
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence[2 + 2] Cycloaddition of phosphaalkenes as a key step for the reductive coupling of diaryl ketones to tetraaryl olefins†
Anna I.
Arkhypchuk
 *,
Nicolas
D'Imperio
,
Jordann A. L.
Wells
and
Sascha
Ott
*
*,
Nicolas
D'Imperio
,
Jordann A. L.
Wells
and
Sascha
Ott
*
Department of Chemistry, Ångström Laboratory, Uppsala University, Box 523, 75120, Uppsala, Sweden. E-mail: Sascha.Ott@kemi.uu.se; Anna.Arkhypchuk@kemi.uu.se
First published on 29th September 2022
Abstract
Procedures for the reductive coupling of carbonyl compounds to alkenes in the literature rely either on a radical coupling strategy, as in the McMurry coupling, or ionic pathways, sometimes catalysed by transition metals, as in more contemporary contributions. Herein, we present the first example of a third strategy that is based on the [2 + 2] cycloaddition of ketone-derived phosphaalkenes. Removal of P-trimethylsilyl groups at the intermediary 1,2-diphosphetane dimer results in its collapse and concomitant release of the tetraaryl-substituted alkene. In fact, the presented strategy is the only alternative to the McMurry coupling in the literature that allows tetraaryl alkene formation from diaryl ketones, with yields as high as 85%. The power of the methodology is illustrated in the reaction of tethered bis-benzophenones which engage in intramolecular reductive carbonyl couplings to form unusual macrocycles without the need for high dilution conditions or templating.
Introduction
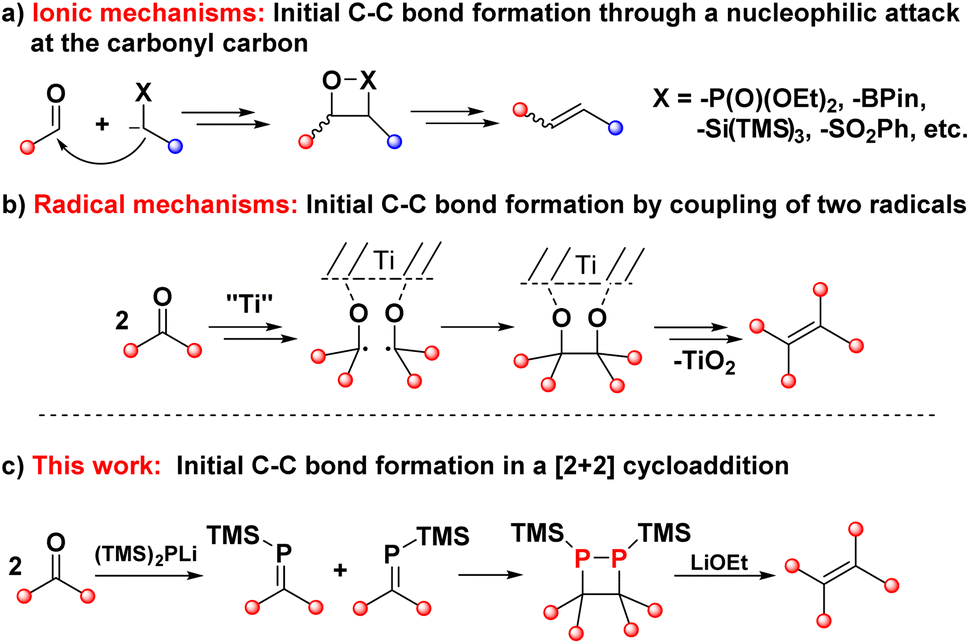
The conversion of carbonyl compounds into olefins has a long history in organic chemistry.1–4 The methodologies that have been developed for this transformation can broadly be divided into two categories: those in which the initial C–C single bond formation proceeds by an ionic mechanism, and those in which this step follows a radical pathway (Fig. 1). Protocols that belong to the first group feature the attack of a carbon-based nucleophile at a carbonyl group to form the C–C σ-bonds (Fig. 1a), followed by an elimination step to establish the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. This category includes all of the classic carbonyl olefinations such as the Wittig,5–8 Horner–Wadsworth–Emmons (HWE)9 or Peterson reactions10,11 as well as more contemporary approaches based on main group element reagents,12–16 or catalysed by transition metals.17
C bond. This category includes all of the classic carbonyl olefinations such as the Wittig,5–8 Horner–Wadsworth–Emmons (HWE)9 or Peterson reactions10,11 as well as more contemporary approaches based on main group element reagents,12–16 or catalysed by transition metals.17
 | ||
| Fig. 1 Current state of the art in olefination reactions where at least one of the substrates is a carbonyl compound. | ||
With the exception of sporadic examples in the Peterson olefination, the nucleophilic attack pathway puts a limit on the substrate scope of these reactions, and tetra-substituted olefins remain largely elusive when using classic olefination protocols. The difficulty arises from the steric congestion in the reaction of ketones with olefination reagents that feature two substituents at the nucleophilic center.
Consequently, access to sterically crowded tetra-substituted olefins from carbonyl compounds is mechanistically vastly different from the above examples. That methodology, known as the McMurry coupling, employs two ketones that are reduced to ketyl radicals and subsequently couple to form the C–C σ-bond in pinacolate intermediates (Fig. 1b).18–21 Further reduction events and deoxygenation give rise to the corresponding olefins. While the McMurry coupling has its place in organic chemistry, it is also described in a textbook as a “tricky” reaction.22 The highly reducing radical nature of the reaction precludes substrates with functional groups that are incompatible with such conditions. There is thus a clear need for mechanistically different and easily applied procedures that can accommodate high steric demand.
