
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAxially chiral indolenine derived chromophore dimers and their chiroptical absorption and emission properties†
Emely
Freytag
a,
Marco
Holzapfel
a,
Asim
Swain
 a,
Gerhard
Bringmann
a,
Matthias
Stolte
ab,
Frank
Würthner
ab and
Christoph
Lambert
*ab
a,
Gerhard
Bringmann
a,
Matthias
Stolte
ab,
Frank
Würthner
ab and
Christoph
Lambert
*ab
aInstitut für Organische Chemie, Universität Würzburg, Am Hubland, Würzburg 97074, Germany. E-mail: christoph.lambert@uni-wuerzburg.de
bCenter for Nanosystems Chemistry, Universität Würzburg, Theodor-Boveri-Weg, Würzburg 97074, Germany
First published on 6th October 2022
Abstract
Yamamoto homocoupling of two chiral oxindoles led to the atropo-diastereoselective formation of an axially chiral oxindole dimer. This building block served as the starting material for the syntheses of axially chiral squaraine and merocyanine chromophore dimers. These dimers show pronounced chiroptical properties, this is, outstandingly high ECD signals (Δε up to ca. 1500 M−1 cm−1) as a couplet with positive Cotton effect for the P-configuration around the biaryl axis and a negative Cotton effect for the M-configuration. All investigated dimers also exhibit pronounced circularly polarised emission with anisotropy values of ca. 10−3 cgs. Time-dependent density functional calculations were used to analyse the three contributions (local one electron, electric–magnetic coupling, and exciton coupling) to the rotational strength applying the Rosenfeld equation to excitonically coupled chromophores. While the exciton coupling term proves to be the dominant one, the electric–magnetic coupling possesses the same sign and adds significantly to the total rotational strength owing to a favourable geometric arrangement of the two chromophores within the dimer.
Introduction
Squaraine dyes possess outstanding optical properties such as a narrow absorption band in the red or NIR spectral region, a high (photo)chemical stability and a high fluorescence quantum yield,1–4 and, thus, are suitable candidates for a number of optoelectronic5–10 and biomedical8,11–16 applications. However, only a few examples of chiral squaraines are known. In most cases, chirality of the dyes was achieved by attaching peripheral groups to the dye, namely cholesterol17,18 and proline,19 or chiral alkyl side chains.20,21 According to Snatzke, bringing a chiral centre closer to the chromophore leads to a more distinct electronic circular dichroism (ECD).22 For this reason, we recently investigated squaraines where the stereogenic centre is in close proximity to the chromophore. In that work, indolenine squaraine monomers with a chiral centre in the 3-position of the indolenine, containing either n-propyl or phenyl groups, were synthesised. Thereby, a significant rise of the ECD effects as compared to monomers containing only chiral sidechains was observed.23In this work, we focus on the synthesis and the chiroptical analysis of two axially chiral squaraine dimers and a merocyanine dimer with the aim to enhance their chiroptical properties beyond those of squaraine monomers with chiral substituents. According to Rosenfeld, the rotational strength R, which is the equivalent for the electric dipole strength of absorption spectroscopy, depends on the scalar product of the electric (μ) and magnetic (m) transition dipole moments24
R = Im(μ·m) = |μ||m|cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ϑ ϑ | (1) |
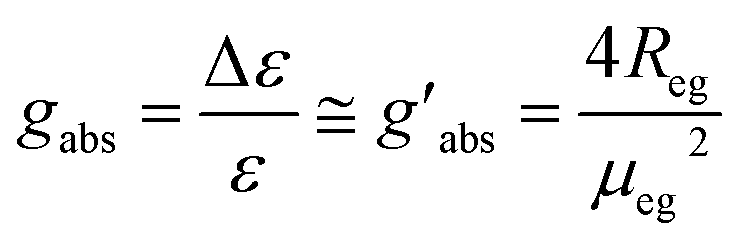 | (2) |
In the case of an excitonically coupled dimer of chromophores such as the squaraine dimers investigated in this work, the Rosenfeld equation expands to
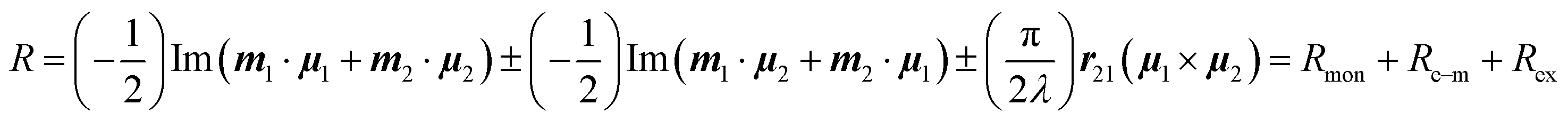 | (3) |
Here, the signs + and − refer to the symmetric and antisymmetric combinations of the two excited-state wavefunctions of the two excitonically coupled monomers. The first term (Rmon) contains the sum of the local (one electron) rotational strengths of the individual monomers. Re–m describes the electric–magnetic coupling of μ of the one with m of the other monomer and vice versa. The third term is the exciton term Rex, which depends on the cross product of the electric transition dipole moments and the distance vector between the centres of the monomers (r21). A chiral orientation of the monomers to each other is mandatory for this term not to be zero. Rex is believed to dominate the rotational strength and is the basis of the so-called exciton chirality method.24,26 This is particularly the case for axially chiral molecules and the reason why molecular dimers with this stereogenic element often possess a high dissymmetry. While the total magnitude of each term is identical for both exciton states, Re–m and Rex possess opposite sign for the two transitions. This leads to the typical exciton couplet with a bisignate signal.
In the case of a chiral orientation between chromophores, the exciton chirality rule can be used to determine the absolute configuration by considering only Rex of eqn (3). Looking at the screw sense of the orientation of both electric transition moments μ with respect to each other, a clockwise arrangement of the chromophores leads to a positive Cotton effect (CE), while a counter-clockwise arrangement is assigned to a negative CE.26
Most axially chiral chromophore arrangements in the literature were derived from axially chiral 1,1-binaphthyl derivatives as the chiral scaffold in combination with a chromophore unit such as BODIPYs,27–30 phthalocyanines,31,32 pentacenes,33 phenylenes,34 triarylboranes,35 spirooxazines,36 methine dyes,37 perylene diimides38,39 or other perylene and pyrene dyes.40 An intriguing but seldom used approach is to connect two chromophores directly via a chiral axis, thus avoiding any chiral scaffold.41,42 In this case additional ortho-substituents next to the axis prevent rotation and therefore provide stability of the thereby formed biaryl axis.43–45 For example, Bruhn et al.46,47 synthesised BODIPY dimers which possess a rotationally hindered biaryl axis formed via oxidative C–C coupling of either the α or β position of the BODIPY. This chromophore dimer appears to be the only directly linked axially chiral cyanine-type chromophore mentioned in the literature so far. For this dimer, using a TDDFT-fragmentation method, a separation of the rotational strength into the contributing terms as in eqn (3) was achieved. Bruhn et al. showed that in their case, the first and the second term are more dominant than Rex. Therefore, the absolute configuration could not be predicted by applying the excition chirality rule.46 The BODIPY dimers showed rather small gabs and glum values of 5 × 10−4 and 4 × 10−4 (α,α dimer) and of 1 × 10−2 and 4 × 10−3 cgs (β,β dimer).47 In a further example by Bringmann et al.,48 the synthesis of atropisomeric porphyrin dimers was achieved by an optimised Suzuki–Miyaura coupling procedure.
However, axially chiral squaraine dyes have to the best of our knowledge not been synthesised and investigated yet despite their promising optical properties such as a very small absorption bandwidth. Thus, in this work we developed a novel strategy to use a stereogenic centre in the indolenine moieties of a squaraine dye to induce axial chirality in a biphenyl-linked squaraine dimer. Therefore, two axially chiral indolenine squaraine dimers dSQA and dSQB (see Fig. 1) were synthesised and investigated in terms of their chiroptical properties. Here, the biaryl axis connecting the two squaraine monomers was formed by a transition metal mediated reaction. The preferred position of the monomers with respect to the axis was directed by the phenyl group at the chiral centre in the 3-position of the indolenine moiety. The steric congestion makes one of the two possible diastereomeric biaryl conformers thermodynamically unfavourable. Thus, starting from an indolenine with an (R)-configurated stereogenic centre leads to a (P)-configuration of the biphenyl axis in the squaraine dimer. To demonstrate the general applicability of this strategy to synthesise indolenine derived dye dimers, the same axial chiral bridging unit was used to synthesise the merocyanine dimer dMC. This merocyanine also possesses a significantly broader absorption bandwidth than the squaraine dyes. For comparison, also reference monomers of all dimers were synthesised and investigated. The chiroptical properties of all chromophores were characterised by ECD and CPL spectroscopy. In order to analyse the individual terms Rmon, Re−m and Rex, which contribute to the overall rotatory strength we performed time-dependent density functional calculations (TD-DFT) calculations.
