
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular flavin catalysts for C–H functionalisation and derivatisation of dehydroamino acids†
Andreas
Rehpenn
,
Alexandra
Walter
and
Golo
Storch
 *
*
School of Natural Sciences and Catalysis Research Center (CRC), Technical University of Munich (TUM), Lichtenbergstr. 4, 85747 Garching, Germany. E-mail: golo.storch@tum.de
First published on 14th November 2022
Abstract
In nature, the isoalloxazine heterocycle of flavin cofactors undergoes reversible covalent bond formation with a variety of different reaction partners. These intermediates play a crucial role inter alia as the signalling states and in selective catalysis reactions. In the organic laboratory, covalent adducts with a new carbon–carbon bond have been observed with photochemically excited flavins but have, so far, only been regarded as dead-end side products. We have identified a series of molecular flavins that form adducts resulting in a new C–C bond at the C4a-position through allylic C–H activation and dehydroamino acid oxidation. Typically, these reactions are of radical nature and a stepwise pathway is assumed. We could demonstrate that these adducts are no dead-end and that the labile C–C bond can be cleaved by adding the persistent radical TEMPO leading to flavin regeneration and alkoxyamine-functionalised substrates. Our method allows for the catalytic oxidation of dehydroamino acids (16 examples) and we show that the acylimine products serve as versatile starting points for diversification. The present results are envisioned to stimulate the design of further catalytic reactions involving intermediates at the flavin C4a-position and their reactivity towards metal complexes or other persistent organic radicals. Our method for dehydrobutyrine derivatisation is orthogonal to the currently used methods (i.e., nucleophilic attack or radical addition) and offers new perspectives for peptide natural product diversification.
Introduction
The isoalloxazine core of flavins has a strong tendency to form covalent adducts with several reagents.1 In nature (Fig. 1A), flavin adenine dinucleotide (FAD, 1) and flavin adenine mononucleotide (FMN) reversibly form covalent C–S bonds in the C4a-position with cysteine in light, oxygen, and voltage (LOV) photoreceptors to yield covalent intermediate 2.2 In a second example from flavoenzymes, the reduced cofactor FADH2 (3) reacts with oxygen to yield C4a-hydroperoxide 4 which is involved in oxygenation reactions.3 Besides these well-studied types of intermediates, the reversible formation of a carbon–carbon single bond at the flavin's C4a-position is less common. In the organic laboratory, Hemmerich et al. (Fig. 1B) have reported the photochemical decarboxylation of phenylacetic acid by quinoid lumiflavin derivative 5, which results in the clean formation of a C4a-benzyl flavin 6.4,5 The authors expanded on these findings and could observe that a number of organic substances undergo photochemical reactions yielding covalent C–C bonds in the C4a-position. One example is compound 7 which resulted from the reaction with 2,5-dimethyl-2,4-hexadiene.6 Related C4a-benzyl adducts have also been observed by photochemical oxidation of benzylamines.7 Such flavin species with a new covalent C–C bond at the C4a-position were often found to be unstable when exposed to air.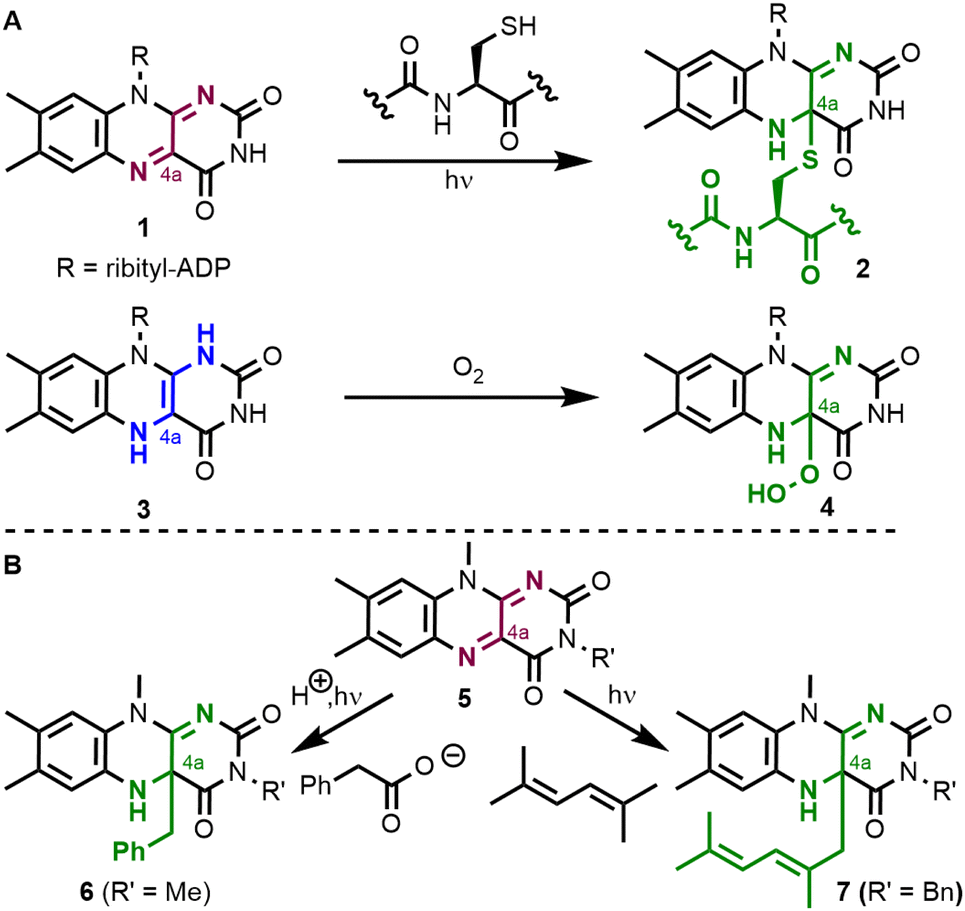 | ||
