
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsight into ortho-boronoaldehyde conjugation via a FRET-based reporter assay†
Nicholas C.
Rose
 ab,
Anaïs V.
Sanchez
ab,
Eve F.
Tipple
ab,
Jason M.
Lynam
a and
Christopher D.
Spicer
*ab
ab,
Anaïs V.
Sanchez
ab,
Eve F.
Tipple
ab,
Jason M.
Lynam
a and
Christopher D.
Spicer
*ab
aDepartment of Chemistry, University of York, Heslington, YO10 5DD, UK. E-mail: chris.spicer@york.ac.uk
bYork Biomedical Research Institute, University of York, Heslington, YO10 5DD, UK
First published on 14th October 2022
Abstract
Ortho-boronoaldehydes react with amine-based nucleophiles with dramatically increased rates and product stabilities, relative to unfunctionalised benzaldehydes, leading to exciting applications across biological and material chemistry. We have developed a novel Förster resonance energy transfer (FRET)-based assay to provide key new insights into the reactivity of these boronoaldehydes, allowing us to track conjugation with unprecedented sensitivity and accuracy under standardised conditions. Our results highlight the key role played by reaction pH, buffer additives, and boronoaldehyde structure in controlling conjugation speed and stability, providing design criteria for further innovations and applications in the field.
Reactions between ortho-boronoaldehydes (oBAs) and amine-based nucleophiles have emerged as powerful tools for bioconjugation and materials chemistry over the last decade, due to a combination of rapid reaction rates and tunable product stability. The ortho-boronoimines, and related derivatives (collectively oBIDs), that form have shown high potential in drug delivery,1in vivo labelling,2 and responsive materials.3 To allow further evolution of these technologies and drive innovative new applications, there is a pressing need for increased understanding of the formation of oBIDs and their responsive behaviour. In this work, we deliver a sensitive and versatile platform to realise such understanding, through a Förster resonance energy transfer (FRET) reporter assay of oBID formation. This assay allows us to probe the rapid kinetics of oBID formation in detail, under complex and biomedically relevant conditions.
The ortho-boronic acid accelerates the rate of oBID formation (k1), relative to unfunctionalised benzaldehyde, by both activating the aldehyde to nucleophilic attack and accelerating the rate-determining dehydration step. Though the rate of hydrolysis (k−1) is also accelerated, stabilising B–N interactions lead to a significant overall shift in equilibrium towards product formation (decrease in dissociation constant, Kd). For example, while benzaldehyde reacts with alkyl amines with k1 ∼ 0.1 M−1 s−1 and Kd ∼ 300 M,4 the ortho-borono analogue ortho-formylphenylboronic acid (FBPA) forms analogous iminoboronates with a k1 ∼ 1000 M−1 s−1 and Kd ∼ 10 mM (Fig. 1a).5 This 5-order of magnitude increase in reaction rate, and 4-order of magnitude decrease in dissociation constant, typifies the unique reactivity of oBAs.
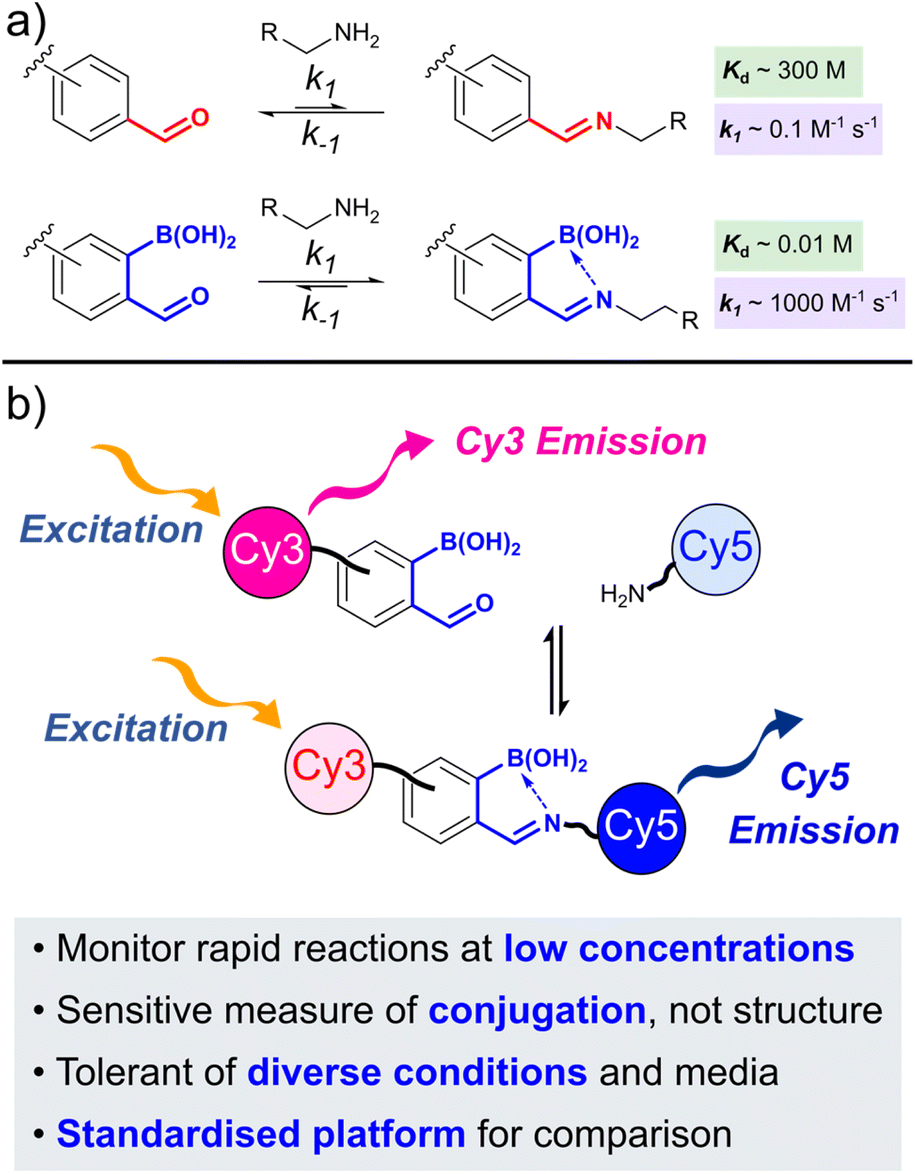 | ||
| Fig. 1 (a) Comparison of benzaldehyde and ortho-boronoaldehyde reactivity, with the ortho-borono group both accelerating and stabilising imine formation; (b) schematic overview of the FRET platform developed in this work, allowing sensitive monitoring of rapid oBID formation (k1 > 105 M−1 s−1). | ||
Through variation of the oBA and nucleophile coupling partners, Kds spanning 10−2–10−9 M have been reported.6 When coupled with high biocompatibility and potential for stimuli-responsive behaviour, this tunability greatly enhances the potential for biomedical applications of oBIDs. Despite this potential, much of our understanding of oBID chemistry comes from simple, unsubstituted model compounds such as FPBA, which poorly reflect the stereo-electronic characteristics of the functionalised oBAs necessary for translational applications.