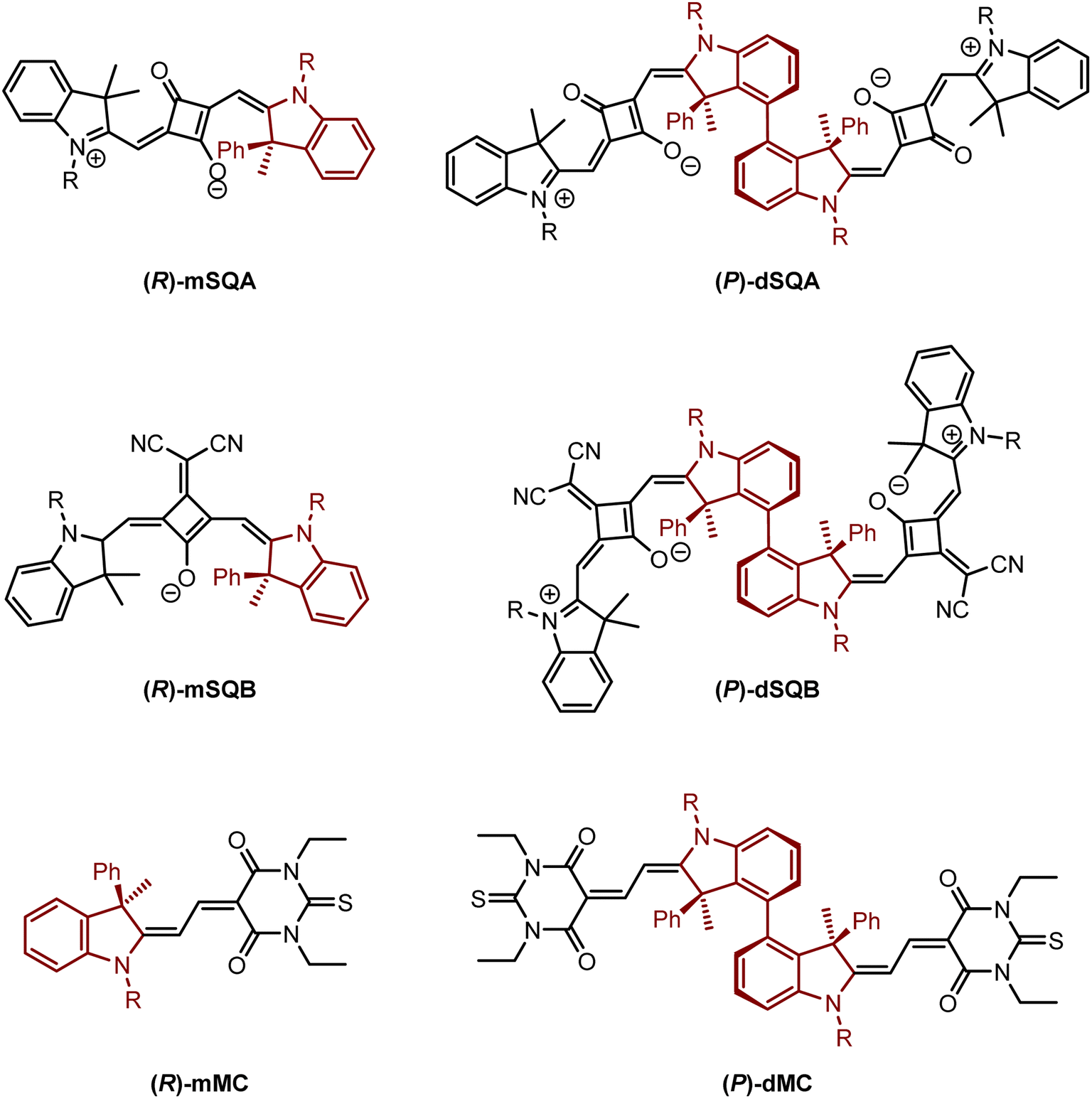 | ||
| Fig. 1 Target molecules: reference monomers (R)-mSQA, (R)-mSQB and (R)-mMC, squaraine dimers dSQA and dSQB and merocyanine dimer dMC with (P)-configuration. R = n-hexyl. | ||
Results and discussion
Synthesis
The key synthetic step of the squaraine dimer preparation is the atropo-diastereoselective synthesis of the oxindole biaryl compound 8 (see Fig. 2). This biaryl was synthesised by direct Ni mediated Yamamoto coupling of enantiopure (R)-oxindole 7 (e.e. > 99%) thereby creating a new stereogenic axis. The oxindole in turn was prepared analogously to a synthesis starting from (D)-serine hydrochloride (see ESI Section 7†), originally developed by Richmond et al.,49 and used by us recently to prepare chiral squaraine monomers.23 Coupling of 7 to give compound 8 was achieved in moderate yield (42%) but excellent d.e. This type of atropo-diastereoselective synthesis appears to be a novel access to atropisomerically pure biaryl systems.44 The bis-oxindole 8 was treated with a methyl Grignard nucleophile followed by acidic elimination to generate the indolinium salt 9, which, because of its instability, was used in situ and condensed with the two semisquaraines 10 or 11 to produce (P)-dSQA and (P)-dSQB, respectively, in reasonable yield. The stereochemical purity was determined by analytic HPLC on a chiral phase to be over 99% d.e. and e.e. for both dimers. The chromatograms can be found in the ESI in Section 5.†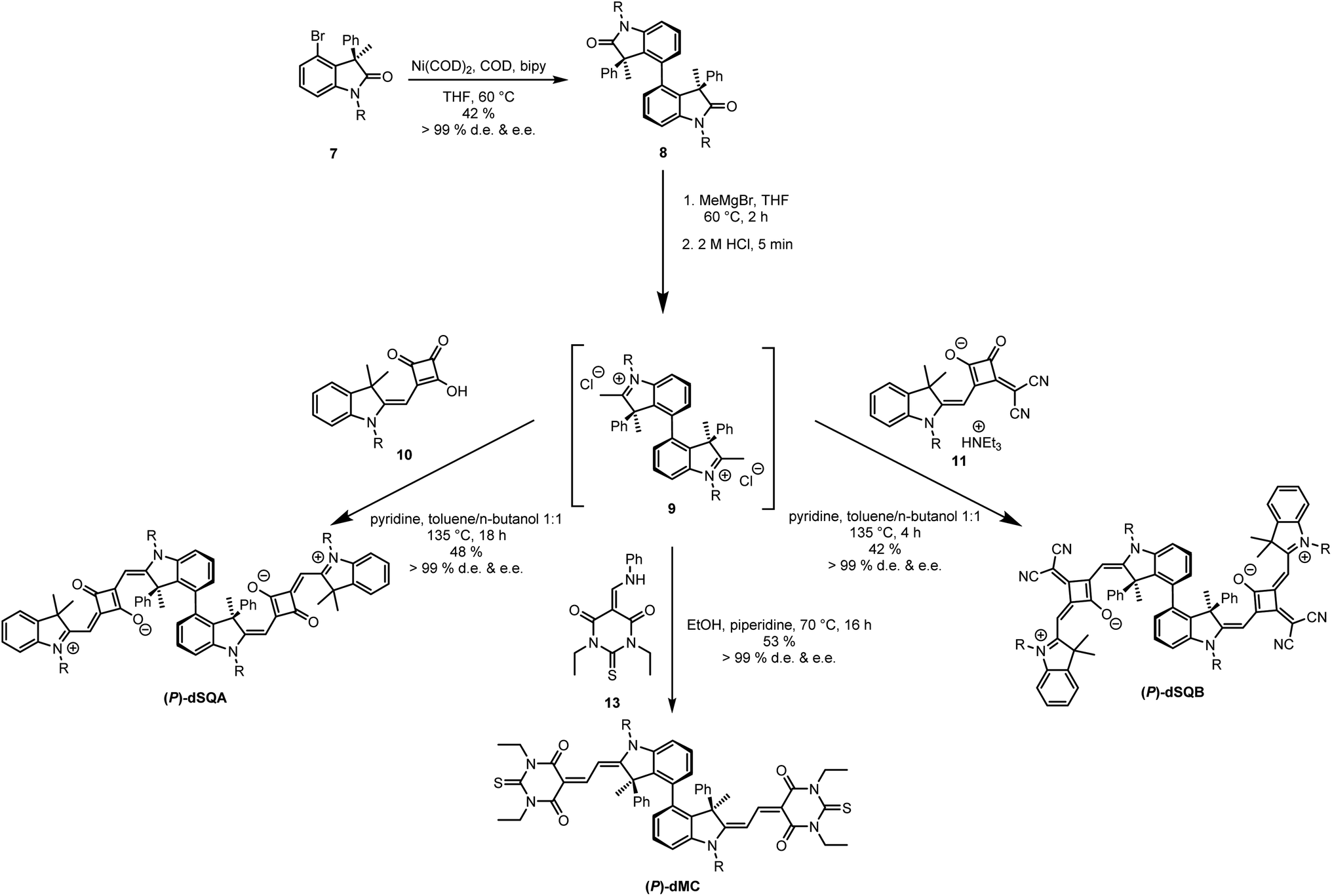 | ||
| Fig. 2 Synthesis of (P)-dSQA, (P)-dSQB and (P)-dMC by Yamamoto coupling of (R)-oxindole 7. R = n-hexyl. | ||
The sketched synthetic pathway via the bis-oxindole has the advantage that this compound can be used as a precursor for the synthesis of diverse axially chiral dyes. As an example, atropisomerically pure merocyanine dimer (P)-dMC was also synthesised. Thus, the indolinium salt 9 was treated with thiobarbituric acid, which was extended to an enamine using triethyl orthoformate and aniline.50,51 A yield of 53% and a stereochemical purity of 99% e.e. were achieved for (P)-dMC.
Analytical HPLC on a chiral phase showed all dimers to be enantiomerically pure (see ESI Fig. S14–S17, S21 and 22†). Rotation around the stereogenic biaryl axis would allow to form two diastereomers. But although we could not find any associated signal in the HPLC, this does not necessarily mean that the barrier between the two diastereomers is too high for an interconversion at room temperature since biaryls with only one ortho-substituent per aryl group are usually configurationally unstable.52 However, thermodynamics could shift a possible dynamic equilibrium completely to one side. In order to assess these issues, we performed DFT optimisations at the B3LYP/6-31G* level of theory on the bis-oxindole 8, which also served as a model for all other chromophore dimers. We optimised the two diastereomers as well as the transition state that interconverts these two structures (see Fig. 3). One diastereomer was found to possess a much higher energy (2396 cm−1 [28.7 kJ mol−1]) than the other, which is caused by steric interactions of the two phenyl groups (see space filling model in Fig. 3). From the energy of the transition state (5288 cm−1 [63.3 kJ mol−1]) it is evident that the higher-energy diastereomer is not stable at room temperature and that the equilibrium will rapidly and completely shift to the side of the lower energy diastereomer.53 Thus, we conclude that the two stereogenic centers of the indolenine units afford diastereoselective control of the biaryl axis and that all chromophore dimers derived from bis-oxindole 8 possess the same axially chirality.
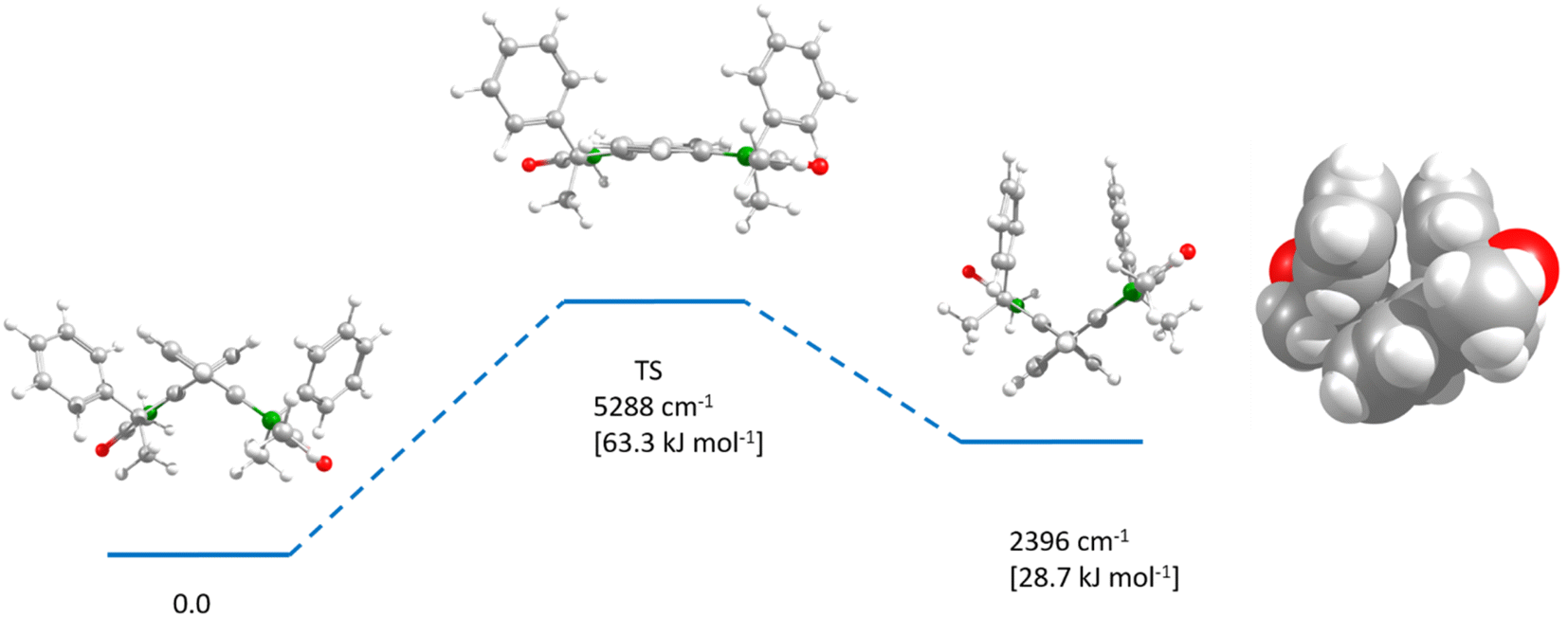 | ||
| Fig. 3 DFT-optimised diastereomers of bis-oxindole 8 at B3LYP/6-31G*. Energies include zero-point correction. The view is along the biphenyl axis. Colour code: oxygen (red), nitrogen (green), carbon (gray), hydrogen (light gray). The flexible hexyl substituents were replaced by methyl groups for the ease of the calculation. | ||
Absorption and fluorescence spectroscopy
The absorption and fluorescence spectra of all discussed compounds in toluene solution are depicted in Fig. 4, the spectroscopic data are summarised in Table 1.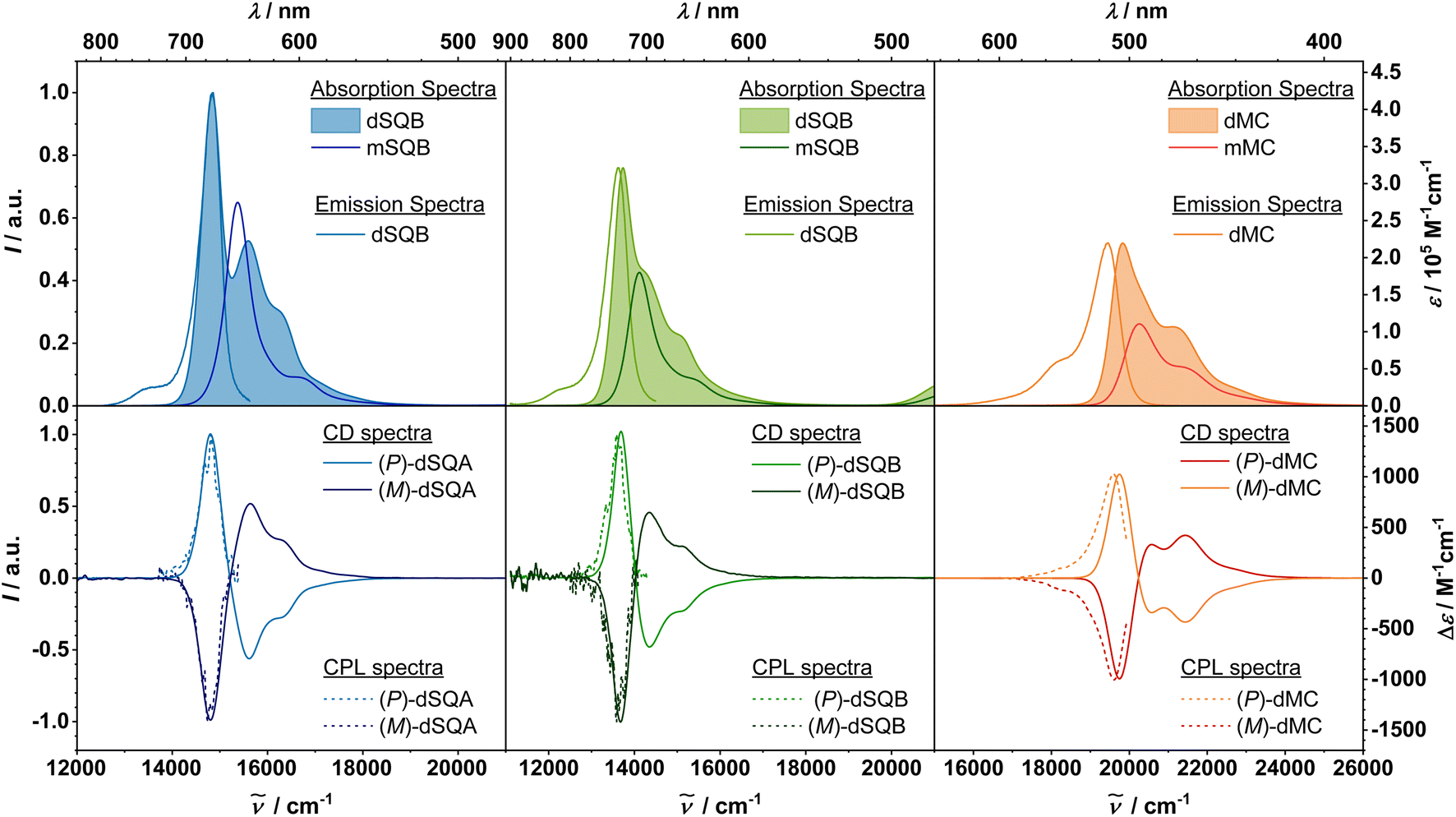 | ||
| Fig. 4 Upper panel: absorption and emission spectra of all monomeric and dimeric chromophores in toluene. Lower panel: CPL and ECD spectra of chromophore dimers. Excitation for the emission spectra was at 16100 cm−1 (620 nm) for dSQA, 14900 cm−1 (670 nm) for dSQB, and 21700 cm−1 (460 nm) for dMC (c ∼5.0 × 10−6 M for absorption and ECD spectra, <10−7 M for fluorescence spectra, c ∼ 5.0 × 10−7 M for CPL spectra). | ||
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) max/cm−1
max/cm−1 |
ε max/cm−1 | μ abs 2/D2 |
fl/cm−1 |
fwhmfl/cm−1 | Φ fl | τ fl/nsb (ampl.) | μ fl 2/D2 | |
|---|---|---|---|---|---|---|---|---|
| a Measured with an integrating sphere. b Measured by TCSPC with excitation at 656 nm (squaraines)/419 nm (merocyanines). c Average lifetime 0.97 ns. d Average lifetime 2.47 ns. | ||||||||
| (P)-dSQA | 14800 |
4.19 × 105 | 222.5 | 14800 |
520 | 0.24 | 1.42 (0.59)c | 85.8 |
| 0.32 (0.41) | ||||||||
| (P)-dSQB | 13700 |
3.21 × 105 | 186.2 | 13600 |
590 | 0.52 | 3.06 (0.63)d | 95.2 |
| 1.47 (0.37) | ||||||||
| (R)-mSQA | 15400 |
2.74 × 105 | 108.8 | 15200 |
0.36 | 1.23 | 94.4 | |
| (R)-mSQB | 14100 |
1.80 × 105 | 88.5 | 13800 |
0.63 | 3.70 | 74.9 | |
| (P)-dMC | 19800 |
2.20 × 105 | 129.3 | 19500 |
940 | 0.07 | 0.16 | 71.7 |
| (R)-mMC | 20200 |
1.11 × 105 | 62.8 | 19600 |
0.05 | 0.10 | 80.3 | |
The two monomeric squaraines (R)-mSQA and (R)-mSQB exhibit a typical narrow absorption band in the red to NIR spectral region with ε = 4.19 × 105 M−1 cm−1 and 3.21 × 105 M−1 cm−1, respectively, along with a vibronic shoulder on the high-energy side.23 The merocyanine monomer (R)-mMC has its main transition band at 20200 cm−1, with an extinction coefficient of 1.11 × 105 M−1 cm−1. A similar literature-known indolenine merocyanine with a barbituric acid acceptor and the same conjugation length shows comparable values of 21600 cm−1 and 8.45 × 104 M−1 cm−1 in toluene.54 Because the exciton coupling strength relates to the magnitude of the dipole strength μabs2 it is important to point at the significantly lower values of the merocyanines used in this study. The reason is that the distribution of the π-electrons along the polymethine chain of these merocyanine dyes is more polyene-like whilst it is cyanine-like for the squaraines.55
Due to the excitonic coupling of the monomeric chromophores states, the excited states of the dimers are split into two states.56 The lower energy transitions of the squaraine dimers are red shifted by 600 cm−1 for (P)-dSQA and 400 cm−1 for (P)-dSQB, with respect to (R)-mSQA and (R)-mSQB. The absorption bands of the higher energy exciton state can be observed at 15600 cm−1 in (P)-dSQA, and, as a shoulder at 14200 cm−1 in (P)-dSQB. On the higher-energy side, there is a vibronic shoulder in both molecules. For (P)-dSQA we can estimate the exciton coupling strength by half of the difference between the two exciton states to be V = 400 cm−1. This coupling is about half as strong as in a dSQA(5-5′) dimer, where the two chromophores are linked at the 5-position rather than at the 4-position of the indolenine.57 For the merocyanine (P)-dMC, the situation is basically identical. The main absorption band can be found at 19800 cm−1, the second exciton state only appears as a weak shoulder at 20300 cm−1. The lowest energy transition is shifted by 400 cm−1 with respect to the corresponding monomer (R)-mMC. The dipole strengths μabs2 for all dimers are approximately twice as high as those of the monomers, satisfying the Thomas–Reiche–Kuhn sum rule by showing an additive behaviour. This proves that there are no other states than the two exciton states in this spectral region.24
The fluorescence spectra of all investigated molecules are mirror images of the lowest exciton state, which is in accordance with Kasha's rule.58,59 For the monomers, the fluorescence quantum yield of (R)-mSQA (36%) is significantly smaller than that of symmetric achiral SQA monomer but that of (R)-mSQB (63%) is similar to the one of achiral SQB monomer.23 The quantum yields of (P)-dSQA and (P)-dSQB (24% and 52%) are reduced compared to the chiral monomers (36% and 63%). For (P)-dMC the fluorescence quantum yield (7%) is rather low as is that of the corresponding monomer (5%). The Stokes shift is rather small for all squaraines (<300 cm−1) and even vanishes for the dimers. The fluorescence maximum of the merocyanine dimer is shifted by 300 cm−1 with respect to the absorption spectrum. Considering the exciton lifetimes, the squaraine dimers (P)-dSQA and (P)-dSQB both show a biexponential decay with a minor short component of <1 ns and longer components of 1.4 and 3.1 ns, respectively. In comparison, the monomers decay monoexponentially but with similar lifetimes as the longer component of the dimers.23 For the merocyanines, the decay is monoexponential with 0.2 ns (dimer) and 0.1 ns (monomer). However, the lifetimes are significantly reduced compared to those of other indolenine barbituric acid merocyanines, which explains the low fluorescence quantum yield.54 From the quantum yield and the lifetime, the radiative rate constant was calculated and from that the squared fluorescence transition moment μfl2via the Strickler–Berg equation (see ESI†). The value of μfl2 of the dimers turns out to be significantly smaller than that of the absorption, which indicates localisation of excitation in the relaxed excited state as observed in other excitonically coupled squaraine dimers recently.60
ECD and CPL spectroscopy
Electronic circular dichroism spectra were measured in toluene for all molecules but circular polarised luminescence was measured for the chromophore dimers only. For all dimers, the spectra are depicted in Fig. 4 and the data are summarised in Table 2. The monomer spectra and data can be found in the ESI in Fig. S6–S8.† While all R-configurated monomers display a negative ECD signal at the position of the lowest energy absorption band with Δε on the order of 10 M−1 cm−1, all dye dimers exhibit a much stronger ECD couplet signal. For the (P)-configurated dimers, the first exciton state comes along with a positive signal, the second one with a negative signal with increasing wavenumber, that is, a positive Cotton effect. The ECD spectra of the enantiomers, the (M)-configurated dimers, correspondingly show a negative Cotton effect. From the peak maxima of the ECD couplet one can estimate the exciton coupling strength which is 417 cm−1 for dSQA, 334 cm−1 for dSQB and 418 cm−1 for dMC. The Δε value of the first exciton state each is outstandingly high61 given the relatively small size of the chromophore dimers,41 with over 1400 M−1 cm−1 for the two squaraine dimers and ca. 1000 M−1 cm−1 for (P)-dMC.|
max/cm−1 |
Δε/M−1 cm−1 | R exp /D μB | |gabs| (max)/cgsb |
CPL/cm−1 |
|glum| (max)/cgsc |
|
|---|---|---|---|---|---|---|
| a Determined by eqn (S5). b g abs = Δε/ε. c g lum = ΔI/I. | ||||||
| (P)-dSQA | 14800 |
1422 | 11.59 | 3.5 × 10−3 | 14800 |
5.3 × 10−3 |
| 15900 |
−795 | −11.83 | 3.6 × 10−3 | |||
| (P)-dSQB | 13700 |
1447 | 11.71 | 4.7 × 10−3 | 13600 |
1.5 × 10−3 |
| 14300 |
−680 | −11.14 | 4.0 × 10−3 | |||
| (P)-dMC | 19700 |
1024 | 8.195 | 4.8 × 10−3 | 19600 |
3.8 × 10−3 |
| 20600 |
−342 | −8.400 | 2.7 × 10−3 | |||
One may ask why the Δε value of the merocyanine dimer is significantly smaller than those of the squaraine dimers despite the same geometrical arrangement of the chromophores (see Fig. 4). Two reasons may account for this observation: the first is the somewhat reduced dipole strength of the merocyanine and the second is the partial mutual cancelation of the negative and the positive ECD signal in the couplet because of the significantly broader bands. Assuming mirror-image relationship of the emission spectra and the unknown absorption profile of the S1 state, the band width of the latter was estimated from the emission spectra and is given in Table 1. Thus, in order to produce large Δε values both a high dipole strength and a small band width compared to the exciton coupling interaction is mandatory.62
The ECD signals of the second exciton states are about half the intensity of the first one, again, because of significant overlap with the vibronic progression of the lower exciton state. Nevertheless, the shift of the maxima of the ECD signals almost correspond to the absorption maxima. Thus, despite the high Δε values, the anisotropy factors gabs are only in the region of 10−3 cgs, which is comparable to literature known axially chiral dyes.46,63 However, in comparison to the corresponding monomers (R)-mSQA, (R)-mSQB, and (R)-mMC, which exhibit only small negative ECD signals with anisotropy factors of 10−5 to 10−4 cgs (ref. 23) (ESI, Fig. S4–S6†), the strong influence of the axial chirality becomes evident. The chiral axis leads to an intrinsic chirality of the chromophore, which leads to much higher ECD effects compared to a chromophore with only central chirality in close proximity.22 Following the exciton chirality method, the positive Cotton effects of all (P)-configurated molecules indicate a clockwise screw sense of the electronic transition dipole moments in the dimers. Since the electric transition dipole moment μ of the monomers are polarised along the long axis (see below), this confirms the predicted (P)-configuration of the atropoisomers, which is generated by the repulsion of the phenyl groups attached to the indolenines.
All dimers exhibit a significant CPL signal, which is positive for the (P)- and negative for the (M)-configurated dimers. For (P)-dSQB, the signal is at 13600 cm−1, which makes it one of only a handful literature known organic NIR CPL emitters.64–66 The glum values are 5.3 × 10−3 cgs for (P)-dSQA, 1.5 × 10−3 cgs for (P)-dSQB, and 3.8 × 10−3 cgs for (P)-dMC. They are therefore similar to glum obtained for axially chiral PDI or BODIPY dimers.46,63 The larger glum compared to gabs in dSQA is probably caused by the relatively bad signal-to-noise (SN) ratio of the CPL spectra. Self-absorption is a common problem when recording emission spectra,67,68 especially for the squaraine dyes, which show a small or even negligible Stokes shift. Although the CPL spectra were corrected for this self-absorption, they nevertheless show a weak SN ratio. On the other hand, the CPL spectrum of the (P)-dMC – which displays a somewhat larger Stokes shift – thus shows a much better resolution and a better agreement of glum and gabs.
Time-dependent density functional calculations
The structures of all chromophores were optimised at B3LYP/6-31G*. Using these structures, for all molecules time-dependent density functional theory (TD-DFT) calculations were performed at the BHandHLYP/6-31G* level of theory because this functional yields slightly more accurate absorption spectra for indolenine squaraines.57 The corresponding calculated optical spectra were shifted for each compound in order to agree with the experimental lowest energy transition. The ECD signal for the lowest-energy transition of the monomeric dyes (R)-mSQB and (R)-mMC are negative in agreement with the experiment (see ESI Fig. S12 and S13†). However, for (R)-mSQA we observed a small bisignate ECD signal. The TD-DFT calculations gave a small but positive ECD signal. However, the angle between the magnetic and electric transition dipole moment is close to 90°. Small changes of the angle thus can lead to a reversal of the sign. We assume that the bisignate experimental ECD signal indeed stems from vibronic coupling, which may change this angle (Fig. 5).69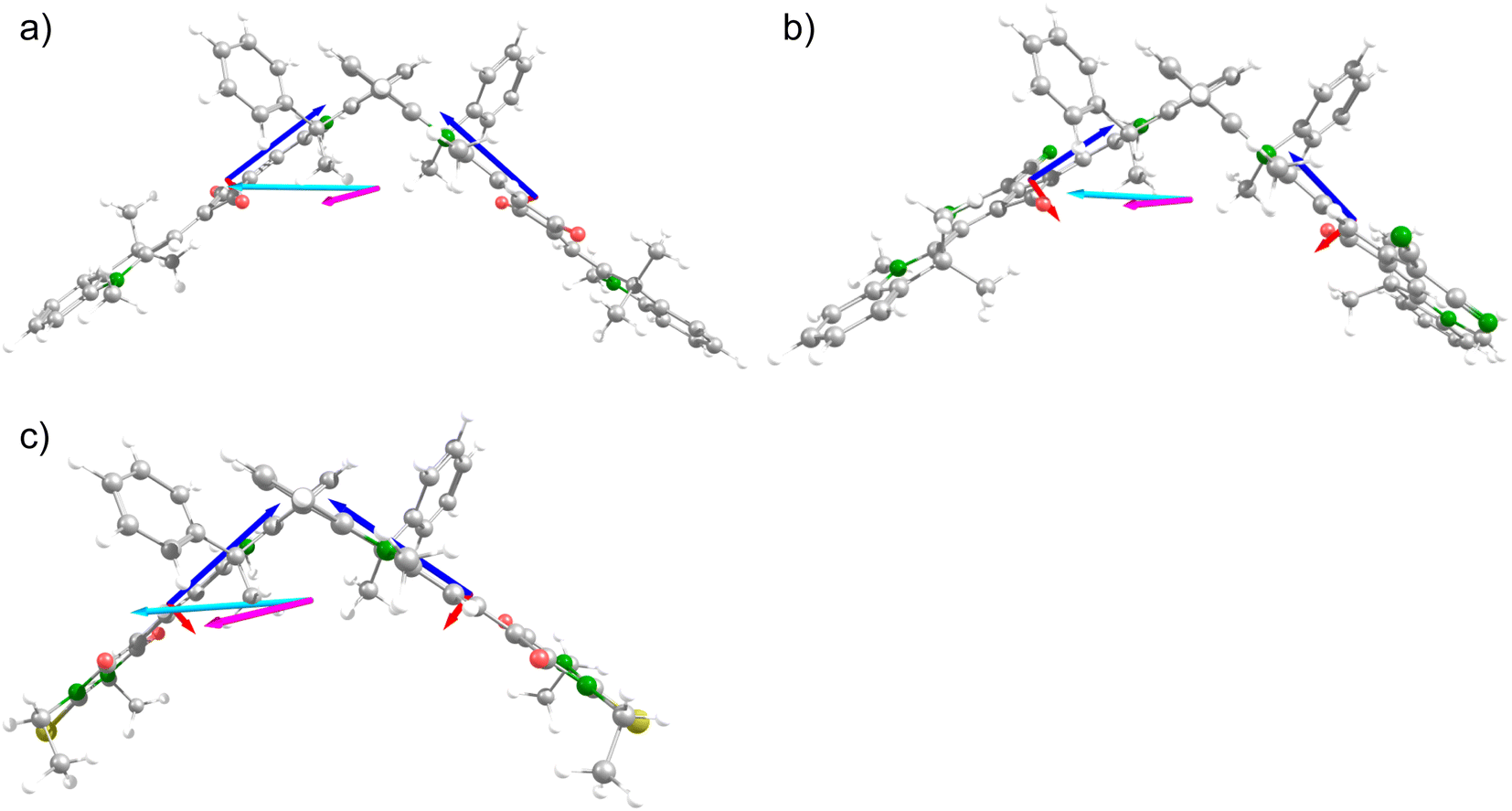 | ||
| Fig. 5 DFT calculated structures (B3LYP/6-31G*) with μ and m of monomer units (blue and red) and resulting μ and m of the dimer (light blue and pink) calculated with TD-DFT (BHandH/6-31G*) of (a) (P)-dSQA, (b) (P)-dSQB, and (c) (P)-dMC. | ||
For the chromophore dimers we calculated the rotational strengths Rtheo for the ECD couplet and the underlying electric and magnetic transition dipole moments μ and m, and the angle ϑ between them, which are collected in Table 3. As expected, both ECD signals of the couplet possess about the same rotational strength. Comparison of the calculated rotational strength with the experimental value (see Table 2) shows that the former is almost three times higher. However, we have to take into account that the experimental rotational strength is a lower limit because the positive and negative ECD signal of the couplet with its final bandwidth cancel each other to some extent. Likewise, when looking at the anisotropy factor gabs, the experimental values are around 1.5 × 10−3 to 5.3 × 10−3 cgs, whereas the calculated ones are between 4.8 × 10−3 cgs and 16 × 10−3 cgs. This overestimation may have three sources: (1) the calculated electric transition dipole moments are too high. For example, the electric transition moment of (R)-mSQA is 10.4 D while the computed one is 12.1 D. A similar difference is found for all chromophores in this work but it is not high enough to account for the enhancement of computed rotational strength. (2) The angle between the electric and magnetic transition moment is wrong. However, for the upper exciton state of the chromophore dimers, this angle is 180° by symmetry, but nevertheless, the calculated rotational strength is much too high for this state. (3) Thus, we are left with the possibility that the calculated magnetic transition moment is too high. In order to estimate the experimental magnetic transition dipole moment of the second exciton state of (P)-dSQA, we need the electric transition moment of this state. This, in turn, was estimated by subtracting the mirror image of the reduced fluorescence spectrum (as an estimate of the spectral distribution of the first exciton state) from the total exciton absorption spectrum. By area integration, this allowed us to estimate μS22 = 85.0 D2 and μS12 = 137.5 D2. Using eqn (1) with ϑ = 180° and RS2 = −11.83 D μB gave mS2 = 1.01 μB, which is half as the computed value (mS2 = 2.26 μB). Thus, it appears that our DFT computations systematically overestimate the magnetic transition moment.70
|
max
/cm−1 |
|μ|/D | |m|/μB | ϑ (μ,m)/Deg | R theo/D·μB | |gtheo|b/cgs | |
|---|---|---|---|---|---|---|
| a (P)-dSQA shifted by 4558 cm−1, (P)-dSQB shifted by 3378 cm−1, (P)-dMC (shifted by 6916 cm−1). b g theo = 4Rtheo/μ2. | ||||||
| (P)-dSQA | 14800 |
14.4 | 3.65 | 59.5 | 26.8 | 4.8 × 10−3 |
| 15900 |
10.5 | 2.26 | 180.0 | −23.6 | 8.0 × 10−3 | |
| (P)-dSQB | 13700 |
12.2 | 3.05 | 34.2 | 30.8 | 7.7 × 10−3 |
| 14400 |
8.02 | 3.48 | 180.0 | −27.9 | 1.6 × 10−2 | |
| (P)-dMC | 19700 |
11.1 | 4.22 | 60.3 | 23.1 | 7.0 × 10−3 |
| 20700 |
8.28 | 2.75 | 180.0 | −22.8 | 1.2 × 10−2 | |
In order to analyse the three contributions to the rotatory strength of eqn (3) we used the electric and magnetic transition dipole moments of the two respective monomer units μ1/μ2 and m1/m2 of the dimer (see ESI Tables S5–S10†), and calculated the angles ϑ1 between μ1 and m1 (=μ2,m2), ϑ2 between μ1 and m2 (=μ2,m1), α between μ1 and μ2 and β between r21 and (μ1 × μ2) Then, eqn (3) was used to calculate the contributing terms Rmon, Re–m und Rex to the overall rotational strength Rsum. As the lower exciton state results from an antisymmetric combination of the vectors in all molecules, the negative signs were used for Re–m and Rex (eqn (3)), and positive signs for the symmetric second exciton state. These values are summarised in Table 4.
|
max/cm−1 |
R mon/D μB | ϑ 1 (μ1,m1)/deg | R e–m/D μB | ϑ 2 (μ1,m2)/deg | R ex/D μB | α (μ1,μ2)/deg | β (r21, (μ1 × μ2))/deg | R sum/D μB | |
|---|---|---|---|---|---|---|---|---|---|
| (P)-dSQA | 14800 |
0.77 | 95.2 | 4.49 | 58.0 | 19.1 | 102.0 | 110.6 | 24.4 |
| 15900 |
0.77 | 95.2 | −4.49 | 58.0 | −19.1 | 102.0 | 110.6 | −22.8 | |
| (P)-dSQB | 13700 |
−0.32 | 88.6 | 11.6 | 30.0 | 15.9 | 106.7 | 122.5 | 26.9 |
| 14400 |
−0.32 | 88.6 | −11.6 | 30.0 | −15.9 | 106.7 | 122.5 | −27.6 | |
| (P)-dMC | 19700 |
−0.11 | 89.3 | 8.91 | 18.1 | 12.3 | 104.4 | 109.8 | 22.0 |
| 20700 |
−0.11 | 89.3 | −8.91 | 18.1 | −12.3 | 104.4 | 109.8 | −22.3 |
Generally, the agreement of Rsum and Rtheo is excellent for all dimer chromophores, highlighting the correctness of exciton coupling theory in the present cases. Analysing the three contributions to the rotatory strength, it is obvious that in all cases Rmon does not play a significant role, which is caused by the almost orthogonal orientation of μ and m. Therefore, the possibly wrong sign of Rmon (see above the discussion for (R)-mSQA) of (P)-dSQA does not have any influence. In all cases, the exciton term Rex is dominating the rotational strength. This is due to the large β(r21, (μ1 × μ2) angle, which is between 110–120° (see Table 4). However, the Re–m term is significant AND possesses the same sign as Rex. This means that both terms add to the total rotational strength, which, thus, is enhanced compared to what was expected by the pure exciton chirality method. This is a consequence of the magnetic transition moments, which are almost perpendicular to the electric transition moment within one monomer. While this leads to an almost vanishing Rmon term it is ideal for the Re–m term provided that the ϑ2 (μ1,m2) angle strongly deviates from orthogonality as is in the present cases. For an axially chiral BODIPY dimer, Bruhn et al. also observed that Rmon + Re–m contributes significantly to the rotatory strength, but in another case the sum of the two terms Rmon and Re–m were even larger than Rex but possessed an opposite sign, which led to a reduction of total rotational strength and an unusual positive Cotton effect for the M-enantiomer.46
The good agreement of Rtheo and Rsum shows that the partitioning of R into the three components appears to be internally consistent for the DFT calculated rotatory strength. However, one should bear in mind that the DFT calculations overestimate m, which leads to too high Rmon and Re–m values, while Rex is independent of m.
Conclusions
An atropo-diastereoselective synthetic route to axially chiral indolenine derived chromophore dimers was worked out and used to synthesise (P)-dSQA and (P)-dSQB as well as an axially chiral merocyanine (P)-dMC. Thus, two C(3)-chiral phenyl-oxindoles were coupled by a Yamamoto-type reaction to give a dimer, where steric interactions of the attached phenyl groups led to a thermodynamically preferred conformational arrangement of the monomers with respect to the formed biaryl axis. The stereochemical configuration was retained in the axially chiral squaraine dimers that resulted from condensation of the oxindole dimer with two semisquaraines. The same oxindole dimer was also used for the synthesis of the merocyanine dimer (P)-dMC and is a potential precursor for other axially chiral dye dimers.In the linear absorption spectra characteristic strong absorption bands were observed for the dimeric squaraine and merocyanine dyes, which, in the case of dSQA possess a pronounced excitonic splitting. The chiroptical properties show significant ECD effects in the region of the main absorption bands with a positive Cotton effect, highlighting the excitonic coupling in all cases. The (P)-configurated compounds exhibit a positive first and a negative second exciton state, and therefore a clockwise screw sense of the electric transition dipole moments according to the exciton chirality method. The ECD spectra of the (M)-configurated dimers are mirror images thereof. Relative to the molecular size, extremely high Δε values of up to 1447 M−1 cm−1 for (P)-dSQB were achieved for the squaraine dimers which is a consequence of the high dipole strength and the small absorption bandwidth in comparison to the excitonic splitting. However, because of the almost exact match of ECD/CPL peak position with absorption/fluorescence signals, this leads to gabs around 10−3 cgs for all dimers, which is typical of chiral biaryls. From the recorded CPL spectra, glum in the same magnitude were determined. TD-DFT calculations of the rotational strengths and the derived gtheo are internally consistent but too high compared to the experimental values, which is caused by a computational overestimation of the magnetic transition moments. Using the Rosenfeld equation, the terms Rmon, Re–m and Rex, which contribute to the total rotational strength, were calculated for (P)-dSQA, (P)-dSQB and (P)-dMC from the electric and magnetic transition dipole moment of the monomeric subunits. As expected for axially chiral compounds, the exciton term Rex is the largest for all dimers. However, the mixed term Re–m possess the same sign and also adds significantly to the total rotational strength, which is due to a favourable arrangement of the monomeric subunits with respect to each other. This provides a strategy to enhance chiroptical properties of chromophore dimers beyond the exciton chirality interactions.
Data availability
All necessary information is included in the ESI.†Author contributions
EF performed all experimental work and data analysis and wrote the manuscript. MH performed the DFT calculations. AS and MS performed the CPL measurements. GB, FW and CL supervised the project and corrected the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (DFG) for financial support of this work (La991/21-1 and CPL/CD hybrid spectrometer Projektnummer 444286426).References
- S. Sreejith, P. Carol, P. Chithra and A. Ajayaghosh, J. Mater. Chem., 2008, 18, 264–274 RSC
.
- S. F. Völker and C. Lambert, Chem. Mater., 2012, 24, 2541–2553 CrossRef
- U. Mayerhöffer, M. Gsänger, M. Stolte, B. Fimmel and F. Würthner, Chem.–Eur. J., 2013, 19, 218–232 (
Chem.–Eur. J.
, 2021
, 27
, 17970–17971
) CrossRef PubMed
- L. Beverina and P. Salice, Eur. J. Org. Chem., 2010, 1207–1225 CrossRef CAS
- G. Chen, H. Sasabe, T. Igarashi, Z. Hong and J. Kido, J. Mater. Chem. A, 2015, 3, 14517–14534 RSC
- Q. Xiao, J. Tian, Q. Xue, J. Wang, B. Xiong, M. Han, Z. Li, Z. Zhu, H.-L. Yip and Z. a. Li, Angew. Chem., Int. Ed., 2019, 58, 17724–17730 CrossRef CAS PubMed
- B. Stender, S. F. Völker, C. Lambert and J. Pflaum, Adv. Mater., 2013, 25, 2943–2947 CrossRef CAS PubMed
- J. He, Y. J. Jo, X. Sun, W. Qiao, J. Ok, T.-i. Kim and Z. a. Li, Adv. Funct. Mater., 2021, 31, 2008201 CrossRef CAS
- J. H. Kim, A. Liess, M. Stolte, A.-M. Krause, V. Stepanenko, C. Zhong, D. Bialas, F. Spano and F. Würthner, Adv. Mater., 2021, 33, 2100582 CrossRef CAS PubMed
- K. Strassel, W.-H. Hu, S. Osbild, D. Padula, D. Rentsch, S. Yakunin, Y. Shynkarenko, M. Kovalenko, F. Nüesch, R. Hany and M. Bauer, Sci. Technol. Adv. Mater., 2021, 22, 194–204 CrossRef CAS PubMed
- S. Sreejith, X. Ma and Y. Zhao, J. Am. Chem. Soc., 2012, 134, 17346–17349 CrossRef CAS PubMed
- M. S. Soumya, K. M. Shafeekh, S. Das and A. Abraham, Chem.-Biol. Interact., 2014, 222, 44–49 CrossRef CAS PubMed
- S. Friães, A. M. Silva, R. E. Boto, D. Ferreira, J. R. Fernandes, E. B. Souto, P. Almeida, L. F. V. Ferreira and L. V. Reis, Bioorg. Med. Chem., 2017, 25, 3803–3814 CrossRef PubMed
- J. R. Johnson, N. Fu, E. Arunkumar, W. M. Leevy, S. T. Gammon, D. Piwnica-Worms and B. D. Smith, Angew. Chem., Int. Ed., 2007, 46, 5528–5531 CrossRef CAS PubMed
- V. Grande, F. Doria, M. Freccero and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 7520–7524 CrossRef CAS PubMed
- L. Yuan, W. Lin, K. Zheng, L. He and W. Huang, Chem. Soc. Rev., 2013, 42, 622–661 RSC
- C. Geiger, M. Stanescu, L. Chen and D. G. Whitten, Langmuir, 1999, 15, 2241–2245 CrossRef CAS
- K. Jyothish, M. Hariharan and D. Ramaiah, Chem.–Eur. J., 2007, 13, 5944–5951 CrossRef CAS PubMed
- R. S. Stoll, N. Severin, J. P. Rabe and S. Hecht, Adv. Mater., 2006, 18, 1271–1275 CrossRef CAS
- P. Chithra, R. Varghese, K. P. Divya and A. Ajayaghosh, Chem.–Asian J., 2008, 3, 1365–1373 CrossRef CAS PubMed
- F. Balzer, M. Schumacher, S. Mattiello, M. Schulz, J. Zablocki, M. Schmidtmann, K. Meerholz, N. Sariciftci, L. Beverina, A. Lützen and M. Schiek, Isr. J. Chem., 2022, 62, e2021000 CrossRef
- G. Snatzke, Angew. Chem., 1979, 91, 380–393 CrossRef CAS
- J. Selby, M. Holzapfel, B. K. Lombe, D. Schmidt, A.-M. Krause, F. Würthner, G. Bringmann and C. Lambert, J. Org. Chem., 2020, 85, 12227–12242 CrossRef CAS PubMed
-
W. W. Parson, Modern Optical Spectroscopy, Springer-Verlag Berlin Heidelberg, Heidelberg, 2009 Search PubMed
- J. A. Schellman, Chem. Rev., 1975, 75, 323–331 CrossRef CAS
-
N. Berova, P. L. Polavarapu, K. Nakanishi and R. W. Woody, Comprehensive Chiroptical Spectroscopy, John Wiley & Sons, Inc., New Jersey, 2012 Search PubMed
- E. M. Sánchez-Carnerero, F. Moreno, B. L. Maroto, A. R. Agarrabeitia, M. J. Ortiz, B. G. Vo, G. Muller and S. d. l. Moya, J. Am. Chem. Soc., 2014, 136, 3346–3349 CrossRef PubMed
- Z. Liu, Z. Jiang, C. He, Y. Chen and Z. Guo, Dyes Pigm., 2020, 181, 108593 CrossRef CAS
- Y. Wu, S. Wang, Z. Li, Z. Shen and H. Lu, J. Mater. Chem. C, 2016, 4, 4668–4674 RSC
- Y. Wu, X. Mao, X. Ma, X. Huang, Y. Cheng and C. Zhu, Macromol. Chem. Phys., 2012, 213, 2238–2245 CrossRef CAS
- N. Kobayashi, Chem. Commun., 1998, 487–488 RSC
- M. Á. Revuelta-Maza, T. Torres and G. d. l. Torre, Org. Lett., 2019, 21, 8183–8186 CrossRef CAS PubMed
- A. Sharma, S. Athanasopoulos, E. Kumarasamy, C. Phansa, A. Asadpoordarvish, R. P. Sabatini, R. Pandya, K. R. Parenti, S. N. Sanders, D. R. McCamey, L. M. Campos, A. Rao, M. J. Y. Tayebjee and G. Lakhwani, J. Phys. Chem. A, 2021, 125, 7226–7234 CrossRef CAS PubMed
- K. Sato, M. Hasegawa, Y. Nojima, N. Hara, T. Nishiuchi, Y. Imai and Y. Mazaki, Chem.–Eur. J., 2021, 27, 1323–1329 CrossRef CAS PubMed
- Z. Jiang, T. Gao, H. Liu, M. S. S. Shaibani and Z. Liu, Dyes Pigm., 2020, 175, 108168 CrossRef CAS
- L.-M. Jin, Y. Li, J. Ma and Q. Li, Org. Lett., 2010, 12, 3552–3555 CrossRef CAS PubMed
- C. Coluccini, M. Caricato, E. Cariati, S. Righetto, A. Forni and D. Pasini, RSC Adv., 2015, 5, 21495–21503 RSC
- C. Kaufmann, D. Bialas, M. Stolte and F. Würthner, J. Am. Chem. Soc., 2018, 140, 9986–9995 CrossRef CAS PubMed
- J. Kumar, T. Nakashima, H. Tsumatori and T. Kawai, J. Phys. Chem. Lett., 2014, 5, 316–321 CrossRef CAS PubMed
- K. Takaishi, S. Murakami, K. Iwachido and T. Ema, Chem. Sci., 2021, 12, 14570–14576 RSC
- S. Kolemen, Y. Cakmak, Z. Kostereli and E. U. Akkaya, Org. Lett., 2014, 16, 660–663 CrossRef CAS PubMed
- K. Takaishi, K. Iwachido, R. Takehana, M. Uchiyama and T. Ema, J. Am. Chem. Soc., 2019, 141, 6185–6190 CrossRef CAS PubMed
- S.-P. Wan, H.-Y. Lu, M. Li and C.-F. Chen, J. Photochem. Photobiol., C, 2022, 50, 100500 CrossRef CAS
- G. Bringmann, A. J. P. Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384–5427 CrossRef CAS PubMed
- G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed
- T. Bruhn, G. Pescitelli, S. Jurinovich, A. Schaumlöffel, F. Witterauf, J. Ahrens, M. Bröring and G. Bringmann, Angew. Chem., Int. Ed., 2014, 53, 14592–14595 CrossRef CAS PubMed
- F. Zinna, T. Bruhn, C. A. Guido, J. Ahrens, M. Bröring, L. Di Bari and G. Pescitelli, Chem.–Eur. J., 2016, 22, 16089–16098 CrossRef CAS PubMed
- G. Bringmann, D. C. G. Götz, T. A. M. Gulder, T. H. Gehrke, T. Bruhn, T. Kupfer, K. Radacki, H. Braunschweig, A. Heckmann and C. Lambert, J. Am. Chem. Soc., 2008, 130, 17812–17825 CrossRef CAS PubMed
- E. Richmond, K. B. Ling, N. Duguet, L. B. Manton, N. Celebi-Olcum, Y.-H. Lam, S. Alsancak, A. M. Z. Slawin, K. N. Houk and A. D. Smith, Org. Biomol. Chem., 2015, 13, 1807–1817 RSC
- D. V. Pestov, V. I. Slesarev, A. I. Ginak and V. I. Slesareva, Chem. Heterocycl. Compd., 1988, 24, 782–786 CrossRef
- A. J. Kay, A. D. Woolhouse, G. J. Gainsford, T. G. Haskell, T. H. Barnes, I. T. McKinnie and C. P. Wyss, J. Mater. Chem., 2001, 11, 996–1002 RSC
- J. Li, R. Seupel, D. Feineis, V. Mudogo, M. Kaiser, R. Brun, D. Brünnert, M. Chatterjee, E.-J. Seo, T. Efferth and G. Bringmann, J. Nat. Prod., 2017, 80, 443–458 CrossRef CAS PubMed
- We also calculated the three structures using a dispersion corrected function at wB97xD/6-31G* level of theory. While the structures change only little, the closed structure is stabilized and is only 979 cm−1 higher in energy than the open structure. The transition state is 6428 cm−1 higher in energy than the open structure. However, dispersion interactions favor the closed structure artificially and would likely be compensated by better solvation of the open structure.
- A. Kulinich, N. Derevyanko and A. Ishchenko, Russ. J. Gen. Chem., 2006, 76, 1441–1457 CrossRef CAS
- S. Beckmann, K.-H. Etzbach, P. Krämer, K. Lukaszuk, R. Matschiner, A. J. Schmidt, P. Schuhmacher, R. Sens, G. Seybold, R. Wortmann and F. Würthner, Adv. Mater., 1999, 11, 536–541 CrossRef CAS
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CAS
- E. Michail, M. H. Schreck, M. Holzapfel and C. Lambert, Phys. Chem. Chem. Phys., 2020, 22, 18340–18350 RSC
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14–19 RSC
- G. Angulo, G. Grampp and A. Rosspeintner, Spectrochim. Acta, Part A, 2006, 65, 727–731 CrossRef PubMed
- H. Marciniak, N. Auerhammer, S. Ricker, A. Schmiedel, M. Holzapfel and C. Lambert, J. Phys. Chem. C, 2019, 123, 3426–3432 CrossRef CAS
- N. J. Schuster, L. A. Joyce, D. W. Paley, F. Ng, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2020, 142, 7066–7074 CrossRef CAS PubMed
- N. Harada, S.-M. L. Chen and K. Nakanishi, J. Am. Chem. Soc., 1975, 97, 5345–5352 CrossRef CAS
- H. Langhals, A. Hofer, S. Bernhard, J. S. Siegel and P. Mayer, J. Org. Chem., 2011, 76, 990–992 CrossRef CAS PubMed
- J. Feng, L. Fu, H. Geng, W. Jiang and Z. Wang, Chem. Commun., 2020, 56, 912–915 RSC
- T. Gao, Z. Jiang, B. Chen, Q. Sun, Y. Orooji, L. Huang and Z. Liu, Dyes Pigm., 2020, 173, 107998 CrossRef CAS
- K. Miki, T. Noda, M. Gon, K. Tanaka, Y. Chujo, Y. Mizuhata, N. Tokitoh and K. Ohe, Chem.–Eur. J., 2019, 25, 9211–9216 CrossRef CAS PubMed
- P. R. Hammond, J. Chem. Phys., 1979, 70, 3884–3894 CrossRef CAS
- T.-S. Ahn, R. O. Al-Kaysi, A. M. Müller, K. M. Wentz and C. J. Bardeen, Rev. Sci. Instrum., 2007, 78, 086105 CrossRef PubMed
- In a recent paper ( J. Selby,
et al.
, J. Org. Chem., 2020, 85, 12227–12242 CrossRef CAS PubMed
- I. Warnke and F. Furche, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 150–166 CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc04600h |
| This journal is © The Royal Society of Chemistry 2022 |