| Fig. 1 Covalent adducts of molecular flavins in the C4a-position. Oxygen and sulfur heteroatom–carbon bonds are formed in LOV domains and oxygenating flavoenzymes (A) while C–C bonds are known from photochemical oxidations (B). | ||
Both the formation of covalent C–C adducts at the C4a-position and presumably also their reaction with O2 are typically radical pathways. We wondered whether the formal homolysis step would allow using flavin C–C adducts as a reservoir for carbon-centred substrate radicals. Following this rationale, the adducts should also react with other radical species besides O2 and we were particularly interested in the reaction with the persistent radical TEMPO. On the substrate side, we focused on the activation of allylic C–H positions in olefins and the oxidation of dehydroamino acids. Radical addition reactions of the latter substrates are a perfect tool for the diversification of peptide natural products,8 which are of high pharmaceutical relevance.9 The typical protocol relies on radical generation and addition to a dehydroamino acid's β-position.10 An iridium-mediated generation of heteroatom-stabilized radicals from dimethylanilines and their subsequent addition to a short peptide 8 yielding conjugate 9 serves as a representative example here (Fig. 2A).11 The orthogonal approach, which relies on the oxidation of a dehydroamino acid and its subsequent intermolecular reaction is, however, much less explored. This reactivity was found in PCET-type12 oxidation of dehydrophenylalanine 10 (BDFE = 456 kJ mol−1) with Ag2O to an intermediate radical 11, which reacts with TEMPO at the β-position yielding alkoxyamine product 12 (Fig. 2B).13 Related TEMPO-functionalisation of peptides has also been accomplished by dechalcogenation.14
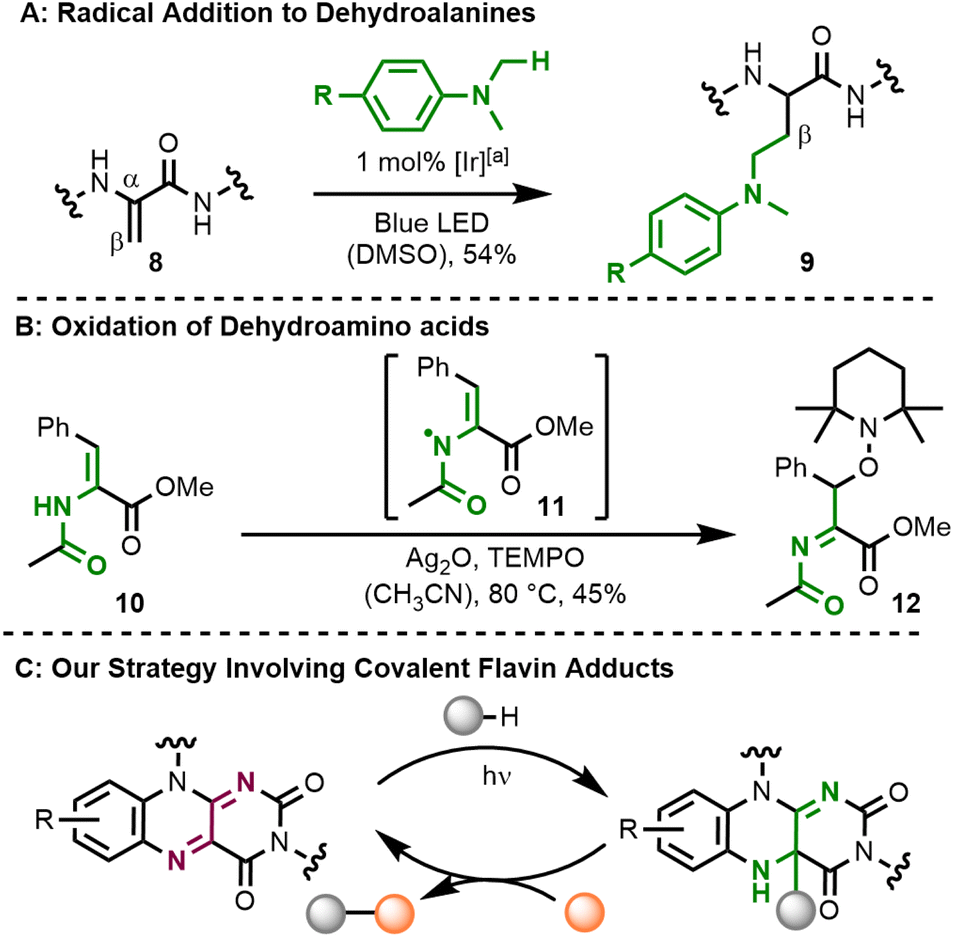 | ||
| Fig. 2 Radical functionalisation of dehydroamino acids. (A) The addition of C-centred radicals to dehydroalanine-containing peptides allows access to conjugates, which contain diverse substituents (in the R-position) such as drug compounds. (B) Silver(I)oxide was found to be a capable base and oxidant for PCET-activation of dehydroamino acids. (C) Our concept for the use of flavin catalysts as substrate oxidants. [a] Iridium catalyst: [Ir(dF(CF3)ppy)2(dtbbpy)](PF6). | ||
We wondered whether such functionalisation of dehydroamino acids could be performed catalytically with molecular flavins.15 While their use as photo-oxidants is well-established,16 we were in particular interested in using covalent intermediates at the flavin's C4a-position as stabilised substrate radicals (Fig. 2C).
Results and discussion
We first prepared a series of molecular flavins which contain electron-withdrawing substituents.17 Flavin synthesis starts with a sequence of SNAr-reaction and nitro group reduction to ortho-diaminoaryls 13 (Fig. 3). The isoalloxazine core is then formed by a two-step reaction involving oxidation of monobutyl barbituric acid followed by its condensation with diamines 13. We prepared flavins with a methyl carboxylate (14) and methyl carboxamide (15) in the C6-position. For comparison reasons we also prepared the C7-methyl carboxylate 16 and the parent alkyl-substituted isoalloxazine 17. This choice of substitution pattern was based on the possibility of forming a stabilising intramolecular hydrogen bond in semiquinone 18, which has been observed in similar cases.18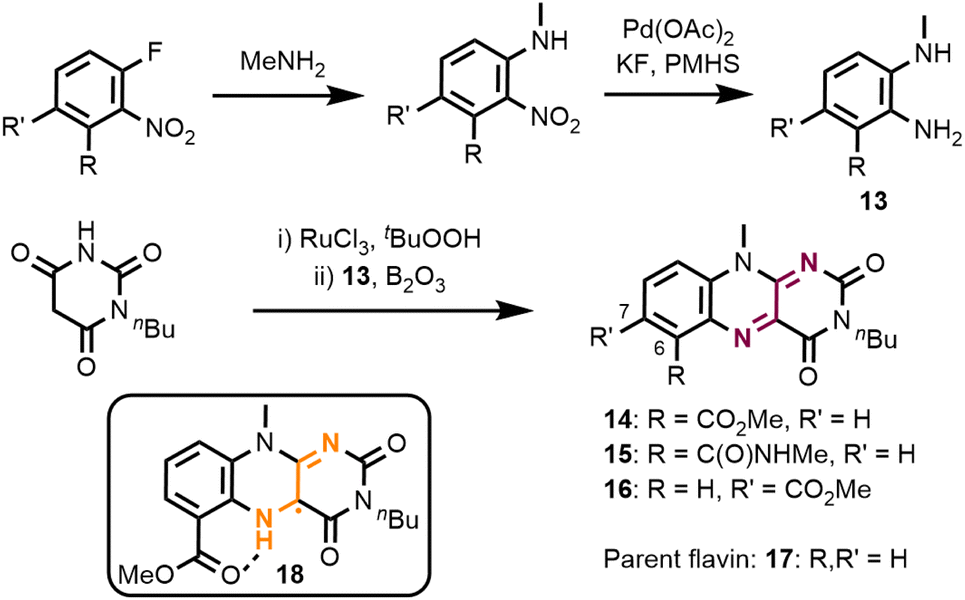 | ||
| Fig. 3 Flavin catalyst synthesis. For reaction conditions and yields of diamines 13, see the ESI.† Flavin synthesis: 2.5 equiv. N-butyl barbituric acid, 3 mol% RuCl3·(H2O)3, 5.0 equiv. tBuOOH (CH2Cl2/H2O), r.t., 22 h; then 1.0 equiv. B2O3 (AcOH), r.t., o/n. Yields for this two-step procedure: 59% (14), 42% (15), and 33% (16). PMHS: poly(methylhydrosiloxane). | ||
Following our design principle, we directly started with ester-modified flavin 14 and interrogated the photochemical allylic activation of cycloheptadiene (BDE for allylic C–H bond is 347.3 kJ mol−1)19 as a model substrate. Indeed, when irradiating a solution of flavin and diene in CD2Cl2 under inert conditions in a J Young NMR tube, we observed the clean formation of covalent adduct 19 (as a set of two diastereomers), which has a newly established C–C bond at the C4a-position (Fig. 4A). This compound was characterized by 2D-NMR and HR-ESI (see ESI† for details). Upon contact with air, quinoid flavin 14 was regenerated. We then studied whether the new C–C bond is weak enough to be cleaved upon the addition of a persistent radical and treated adduct 19 with TEMPO under inert conditions (Fig. 4B). Again, we observed the formation of quinoid flavin 14 but we could also detect the stoichiometric release of alkoxyamine-functionalised diene 20 in the NMR tube (Fig. 4C).
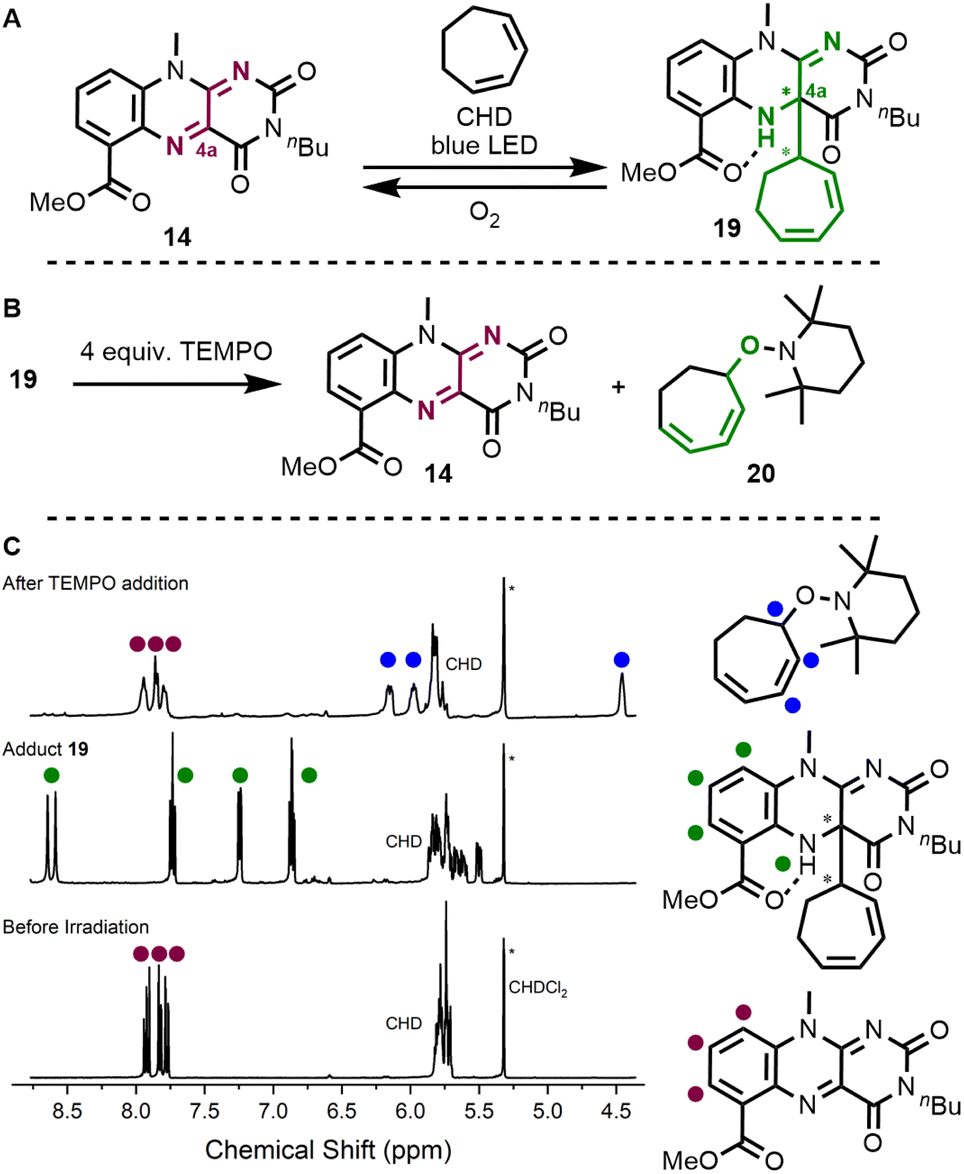 | ||
| Fig. 4 Studies of covalent adduct formation. The reaction of flavin 14 with cycloheptadiene (CHD) was found to liberate quinoid flavin when the adduct is exposed to air (A). When conducted under inert conditions, the weak C–C bond at the C4a-position can also be cleaved by addition of TEMPO (B and C). | ||
From a synthetic standpoint, the formation of alkoxyamine 20 reminded us of the known reactivity of alkenes towards nitroxyl radicals20 and oxoammonium cations,21 which is typically very low with disubstituted olefins. We wondered, whether our flavin activation method might offer a way to catalyse these reactions and found that a flavin-mediated method with TEMPO as a reactant and oxidant indeed leads to product formation (Table 1, entry #1). When using Bobbitt's salt (21) instead of TEMPO, only one equivalent of the reagent is required since the oxoammonium salt not only acts as an oxidant (Emp [nitroxo+/nitroxyl˙] = +0.65 V vs. Ag/AgCl (+0.61 V vs. SCE)),22 but also liberates the persistent radical reaction partner. Potassium phosphate is added as a base to trap the released fluoroboric acid (entry #2). The parent flavin RFTA(Me) performed worse (entry #3) and all control experiments verified that a photochemically excited flavin is required (entries #4 and #5).
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Change of conditions | Yielda,b |
| a Determined by NMR vs. internal standard. b n.d. = no product formation was detected. c TEMPO (without a base) was used instead of Bobbitt's salt 21 leading to product 20. d Isolated yield. e (−)-N-3-Methyl riboflavin tetraacetate. | |||
| #1 | 14 | TEMPO instead of 21 | 27%c |
| #2 | 14 | — | 42% (37%)d |
| #3 | RFTA(Me)e | — | 17% |
| #4 | 14 | No irradiation | n.d./n.d.c |
| #5 | None | — | n.d./n.d.c |

With these encouraging results in hand, we investigated whether flavin catalysis would also allow the oxidative TEMPO-functionalisation of more complex substrates such as dehydroamino acids (c.f.Fig. 2). We chose dehydrobutyrine 23 as a model substrate and indeed, the formation of acylimine 24 was achieved.
Here, the use of TEMPO (3.0 equiv.) was found to result in better yields when compared to Bobbitt's salt 21 (Table 2, entries #1 and #2). The carboxamide flavin 15, C7-substituted ester 16, and parent flavin 17 all resulted in lower yields (entries #3–#5). Since catalysis product 24 is sensitive towards hydrolysis, the addition of water (10 equiv.) to the reaction mixture resulted in only a trace amount of product (entry #6). The control reactions (entries #7 and #8) confirmed that the reaction is driven by a photochemically excited flavin catalyst.
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Change of conditions | Yielda,b |
| a Determined by NMR vs. internal standard. b n.d. = no product formation was detected. c Acylimine 24 slowly hydrolyses during purification by column chromatography and a significantly reduced isolated yield (14%) was obtained. d Reagent 21 (1.0 equiv. together with K3PO4 as a base) was used instead of TEMPO leading to product 24NHAc with an acetamide substituent in the piperidine's 4-position. | |||
| #1 | 14 | — | 60%c |
| #2 | 14 | 21 instead of TEMPO | 6%d |
| #3 | 15 | — | 15% |
| #4 | 16 | — | 35% |
| #5 | 17 | — | 43% |
| #6 | 14 | With 10 equiv. H2O | <5% |
| #7 | 14 | No irradiation | n.d./n.d.d |
| #8 | None | — | n.d./n.d.d |
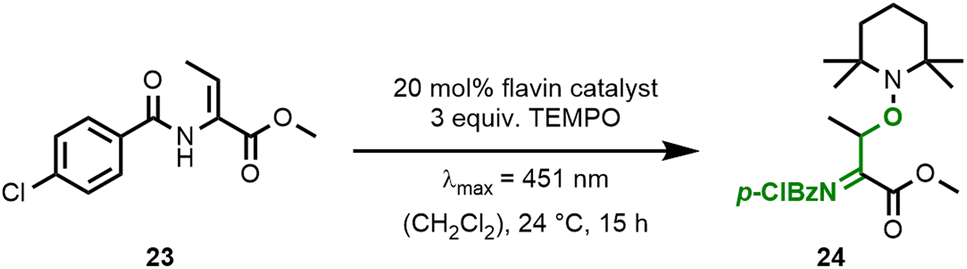
We were intrigued by the improved activity of ester-modified flavin 14 and went on to study similarities and differences when compared to RFTA(Me). Both quinoid flavins show relatively similar spectroscopic properties (Fig. 5A, see ESI† for details): λabsmax = 442 nm, E(S0 ← S1) = 246 kJ mol−1 (EtOH, r.t.), and E(S0 ← T1) = 205 kJ mol−1 (EtOH, 77 K) (14) vs. λabsmax = 448 nm, E(S0 ← S1) = 244 kJ mol−1 (EtOH, r.t.), and E(S0 ← T1) = 205 kJ mol−1 (EtOH, 77 K) (RFTA(Me)). Cyclic voltammetry, however, revealed characteristic differences: The N3-alkylated flavin RFTA(Me) shows a simple cyclic voltammogram with a reversible reduction process (E1/2 = −0.86 V vs. SCE, see ESI† for details).23 Under analogous conditions, flavin 14 shows two separate oxidation waves (waves 2 and 3) and a half-wave potential of E1/2 = −0.72 V vs. SCE for the reversible reduction. We rationalise this observation by a significantly faster protonation of the immediately formed anion 14˙− to the neutral radical 18, which is reasonable based on the favoured intramolecular hydrogen bonding. This neutral semiquinone 18 is easier to reduce than quinoid flavin 14 and, therefore, it is converted to hydroquinoid 18− in wave 1. Reoxidation of 14˙− (wave 2) and 18− (wave 3) then occur separately. In order to prove this hypothesis, we accelerated the protonation step by measuring cyclic voltammograms in the presence of acetic acid.24 Indeed, this resulted in the detection of only one oxidation wave, which corresponds to the oxidation of 18− (Fig. 5B, see ESI† for details).
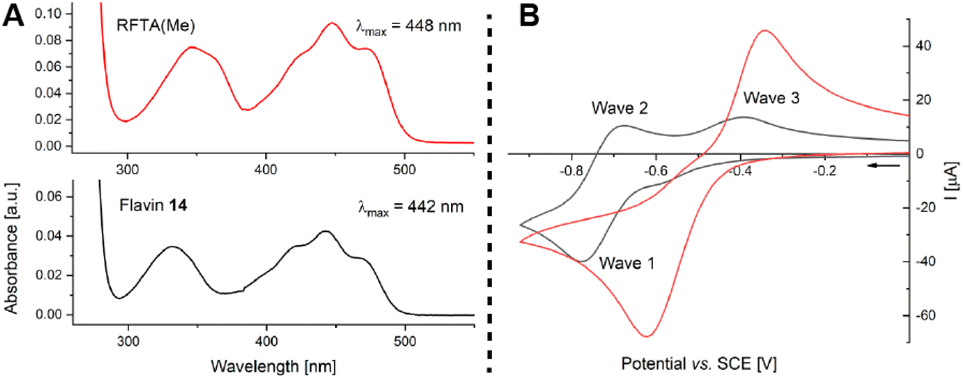 | ||
| Fig. 5 Studies of flavin 14 by UV/Vis spectroscopy and cyclic voltammetry. (A) Absorption spectra of RFTA(Me) (top, red) and flavin 14 (bottom, black) in CH2Cl2 (0.1 mM). (B) Cyclic voltammetry of flavin 14 in 0.1 M TBAPF6/CH3CN with a scan rate of 0.5 V s−1 (in black) and in the presence of 0.4 equiv. AcOH (in red). | ||
The flavin-catalysed method was successfully applied to the oxidative modification of a variety of dehydroamino acid substrates (Fig. 6), which also do not show any uncatalysed reactivity under our conditions with either TEMPO or Bobbitt's salt (see ESI† for attempted conversion of a dehydrobutyrine substrate to acylimine 25). Owing to the decreased electrophilicity of the acylimine amides compared to ester-derived product 24, the former are significantly less reactive towards water hydrolysis and their isolation is straightforward. Similar results were observed when increasing the size of the alkyl chain on the substrate's C–C double bond or the amide (products 25–28). The acylamides 29–31 from amides consisting of two amino acid residues were also successfully formed. We found that our method is not limited to benzamide substrates and the readily available Boc- and Cbz-protected products 32–34 were obtained in reasonable yields as well. Aromatic amino acid side chains such as phenylalanine (35) or protected histidine (36) and tyrosine (37) are also tolerated. Dehydroamino acid amides are significantly more reactive compared to the analogous esters, which allowed the selective formation of acylimine 25 with only negligible amounts of ester 38 in a competition experiment. The same preference was observed in an intramolecular setting, where mono-functionalised acylimine 39 was the only species we observed when using a substrate with two dehydrobutyrine residues.
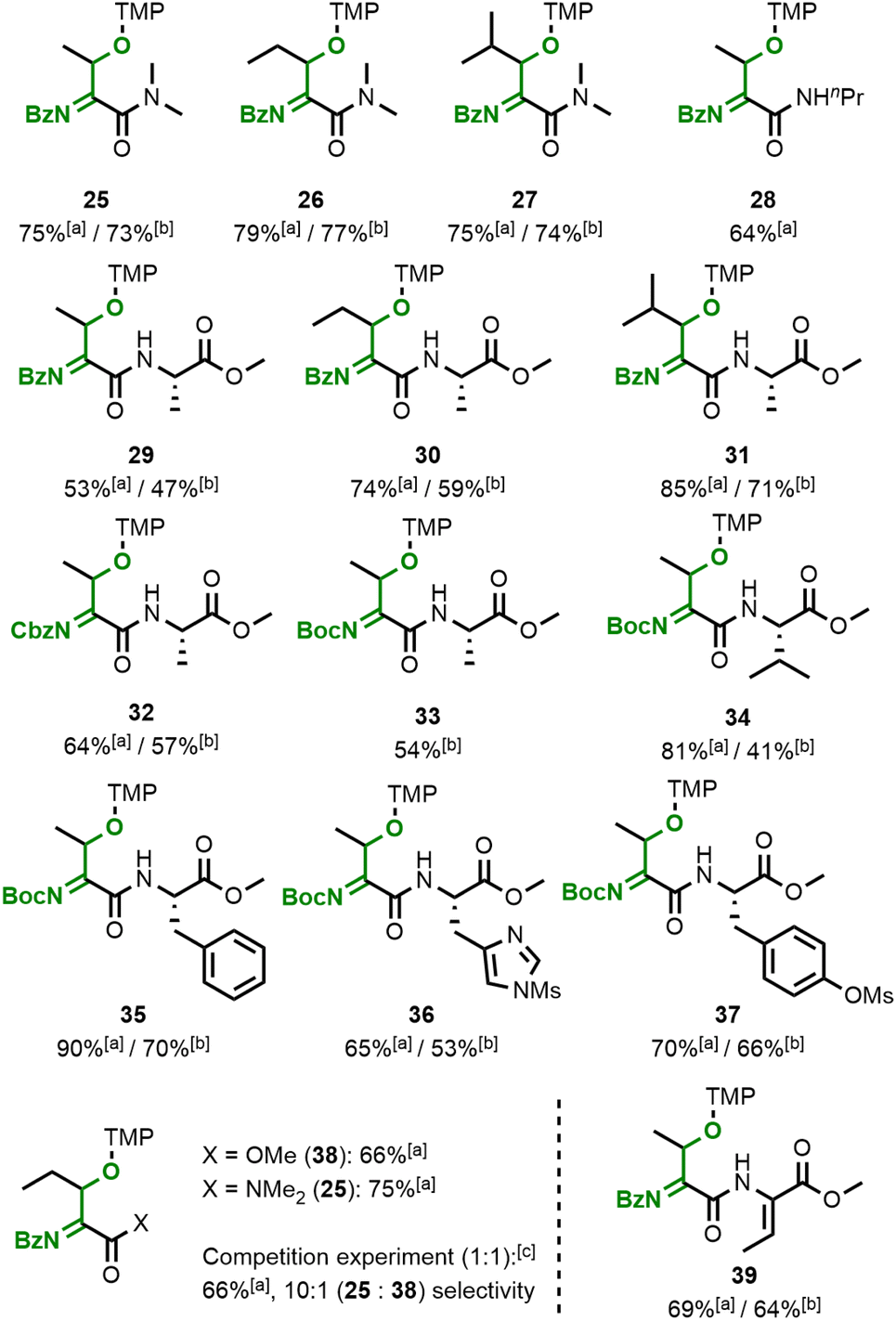 | ||
| Fig. 6 Scope of the flavin-catalysed oxidative functionalisation of dehydroamino acids. Conditions: 20 mol% flavin 14, 3.0 equiv. TEMPO, λmax = 451 nm (CH2Cl2), 15 °C, 15 h. Compound 28 evaded isolation due to hydrolysis and the NMR yield of 33 could not be obtained due to overlapping signals. [a]: Determined by NMR vs. internal standard. [b]: Isolated yield. [c]: 2.0 equiv. TEMPO used. TMP = tetramethylpiperidine. | ||
We subsequently interrogated the mechanism of dehydroamino acid activation. Consistent with initial radical formation at the amide nitrogen position yielding an N-centered radical, we did not observe any conversion of N-methylated substrate (E)-41 under standard catalysis conditions (see ESI†). When irradiating this amino acid substrate together with flavin 14 in a J Young NMR tube under the exclusion of air, we confirmed that neither E/Z-isomerisation25 nor substrate conversion takes place (Fig. 7). This also holds true for the isomeric (Z)-41, which was also not converted under these conditions. In contrast, oxidation of both (E)- and (Z)-40 results in radical formation at the dehydroamino acid's β-position. When we conducted such experiments in J Young NMR tubes with the exclusion of air and without TEMPO, we observed the formation of covalent flavin adducts within minutes and confirmed this by HR-ESI measurements. Careful inspection of 2D-NMR spectra and NOE-contacts corroborated our assignment of a covalent C–C adduct at the C4a-position (see ESI† for details). Flavin adduct 42 is formed as an almost equal mixture of two diastereomers. When adduct 42 is irradiated in the presence of TEMPO, quinoid flavin 14 is regenerated and acylimine 25 is released.
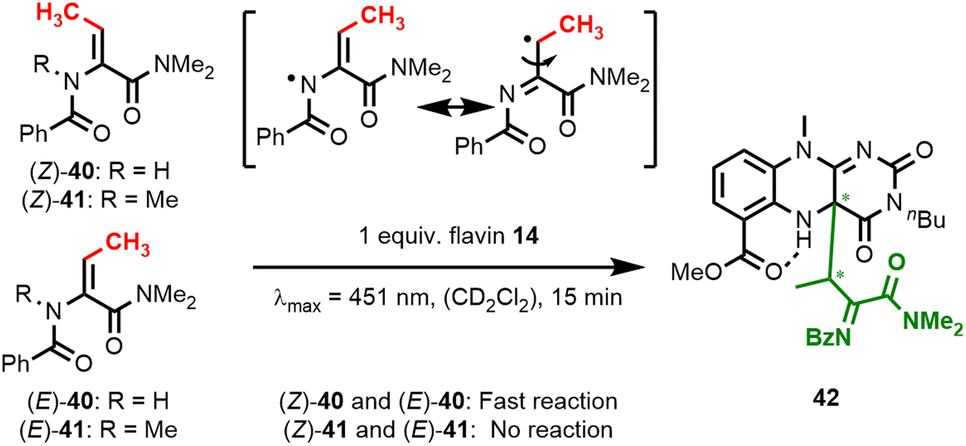 | ||
| Fig. 7 Irradiation experiments with dehydrobutyrine substrates 40 and 41 in J Young NMR tubes under the exclusion of air. | ||
Acylimines are easily reduced to the corresponding amides by sodium borohydride, which leaves the TEMPO-functionalisation unaltered (Fig. 8A). Also, the relatively weak C–O bond in TEMPO-containing organic products allows subsequent transformations,26,27 and we demonstrated the oxidation of alkoxyamine 43 to ketone 44 by mCPBA. All of the obtained acylimine products resemble reactive electrophiles and, therefore, offer the possibility for one-pot reactions leading to diversified amino acid products. Acylimine 28 was found to be unstable during chromatography with silica gel, but when subjected to a one-pot malonate addition sequence,28 the formation of stable pyrrolidine-2,5-dione 45 was observed (Fig. 8B). We confirmed the relative stereochemistry in the five-membered ring by NOE-contacts. There are also literature protocols for the reduction of acylimines to the corresponding amides.29 As a representative example for transforming the weak C–O bond in the alkoxyamine products, we performed a one-pot three-step sequence starting from dehydrobutyrine 23 with subsequent malonate addition and mCPBA oxidation to ketone 46 (Fig. 8C). These examples shall demonstrate that flavin catalysis is a powerful tool for the functionalisation of dehydroamino acid derivatives.
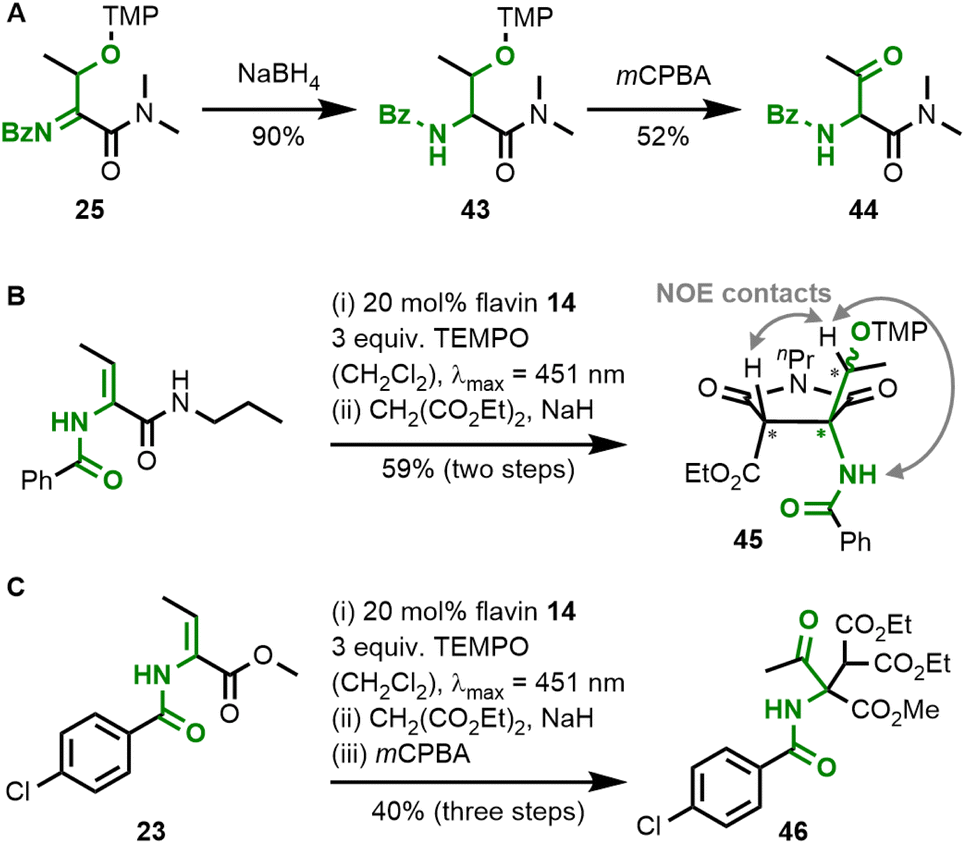 | ||
| Fig. 8 Examples of synthetic one-pot modifications of dehydrobutyrine substrates in the β-position mediated by flavin catalysts: (A) reduction of acylimine 25 and subsequent TEMPO cleavage. (B) Two-step TEMPO-functionalisation and subsequent malonate addition. (C) Three-step reaction with oxidation of the alkoxyamine product to ketone 46. | ||
Conclusions
This proof-of-concept study of the reversible formation of covalent C–C bonds at the flavin's C4a-position demonstrates that such compounds can indeed be intermediates in catalytic cycles rather than only dead-end structures. In the presented transformations, flavin catalysis allows the functionalisation of dehydroamino acid substrates in the β-position. Such acylimine products offer a variety of different follow-up reactions and, therefore, our method is envisioned to be a valuable tool for amino acid diversification. Building on the general reactivity of flavin adducts at the C4a-position, we also expect other transformations to make use of such intermediates in the future. The dehydroamino acid derivatisation strategies may add to the toolbox of useful strategies in peptide natural product diversification.Data availability
Original NMR datasets (FIDs) are available at Open ScienceFramework at https://osf.io/jyf9p/.Author contributions
A. R. performed and analysed the experiments. A. R. and G. S. designed the experiments and wrote the manuscript. G. S. supervised the project. A. W. performed and analysed the cyclic voltammetry measurements. All authors contributed to the analysis of data and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Fonds der Chemischen Industrie (FCI, PhD Fellowship to A. W. and Liebig Fellowship to G. S.) is gratefully acknowledged. The project was funded by the Deutsche Forschungsgemeinschaft (Emmy Noether Programme, STO 1175/3-1). We thank J. Groβkopf for spectroscopic measurements. Our group is supported by the Technical University of Munich through the Junior Fellow Programme. G. S. is very grateful to Prof. T. Bach for his continuous support.References
- V. Piano, B. A. Palfey and A. Mattevi, Trends Biochem. Sci., 2017, 42, 457 CrossRef CAS.