Moreover, much of this understanding is pieced together from a large number of independent studies, performed and analysed under varying conditions, making comparison challenging. This is particularly important given the highly dynamic nature of oBID linkages, leading to a high sensitivity to environmental conditions and choice of analytical technique. Ultimately, this leads to significant discrepancies in reported rates of formation and stabilities of oBIDs, due to subtle differences in substrate choice, reaction conditions, or analysis method.7,8
To address this, we herein describe a versatile and sensitive FRET-assay for studying oBID formation, stability, and cleavage. This assay is highly tolerant of environmental conditions, allowing us to provide critical new insights into the effects of pH, additives, media, and oBA structure on conjugation. We therefore anticipate that this assay will find future use for the high-throughput screening of novel oBIDs and their stimuli-responsive behaviour.
Results and discussion
Platform design
To develop a platform for studying oBID chemistry, we took three key considerations into account:(i) The platform should be operational at low concentrations. Many oBIDs form with k1 > 103 M−1 s−1, and at the substrate concentrations required for UV-vis/NMR analysis reactions are complete prior to the first measurements, preventing accurate quantification.9–11 Sensitive detection of oBID formation at low concentrations is therefore essential to allow a suitable number of time points to be recorded before equilibrium is reached;
(ii) The platform should function across a broad range of reaction conditions representative of the end applications, including the presence of complex additives, allowing us to study oBID formation under conditions relevant to biological applications;
And (iii) a readout of conjugation, rather than structure, should be provided. Following conjugation of an oBA and nucleophile, the oBID formed remains highly dynamic. Often, complex mixtures of rapidly interchanging species exist in equilibrium, in a manner that is highly sensitive to environmental conditions (e.g. pH dependent equilibrium between ortho-boronohydrazones and diazoborines;12 diastereomeric mixtures formed during thiazolidino boronate formation8). By focussing on conjugation, we would be able to gain an overall view of all species that contribute positively to oBID formation, which would not be provided by focussing on the formation of specific structures. UV-vis and NMR studies are again poorly suited to providing such an overview.
We reasoned that a FRET-based reporter system would fulfil these criteria (Fig. 1b).13 Upon conjugation of complementary dyes bearing either oBA or nucleophile reactive handles, the proximity of the dyes would allow energy transfer to take place under donor excitation. The donor![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acceptor emission ratio (FRET ratio) would then directly correlate to reaction conversion, and would be largely independent of oBID structure. Notably, Schmidt et al. previously exploited a fluorescence quenching assay to monitor borono-oxime formation, but such experiments can be prone to interference from environmental conditions. In contrast, the ratiometric nature of FRET minimises such complications, and provides an extra level of robustness to kinetic measurements.14
:acceptor emission ratio (FRET ratio) would then directly correlate to reaction conversion, and would be largely independent of oBID structure. Notably, Schmidt et al. previously exploited a fluorescence quenching assay to monitor borono-oxime formation, but such experiments can be prone to interference from environmental conditions. In contrast, the ratiometric nature of FRET minimises such complications, and provides an extra level of robustness to kinetic measurements.14
The cyanine dyes Cy3 and Cy5 were chosen as a suitable FRET pair due to their chemical-, thermal-, and photo-stability, emission across a wide pH range, and known ability to undergo energy transfer.15 Due to the strong distance dependence of FRET (1/r6), we aimed to minimise the linker length from the dyes to the oBA/nucleophile functional handles, leading us to synthesise novel amino-cyanines 1 and 2 (Scheme 1). These derivatives, bearing a pendant sulfonate group that enhanced water solubility without affecting fluorescence,15 could be easily derivatised via amide coupling. Phthalimide protection of the amine (3 and 4) was required due to the harsh conditions needed for synthesis of the dye cores. Though traditional hydrazinolysis of the phthalimide was found to be problematic, due to the reaction of hydrazine with the conjugated backbone of Cy5, deprotection could be successfully achieved with high concentrations of methylamine to deliver 1 and 2 on a gram scale.
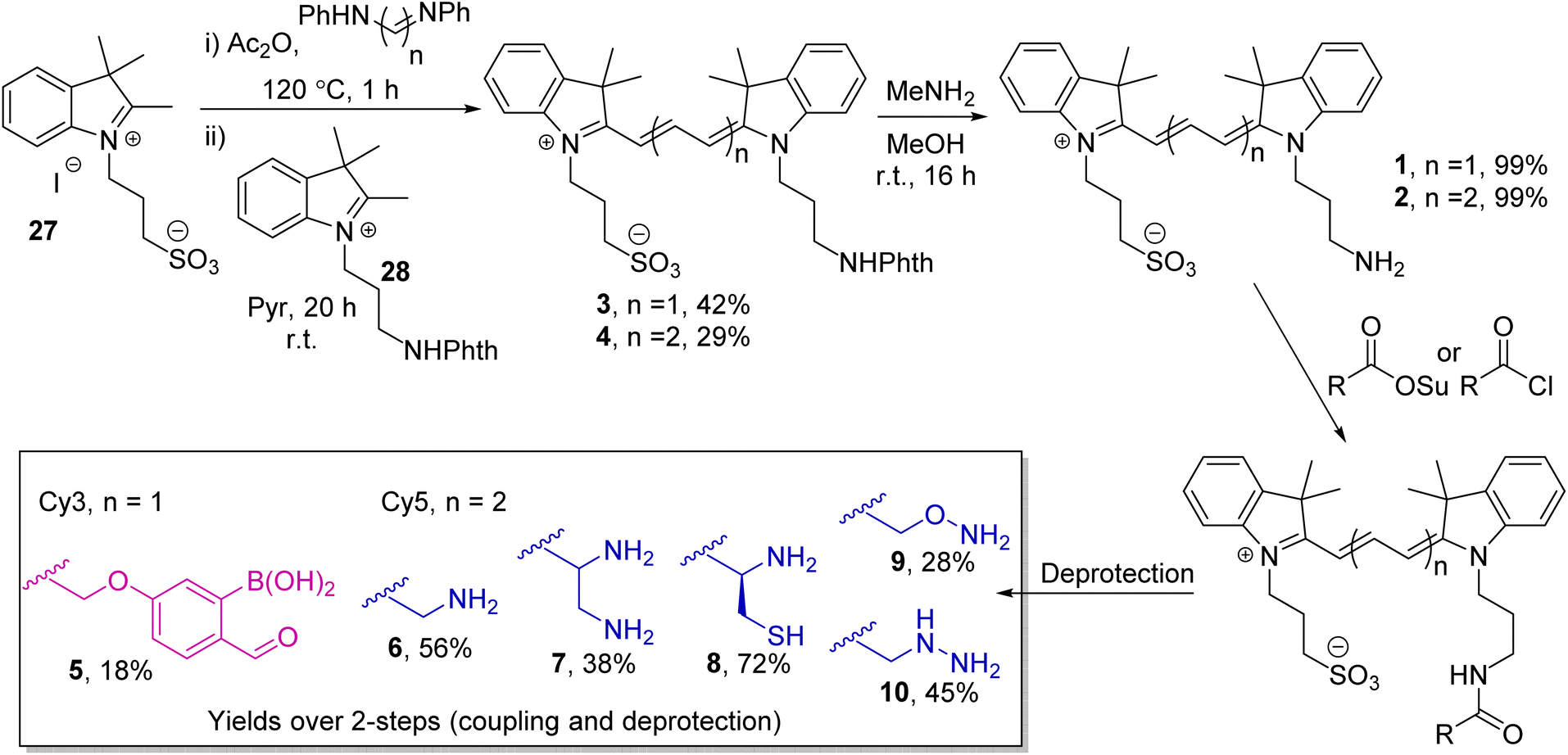 | ||
| Scheme 1 Synthesis of oBA-functionalised Cy3 dye 5 and nucleophile-functionalised Cy5 dyes 6–10. | ||
With the parent dyes in hand, they were functionalised with reactive handles for oBID formation. A 2-borono-4-alkoxy-benzaldehyde handle (5) was chosen as a representative oBA for attachment to Cy3 1. This choice was based on the ease with which 5 could be synthesised, relative to other oBA analogues, and therefore its position as a widely used functional oBA within the community.16–18 However, as discussed later, this choice of oBA plays a role in dictating conjugation rate and stability, and therefore has significant implications.