At the heart of the problem is the question of how to create an initial C–C σ-bond between two carbon centers with the same polarity and low reactivity. In the McMurry coupling, this problem is resolved by the radical coupling mechanism, while the vast majority of other methods, be they classic or more recent olefination strategies, fail in this respect. A reaction type that has been overlooked in this context is the [2 + 2] cycloaddition reaction. While dimerisation will not occur between ketones for obvious reasons, the dimerisation of phosphaalkenes — the phosphorus-congeners of alkenes — has some precedence in the literature.22–24 Phosphalkenes, in turn, can easily be produced from ketones in multiple ways, presenting opportunities for new reductive ketone coupling pathways.25–29
Results and discussion
The [2 + 2] dimerisation of triphenylphosphaalkene was first reported by Becker in 1981, although the product was falsely assigned as the 1,3-diphosphetane.24 In 2016, Gates and co-workers crystallised the dimerisation product to prove unambiguously that dimerisation proceeds in a head-to-head fashion, giving rise to hexaphenyl-1,2-diphosphetane.30 In both reports, traces of tetraphenylethene were observed either after pyrolysis of the diphosphetane in the former or as side product of unknown origin in the latter report. While hexaphenyl-1,2-diphosphetane is a stable compound under inert atmosphere, the dimerisation of the parent phosphaalkene has produced the initial C–C σ-bond desired for alkene formation, raising the tantalising question of whether 1,2-diphosphetanes with different P-substituents could be collapsed to form tetra-substituted alkenes. We hypothesised that this reactivity could be promoted by the on-demand cleavage of P-substituents, i.e. after the formation of the 1,2-diphosphetane.The choice of a suitable P-substituent fell on the trimethylsilyl (TMS) group, as it has previously been shown that it can be cleaved smoothly from phosphorus centers using LiOEt.14,31 Hence, the preparation of P,P-bis(trimethylsilyl)-1,2-diphosphetane 4a from two equivalents of benzophenone was explored (Scheme 1). Lithium bis(trimethylsilyl)phosphide 2 can be conveniently prepared from commercially available tris(trimethylsilyl)phosphine by treatment with a LiOEt solution in THF. Prepared in this way, the lithium phosphide 2 can be isolated as an off-white powder which contains 0.5 molecules of THF per phosphorus centre and can be stored in a glove box for several months with no change in activity. Treatment of ethereal solutions of benzophenone 1a with one equivalent of 2 results in the formation of phosphaalkene 3a (δ(31P, in Et2O) = +289.1 ppm) as a single product. As expected for a phosphaalkene with little kinetic stabilisation, the corresponding dimer 4a starts to form immediately, as evidenced by the emergence of a new signal in the 31P NMR spectrum (δ(31P, in Et2O) = −51 ppm). Layering of the reaction mixture with pentane allows the isolation of 1,2-diphosphetane 4a, yielding single crystals suitable for X-ray diffraction analysis (Fig. 2). Most importantly for the overall alkene formation pathway, single-crystal diffraction experiments on 4a confirm that dimerisation occurs in a head-to-head fashion. The asymmetric unit consists of one and a half molecules of 4a, with one near-symmetric molecule and another symmetric molecule of 4a organised along a crystallographic axis. In both, the cyclic P2C2 core is not planar, with an average torsion angle of 26.0°. The two P-TMS groups reside above and below the diphosphetane ring plane. The C–C, C–P, and P–P bond distances of 1.628(4), 1.938(3), and 2.202(1) Å, respectively, are comparable to those found in previously reported examples.30,32
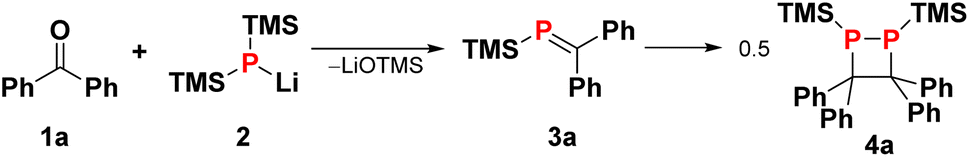 | ||
| Scheme 1 Reaction of lithium bis(trimethylsilyl)phosphide 2 with benzophenone 1a to form phosphaalkene intermediate 3a that dimerises to 1,2-diphosphetane 4a. | ||
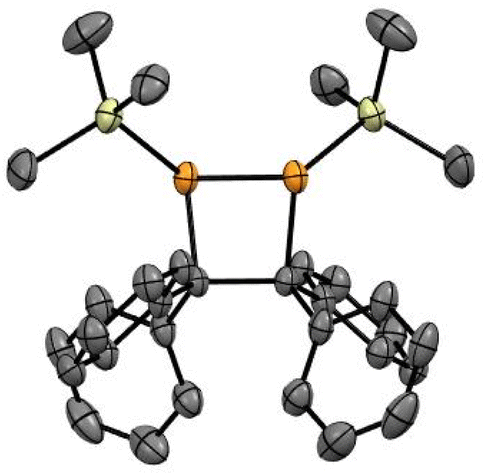 | ||
| Fig. 2 Solid-state structure of 4a. Selected bond lengths (Å) and angles (°): P1–P2: 2.202 (1); C1–C2: 1.628 (4); P1–C1: 1.936 (3); P2–C2: 1.972 (3); C1–C2–P2: 95.3 (2); C1–P1–P2: 79.20 (9); P1–C1–C2–P2: 26.0 (2). | ||
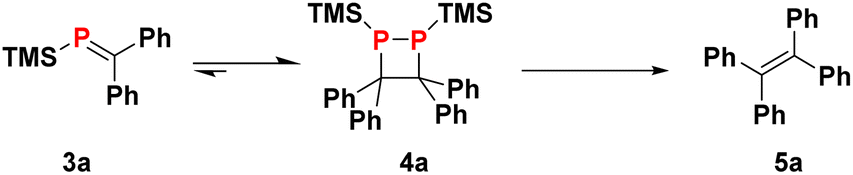
Finding optimal conditions for the collapse of 1,2-diphosphetane 4a to release the corresponding alkene product is complicated by the chemical equilibrium between 4a and phosphaalkene 3a, which has implications for the overall reaction yield. This equilibrium can be monitored by 31P NMR by dissolving crystalline 4a in d8-THF which results in the emergence of a signal for monomeric 3a (see ESI† for details). The equilibrium requires several hours to establish at room temperature, and the dimer is the thermodynamic product. Together, these observations underline that time is a crucial parameter to be taken into consideration when optimising the protocol.
As suggested by previous work,15 THF solutions of LiOEt may be able to selectively desilylate 4a, and indeed, addition of LiOEt to 4a solutions induced diphosphetane collapse to provide tetraphenyl-ethylene 5a in 71% isolated yield.
With the formation of the tetrasubstituted olefin product 5a from crystalline 4a confirmed, we set out to optimise the conditions in order to obtain 5a directly from 2 and benzophenone 1a without the need to isolate the diphosphetane intermediate. As discussed above, the yield of alkene formation will most likely depend on the time that the intermediate phosphaalkene 3a is given to dimerise. Indeed, addition of LiOEt three hours after reaction initiation resulted in a moderate alkene formation yield of 47% (Table 1, entry 1). Allowing more time (24 hours) for the equilibrium to reach maximum 4a concentrations before LiOEt addition resulted in a remarkable 85% isolated yield of 5a, accompanied by 9% unreacted 1a. Longer time periods (up to 3 days) resulted in somewhat decreased yields (65%), presumably due to unwanted decomposition pathways.
|
|
|||
|---|---|---|---|
| Dimerisation time | Conditions for conversion of 4a to 5a | Yield | |
| 1 | 3 h | LiOEt in THF | 47% |
| 2 | 24 h | LiOEt in THF | 85% |
| 3 | 3 days | LiOEt in THF | 64% |
| 4 | 24 h | THF | ≤5% |
The 85% yield for alkene formation from benzophenone 1a is higher than that obtained from isolated 4a, which can be attributed to the different solvent compositions in the two experiments. In the former case, the main solvent is diethyl ether which is only slightly diluted with THF during LiOEt addition, while the latter is conducted entirely in THF. It thus seems that LiOEt is more selective as a desilylation agent in the less polar Et2O/THF mixture, consistent with previous observations in the group.15
Further information on the reactivity of the system was obtained by following the reductive coupling of 4,4′-difluorobenzophenone 1b by 31P and 19F-NMR spectroscopy (Fig. 3). From the 31P-NMR spectra in Fig. 3a (see ESI† for full spectra), it is apparent that the lithium phosphide starting material 2 (δ = −303.8) is consumed after 10 minutes, giving rise to two new signals corresponding to the phosphaalkene (δ = +291.8 ppm) and its dimer (δ = −50.6 ppm) with a combined (NMR) yield of 81%. After 60 minutes, the relative integrals for the signals indicate a yield increase to 87% (64% dimer, 23% phosphaalkene). After 6 hours, only traces of phosphaalkene are present, and the desired 1,2-diphosphetane product is the dominant species.
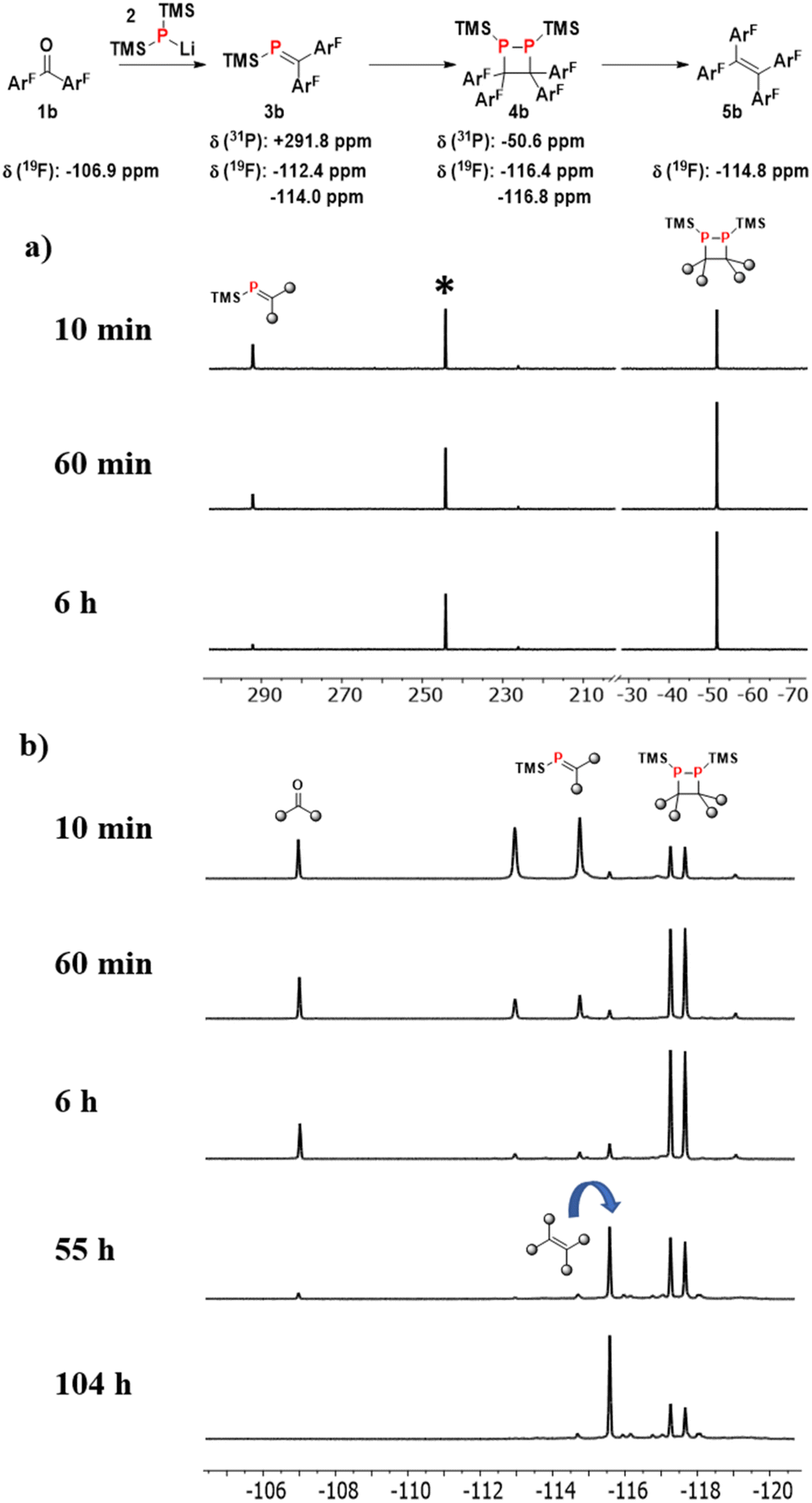 | ||
| Fig. 3 31P (a) and 19F (b) NMR spectroscopic monitoring of the reaction between 4,4′-difluorobenzophenone and LiP(TMS)2 (2). The signal marked with a “*” at 244.5 ppm corresponds to (E)-(4-methoxybenzylidene)(2,4,6-tri-tert-butylphenyl)phosphane which is added as a standard for quantification. | ||
The fluoro-groups in 1b also allow monitoring of the ketone starting material. Somewhat unexpectedly, circa 10% of the starting ketone (δ = −106.9 ppm) is still present in the reaction mixture at the point when all phosphide 2 is consumed (after 10 minutes). This is despite of the fact that the ketone and 2 were added to the reaction in equimolar amounts. We ascribe this observation to the presence of relatively stable clusters of TMS2PLi that are formed during the synthesis of 2. Different molecular compositions of these clusters, their low concentrations as well as multiple NMR P-coupling patterns explain that they escape detection by 31P-NMR spectroscopy. Clusters with different ratios of phosphorus and lithium do have precedence in the literature.33 The reactivity of these clusters is greatly reduced compared to that of 2, but sufficient to react with the ketone on the timescale of days, leading to complete consumption of the latter after circa 2.5 days. Nevertheless, this observation explains the recovery of 9% of 2a alongside alkene product 5a, as described above.
Another unexpected observation is that traces of the final alkene product (δ = −114.8 ppm) can be observed within the first 10 minutes. We do not believe that the alkene is formed by a different pathway, but that the LiOTMS by-product from phosphaalkene formation (Scheme 1) can also act as a desilylation reagent, albeit at a slower rate compared to LiOEt. This is consistent with the observation that alkene concentration increases with time over the course of reaction monitoring, even in the absence of LiOEt. In contrast, solutions of purified 1,2-diphosphetate do not yield alkene product, but do re-establish the equilibrium with the phosphaalkene. In summary, these NMR studies confirm that the concentrations of product and diphosphetane are at their maximum after 24 hours, which is thus the optimal time for LiOEt addition.
The reductive coupling protocol was tested on a variety of diarylketones (Schemes 2 and 3). Both electron-deficient and -rich benzophenones, as well as fluorenone, produce the corresponding alkenes in good yields ranging from 50–85%. Unsurprisingly, acetophenones are not compatible with the protocol, as their acidic α-protons quench the lithium phosphide 2.
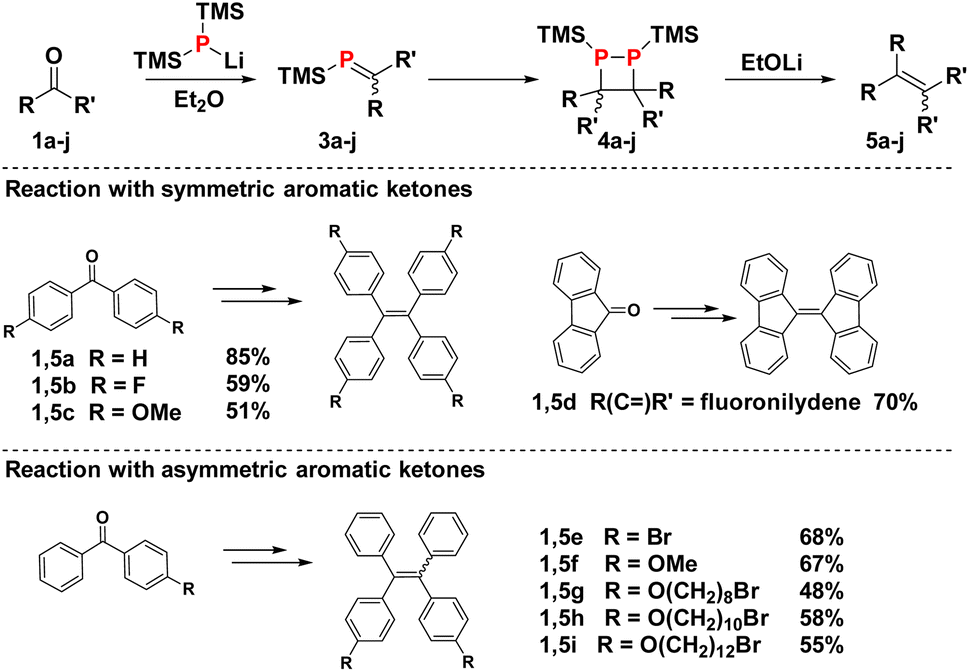 | ||
| Scheme 2 Reaction scope using substituted benzophenones and fluorenone. | ||
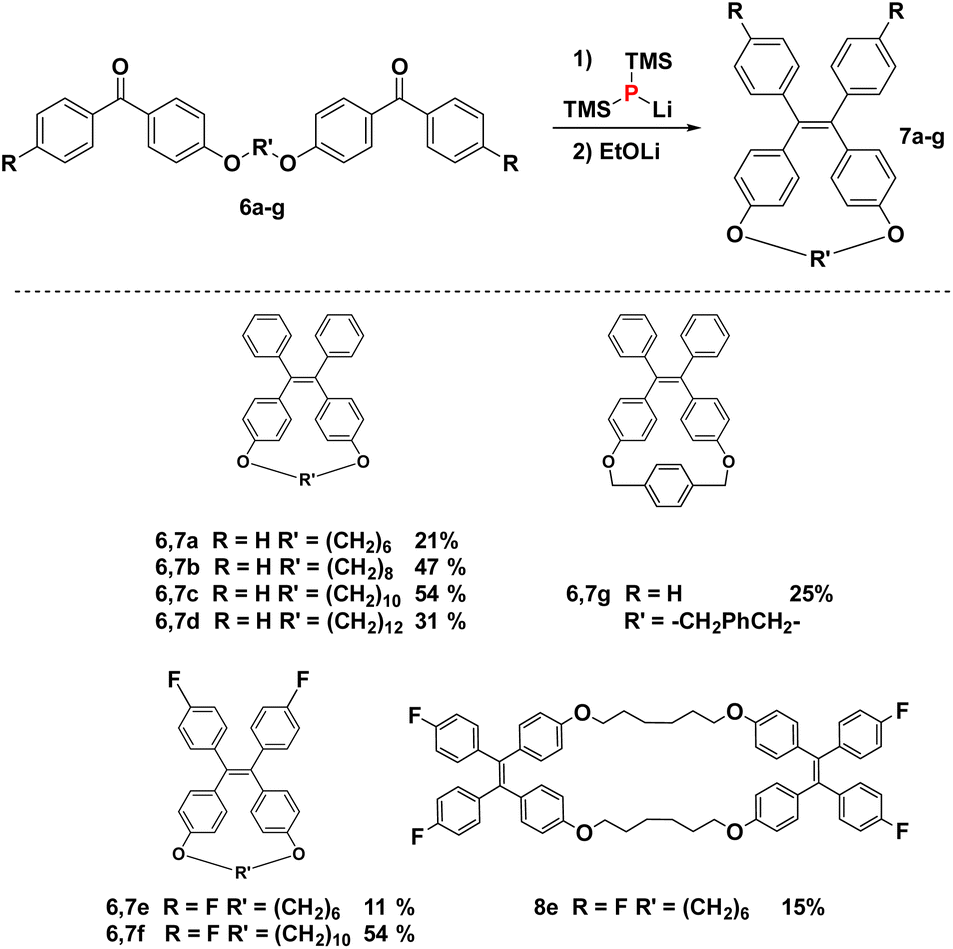 | ||
| Scheme 3 Macrocyclisations by the reaction of tethered bis-benzophenones. | ||
Small differences in reactivity can be observed within the family of substituted benzophenones. In the presence of electron donating OMe groups as in 1c, the equilibrium between 3 and 4 tend to lie further on the side of the monomeric phosphaalkenes, leading to somewhat lower alkene yields. On the other hand, phosphaalkenes with electron withdrawing fluoride groups as in 1b dimerise faster but are generally more reactive, giving rise to unwanted side reactions that compromise the yield of 5b. These stability trends are consistent with previous observations on phosphaalkenes with P-phenyl substituents.31 In the case of asymmetric benzophenones, mixtures of the E and Z isomers are obtained in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (5e–i). Interestingly, ketones with terminal bromoalkyl chains (1g–1i) are compatible with the procedure, and debromination by the lithium phosphide reagent is not observed, consistent with fast phosphaalkene formation. The P-TMS substituted phosphaalkenes are unreactive towards remaining phosphide salt 2, making the reaction compatible with ketones of low solubility in diethyl ether. In contrast, P-phenyl-phosphaalkenes react with excess lithium bis(trimethylsilyl)phosphide, giving rise to the formation of diphosphirane addition products.30,31
:1 ratio (5e–i). Interestingly, ketones with terminal bromoalkyl chains (1g–1i) are compatible with the procedure, and debromination by the lithium phosphide reagent is not observed, consistent with fast phosphaalkene formation. The P-TMS substituted phosphaalkenes are unreactive towards remaining phosphide salt 2, making the reaction compatible with ketones of low solubility in diethyl ether. In contrast, P-phenyl-phosphaalkenes react with excess lithium bis(trimethylsilyl)phosphide, giving rise to the formation of diphosphirane addition products.30,31
With diphosphetane formation being crucial for the mechanism, the reaction offers the opportunity to create macrocyclic structures with internal double bonds by employing the tethered bis-benzophenones 6a–g. Such macrocycles are interesting as components in supramolecular entities such as catenanes or rotaxanes. Macrocycles 7a–g could be obtained in all cases without employing commonly used strategies (e.g. high dilution, templates…) for the preparation of this type of compounds.34 The yields of macrocyclisation are influenced by the chain length between the two benzophenone moieties. The best results are obtained for compounds with a ten-membered saturated tether (54% for both 7c and 7f) followed by the eight-membered chain analogue (7b, 47%). Much lower yields are obtained for 7a (21%), 7e (24%) and the largest macrocycle 7d (31%). Bis-ketone 6g with the more rigid xylene-based linker is only sparingly soluble in the reaction medium, but can still be dimerized to the cyclic alkene 7g, however only in low yields of 25% (accompanied with quantitative recovery of unreacted 6g). Bis-benzophenone 6e is an interesting substrate as its fluoride substituents promote fast dimerization, while the short tether disfavors the intramolecular reaction. Consequently, this bis-ketone engages in intermolecular diphosphetane formation, and forms the bis-alkene 8e (see Fig. 4). The structures of all products were assigned by NMR spectroscopy, and in the case of 7a, 7f, 7g and 8e also by single-crystal X-ray diffraction analysis (Fig. 4), exhibiting the expected bond metrics (see ESI†).
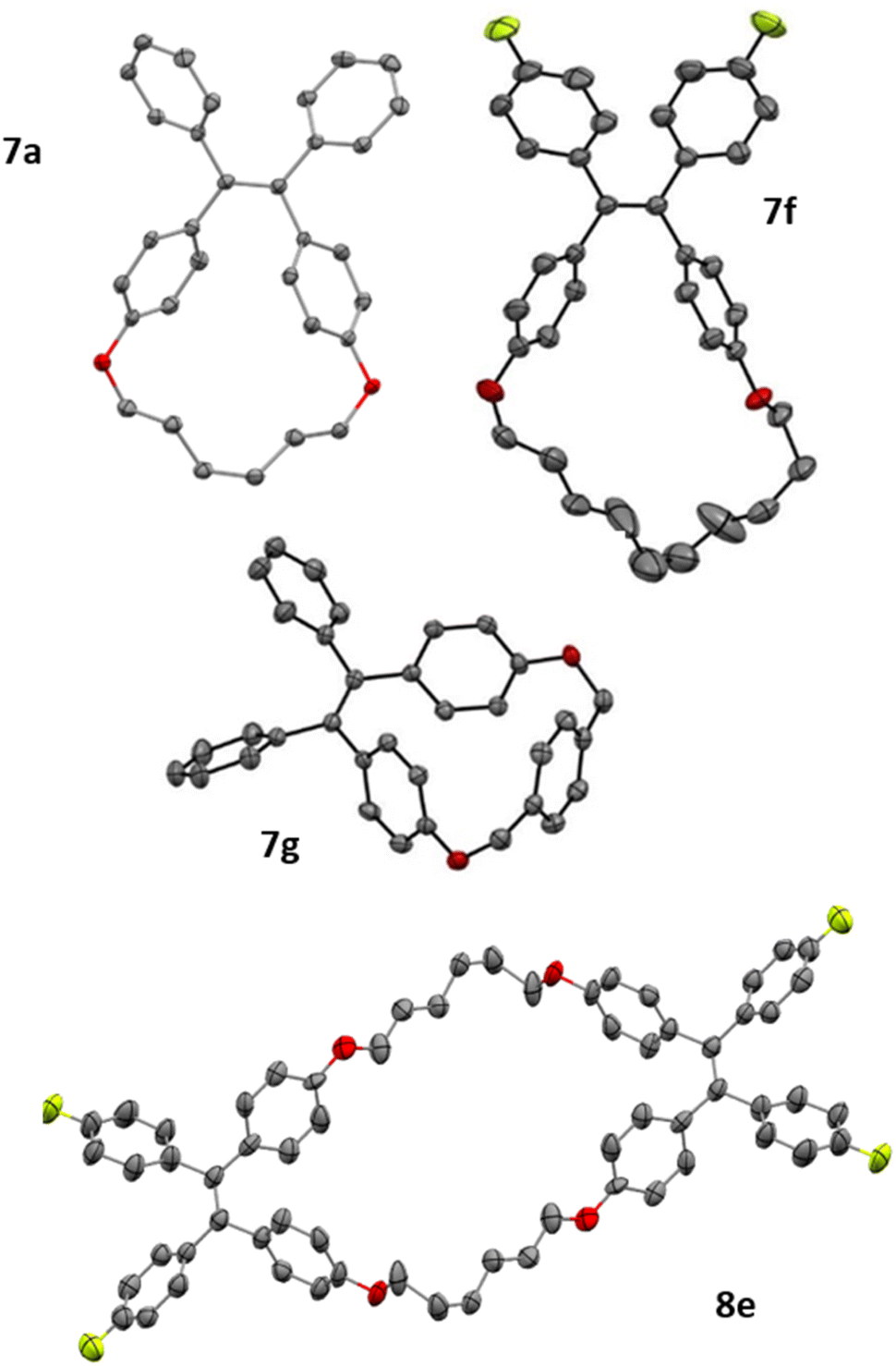 | ||
| Fig. 4 Solid-state structures of 7a, 7f, 7g, and 8e. | ||
Conclusions
This work has established a [2 + 2] cycloaddition strategy for the reductive coupling of two benzophenones to tetra-substituted alkenes. As such, it is the first alternative to the McMurry reaction which has been used for this purpose for over 50 years. The reaction proceeds under mild conditions, and can tolerate functional groups (e.g. bromides) which are incompatible with the radical character of the McMurry coupling. The reaction is driven by a simple (TMS)2PLi reagent that converts the ketone to a P-TMS-phosphaalkene. The latter spontaneously dimerises to a 1,2-diphosphetane, thereby establishing the critical C–C σ-bond, impossible in most ionic mechanisms, whether through classic reactions (e.g. Wittig), or by transition metal-catalysed means, due to steric crowding. The diphosphetane P-TMS groups can be cleaved by the addition of LiOEt, leading to the collapse of the heterocycle and elimination of the tetraarylethene product in yields up to 85%. Intramolecular tethering of two benzophenones allows the production of unprecedented carbocycles in acceptable yields without the need for templating or high dilution strategies. The developed method is complementary to our previous works on reductive aldehyde couplings to stilbenes,13–16 and mechanistically distinct from N-heterocyclic carbene-catalysed metathesis of phosphaalkenes to tetraarylalkenes.35On a broader perspective, the present report shows a new avenue for the construction of CC double bonds by involving cyclic intermediates that stem from dimerisation reactions using non-ionic pathways. We believe that this reactivity is not exclusive to phosphaalkenes, as has recently been demonstrated by the first example of a related phospha-bora-Wittig reaction.36 Investigations along those lines using heavy alkenes are currently on-going in our labs.
Data availability
There is no computational data associated with the article. All relevant experimental data are included in the ESI.†Author contributions
S. O. and A. I. A. designed the study. A. I. A. and N. D. performed all experimental work except for all crystal structures which were contributed by J. A. L. W. Funding was acquired by S. O. The manuscript was written through contributions from all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this work through the Swedish Research Council and Uppsala University is gratefully acknowledged. Swedish Research Council (grant no. 2015-04640 & 2019-04415).References
- S. E. Kelly, in Comprehensive Organic Synthesis, ed. Trost B. M. and Fleming I., Pergamon, Oxford, 1991, pp. 729–817 Search PubMed.
- T. Takeda, Modern Carbonyl Olefination, Wiley-VCH, Weinheim, Germany, 2004, pp. 1–150 Search PubMed.
- W.-Y. Siau, Y. Zhang and Y. Zhao, in Stereoselective Alkene Synthesis: Stereoselective Synthesis of Z-Alkenes, ed. Wang, J., Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 33–58 Search PubMed.
- Y. Gu and S.-K. Tian, in Stereoselective Alkene Synthesis: Olefination Reactions of Phosphorus-Stabilized Carbon Nucleophiles, Springer-Verlag, Berlin Heidelberg, 2012, pp. 197–238 Search PubMed.
- G. Wittig and G. Geissler, Zur Reaktionsweise des Pentaphenyl-phosphors und einiger Derivate, Liebigs Ann. Chem., 1953, 580(1), 44–57 CrossRef.
- G. Wittig and U. Schöllkopf, Über Triphenyl-phosphin-methylene als olefinbildende Reagenzien, Chem. Ber., 1954, 87(9), 1318–1330 CrossRef.
- G. Wittig, From Diyls to Ylides to My Idyll, Science, 1980, 210(4470), 600–604 CrossRef PubMed.
- B. E. Maryanoff and A. B. Reitz, The Wittig olefination reaction and modifications involving phosphoryl-stabilized carbanions. Stereochemistry, mechanism, and selected synthetic aspects, Chem. Rev., 1989, 89(4), 863–927 CrossRef.
- W. S. Wadsworth and W. D. Emmons, The Utility of Phosphonate Carbanions in Olefin Synthesis, J. Am. Chem. Soc., 1961, 83(7), 1733–1738 CrossRef.
- D. J. Peterson, Carbonyl olefination reaction using silyl-substituted organometallic compounds, J. Org. Chem., 1968, 33(2), 780–784 CrossRef.
- L. F. V. Staden, D. Gravestock and D. J. Ager, New developments in the Peterson olefination reaction, Chem. Soc. Rev., 2002, 31(3), 195–200 RSC.
- S. Wang, N. Lokesh, J. Hioe, R. M. Gschwind and B. König, Photoinitiated carbonyl-metathesis: deoxygenative reductive olefination of aromatic aldehydes via photoredox catalysis, Chem. Sci., 2019, 10(17), 4580–4587 RSC.
- A. I. Arkhypchuk, N. D'Imperio and S. Ott, Triarylalkenes from the site-selective reductive cross-coupling of benzophenones and aldehydes, Chem. Commun., 2019, 55, 6030–6033 RSC.
- K. Esfandiarfard, J. Mai and S. Ott, Unsymmetrical E-Alkenes from the Stereoselective Reductive Coupling of Two Aldehydes, J. Am. Chem. Soc., 2017, 139(8), 2940–2943 CrossRef CAS PubMed.
- A. I. Arkhypchuk, N. D'Imperio and S. Ott, One-Pot Intermolecular Reductive Cross-Coupling of Deactivated Aldehydes to Unsymmetrically 1,2-Disubstituted Alkenes, Org. Lett., 2018, 20(17), 5086–5089 CrossRef CAS PubMed.
- J. Mai, A. I. Arkhypchuk, S. Wagner, A. Orthaber and S. Ott, Z-selective alkene formation from reductive aldehyde homo-couplings, Eur. J. Org. Chem., 2022 DOI:10.1002/ejoc.202200365.
- W. Wei, X.-J. Dai, H. Wang, C. Li, X. Yang and C.-J. Li, Ruthenium(II)-catalyzed olefination via carbonyl reductive cross-coupling, Chem. Sci., 2017, 8(12), 8193–8197 RSC.
- J. E. McMurry and M. P. Fleming, New method for the reductive coupling of carbonyls to olefins. Synthesis of β-carotene, J. Am. Chem. Soc., 1974, 96(14), 4708–4709 CrossRef CAS PubMed.
- J. E. McMurry, Organic chemistry of low-valent titanium, Acc. Chem. Res., 1974, 7(9), 281–286 CrossRef CAS.
- J. E. McMurry, Carbonyl-coupling reactions using low-valent titanium, Chem. Rev., 1989, 89(7), 1513–1524 CrossRef CAS.
- M. Ephritikhine, A new look at the McMurry reaction, Chem. Commun., 1998, 23, 2549–2554 RSC.
- L. Kurti and B. Czako, Strategic Applications of Named Reactions in Organic Synthesis, Elsevier Academic Press, San Diego, 2005 Search PubMed.
- M. Yam, J. H. Chong, C.-W. Tsang, B. O. Patrick, A. E. Lam and D. P. Gates, Scope and Limitations of the Base-Catalyzed Phospha-Peterson PC Bond-Forming Reaction, Inorg. Chem., 2006, 45(13), 5225–5234 CrossRef CAS PubMed.
- G. Becker, W. Uhl and H.-J. Wessely, Acyl- und Alkylidenphosphane. XVI. (Dimethylaminomethyliden)- und (Diphenylmethyliden)phosphane, Z. Anorg. Allg. Chem., 1981, 479(8), 41–56 CrossRef CAS.
- R. Pietschnig, E. Niecke, M. Nieger and K. Airola, Synthesis and reactivity of P-ferrocenylalkylidenephosphanes and related iminophosphanes and diphosphenes, J. Organomet. Chem., 1997, 529(1), 127–133 CrossRef CAS.
- G. Becker, W. Becker and G. Uhl, Acyl- und Alkylidenphosphane. XXIV. (N,N-Dimethylthiocarbamoyl)trimethylsilylphosphane und 1,2-Di(tert-butyl)-3-dimethylamino-1-thio-4-trimethylsilylsulfano-1λ5,2λ3-diphosphet-3-en, Z. Anorg. Allg. Chem., 1984, 518(11), 21–35 CrossRef CAS.
- S. Shah and J. D. Protasiewicz, ‘Phospha-variations’ on the themes of Staudinger and Wittig: phosphorus analogs of Wittig reagents, Coord. Chem. Rev., 2000, 210(1), 181–201 CrossRef CAS.
- C. C. Cummins, R. R. Schrock and W. M. Davis, Phosphinidenetantalum(V) Complexes of the Type [(N3N)Ta=PR] as Phospha-Wittig Reagents, Angew. Chem., Int. Ed., 1993, 32(5), 756–759 CrossRef.
- T. L. Breen and D. W. Stephan, Phosphinidene Transfer Reactions of the Terminal Phosphinidene Complex Cp2Zr(:PC6H2-2,4,6-t-Bu3)(PMe3), J. Am. Chem. Soc., 1995, 117(48), 11914–11921 CrossRef CAS.
- S. Wang, K. Samedov, S. C. Serin and D. P. Gates, PhP=CPh2 and Related Phosphaalkenes: A Solution Equilibrium between a Phosphaalkene and a 1,2-Diphosphetane, Eur. J. Inorg. Chem., 2016, 26, 4144–4151 CrossRef.
- N. D'Imperio, A. I. Arkhypchuk, J. Mai and S. Ott, Triphenylphosphaalkenes in Chemical Equilibria, Eur. J. Inorg. Chem., 2019, 11–12, 1562–1566 CrossRef.
- M. Yam, J. H. Chong, C. W. Tsang, B. O. Patrick, A. E. Lam and D. P. Gates, Scope and limitations of the base-catalyzed phospha-peterson P=C bond-forming reaction, Inorg. Chem., 2006, 45(13), 5225–5234 CrossRef CAS.
- G. Fritz, J. Härer and K. H. Scheider, Untersuchungen zur Reaktionsfähigkeit der höheren Silylphosphane (me3Si)4P2, [(me3Si)2P]2PH, [(me3Si)2P]2P–Sime3 und (me3Si)3P7, Z. Anorg. Allg. Chem., 1982, 487(1), 44–58 CrossRef CAS.
- H.-B. Chen, J. Yin, Y. Wang and J. Pei, Unsaturated Strained Cyclophanes Based on Dibenz[a,j]anthracene by an Intramolecular McMurry Olefination, Org. Lett., 2008, 10(14), 3113–3116 CrossRef CAS PubMed.
- Z. Han and D. P. Gates, Metathesis of P=C Bonds Catalyzed by N-Heterocyclic Carbenes, Chem.–Eur. J., 2021, 27(59), 14594–14599 CrossRef CAS PubMed.
- A. M. Borys, E. F. Rice, G. S. Nichol and M. J. Cowley, The Phospha-Bora-Wittig Reaction, J. Am. Chem. Soc., 2021, 143(35), 14065–14070 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterisation of all compounds. CCDC 2162400–2162404. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03073j |
| This journal is © The Royal Society of Chemistry 2022 |