J. Banik and R. Akolkar, J. Electrochem. Soc., 2013, 160, D519–D523 CrossRef CAS
- S. J. Banik and R. Akolkar, Electrochim. Acta, 2015, 179, 475–481 CrossRef CAS
- M. H. Lin, C. J. Huang, P. H. Cheng, J. H. Cheng and C. C. Wang, J. Mater. Chem. A, 2020, 8, 20637–20649 RSC
- L. Godeffroy, I. Aguilar, J. M. Medard, D. Larcher, J. M. Tarascon and F. Kanoufi, Adv. Energy Mater., 2022, 12, 2200722 CrossRef CAS
- X. Q. Ye, M. Saqib, J. X. Mao, G. P. Li and R. Hao, Cell Rep. Phys. Sci., 2021, 2, 100420 CrossRef CAS
- K. L. Wang, P. C. Pei, Z. Ma, H. C. Chen, H. C. Xu, D. F. Chen and X. Z. Wang, J. Mater. Chem. A, 2015, 3, 22648–22655 RSC
- J. Wan, Y. Hao, Y. Shi, Y. X. Song, H. J. Yan, J. Zheng, R. Wen and L. J. Wan, Nat. Commun., 2019, 10, 3265 CrossRef
- X. Z. Zhou, Y. Lu, Q. Zhang, L. C. Miao, K. Zhang, Z. H. Yan, F. J. Li and J. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 55476–55482 CrossRef CAS
- A. Sarkar, P. Shrotriya and I. C. Nlebedim, ACS Appl. Energy Mater., 2022, 5, 3179–3188 CrossRef CAS
- W. J. Lu, C. K. Zhang, H. M. Zhang and X. F. Li, ACS Energy Lett., 2021, 6, 2765–2785 CrossRef CAS
- Z. L. Li, S. Ganapathy, Y. L. Xu, Z. Zhou, M. Sarilar and M. Wagemaker, Adv. Energy Mater., 2019, 9, 1900237 CrossRef
- B. Lee, H. R. Seo, H. R. Lee, C. S. Yoon, J. H. Kim, K. Y. Chung, B. W. Cho and S. H. Oh, ChemSusChem, 2016, 9, 2948–2956 CrossRef CAS
- Y. Wu, Z. H. Zhu, D. Shen, L. A. Chen, T. Y. Song, T. X. Kang, Z. Q. Tong, Y. B. Tang, H. Wang and C. S. Lee, Energy Storage Mater., 2022, 45, 1084–1091 CrossRef
- R. Hao, Y. Fan, C. Han and B. Zhang, Anal. Chem., 2017, 89, 12652–12658 CrossRef CAS
- M. Wood and B. Zhang, ACS Nano, 2015, 9, 2454–2464 CrossRef CAS PubMed
- S. E. Fosdick, K. N. Knust, K. Scida and R. M. Crooks, Angew. Chem., Int. Ed. Engl., 2013, 52, 10438–10456 CrossRef CAS PubMed
- R. M. Crooks, ChemElectroChem, 2016, 3, 357–359 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc04877a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |