
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tyrosine bioconjugation with hypervalent iodine†
Nina
Declas
a,
John R. J.
Maynard
b,
Laure
Menin
c,
Natalia
Gasilova
c,
Sebastian
Götze
 d,
Jakob L.
Sprague
e,
Pierre
Stallforth
d,
Stefan
Matile
b and
Jerome
Waser
*a
d,
Jakob L.
Sprague
e,
Pierre
Stallforth
d,
Stefan
Matile
b and
Jerome
Waser
*a
aLaboratory of Catalysis and Organic Synthesis, Institut des Sciences et Ingénierie Chimique, Ecole Polytechnique Fédérale de Lausanne, CH-1015, Lausanne, Switzerland. E-mail: jerome.waser@epfl.ch
bDepartment of Organic Chemistry, University of Geneva, 1211 Geneva, Switzerland. E-mail: stefan.matile@unige.ch
cInstitut des Sciences et Ingénierie Chimique, Ecole Polytechnique Fédérale de Lausanne, EPFL, 1015 Lausanne, Switzerland
dDepartment of Paleobiotechnology, Leibniz Institute for Natural Product Research and Infection Biology, Hans Knöll Institute (HKI), 07745 Jena, Germany
eDepartment of Microbial Pathogenicity Mechanisms, Leibniz Institute for Natural Product Research and Infection Biology, Hans Knöll Institute (HKI), 07745 Jena, Germany
First published on 12th October 2022
Abstract
Hypervalent iodine reagents have recently emerged as powerful tools for late-stage peptide and protein functionalization. Herein we report a tyrosine bioconjugation methodology for the introduction of hypervalent iodine onto biomolecules under physiological conditions. Tyrosine residues were engaged in a selective addition onto the alkynyl bond of ethynylbenziodoxolones (EBX), resulting in stable vinylbenziodoxolones (VBX) bioconjugates. The methodology was successfully applied to peptides and proteins and tolerated all other nucleophilic residues, with the exception of cysteine. The generated VBX were further functionalized by palladium-catalyzed cross-coupling and azide–alkyne cycloaddition reactions. The method could be successfully used to modify bioactive natural products and native streptavidin to enable thiol-mediated cellular uptake.
Introduction
Site-specific chemical modification of peptides and proteins is becoming increasingly important in research and industry for monitoring cellular events or designing therapeutics, targeting ligands and molecular probes.1 However the number of chemical transformations that are suitable for efficient biomolecule functionalization is limited by the stringent conditions required (mild and aqueous conditions, high selectivity, low toxicity) and developing chemo- and site-selective transformations remains a prominent challenge.2 Traditional methods for bioconjugation target nucleophilic lysine and cysteine residues (Scheme 1a).2 However, functionalization of the abundant lysine tends to lack selectivity. Modification of the rarer cysteine often requires cleavage of disulfide bonds, which play an important role in the folding and stability of proteins,3 or the expression of non-natural proteins with selectively introduced cysteines. To overcome these limitations and expand the functionalization toolbox, less exploited amino acids such as methionine,4 tryptophan,5 histidine6 and tyrosine7 have been investigated.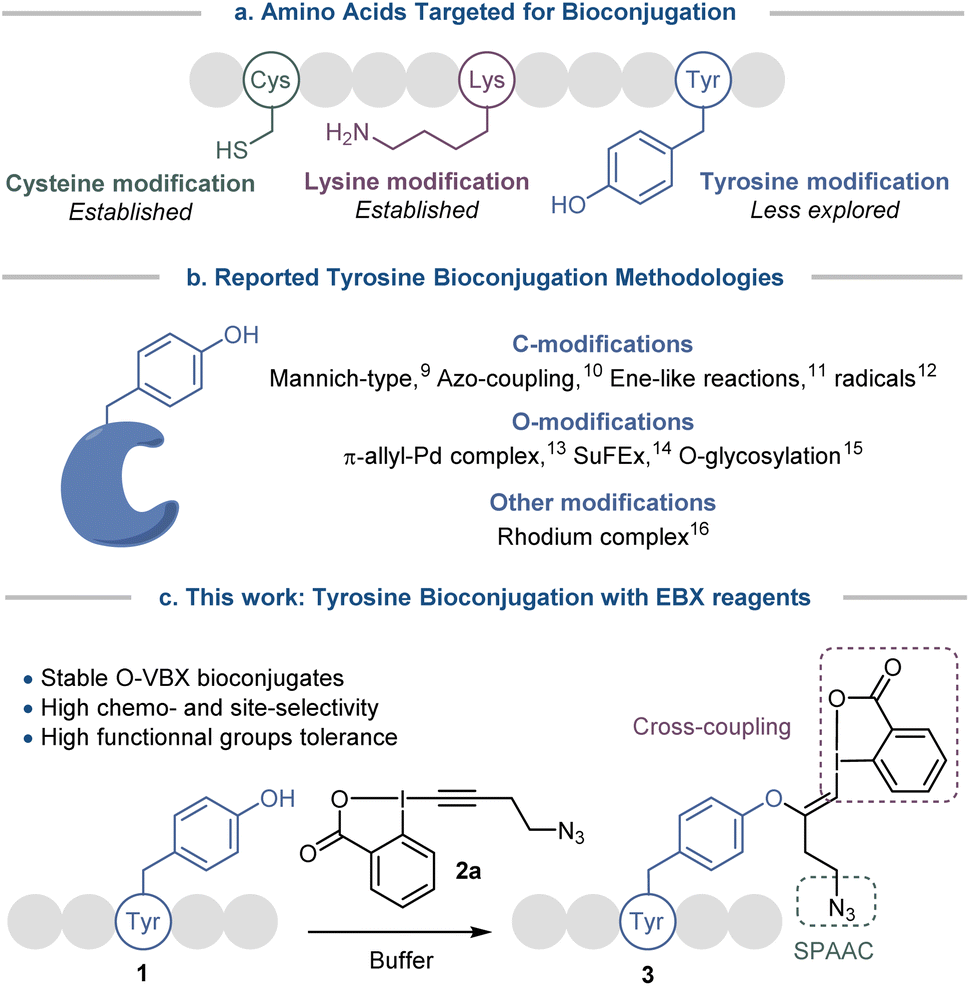 | ||
| Scheme 1 (a) Amino acids targeted for bioconjugation. (b) Reported tyrosine bioconjugation methodologies. (c) Development of a tyrosine-selective bioconjugation method with hypervalent iodine reagents. | ||
Tyrosine bioconjugation in particular shows promising results (Scheme 1b).7 Among natural amino acids, it is an interesting residue for site-selective modification, as it is relatively rare on protein surfaces,8 and is often buried within the protein structure due to the amphiphilicity of the phenol moiety. Several elegant methods have been reported for C-modification of tyrosine, such as three-component Mannich-type reactions,9 or reactions with aryldiazonium salts,10 diazodicarboxamides11 and radicals.12 O-functionalization has also been described, with for example reaction with electrophilic π-allyl palladium complexes,13 SuFEx reagents14 and glycosylation reactions.15 Functionalization has also been achieved via dearomatization−rearomatization strategies,16 or formation of a rhodium arene complex.17
All these strategies have advantages, but also inherent limitations, such as low site- or chemo-selectivity with competitive amino acids, side reactions, the use of expensive or toxic transition metals or the stability of the reagents and the bioconjugates. Therefore the development of alternative methods is still highly desirable. With their low toxicity, high functional group tolerance and stability in biocompatible media, hypervalent iodine reagents have recently emerged as powerful tools for late-stage peptide and protein functionalization.18 In 2019, our group reported the synthesis of vinylbenziodoxolone reagents (VBX) by stereoselective addition of phenols, sulfonamides and thiols nucleophiles onto ethynylbenziodoxolones (EBX).19,20 The VBX were shown to be bench-stable and the enhanced reactivity of the hypervalent bond allowed their use as electrophiles in palladium-catalyzed cross-couplings at room temperature. During this work,19 an N- and C-protected tyrosine could be used as a nucleophile, but the reaction conditions developed for small organic molecules were not compatible with peptides and proteins.
Moving from protected amino-acids to native peptides is challenging, as they contain multiple reactive nucleophilic functionalities and require aqueous buffers for solubilization, which are attenuating the reactivity of the residues and could degrade the reagents. Formation of S-VBX reagents from cysteine under physiological conditions was previously reported by our group.20 As a harder and less nucleophilic residue, tyrosine usually displays a lower reactivity with electrophiles than the soft and highly nucleophilic cysteine. Indeed, no tyrosine functionalization had been observed under the conditions developed for S-VBX formation. Nevertheless, free cysteine residues are occurring only rarely in peptides and proteins, most of them form disulfide bonds, which usually do not react with EBX reagents.20 It should be therefore possible to selectively target tyrosine if adequate conditions could be developed and other nucleophilic amino acids would not interfere. In this work, we report a tyrosine-selective modification of peptides with EBX 2a under mild and biocompatible conditions (Scheme 1c). With the exception of cysteine, all nucleophilic residues present in peptides were tolerated, and the method could be used for both synthetic peptides and natural products. Preliminary results indicate that the method can also be used with proteins with surface accessible tyrosine residues. The stable O-VBX conjugates were then used as a platform for further orthogonal modifications via a palladium-catalyzed Suzuki–Miyaura cross-coupling and a metal-free Strain-Promoted Alkyne–Azide Cycloaddition (SPAAC), providing a wide range of labelled biomolecules. Using this methodology, native streptavidin was modified to enable thiol-mediated cellular uptake.
Results and discussion
Optimization of the labelling on peptide tetramer 1a
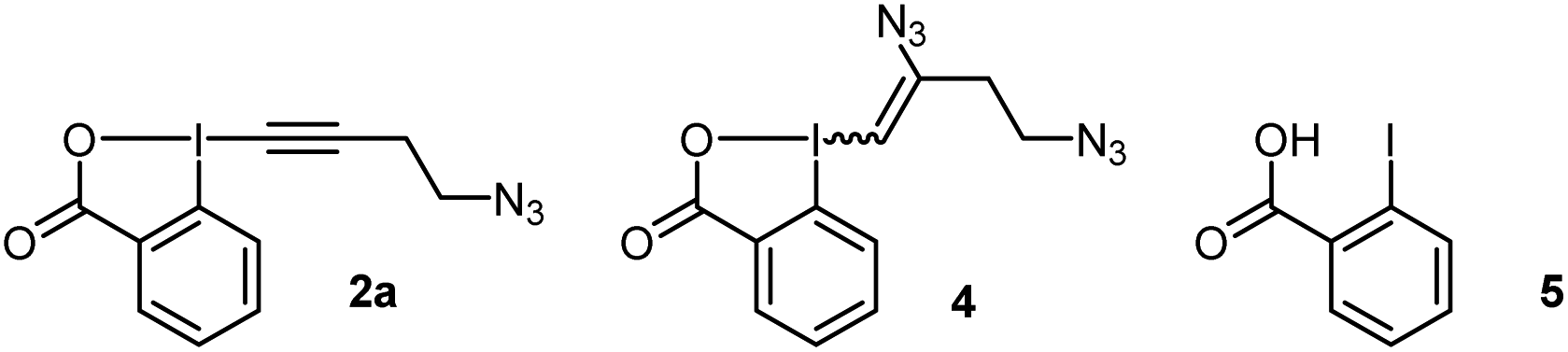
We started our study with the functionalization of tetramer AFYA-NH2 (1a) as model substrate. AFYA-NH2 (1a) was easily obtained by solid-phase peptide synthesis (SPPS) and allowed a good detection by HPLC. Although 1a did not contain nucleophilic side chains, the presence of the free N-terminus was already expected to give an impression of the selectivity of the method towards other nucleophiles. We chose the azide-functionalized EBX JW-RF-010 (2a) as labeling reagent,21 as the azide group can be used for further functionalization via SPAAC.20 By simple dissolution of the peptide in non-degassed Tris buffer (10 mM, pH 8.2), and then addition of a solution of EBX 2a in DMSO, the O-VBX bioconjugate 3a could be obtained in 76% HPLC-yield after 24 hours of reaction at 37 °C (Table 1, entry 1). The reaction was clean and the only side products observed were N3-VBX 4 (ref. 22) and 2-iodobenzoic acid (5), both originating from EBX decomposition. No reaction on the potentially nucleophilic N-terminus was observed.|
|
||||
|---|---|---|---|---|
| Entry | Buffer (Molarity) | pH | T (°C) | Yieldb |
| a Conditions: AFYA-NH21a (1.0 μmol), N3-EBX 2a (3.0 equiv), Buffer (2% v/v DMSO), 2 mM, 24 h. b HPLC-MS yields are given. The yields were approximated as the ratio of Aprod/Atotal where Aprod = area in mAU of the product peak and Atotal = area in mAU of all peptides products (product, starting material, and side products if present). c Reaction time: 1 h. d Reaction time: 4 h. | ||||
| 1 | Tris (10 mM) | 8.2 | 37 | 76% |
| 2 | Tris (1 mM) | 8.2 | 37 | 52% |
| 3 | Tris (100 mM) | 8.2 | 37 | 96% |
| 4 | Tris (1 M) | 8.2 | 37 | 90% |
| 5 | PB (100 mM) | 8.2 | 37 | 86% |
| 6 | HEPES (100 mM) | 8.2 | 37 | 87% |
| 7 | 10× PBS | 8.2 | 37 | 86% |
| 8 | Tris (100 mM) | 7.0 | 37 | 24% |
| 9 | Tris (100 mM) | 9.0 | 37 | >99% |
| 10 | Tris (100 mM) | 9.0 | rt | 98% |
| 11 | Tris (100 mM) | 9.0 | 50 | 70% |
| 12 | Tris (100 mM) | 9.0 | 90 | 50% |
| 13c | Tris (100 mM) | 9.0 | 37 | 58% |
| 14d | Tris (100 mM) | 9.0 | 37 | 80% |
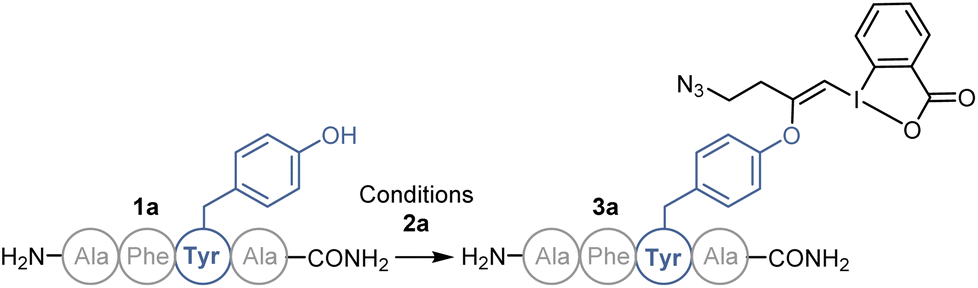
Scope of peptides
In order to investigate the functional group tolerance of the labelling method, tetrapeptides 1 containing various unprotected natural amino acids were then engaged in the reaction (Scheme 2a). Reproducibility of the reaction on larger scale (20 instead of 1 μmol) was first confirmed by full conversion of AFYA-NH2 (1a) to the corresponding O-VBX 3a, which was isolated in 64% yield following HPLC purification and characterized by 1H, 13C and 2-dimensional NMR spectroscopy.24 A sterically hindered leucine residue did not affect the reactivity and O-VBX bioconjugate 3b was obtained in 65% isolated yield. High chemoselectivity was achieved in the presence of tryptophan, however peptide 3c could only be isolated as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mixture with N3-VBX 4. No side reactions occurred in the presence of the nucleophilic and basic arginine (3d), histidine (3e) and lysine (3f) residues.
:2 mixture with N3-VBX 4. No side reactions occurred in the presence of the nucleophilic and basic arginine (3d), histidine (3e) and lysine (3f) residues.
 | ||
| Scheme 2 (a) Scope of tetramers on 20.0 μmol. (b) Scope of bioactive peptides on 1.0 μmol or 1.0 mg. Tyr* = Tyr modified with EBX. HPLC-MS yields are given, determined as indicated in Scheme 1. aIsolated yield. bObtained as a 1:2 mixture with 4. | ||
When cysteine-containing tetramer 1g was submitted to the reaction conditions, fast addition of the cysteine residue was observed,20 followed by reaction of the tyrosine, leading to a doubly labelled S- and O-VBX product 3g in 40% isolated yield. Due to the inherent higher nucleophilicity of thiols, the functionalization of tyrosine in the presence of free cysteine has usually not been reported,9,12c,15,25 except for a few exceptions with selected biomolecules.13 Serine (3h), methionine (3i) and aspartic acid (3j) did not affect the reactivity, whereas a slightly lower yield due to incomplete conversion was observed for asparagine (3k). Finally, the position of tyrosine in the tetramer was varied (1l–1n). All peptides were successfully labelled to give products 3l–n, including N- and C-terminal tyrosine residues (3m and 3n).
The promising functional group tolerance and high chemoselectivity observed on tetramers drove us to apply the methodology to more complex peptides (Scheme 2b). The chemoselectivity of the peptides labelling was confirmed by MS/MS analysis (see ESI† for details). The neurohypophysial hormones vasopressin (1o) and oxytocin (1p), both containing a disulfide bond, were successfully functionalized at the tyrosine position (3o and 3p) with high conversion and no damage to the macrocyclic structure. High labelling conversion was also obtained at the N-terminal tyrosine of HIV-1-tat (1q) and β-casomorphin human (1r) peptides (VBXs 3q and 3r). Lower efficiency was observed on angiotensin II (1s), as well as on bigger amyloid β-proteins 1t and 1u, with however still high chemoselectivity towards the tyrosine residue (VBXs 3s, 3t and 3u). The cyclopentapeptide RGD (1v), containing an Arg–Gly–Asp motif and commonly used in targeted therapy for its property to bind specifically to integrin receptor on cell surface,26 was successfully labelled to give VBX 3v in 76% yield. Jagaricin (1w) is a cyclic lipopeptide natural product with promising antifungal properties.27 Despite the diversity of chemical structures present including threonine, histidine, glutamine and sensitive dehydrothreonine units, jagaricin (1w) was selectively labelled on the tyrosine residue with EBX 2a in 61% HPLC-MS yield and could be isolated in 30% yield (3wa).28
Preliminary results on proteins
Next, the capacity of EBX 2a to label tyrosine on proteins was assessed (Scheme 3). When the small regulatory native protein Ubiquitin (8.6 kDa) was submitted to the reaction conditions, nearly no reaction was observed. This confirmed that only accessible tyrosine can be functionalized and no other residues react. However, with denaturing conditions (6.0 M GdmHCl) to gain access to the buried tyrosine together with a higher excess of EBX 2a (10 equiv) and a longer reaction time (72 hours) (conditions Aa), modified ubiquitin 6 was obtained in 24% yield.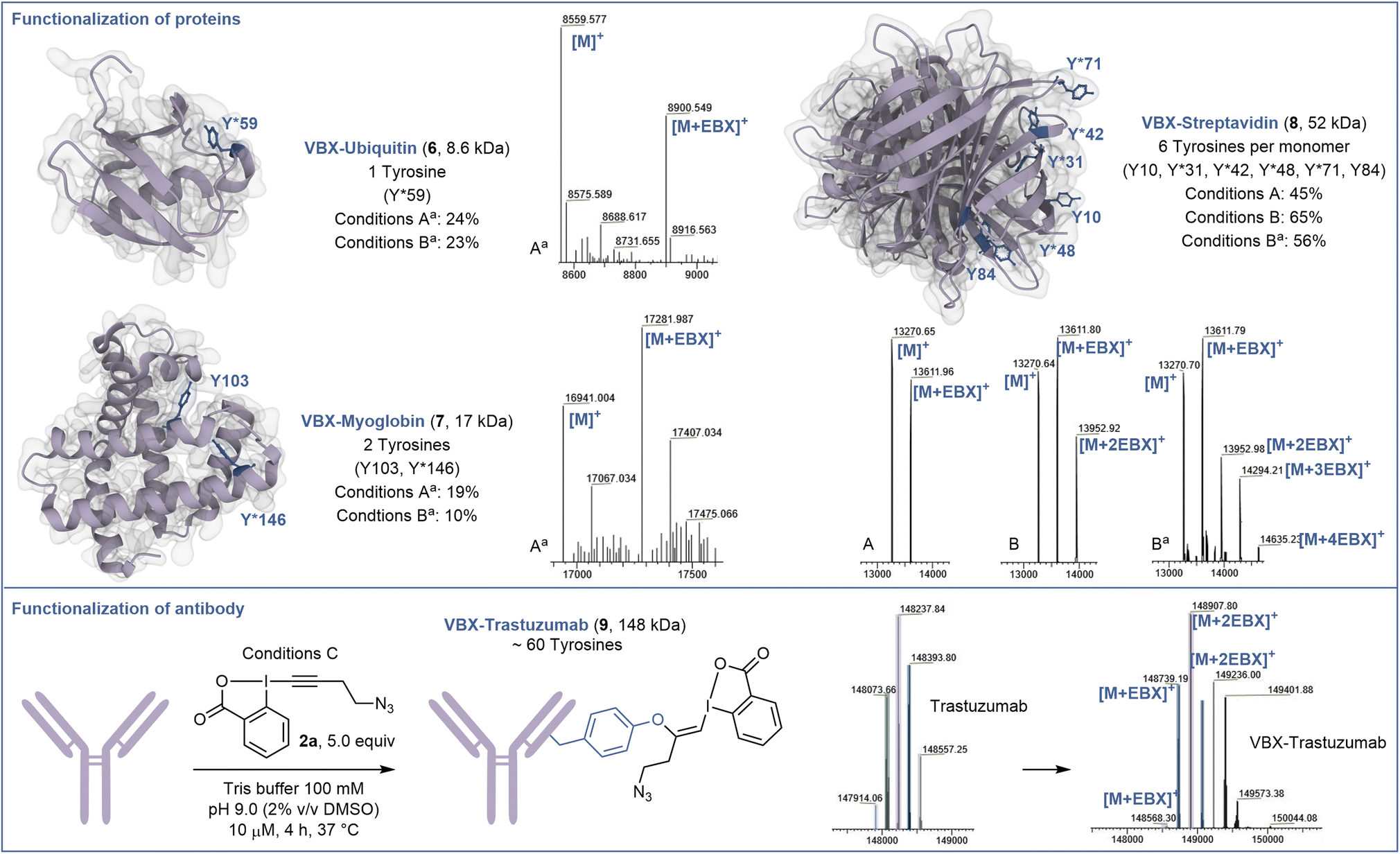 | ||
| Scheme 3 Functionalization of proteins and antibody. Conditions A: Protein (10.0 nmol), N3-EBX 2a (10.0 equiv), 100 μM in Tris buffer (100 mM, pH 9.0), 72 h. Conditions B: Protein (10.0 nmol), N3-EBX 2a (50.0 equiv), 100 μM in Tris buffer (100 mM, pH 9.0), 24 h. Conditions C: Trastuzumab (1.0 nmol), N3-EBX 2a (5.0 equiv), 10 μM in Tris buffer (100 mM, pH 9.0), 4 h. The ratio indicated corresponds to the signal intensity of all the functionalized products mass over the sum of the intensities of all other products with a threshold of 5% intensity. aDenaturing conditions: Tris buffer (100 mM, pH 9.0, 6.0 M GdmHCl). | ||
Selective modification of residue Y59 was confirmed by targeted top-down mass spectrometry analysis (see ESI† for details). Higher conversion could be obtained using 50 equiv of EBX 2a, but without increase in yield of 6 due to side reactions (conditions Ba). The same conditions Aa were then applied to the hemeprotein myoglobin (17 kDa) containing 2 tyrosine residues. Chemo- and site-selectivity was achieved on the more reactive tyrosine Y146 to give 7 in 19% yield. Finally, we studied the functionalization of the tetrameric recombinant protein streptavidin (52 kDa), which contains 6 tyrosine residues per monomer (13 kDa), which are more accessible. Under native conditions (conditions A), 10 equiv of EBX 2a allowed to mono-label streptavidin on the most accessible tyrosine Y31, Y42, Y48 or Y71 to give 8. This result reflects the reactivity and accessibility of these amino acids compared to Y10 and Y84, which were not modified. Increasing the amount of EBX 2a to 50 equiv allowed to doubly modify the protein after 24 hours of incubation (conditions B). Again, only Y31, Y42, Y48 and Y71 were functionalized. Finally, under denaturing conditions, the 4 reactive tyrosine residues could be labelled (conditions Ba). Interestingly, when another mutant streptavidin (66 kDa) was used, a better conversion was observed (see ESI†).
The incomplete conversion observed indicated that the tyrosines of the protein investigated so far were not fully accessible. We therefore decided to investigate the functionalization of trastuzumab (148 kDa), a monoclonal antibody used to treat breast cancer,29 containing a high number of tyrosine residues including some with high accessibility and reactivity.30 Gratefully, and despite the glycans present on the heavy chains, under mild conditions without denaturation, trastuzumab could be efficiently labelled with an average of 2 EBX and complete conversion after 4 hours to give 9. A longer reaction time and higher excess of EBX allowed to reach a higher degree of bioconjugation (see ESI† for details).
Scope of EBX and further functionalization
Beside azide-bearing 2a, other hypervalent iodine reagents could be used as labelling reagents under the same reaction conditions (Scheme 4). EBX with alkyne (2b), chlorine (2c) and alcohol (2d) functional groups as well as a rhodamine fluorophore (2e), were successfully added to the N-terminal tyrosine of β-casomorphin human peptide (3r) to give VBXs 3rb–3re. In addition, jagaricin 3w, was labeled with a fluorescent rhodamine 2e and the isolated O-VBX (3we) could be used as a fluorescent stain in Candida albicans cells (see ESI† for microscopy images). Interestingly, TMS-EBX (2f) led to unsubstituted bioconjugate 3rg. Such a result was previously observed on cysteine,31 and is due to a fast and complete desilylation of TMS-EBX (2f) to give the free EBX (2g). However, unlike with cysteine, where the generated S-VBX underwent a slow rearrangement into the ethynylated product, the unsubstituted O-VBX 3rg remained stable over time. During screening of EBX reagents, some limitations were observed. TIPS- and Ph-EBX (2h and 2i) did not provide the expected labelled product 3rh and 3ri. No reaction occurred, which could be due to the low solubility of these reagents in aqueous media. Similarly, low conversion was obtained with p-NO2 substituted EBX 2j to give product 3rj. When the water soluble EBX 2k was used,32 no improvement was observed and less than 5% of bioconjugate 3rk was formed.33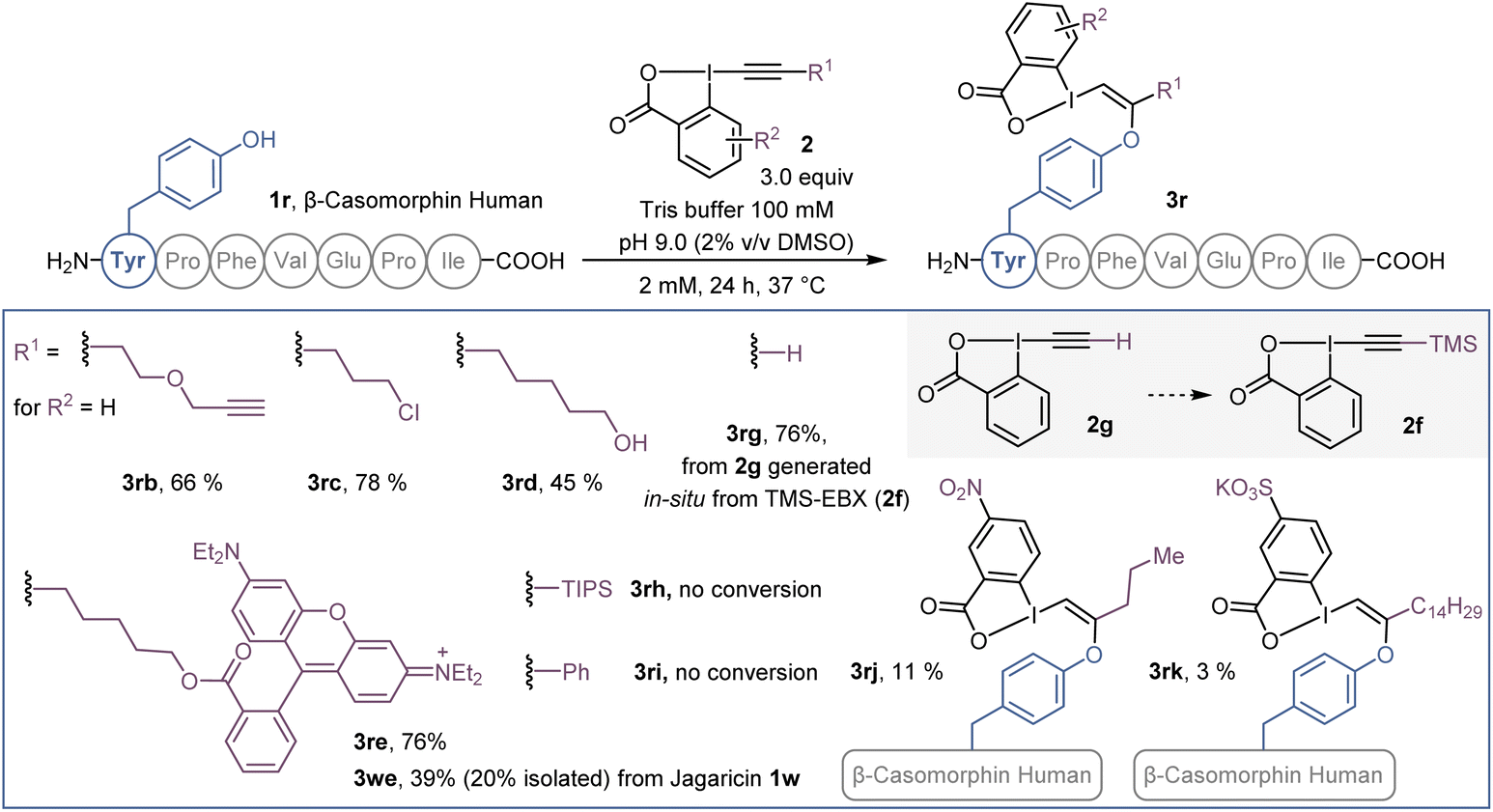 | ||
| Scheme 4 Scope of EBX reagents on β-casomorphin human peptide 1r and jagaricin (1w). HPLC-MS yields are given as indicated in Scheme 1. | ||
The potential of the obtained bioconjugates for further functionalization was then investigated. We first optimized a copper-free SPAAC,34 taking advantage of the azide moiety on the labelled AFYA-NH2 tetramer 3a (Scheme 5a). In Tris buffer, an excess of PEG-functionalised bicyclononyne (BCN) 10a efficiently reacted with the azide functional group of the O-VBX 3a, leading to 11a in 51% isolated yield. With this result in hand, we then extended the click reaction to more complex peptides using a one-pot two-step procedure. β-Casomorphin human (1r) was successfully labelled with 10a to give 12a in 86% HPLC-yield. Both β-casomorphin human (1r) and vasopressin (1o) peptides were successfully labelled with N3-EBX (2a), followed by cycloaddition with BCN 10b–d functionalized with biotin, a fluorophore or a carbohydrate derivative (bioconjugates 12b–d and 13b–d).
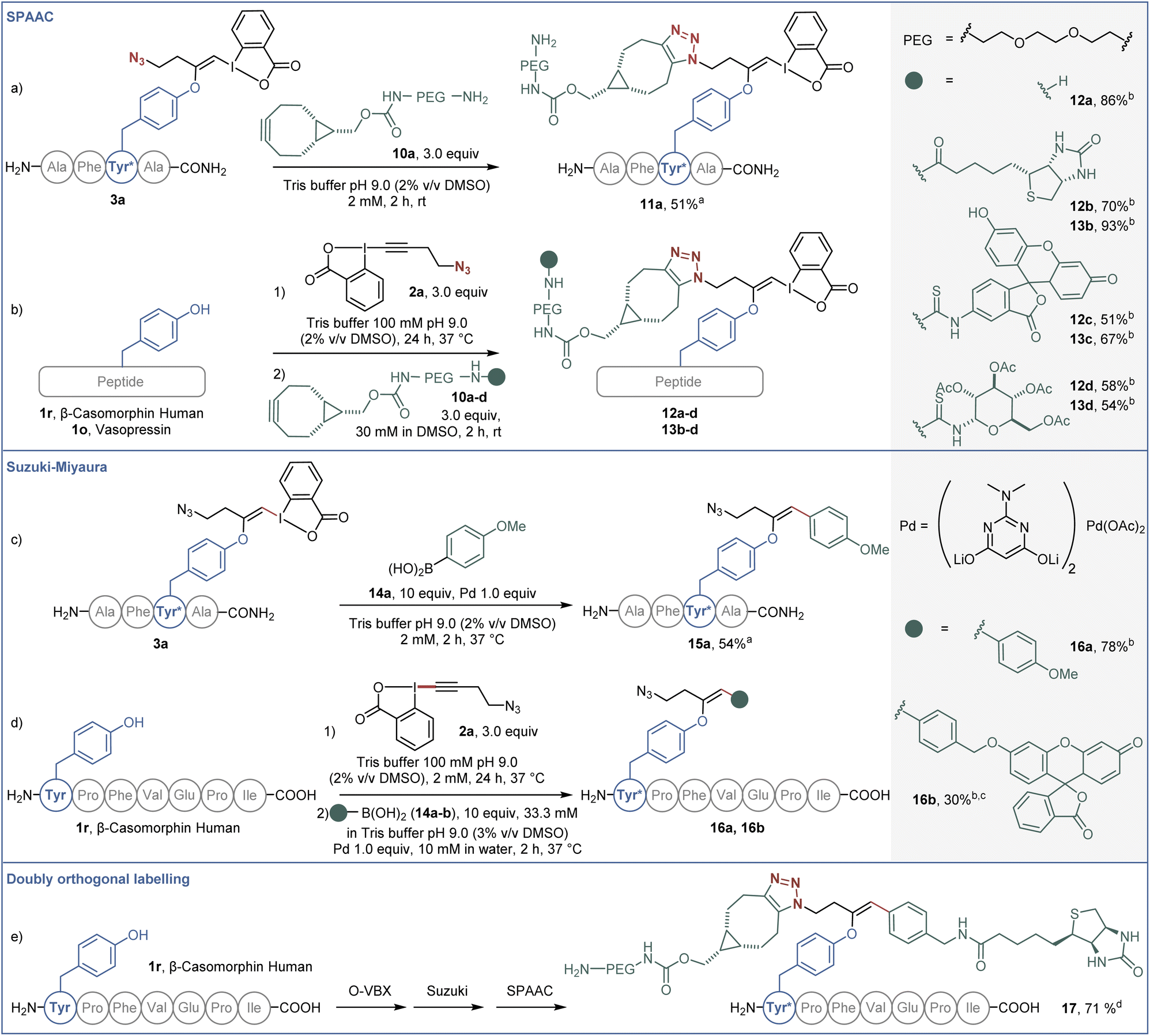 | ||
| Scheme 5 (a) SPAAC functionalization on O-VBX 3a, 20.0 μmol. (b) One-pot two-step labeling/SPAAC functionalization of peptides. Reaction on vasopressin (1o) (0.762 μmol) and β-casomorphin human peptide (1r) (1.0 μmol). (c) Suzuki–Miyaura functionalization on O-VBX 3a, 20.0 μmol. (d) One-pot two-step labeling/Suzuki–Miyaura functionalization of β-casomorphin human peptide (1.0 μmol). (e) One-pot three-step labeling/Suzuki–Miyaura/SPAAC functionalization of β-casomorphin human peptide (1.0 μmol). aIsolated yield is given. bHPLC-UV yield over two steps is given. c10.0 equiv of Pd and 20% of DMSO were used. The reaction time was 4 hours. dHPLC-UV yield over three steps is given. | ||
In addition, the hypervalent iodine handle displayed high reactivity for cross-coupling reactions under mild conditions. Our group previously reported a Suzuki–Miyaura cross-coupling of the hypervalent iodine bond of S-VBX with boronic acids in aqueous media.20 Under similar conditions, labelled AFYA-NH2 tetramer 3a was successfully coupled with boronic acid 14a in 54% isolated yield (Scheme 5b). Coupling of p-methoxyphenyl boronic acid (14a) was also successful with β-casomorphin peptide (1r) using a one-pot two-step procedure. A fluorescein-substituted boronic acid 14b also reacted with the in situ generated O-VBX 3r, but a higher loading of palladium was required, as well as a higher ratio of DMSO to allow solubilization of the coupling partner.
Finally, both reactive handles were successfully employed in a labeling/Suzuki–Miyaura/SPAAC one-pot three-step process to afford doubly-functionalized β-casomorphin peptide 17. A biotin unit was installed using the palladium catalyzed reaction on the hypervalent iodine moiety, whereas the PEG unit was introduced by the click reaction.
Application to the thiol-mediated cellular uptake of streptavidin
The cellular membrane provides a barrier to the passage of large, hydrophilic molecules, such as proteins, into the cytosol. Thiol-mediated uptake, postulated to function by transporter units engaging in dynamic covalent exchange cascades with cellular thiols and disulfides, has been employed previously for the efficient cytosolic delivery of streptavidin.35–39 Using the orthogonal reactivities offered by the EBX modification developed here, we were able to functionalize streptavidin (66 kDa) with both a transporter for thiol-mediated uptake, via CuAAC ‘click’ chemistry with the azide handle, and a lipophilic CF3 functionalized aromatic unit,40via Suzuki coupling with the hypervalent iodine motif. The functionalized proteins were still easily complexed with 2 equiv of a red fluorescent TAMRA-biotin derivative (Fig. 1a), and uptake of the complexes was studied in HeLa MZ cells. Compared with the protein after Suzuki coupling, but prior to click functionalization (18, R = N3), streptavidin functionalized with asparagusic acid (19, R = AspA) showed ∼3-fold increase in cellular uptake whereas streptavidin functionalized with a cyclic thiosulfonate (20, R = CTO), gave ∼28-fold increase in cellular uptake versus the N3 control (Fig. 1b). These results were in accordance with the relative reactivities of the two transporter motifs in thiol-disulfide exchange, and previous studies with small molecule fluorophore cargoes.41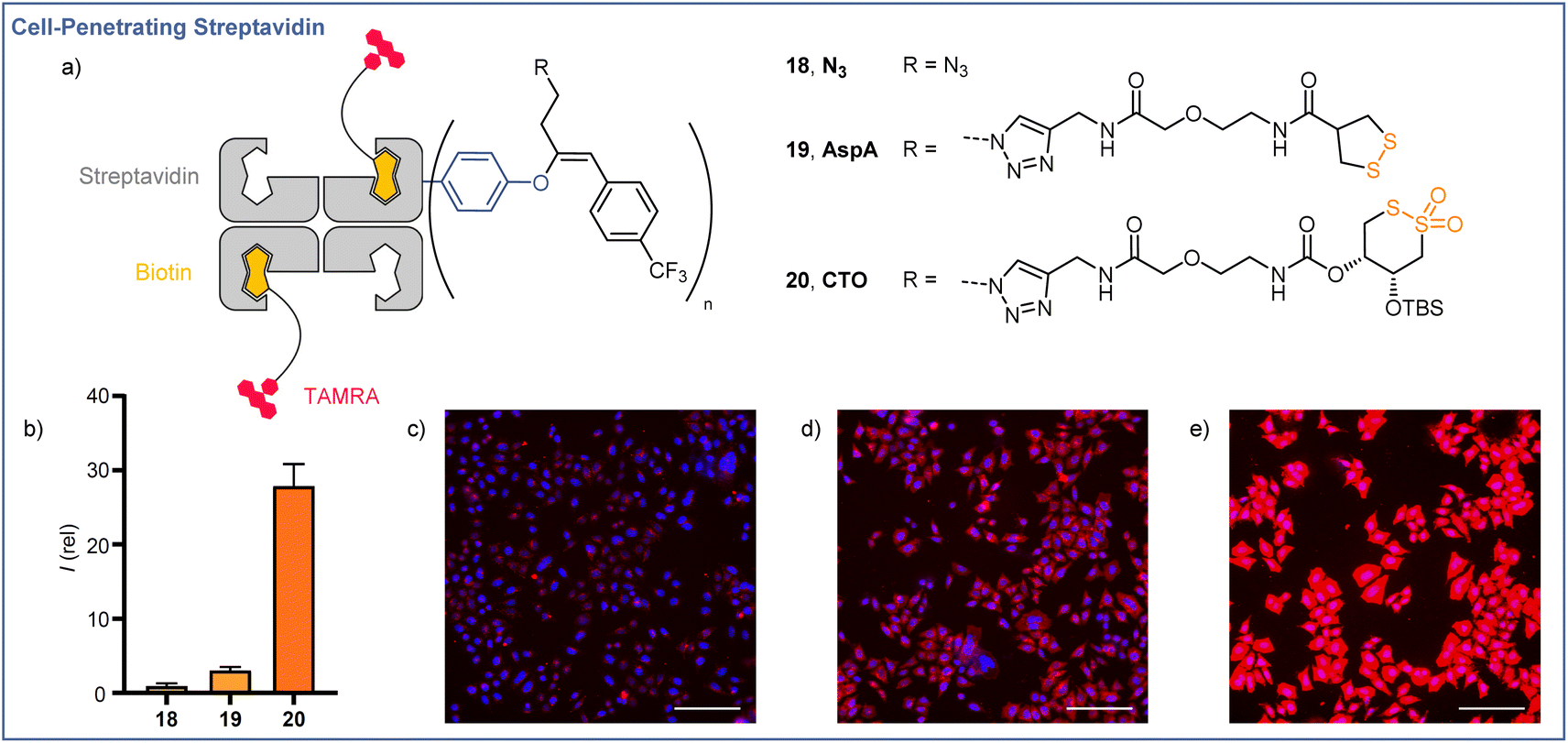 | ||
| Fig. 1 Cellular uptake of streptavidin using O-VBX. (a) Schematic representations of fluorescent biotin-streptavidin complexes used in uptake studies. (b) Relative fluorescence intensities of live HeLa MZ cells in SDCM images following incubation with 10 μM N3, AspA, or CTO for 6 h. Images of HeLa cells incubated with 18 N3 (c), 19 AspA (d), and 20 CTO (e) at 10 μM for 6 h (Blue = Hoechst 33342, Red = TAMRA). Scale bars: 150 μm. Brightness and contrast were adjusted equally in all images. | ||
Structure–activity relationships for cyclic thiosulfonate transporters and thiol-mediated uptake inhibitors revealed a strong dependence in either case on the presence of hydrophobic directing groups to target aprotic environments such as the cellular membrane and proteins, in which the proticity-dependent cascade exchange of cyclic thiosulfonates is enhanced.41 The ∼9-fold increase in uptake efficiency observed between AspA and CTO complexes exceeds the ∼3-fold difference seen with small molecule fluorophore cargoes, the greater differential perhaps resulting from the additional hydrophobic directing group provided by the CF3-functionalised aromatic unit, which is known to be more important for CTO transporters.41
In the case of AspA and CTO, a diffuse pattern of fluorescence was observed in spinning disk confocal microscopy (SDCM) images (Fig. 1d and e). In line with previous reports, this implied delivery to the cytosol rather than entrapment within endosomes following endocytosis, whereas N3 complex 18 showed a punctate fluorescence indicative of endocytosis (Fig. 1c).42,43 These results indicate the potential of readily accessible, broadly stable cyclic thiosulfonates as transporters for the efficient cytosolic delivery of proteins via thiol-mediated uptake, and highlight the possibilities offered by the dual, orthogonal functionalizations available following EBX-tyrosine modification.
Conclusions
In summary, we have reported a chemo- and site-selective tyrosine bioconjugation methodology using hypervalent iodine reagents. Under mild and biocompatible conditions, tyrosine residues reacted with EBX reagents to generate stable O-VBX bioconjugates. The high functional group tolerance of the conjugation method allowed to selectively label a broad range of bioactive peptides, including the natural product jagaricin (3w). Preliminary results were obtained with selected proteins and an antibody. The generated O-VBX conjugates could be further functionalized orthogonally by palladium-catalyzed Suzuki–Miyaura cross-coupling and azide–alkyne cycloaddition, providing a wide range of labelled biomolecules. The potential of the doubly-orthogonal functionalization was demonstrated in a cellular uptake experiment, which further establishes that the functionalized streptavidin retains its structural integrity and activity to bind biotin, a crucial feature for applications in chemical biology. This work further highlights the potential of hypervalent iodine reagents in biomolecule labeling. Moreover, the high tyrosine selectivity described compares well with existing methods for O-functionalization of tyrosine residues, in which labelling of other nucleophilic amino acids such as lysine and histidine has often been reported.14Data availability
Experimental procedures, compound characterization and microscopy images are provided within the ESI† and raw data is available at zenodo.org: https://doi.org/10.5281/zenodo.7074420.Author contributions
N. D. developed and optimized the bioconjugation methodology, examined the scope of the reaction on peptides and proteins, investigated functionalization of the bioconjugates and prepared the related part of the manuscript and experimental parts. J. R. J. M. performed the cellular uptake experiments and prepared the related part of the manuscript and experimental parts. L. M. and N. G. performed the mass analysis and prepared the related experimental part. S. G. performed structural analysis studies on the jagaricin conjugates 3w and 3we and determined the biological activity of 3w. J. L. S. performed the microscopy experiments with 3we. P. S. supervised the experiments on the jagaricin conjugates and proofread the manuscript. S. M. supervised the cellular uptake experiments and proofread the manuscript. J. W. supervised the bioconjugation work, proofread the manuscript and experimental part and coordinated the research project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the ACCESS Geneva platform for services, the Swiss National Science Foundation (SNSF, grant no. 200020_182798, J. W.; 200020_204175, S. M.), the National Centre Chemical Biology (NCCR) Chemical Biology, EPFL and University of Geneva for financial support. P. S. acknowledges the Werner Siemens-Stiftung for financial support. The research of J. L. S. is funded by the German Research Foundation (Deutsche Forschungsgemeinschaft–DFG) within the Collaborative Research Center (CRC)/Transregio (TRR) 124 “FungiNet” project C1 (DFG project number 210879364). We thank Dr Lars Regestein and Nicolas Schlosser (Leibniz-HKI) for providing jagaracin.Notes and references
- (a) K. Fosgerau and T. Hoffmann, Drug Discovery Today, 2015, 20, 122 CrossRef CAS PubMed; (b) Q.-Y. Hu, F. Berti and R. Adamo, Chem. Soc. Rev., 2016, 45, 1691–1719 RSC; (c) S. B. Gunnoo and A. Madder, Org. Biomol. Chem., 2016, 14, 8002–8013 RSC; (d) J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS PubMed.
- (a) C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed; (b) O. Koniev and A. Wagner, Chem. Soc. Rev., 2015, 44, 5495–5551 RSC; (c) O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed; (d) E. A. Hoyt, P. M. S. D. Cal, B. L. Oliveira and G. J. L. Bernardes, Nat. Rev. Chem., 2019, 3, 147–171 CrossRef CAS; (e) T. Tamura and I. Hamachi, J. Am. Chem. Soc., 2019, 141, 2782–2799 CrossRef CAS PubMed; (f) A. Boto, C. C. González, D. Hernández, I. Romero-Estudillo and C. J. Saavedra, Org. Chem. Front., 2021, 8, 6720–6759 RSC; (g) S. J. Walsh, J. D. Bargh, F. M. Dannheim, A. R. Hanby, H. Seki, A. J. Counsell, X. Ou, E. Fowler, N. Ashman and Y. Takada, et al., Chem. Soc. Rev., 2021, 50, 1305–1353 RSC; (h) C. Sornay, V. Vaur, A. Wagner and G. Chaubet, R. Soc. Open Sci., 2022, 9, 211563 CrossRef CAS PubMed.
- C. S. Sevier and C. A. Kaiser, Nat. Rev. Mol. Cell Biol., 2002, 3, 836–847 CrossRef CAS PubMed.
- (a) S. Lin, X. Yang, S. Jia, A. M. Weeks, M. Hornsby, P. S. Lee, R. V. Nichiporuk, A. T. Iavarone, J. A. Wells and F. D. Toste, et al., Science, 2017, 355, 597–602 CrossRef CAS PubMed; (b) M. T. Taylor, J. E. Nelson, M. G. Suero and M. J. Gaunt, Nature, 2018, 562, 563–568 CrossRef CAS PubMed; (c) J. Kim, B. X. Li, R. Y.-C. Huang, J. X. Qiao, W. R. Ewing and D. W. C. MacMillan, J. Am. Chem. Soc., 2020, 142, 21260–21266 CrossRef CAS PubMed.
- (a) J. M. Antos and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 10256–10257 CrossRef CAS PubMed; (b) Y. Seki, T. Ishiyama, D. Sasaki, J. Abe, Y. Sohma, K. Oisaki and M. Kanai, J. Am. Chem. Soc., 2016, 138, 10798–10801 CrossRef CAS PubMed; (c) S. J. Tower, W. J. Hetcher, T. E. Myers, N. J. Kuehl and M. T. Taylor, J. Am. Chem. Soc., 2020, 142, 9112–9118 CrossRef CAS PubMed.
- (a) J. Ohata, M. B. Minus, M. E. Abernathy and Z. T. Ball, J. Am. Chem. Soc., 2016, 138, 7472–7475 CrossRef CAS PubMed; (b) S. Jia, D. He and C. J. Chang, J. Am. Chem. Soc., 2019, 141, 7294–7301 CrossRef CAS PubMed.
- Recent reviews: (a) P. A. Szijj, K. A. Kostadinova, R. J. Spears and V. Chudasama, Org. Biomol. Chem., 2020, 18, 9018–9028 RSC; (b) D. A. Dorta, D. Deniaud, M. Mével and S. G. Gouin, Chem. - Eur. J., 2020, 26, 14257–14269 CrossRef PubMed.
- N. Echols, P. Harrison, S. Balasubramanian, N. M. Luscombe, P. Bertone, Z. Zhang and M. Gerstein, Nucleic Acids Res., 2002, 30, 2515–2523 CrossRef CAS PubMed.
- N. S. Joshi, L. R. Whitaker and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 15942–15943 CrossRef CAS PubMed.
- (a) J. M. Hooker, E. W. Kovacs and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 3718–3719 CrossRef CAS PubMed; (b) F. W. Kimani and J. C. Jewett, Angew. Chem., Int. Ed., 2015, 54, 4051–4054 CrossRef CAS PubMed.
- (a) H. Ban, J. Gavrilyuk and C. F. Barbas, J. Am. Chem. Soc., 2010, 132, 1523–1525 CrossRef CAS PubMed; (b) H. Ban, M. Nagano, J. Gavrilyuk, W. Hakamata, T. Inokuma and C. F. Barbas, Bioconjugate Chem., 2013, 24, 520–532 CrossRef CAS PubMed; (c) M. Bauer, I. Ahmed, A. Vigovskaya and L. Fruk, Bioconjugate Chem., 2013, 24, 1094–1101 CrossRef PubMed.
- (a) K. Kim, D. A. Fancy, D. Carney and T. Kodadek, J. Am. Chem. Soc., 1999, 121, 11896–11897 CrossRef CAS; (b) K. L. Seim, A. C. Obermeyer and M. B. Francis, J. Am. Chem. Soc., 2011, 133, 16970–16976 CrossRef CAS PubMed; (c) K. Maruyama, T. Ishiyama, Y. Seki, K. Sakai, T. Togo, K. Oisaki and M. Kanai, J. Am. Chem. Soc., 2021, 143, 19844–19855 CrossRef CAS PubMed; (d) A. M. Marmelstein, M. J. Lobba, C. S. Mogilevsky, J. C. Maza, D. D. Brauer and M. B. Francis, J. Am. Chem. Soc., 2020, 142, 5078–5086 CrossRef CAS PubMed; (e) B. X. Li, D. K. Kim, S. Bloom, R. Y.-C. Huang, J. X. Qiao, W. R. Ewing, D. G. Oblinsky, G. D. Scholes and D. W. C. MacMillan, Nat. Chem., 2021, 1–7 Search PubMed; (f) C. S. Mogilevsky, M. J. Lobba, D. D. Brauer, A. M. Marmelstein, J. C. Maza, J. M. Gleason, J. A. Doudna and M. B. Francis, J. Am. Chem. Soc., 2021, 143, 13538–13547 CrossRef CAS PubMed.
- S. D. Tilley and M. B. Francis, J. Am. Chem. Soc., 2006, 128, 1080–1081 CrossRef CAS PubMed.
- (a) E. C. Hett, H. Xu, K. F. Geoghegan, A. Gopalsamy, R. E. Kyne, C. A. Menard, A. Narayanan, M. D. Parikh, S. Liu and L. Roberts, et al., ACS Chem. Biol., 2015, 10, 1094–1098 CrossRef CAS PubMed; (b) H. S. Hahm, E. K. Toroitich, A. L. Borne, J. W. Brulet, A. H. Libby, K. Yuan, T. B. Ware, R. L. McCloud, A. M. Ciancone and K.-L. Hsu, Nat. Chem. Biol., 2020, 16, 150–159 CrossRef CAS PubMed; (c) J. W. Brulet, A. L. Borne, K. Yuan, A. H. Libby and K.-L. Hsu, J. Am. Chem. Soc., 2020, 142, 8270–8280 CrossRef CAS PubMed; (d) T. Huang, S. Hosseinibarkooie, A. L. Borne, M. E. Granade, J. W. Brulet, T. E. Harris, H. A. Ferris and K.-L. Hsu, Chem. Sci., 2021, 12, 3295–3307 RSC.
- T. J. Wadzinski, A. Steinauer, L. Hie, G. Pelletier, A. Schepartz and S. J. Miller, Nat. Chem., 2018, 10, 644–652 CrossRef CAS PubMed.
- (a) P. Wang, Y. Cheng, C. Wu, Y. Zhou, Z. Cheng, H. Li, R. Wang, W. Su and L. Fang, Org. Lett., 2021, 23, 4137–4141 CrossRef CAS PubMed; (b) M. J. Lobba, C. Fellmann, A. M. Marmelstein, J. C. Maza, E. N. Kissman, S. A. Robinson, B. T. Staahl, C. Urnes, R. J. Lew and C. S. Mogilevsky, et al., ACS Cent. Sci., 2020, 6, 1564–1571 CrossRef CAS PubMed.
- J. Ohata, M. K. Miller, C. M. Mountain, F. Vohidov and Z. T. Ball, Angew. Chem., Int. Ed., 2018, 57, 2827–2830 CrossRef CAS PubMed.
- E. M. D. Allouche, E. Grinhagena and J. Waser, Angew. Chem., Int. Ed., 2022, 60, 2–17 Search PubMed.
- P. Caramenti, N. Declas, R. Tessier, M. D. Wodrich and J. Waser, Chem. Sci., 2019, 10, 3223–3230 RSC.
- R. Tessier, J. Ceballos, N. Guidotti, R. Simonet-Davin, B. Fierz and J. Waser, Chem, 2019, 5, 2243–2263 CAS.
- D. Abegg, R. Frei, L. Cerato, D. Prasad Hari, C. Wang, J. Waser and A. Adibekian, Angew. Chem., Int. Ed., 2015, 54, 10852–10857 CrossRef CAS PubMed.
- VBX 4 was probably formed through the nucleophilic addition of an azide to the alkynyl triple bond of EBX. A. Yoshimura, C. D. Huss, M. Liebl, G. T. Rohde, S. M. Larson, G. B. Frahm, M. W. Luedtke, T. J. Schumacher, Z. S. Gardner, V. V. Zhdankin, P. S. Postnikov, M. S. Yusubov, T. Kitamura and A. Saito, Adv. Synth. Catal., 2021, 363, 3365–3371 CrossRef CAS . The azide anion would probably origin from a non-yet identified degradation pathway of EBX 2a..
- Addition of the phosphate or sulfonate salt of the buffer on the EBX..
- O-VBX 3a displays a high stability at low and high pH and temperature and in presence of a low and high concentration of GSH (see ESI† for details)..
- Additional selected examples: (a) D. W. Romanini and M. B. Francis, Bioconjugate Chem., 2008, 19, 153–157 CrossRef CAS PubMed; (b) J. M. McFarland, N. S. Joshi and M. B. Francis, J. Am. Chem. Soc., 2008, 130, 7639–7644 CrossRef CAS PubMed; (c) H.-M. Guo, M. Minakawa, L. Ueno and F. Tanaka, Bioorg. Med. Chem. Lett., 2009, 19, 1210–1213 CrossRef CAS PubMed; (d) J. Gavrilyuk, H. Ban, M. Nagano, W. Hakamata and C. F. Barbas, Bioconjugate Chem., 2012, 23, 2321–2328 CrossRef CAS PubMed.
- M. Alipour, M. Baneshi, S. Hosseinkhani, R. Mahmoudi, A. Jabari Arabzadeh, M. Akrami, J. Mehrzad and H. Bardania, J. Biomed. Mater. Res., 2020, 108, 839–850 CrossRef CAS PubMed.
- K. Graupner, K. Scherlach, T. Bretschneider, G. Lackner, M. Roth, H. Gross and C. Hertweck, Angew. Chem., Int. Ed., 2012, 51, 13173–13177 CrossRef CAS PubMed.
- The bioactivity of the isolated lipopetide 3wa against the pathogenic yeast Candida albicans was tested. Compared to the natural product jagaricin (3w), which displayed a minimal inhibition concentration (MIC) of 1.6 μg/mL, the derivative exhibited a MIC of 50 μg/mL..
- R. Nahta and F. J. Esteva, Oncogene, 2007, 26, 3637–3643 CrossRef CAS PubMed.
- S. Sato, M. Matsumura, T. Kadonosono, S. Abe, T. Ueno, H. Ueda and H. Nakamura, Bioconjug. Chem., 2020, 31, 1417–1424 CrossRef CAS PubMed.
- R. Tessier, R. K. Nandi, B. G. Dwyer, D. Abegg, C. Sornay, J. Ceballos, S. Erb, S. Cianférani, A. Wagner, G. Chaubet, A. Adibekian and J. Waser, Angew. Chem., Int. Ed., 2020, 59, 10961–10970 CrossRef CAS PubMed.
- A. K. Mishra, R. Tessier, D. P. Hari and J. Waser, Angew. Chem., Int. Ed., 2021, 60, 17963–17968 CrossRef CAS PubMed.
- (a) In aqueous media, EBX 2k bearing a sulfonate on the aromatic ring was reported to preferably undergo alkynylation instead of VBX formation with cysteine nucleophiles (ref. 29). The S-VBX products could be observed as a minor product, which could suggest either that an α-addition mechanism was favored or that 1,2-sulfur shift was accelerated (ref. 30b). On the contrary, neutral alkyl-substituted-EBXs such as 2a led to S-VBX products exclusively via a β-addition pathway (ref. 20). If an α-addition would occur with tyrosine, an O-alkynylation reaction would be expected. However, to our knowledge, such reactivity was not reported with EBX reagents and phenol nucleophiles, and the corresponding ynol ether would be expected to be unstable. The lower yield observed with EBX 2j are more difficult to rationalize at this stage.; (b) R. Frei, M. D. Wodrich, D. P. Hari, P.-A. Borin, C. Chauvier and J. Waser, J. Am. Chem. Soc., 2014, 136, 16563–16573 CrossRef CAS PubMed.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- G. Gasparini and S. Matile, Chem. Commun., 2015, 51, 17160–17162 RSC.
- J. López-Andarias, J. Saarbach, D. Moreau, Y. Cheng, E. Derivery, Q. Laurent, M. González-Gaitán, N. Winssinger, N. Sakai and S. Matile, J. Am. Chem. Soc., 2020, 142, 4784–4792 CrossRef PubMed.
- Y. Okamoto, R. Kojima, F. Schwizer, E. Bartolami, T. Heinisch, S. Matile, M. Fussenegger and T. R. Ward, Nat. Commun., 2018, 9, 1943 CrossRef PubMed.
- J. Fu, C. Yu, L. Li and S. Q. Yao, J. Am. Chem. Soc., 2015, 137, 12153–12160 CrossRef CAS PubMed.
- B. Lim, Y. Cheng, T. Kato, A.-T. Pham, E. Le Du, A. K. Mishra, E. Grinhagena, D. Moreau, N. Sakai, J. Waser and S. Matile, Helv. Chim. Acta, 2021, 104, e2100085 CrossRef CAS.
- A. D. de Araujo, H. N. Hoang, J. Lim, J. Mak and D. P. Fairlie, Angew. Chem., Int. Ed., 2022, anie.202203995 Search PubMed.
- T. Kato, B. Lim, Y. Cheng, A.-T. Pham, J. Maynard, D. Moreau, A. I. Poblador-Bahamonde, N. Sakai and S. Matile, JACS Au, 2022, 2, 839–852 CrossRef CAS PubMed.
- D. C. Luther, T. Jeon, R. Goswami, H. Nagaraj, D. Kim, Y.-W. Lee and V. M. Rotello, Bioconjug. Chem., 2021, 32, 891–896 CrossRef CAS PubMed.
- S. Du, S. S. Liew, L. Li and S. Q. Yao, J. Am. Chem. Soc., 2018, 140, 15986–15996 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterization and microscopy images. See DOI: https://doi.org/10.1039/d2sc04558c |
| This journal is © The Royal Society of Chemistry 2022 |