As nucleophiles, Cy5 analogues 6–10 were synthesised to represent the most common reagents for oBID formation. These analogues encompass the archetypal amine nucleophile (6) associated with iminoboronate formation (11),19 1,2-diamine (7)18 and 1,2-aminothiol (8)8 based nucleophiles which form cyclic imidazolidino-(IzB, 12) and thiazolidino-(TzB, 13) boronates respectively, and the ‘α-effect’ hydroxylamine (9)14 and hydrazine (10)20 handles that react to form stabilised oximes (14) and hydrazones/diazoborines (DAB, 15). A consistent linker structure was used to minimise any distance-dependent changes in FRET efficiency.
Preliminary platform validation
oBID formation was initially screened in a 96-well format under pseudo-first order conditions, to allow optimal timeframes for reaction monitoring to be established. Significant drops in Cy3 emission (580 nm) were observed for mixtures of Cy3-oBA 5 (5 μM) and nucleophiles 7–11 (50 μM) over a 30 minutes timeframe, with amine 6 leading to little change (See ESI Fig. S1†). Importantly, control reactions between 5 and nucleophiles that were not functionalised with the Cy5 acceptor, did not lead to a change in Cy3 emission (See ESI Fig. S2†). This confirmed that energy transfer was taking place between the two dyes specifically upon conjugation, and ruled out non-radiative quenching by the oBID. Similarly, no drop in emission was seen for control reactions using Cy3 substrates lacking either the boronic acid or aldehyde functional groups, confirming the importance of B–N stabilisation. Though benzaldehyde can form non-stabilised thiazolidines, oximes, and hydrazones under these conditions, reactions are slow at neutral pH in the absence of catalysts (k1 < 0.01 M−1 s−1) and therefore would not be expected to form over the timeframes of these studies.21,22 Cumulatively, these experiments validate the platform design.oBID formation at pH 7.4
Following these preliminary studies, we went on to study the formation of 11–15 under second-order conditions. Cy3-oBA donor 5 was mixed with nucleophiles 6–10 at a concentration of 2.5 μM in phosphate buffered saline (pH 7.4, experiments run in triplicate, Fig. 2a). Emission spectra (520–700 nm) following Cy3 excitation were then recorded every 15 seconds over a period of 25 min. To correlate Cy3:Cy5 emission ratios to conversions, reactions using an acetyl-capped Cy5-amine 16 (negative ‘0% conjugation’ control) and a covalently linked Cy3-Cy5 pair 17 (positive ‘100% conjugation’ control) were used as references (Fig. 2b and Table 1, see ESI Section 9† for details of data processing).
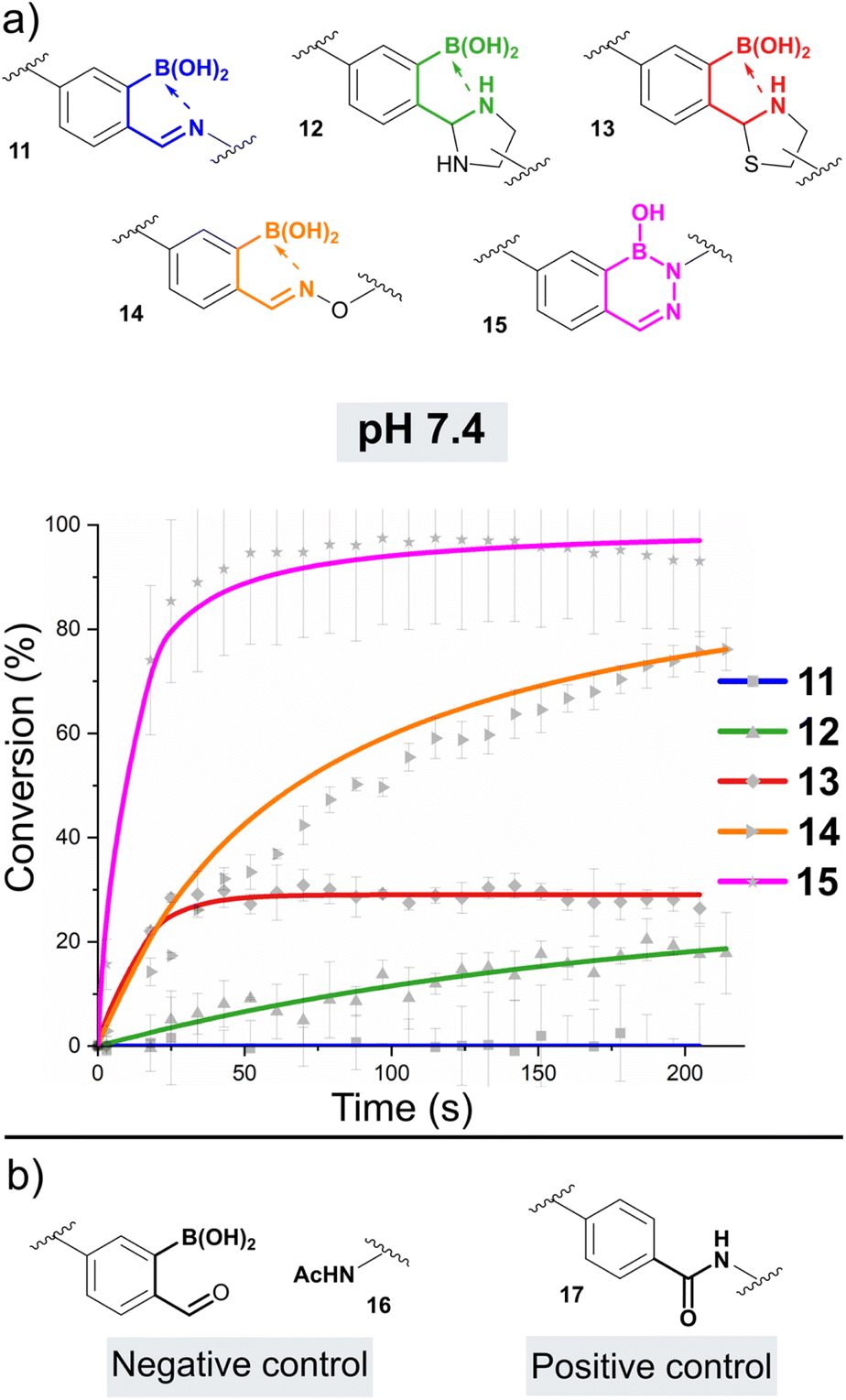 | ||
| Fig. 2 (a) Plot of reaction conversion against time for the formation of oBIDs 11–15 from Cy3-oBA 5 and the relevant Cy5-nucleophile. Reactions were run at a concentration of 2.5 μM under second-order conditions at pH 7.4. Fits are based on second-order irreversible (14, 15) or reversible models (11–13), with errors based on the standard deviation of experiments run in triplicate; (b) control substrates used to provide reference measurements for the calculation of conversions. Nb. data for 11 tracks the baseline (0% conversion) across all time points. | ||
| pH | k 1/M−1 s−1 | k −1/s−1 | K d | |
|---|---|---|---|---|
| 11 | 6 | — | — | — |
| 12 | 6 | — | — | — |
| 13 | 6 | 250 ± 3 | 1.13 ± 0.04 × 10−3 | 4.5 μM |
| 14 | 6 | 3034 ± 27 | 6.04 ± 1.08 × 10−5 | 19 nM |
| 15 | 6 | 22920 ± 839 |
8.07 ± 0.29 × 10−6 | 352 pM |
| 11 | 7.4 | — | — | — |
| 12 | 7.4 | 9882 ± 978 | 4.02 ± 0.45 × 10−2 | 4.3 μM |
| 13 | 7.4 | 613 ± 16 | 3.01 ± 0.11 × 10−3 | 4.9 μM |
| 14 | 7.4 | 4370 ± 164 | 5.61 ± 1.28 × 10−5 | 12 nM |
| 15 | 7.4 | 169030 ± 6962 |
2.60 ± 0.55 × 10−6 | 15 pM |
| 11 | 8 | — | — | — |
| 12 | 8 | 17521 ± 1211 |
2.72 × 10−2 | 1.5 μM |
| 13 | 8 | 21261 ± 591 |
1.22 × 10−3 | 57 nM |
| 14 | 8 | 1528 ± 10 | 3.56 ± 0.11 × 10−4 | 233 nM |
| 15 | 8 | 37471 ± 1238 |
8.05 ± 0.26 × 10−6 | 215 pM |
| 14 | 4 | 1517 ± 81 | 5.23 ± 0.56 × 10−5 | 34 nM |
| 15 | 4 | 876 ± 163 | 3.36 ± 0.70 × 10−2 | 38 μM |
As expected given reports of Kd ∼ 10 mM, iminoboronate 11 was not found to form to an appreciable extent at the low concentrations of these experiments.6 However, for the formation of IzB 12 and TzB 13 the data was found to fit a reversible second-order kinetic model, allowing us to extrapolate k1 and k−1, and thus calculate Kd.23 Interestingly, though the overall position of equilibrium for each reaction was similar (Kd ∼ 4–5 μM), IzB was found to form at an order of magnitude higher rate (∼9900 M−1 s−1vs. 610 M−1 s−1), highlighting thiol cyclisation as rate-limiting for the formation of a stable conjugate. Though our results are in line with previous reports on TzB formation,5 the rate of IzB formation was found to be accelerated by a factor of 10 relative to literature precedent.18 This difference can be attributed to the use of a functionalised 2-borono-4-alkoxy-benzaldehyde handle within our studies, in contrast to the use of FPBA models in the previous reports. This result therefore highlights the critical importance of studying derivatised oBIDs which more accurately reflect the substrates used in real-world applications. Moreover, our measurements were insensitive to the mixtures of diastereomers which complicate NMR analysis of IzB and TzB formation, providing further benefit over previous analyses.18,22
The formations of oxime 14 and hydrazone/DAB 15 were found to fit a second-order irreversible model, and so k1 was calculated from plots of inverse reagent concentration against time. Due to the fast kinetics with which 15 was formed, reactions were performed at high dilutions (750 nM) to provide sufficient data points within the linear region of the plot (See ESI Fig. S3†). Attempts to cleave 14 or 15 using an excess of a competitive nucleophile were unsuccessful, due to slow side-reactions with the conjugated framework of the dyes which became significant over the long timeframes of the equilibration. We therefore monitored the equilibration between propyl-amine functionalised oxime 18 or DAB 19 and an excess of a competitive nucleophile (20 or 21) via liquid chromatography analysis, allowing us to determine the rate of hydroxylamine/hydrazine exchange and thus calculate k−1 and Kd (Scheme 2, see ESI Fig. S4† for data).
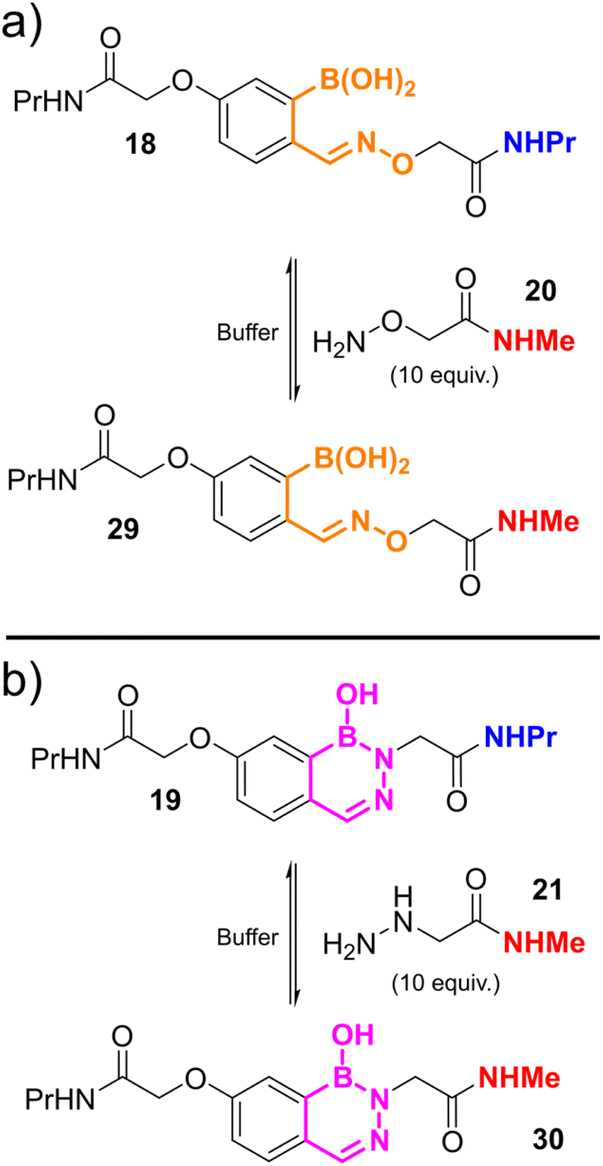 | ||
| Scheme 2 Equilibration of (a) propylamine-oxime 18 with and excess of methylamine-functionalised hydroxylamine 20; and (b) propylamine-DAB 19 with an excess of methylamine-functionalised hydrazine 21; allowing k−1 to be calculated over extended periods of time via LC-MS analysis. | ||
The rate of oxime formation and dissociation constant (k1 ∼ 5000 M−1 s−1; Kd ∼ 10 nM) is in line with previous reports by Schmidt et al. (k1 ∼ 11000 M−1 s−1; Kd ∼ 25 nM). Interestingly, this previous work studied the reactivity of 2-borono-5-alkoxy benzaldehydes, in contrast to the 4-alkoxy analogues used in this work, again highlighting the contributions of stereo-electronic arrangements to oBA reactivity.14
The reactivity of hydrazines with FPBA has been studied extensively by the Bane and Gillingham groups. They have shown that following the formation of an intermediate hydrazone, a second intramolecular cyclisation and dehydration step results in the eventual formation of an aromatic DAB at neutral pH. Though the rates of DAB formation have been determined (∼0.01 s−1), previous studies have had insufficient kinetic resolution to provide insight into the initial hydrazone formation.10,12,20 Our results indicate that this step is remarkably rapid, with k1 ∼ 105 M−1 s−1. Within the timeframe of our FRET experiments, it is unlikely that DAB formation makes a significant contribution, but would not be expected to significantly affect our FRET results, which report on conjugation not structure, regardless. This result again highlights the benefit of analysing all conjugated products collectively, rather than individual species which may otherwise be undergoing exchange or serving as intermediates towards an eventual thermodynamic product. Equilibration studies over the course of 1 week indicated the reaction remained dynamic, with 19 undergoing hydrolysis with a rate of ∼10−6 s−1 leading to a Kd for hydrazone/DAB formation of ∼10 pM (see ESI Fig. S4†). To the best of our knowledge, this represents the lowest Kd recorded for a dynamic oBID to date, in contrast to the nopoldiol-stabilised thiosemicarbazone reagents developed by the Hall group which show no reversibility.24
pH-dependent oBID formation
We next went on to study the pH dependence of oBID formation (Fig. 3 and Table 1, see ESI Fig. S5† for data arranged by nucleophile rather than pH). The formation of IzB 12 was accelerated at pH 8 (k1 ∼ 17500 M−1 s−1), with a corresponding small increase in stability, but at pH 6 no conversion was observed. Accounting for error and noise within our calculations, this means Kd > 45 μM for IzB formation under such conditions. Notably, Li et al. previously reported that the carboxylic acid of diaminopropionic acid was able to cyclise to form a mixed boron-carbon anhydride structure which was favoured at pH 6 and stabilised IzB formation. However, when the 1,2-diamine is conjugated to a functional cargo, as in our system, no such interactions are possible demonstrating the importance of studying models relevant to functional applications.18
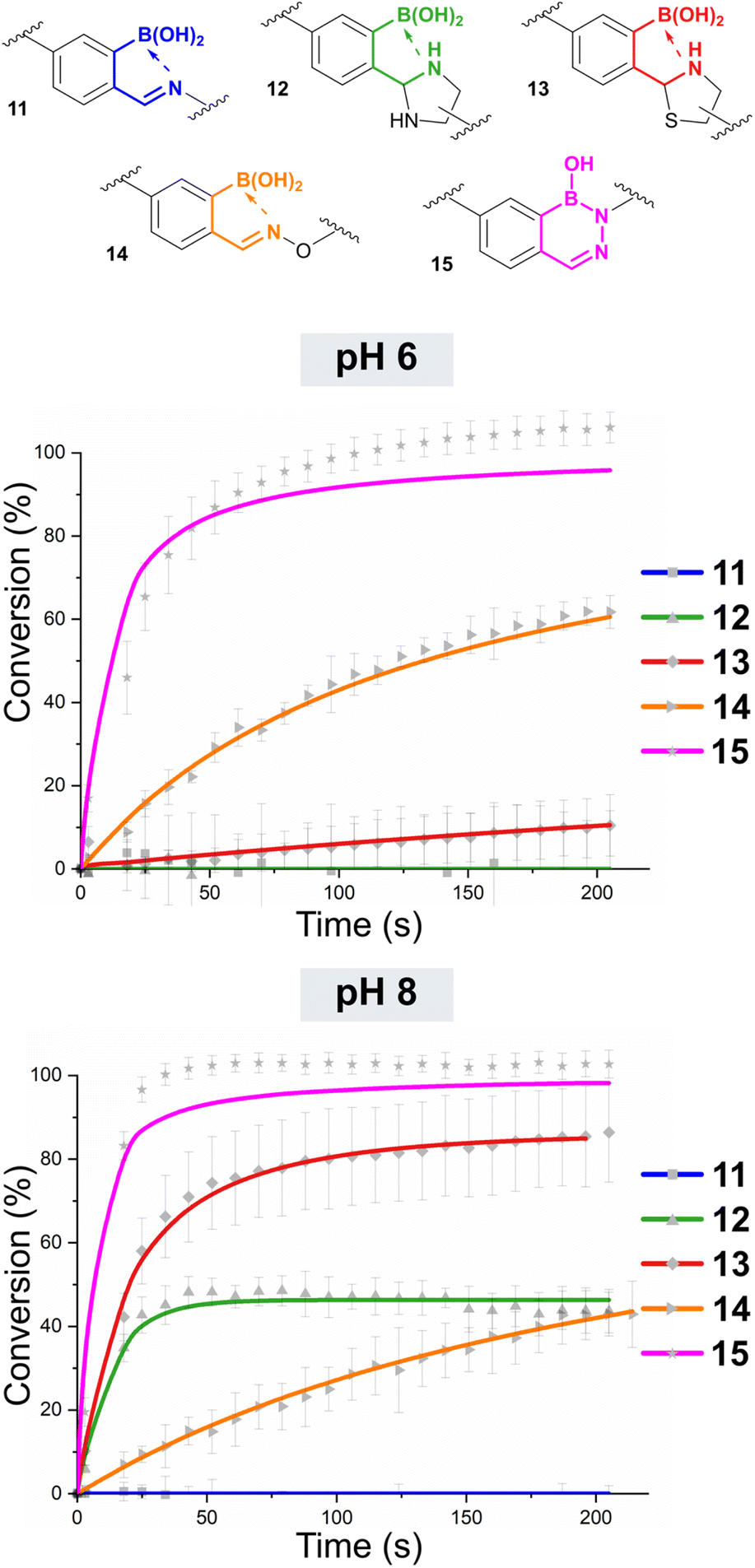 | ||
| Fig. 3 Plot of reaction conversion against time for the formation of oBIDs 11–15 from Cy3-oBA 5 and the relevant Cy5-nucleophile. Reactions were run at a concentration of 2.5 μM under second-order conditions at pH 6 or 8. Fits are based on second-order irreversible (15) or reversible models (11–14), with errors based on the standard deviation of experiments run in triplicate. Nb. data for 11 tracks the baseline (0% conversion) across all time points. | ||
A similar increase in reaction was observed for the formation of TzB 13 at pH 8. In contrast to the results obtained at pH 7.4, TzB formation was actually faster than IzB formation at this pH (k1 ∼ 37000 M−1 s−1vs. 21000 M−1 s−1), presumably due to the accelerated cyclisation of the deprotonated thiolate that can form at higher pH. The stability of the TzB was also increased by 2-orders of magnitude (Kd ∼ 50 nM). At pH 6, the rate of formation and stability of 13 was comparable to the results obtained at pH 7.4, lending further support to the hypothesis that the protonation state of the thiol plays a key role in TzB formation.
For the formation of oxime 14, pH 7.4 was found to provide optimal stability within the conditions tested (Kd ∼ 10 nM). At pH 6 or 8, k1 was reduced slightly, but contributions from accelerated oxime hydrolysis to Kd were far more significant leading to Kds ∼ 20 nM and 200 nM at pH 6 and 8 respectively. These results are in line with recent work by Han and Domaille,25 as well as a prior study by Schmidt et al.,14 showing that borono-oxime stability is highly pH dependent. Interestingly, at pH 4 Kd ∼ 34 nM, indicating that acidic pH did not have a large effect on borono-oxime stability.
At both pH 6 and 8, the rate of reaction between oBA 5 and hydrazine 10 was reduced relative to the reaction at pH 7.4, with a corresponding increase in k−1 leading to an overall increase in Kd from ∼10 pM at pH 7.4 to ∼300 pM and ∼200 pM at pH 6 and 8 respectively. Overall, the reactions were still found to proceed very rapidly at both pHs though, with k1 22000–37000 M−1 s−1 necessitating measurements at lowered concentrations (750 nM) to allow sufficient data points for analysis. At pH 4, the rate of formation of 15 was greatly decreased (∼850 M−1 s−1), with conjugate stability showing an even more dramatic 6-orders of magnitude drop (Kd ∼ 40 μM). It is noteworthy that at pH 4 Gu et al. reported that for the reaction of oBAs with related hydrazides, the open chain hydrazone oBID is dominant over the cyclic DAB.12 However, we found that the products formed from the hydrazine nucleophiles used in this study existed solely as the ring-closed DAB, with no evidence of the ring-open hydrazone by NMR, even at pH 4 (see ESI Section 11†). A switch between ring-closed and ring-open structure is therefore not responsible for the altered stability at lower pH.
Reaction additives
The ability to study oBID formation in the presence of additives, which would otherwise complicate absorbance measurements, is a major benefit of our FRET-based approach. Monosaccharides and more complex sugars are present at high concentrations in biological settings (glucose ∼ 5 mM in blood) and are known to dynamically complex boronic acids, and are therefore likely to play a significant role in oBID chemistry. In a number of reports it has been shown that pre-formed oBIDs are relatively stable to the addition of both glucose (Kd ∼ 10 mM with phenylboronic acid) and fructose (Kd ∼ 500 μM), but less is known about how these sugars affect oBID formation.26 The formations of 14 and 15 were therefore monitored in PBS containing 100 mM of either monosaccharide (Table 2, see ESI Fig. S6†). In all cases, rates of conjugation were found to be slowed, with fructose having a more significant influence in accordance with its higher boronic acid binding ability (∼ ×10 reduction in k1 for both hydroxylamine and hydrazine reactivity).| Additive | k 1/M−1 s−1 | k −1/s−1 | K d | |
|---|---|---|---|---|
| 14 | — | 4370 ± 164 | — | — |
| 14 | Glucose | 1340 ± 10 | 2.04 ± 0.12 × 10−4 | 152 nM |
| 14 | Fructose | 446 ± 4 | 2.54 ± 0.20 × 10−4 | 571 nM |
| 14 | 10% Serum | 64.0 ± 4.4 | 9.03 ± 2.22 × 10−4 | 14.1 μM |
| 15 | — | 169030 ± 6962 |
2.60 ± 0.55 × 10−6 | 15 pM |
| 15 | Glucose | 45750 ± 6614 |
2.36 ± 0.50 × 10−6 | 51 pM |
| 15 | Fructose | 16549 ± 1338 |
2.35 ± 0.50 × 10−6 | 142 pM |
| 15 | 10% Serum | 153 ± 6 | 2.93 ± 0.20 × 10−3 | 19.2 μM |
In the case of hydrazones/DABs, the decrease in k1 led to an overall small increase in Kd (50–150 pM, vs. 15 pM in the absence of additives), despite there being no effect of either sugar on k−1. This may suggest that monosaccharides do not complex hydrazones/DABs once formed, nor does pre-complexation lead to a stable tri-component complex, as such scenarios would lead to differing k−1 values across the three samples. In contrast, the stability of oxime 14 was significantly decreased (Kd ∼ 100–500 nM), in contrast to previous reports that suggest minimal influence of sugar additives on the stability of pre-formed oximes.14 There are two possible origins for this effect: (i) The increased sensitivity of our platform may have given insight into reversibility that was not previously observed due to the use of high reagent concentrations; or (ii) The presence of glucose/fructose during the reaction altered the structure of the product formed. For example, it is possible that sugar complexation after oxime formation is no longer possible/favoured, and so tests of stability could give contrasting results to reactions that study formation in the presence of the additive. To investigate this, oxime 18 or DAB 19 were incubated at pH 7.4 with either glucose or fructose at a concentration of 9 mM and binding was assessed via NMR. In all cases, no binding was observed, suggesting sugar complexation is not a major contributing factor to oxime or DAB stability at concentrations relevant to this study (see ESI Section 12†). Further investigations are currently ongoing in our lab to study the interplay between oBA-sugar binding and reaction kinetics, and to elucidate the effects of other relevant additives on oBID formation, structure, and stability, exploiting the platform reported here.
To further demonstrate the benefits of our approach, we next studied the formation and stability of oxime and hydrazone/DAB constructs in serum (Table 2, see ESI Fig. S6†). Understanding oBID chemistry in this environment is essential for future in vivo applications, particularly for potential within drug delivery to be realised. However, studying conjugation chemistry within serum is highly challenging due to its complex composition. The limited number of previous studies into oBID formation have relied on high concentrations of reagents that may not truly reflect the end application.11,14 In contrast, as background fluorescence within the 520–700 nm window is minimal and can be accounted for, FRET provides us with an ideal means to monitor oBID formation in serum at low μM concentrations.
The rate of oxime formation was slowed significantly in the presence of 10% bovine serum (k1 ∼ 60 M−1 s−1vs. 4000 M−1 s−1 in PBS), and the conjugates were also found to be highly dynamic, with a Kd ∼ 15 μM being observed. The decrease in reaction rate was even more significant for hydrazine 15, with a three order of magnitude reduction in k1 being recorded (∼150 M−1 s−1). Furthermore, conjugation was also found to be dynamic with a Kd ∼ 20 μM. Since the rate of DAB formation is significantly slower than initial hydrazone formation, this data may indicate that this intermediate species is dynamic in the presence of serum, with implications for oBID formation in complex medias. In contrast, when DAB 15 was pre-formed in PBS prior to dilution in 10% serum, the FRET ratio was found to stay unchanged over a period of 25 min (see ESI Fig. S7†). This supports previous reports that DABs are stable in serum, with exciting opportunities for future in vivo applications. Interestingly, oximes pre-formed in PBS were also observed to undergo minimal change in FRET ratio upon addition of 10% serum, pointing towards a potential role for serum complexation to the oBA precursors, leading to the observed slow formation of 14.
Cumulatively, these results highlight the potential of our platform to give far greater insight into oBID chemistry under relevant conditions to their translational applications, and efforts to explore such factors in high throughput are currently underway in our group.
Alternative oBA architectures
The studies described above provided us with detailed insight into the chemistry of the 2-borono-4-alkoxy-benzaldehyde handles that are widely used in the community. However, other handles, most commonly the 3-alkoxy (22) and 4-alkyl (23) derivatives are also highly relevant. Despite the greatly differing stereo-electronic configurations of these handles, the effect of this on oBID formation has not previously been studied. Basic density functional theory (DFT) calculations suggested that the relative contributions of N–B interactions to oBID stability across these analogues, as well as the previously unreported 4-carboxy analogue (24), were likely to be significant (up to 30 kJ mol−1 difference in stabilisation, see ESI Section 14†). Due to the significant synthetic effort needed to derive a series of dye-labelled constructs and controls for each distinct oBA, we therefore set out to exploit the data obtained through our FRET study to quantify parameters for each oBA in NMR competition studies.Reactions were run at a 1:1:1 ratio of 4-alkoxy oBA (25), oBA analogue, and the relevant nucleophile, with product ratios used to determine reaction parameters (Fig. 4). When 1,2-diamine 26 was used as a nucleophile, the rapid equilibration of the system (i.e. high k1 AND k−1) allowed us to calculate relative Kds, as described in the SI. The results obtained show a clear trend of increasing oBID stability as the substituent at the 4-position moved from electron donating (25) through to electron withdrawing (24). Unfortunately, efforts to perform analogous experiments using a hydrazine nucleophile as a kinetic trap proved unsuccessful, with the rapid rates of reaction leading insufficient resolution to distinguish differences in product distribution.
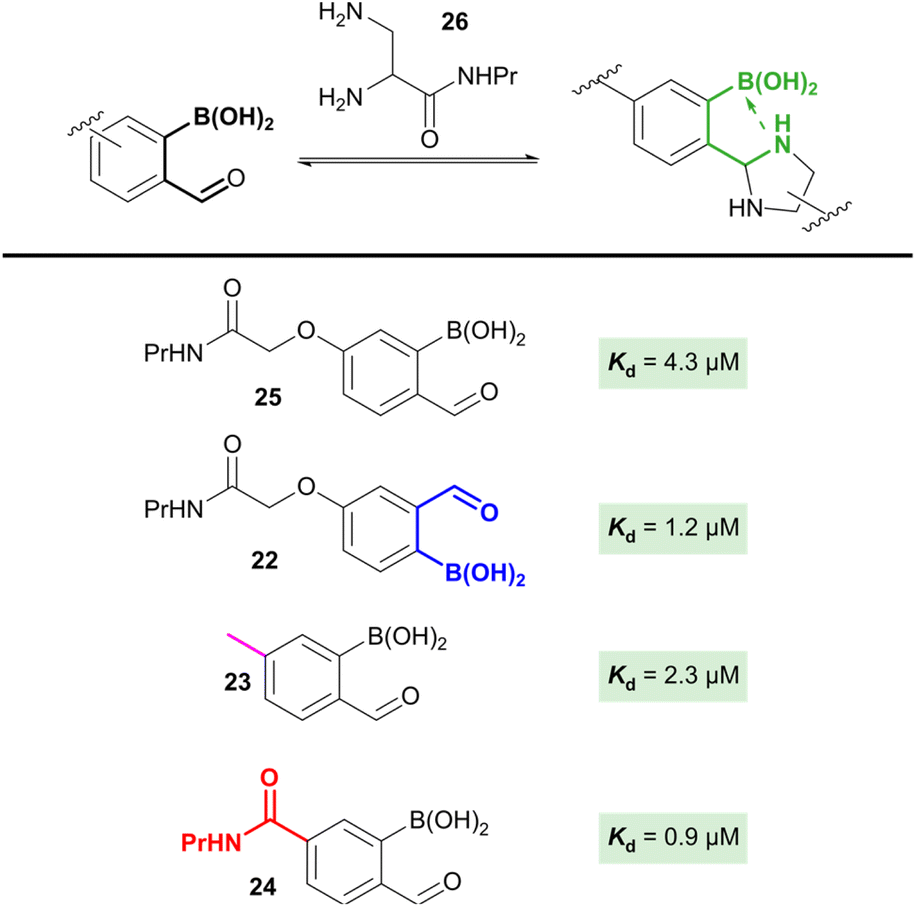 | ||
| Fig. 4 Competition experiments between 4-alkoxy-oBA 25 and analogues 22–24. A 1:1 ratio of 25 and the relevant analogue were combined with 1 equiv. of nucleophile 26 and the product distribution observed by NMR used to calculate Kd. | ||
The intriguing insights provided by these studies of relative oBID stability open the door to the design of new oBA derivatives with tuneable properties. For example, rational design of highly electron-deficient oBAs may lead to the production of oBIDs with greatly increased stability relative to the current state of the art. Further efforts to explore these possibilities are currently being undertaken.
Conclusions
In this study we have developed a novel FRET-based assay that allows the formation of oBIDs to be probed with unprecedented levels of detail. In particular, the ability to monitor the coupling of oBAs and amine-based nucleophiles at low concentrations with high sensitivity provides a direct way to measure conjugation. In turn, this allows us to cumulatively monitor the formation of all species which can contribute to the end applications of oBIDs, rather than relying on measurements of individual species which may undergo structural exchange and therefore do not represent the full picture.Using this platform, we were able to calculate k1,k−1, and Kd for the most widely used oBA-nucleophile pairs used in the literature across a range of conditions, allowing us to identify optimal pH ranges and interesting behaviour that provides new insight into oBID chemistry. The ability of our platform to function in the presence of buffer additives is particularly powerful, allowing us to study conjugation chemistry in complex environments such as serum. We therefore provide insight into the rate of formation of ortho-borono oximes and hydrazones/DABs for the first time. Our results also suggest that further tuning and optimization of oBID chemistry is possible through even small changes to the stereo-electronic configurations of the oBA and nucleophile precursors, and our platform provides an ideal means to test such structures. Efforts to do so, and deliver new functional methods relevant to bioconjugation and biomaterials chemistry, are currently underway in our lab, and we hope that our study will serve to stimulate further exploration and optimisation of the underlying chemistry of oBIDs in the future.
Data availability
Data associated with the study is availabe on the University of York Research Database via the https://doi.org/10.15124/5dfbc414-7af6-4377-9c9f-1ce65103b84e.Author contributions
N. C. R. performed all synthetic studies and FRET measurements, and data analysis and curation. A. V. S. performed preliminary studies which led to this work. E. F. T. performed DFT calculations, supervised by J. M. L., C. D. S. developed the project concept, performed data analysis, supervised and managed the study, and wrote the manuscript. All authors contributed to the editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Prof. Dave Smith and Dr Will Unsworth are thanked for insightful discussions throughout this work. N. C. R. and C. D. S. acknowledge PhD Studentship support from the EPSRC Doctoral Training Partnership and the University of York. A. V. S. acknowledges support from the Erasmus++ Programme. E. F. T. is grateful to the Royal Society of Chemistry for funding an Undergraduate Research Bursary. DFT calculations were undertaken on the Viking Cluster, which is a high-performance computer facility provided by the University of York. We are grateful for computational support from the University of York High Performance Computing service, Viking and the Research Computing team. J. M. L. is supported through a Royal Society Industrial Fellowship (2022–24). C. D. S. acknowledges support from the Royal Society of Chemistry Research Fund (RF19-8128) and a Royal Society Research Grant (RGS\R1\19120).References
- J. P. M. António, J. I. Carvalho, A. S. André, J. N. R. Dias, S. I. Aguiar, H. Faustino, R. M. R. M. Lopes, L. F. Veiros, G. J. L. Bernardes, F. A. Silva and P. M. P. Gois, Angew. Chem., Int. Ed. Engl., 2021, 60, 25914–25921 CrossRef.
- S. Palvai, J. Bhangu, B. Akgun, C. T. Moody, D. G. Hall and Y. Brudno, Bioconjugate Chem., 2020, 31, 2288–2292 CrossRef CAS PubMed.
- X. Ding, G. Li, P. Zhang, E. Jin, C. Xiao and X. Chen, Adv. Funct. Mater., 2021, 31, 2011230 CrossRef CAS.
- T. C. French and T. C. Bruice, Biochem. Biophys. Res. Commun., 1964, 15, 403–408 CrossRef CAS.
- S. Cambray and J. Gao, Acc. Chem. Res., 2018, 51, 2198–2206 CrossRef CAS PubMed.
- S. Chatterjee, E. V. Anslyn and A. Bandyopadhyay, Chem. Sci., 2021, 12, 1585–1599 RSC.
- H. Faustino, M. J. S. A. Silva, L. F. Veiros, G. J. L. Bernardes and P. M. P. Gois, Chem. Sci., 2016, 7, 5052–5058 RSC.
- A. Bandyopadhyay, S. Cambray and J. Gao, Chem. Sci., 2016, 7, 4589–4593 RSC.
- A. Bandyopadhyay and J. Gao, Chem.–Eur. J., 2015, 21, 14748–14752 CrossRef CAS.
- C. J. Stress, P. J. Schmidt and D. G. Gillingham, Org. Biomol. Chem., 2016, 14, 5529–5533 RSC.
- A. Bandyopadhyay, S. Cambray and J. Gao, J. Am. Chem. Soc., 2017, 139, 871–878 CrossRef CAS.
- H. Gu, T. I. Chio, Z. Lei, R. J. Staples, J. S. Hirschi and S. Bane, Org. Biomol. Chem., 2017, 7543–7548 RSC.
- E. Herbst and D. Shabat, Org. Biomol. Chem., 2016, 14, 3715–3728 RSC.
- P. Schmidt, C. Stress and D. Gillingham, Chem. Sci., 2015, 6, 3329–3333 RSC.
- M. Levitus and S. Ranjit, Q. Rev. Biophys., 2011, 44, 123–151 CrossRef CAS PubMed.
- A. Bandyopadhyay, K. A. McCarthy, M. A. Kelly and J. Gao, Nat. Commun., 2015, 6, 6561 CrossRef CAS PubMed.
- P. M. S. D. Cal, R. F. M. Frade, C. Cordeiro and P. M. P. Gois, Chem.–Eur. J., 2015, 21, 8182–8187 CrossRef CAS PubMed.
- K. Li, C. Weidman and J. Gao, Org. Lett., 2018, 20, 20–23 CrossRef CAS PubMed.
- P. M. S. D. Cal, J. B. Vicente, E. Pires, A. V. Coelho, L. F. Veiros, C. Cordeiro and P. M. P. Gois, J. Am. Chem. Soc., 2012, 134, 10299–10305 CrossRef CAS PubMed.
- O. Dilek, Z. Lei, K. Mukherjee and S. Bane, Chem. Commun., 2015, 51, 16992–16995 RSC.
- D. K. Kölmel and E. T. Kool, Chem. Rev., 2017, 117, 10358–10376 CrossRef PubMed.
- D. Bermejo-Velasco, G. N. Nawale, O. P. Oommen, J. Hilborn and O. P. Varghese, Chem. Commun., 2018, 54, 12507–12510 RSC.
- A. Dirksen, S. Dirksen, T. M. Hackeng and P. E. Dawson, J. Am. Chem. Soc., 2006, 128, 15602–15603 CrossRef CAS PubMed.
- B. Akgun, C. Li, Y. Hao, G. Lambkin, R. Derda and D. G. Hall, J. Am. Chem. Soc., 2017, 139, 14285–14291 CrossRef CAS PubMed.
- G. S. Han and D. W. Domaille, Org. Biomol. Chem., 2021, 19, 4986–4991 RSC.
- X. Wu, Z. Li, X.-X. Chen, J. S. Fossey, T. D. James and Y.-B. Jiang, Chem. Soc. Rev., 2013, 42, 8032–8048 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The supporting information contains all experimental details, including reagent synthesis, FRET measurements, and studies of reversibility. See DOI: https://doi.org/10.1039/d2sc04574e |
| This journal is © The Royal Society of Chemistry 2022 |