- (a) Y. Sato, T. Iwata, S. Tokutomi and H. Kandori, J. Am. Chem. Soc., 2005, 127, 1088 CrossRef CAS; (b) K. Magerl, I. Stambolic and B. Dick, Phys. Chem. Chem. Phys., 2017, 19, 10808 RSC; (c) A. Losi, K. H. Gardner and A. Möglich, Chem. Rev., 2018, 118, 10659 CrossRef CAS PubMed; (d) R. N. A. Maia, D. Ehrenberg, S. Oldemeyer, E. Knieps-Grünhagen, U. Krauss and J. Heberle, J. Am. Chem. Soc., 2021, 143, 12535 CrossRef CAS PubMed.
- (a) V. Massey, J. Biol. Chem., 1994, 269, 22459 CrossRef CAS; (b) E. Romero, J. R. Gómez Castellanos, G. Gadda, M. W. Fraaije and A. Mattevi, Chem. Rev., 2018, 118, 1742 CrossRef CAS.
- P. Hemmerich, V. Massey and G. Weber, Nature, 1967, 213, 728 CrossRef CAS PubMed.
- For related decarboxylative adduct formation, see: M. Novak, A. Miller, T. C. Bruice and G. Tollin, J. Am. Chem. Soc., 1980, 102, 1465 CrossRef CAS.
- (a) W. R. Knappe and P. Hemmerich, Z. Naturforsch., B: Anorg. Chem., Org. Chem., Biochem., Biophys., Biol., 1972, 27, 1032 CrossRef CAS PubMed; (b) W.-R. Knappe and P. Hemmerich, Liebigs Ann. Chem., 1976, 2037 CrossRef CAS.
- J. M. Kim, I. S. Cho and P. S. Mariano, J. Org. Chem., 1991, 56, 4943 CrossRef CAS.
- (a) J. W. Bogart and A. A. Bowers, Org. Biomol. Chem., 2019, 17, 3653 RSC; (b) J. R. Immel, M. Chilamari and S. Bloom, Chem. Sci., 2021, 12, 10083 RSC; (c) X. Peng, K. Xu, Q. Zhang, L. Liu and J. Tan, Trends Chem., 2022, 4, 643 CrossRef CAS.
- (a) J. A. McIntosh, M. S. Donia and E. W. Schmidt, Nat. Prod. Rep., 2009, 26, 537 RSC; (b) T. Dang and R. D. Süssmuth, Acc. Chem. Res., 2017, 50, 1566 CrossRef CAS PubMed.
- (a) J. R. Immel, M. Chilamari and S. Bloom, Chem. Sci., 2021, 12, 10083 RSC; (b) J. A. C. Delgado, J. T. M. Correia, E. F. Pissinati and M. W. Paixão, Org. Lett., 2021, 23, 5251 CrossRef CAS.
- R. A. Aycock, C. J. Pratt and N. T. Jui, ACS Catal., 2018, 8, 9115 CrossRef CAS.
- L. Q. Nguyen and R. R. Knowles, ACS Catal., 2016, 6, 2894 CrossRef CAS.
- H.-Q. Cao, H.-N. Liu, Z.-Y. Liu, B. Qiao, F.-G. Zhang and J.-A. Ma, Org. Lett., 2020, 22, 6414 CrossRef CAS PubMed.
- R. C. Griffiths, F. R. Smith, J. E. Long, H. E. L. Williams, R. Layfield and N. J. Mitchell, Angew. Chem., Int. Ed., 2020, 59, 23659 CrossRef CAS.
- For a review on molecular flavin catalysis, see: A. Rehpenn, A. Walter and G. Storch, Synthesis, 2021, 53, 2583 CrossRef CAS.
- For selected examples, see: (a) B. Mühldorf and R. Wolf, ChemCatChem, 2017, 9, 920 CrossRef; (b) M. Lesieur, C. Genicot and P. Pasau, Org. Lett., 2018, 20, 1987 CrossRef CAS; (c) S. Bloom, C. Liu, D. K. Kölmel, J. X. Qiao, Y. Zhang, M. A. Poss, W. R. Ewing and D. W. C. MacMillan, Nat. Chem., 2018, 10, 205 CrossRef CAS PubMed.
- Y.-M. Legrand, M. Gray, G. Cooke and V. M. Rotello, J. Am. Chem. Soc., 2003, 125, 15789 CrossRef CAS PubMed.
- T. Akiyama, F. Simeno, M. Murakami and F. Yoneda, J. Am. Chem. Soc., 1992, 114, 6613 CrossRef CAS.
- Y.-R. Luo, Handbook of Bond Dissociation Energies in Organic Compounds, CRC Press, Boca Raton, FL, 2003 Search PubMed.
- J. E. Babiarz, G. T. Cunkle, A. D. DeBellis, D. Eveland, S. D. Pastor and S. P. Shum, J. Org. Chem., 2002, 67, 6831 CrossRef CAS.
- P. P. Pradhan, J. M. Bobbitt and W. F. Bailey, Org. Lett., 2006, 8, 5485 CrossRef CAS.
- M. Rafiee, K. C. Miles and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 14751 CrossRef CAS.
- M. März, M. Kohout, T. Neveselý, J. Chudoba, D. Prukała, S. Niziński, M. Sikorski, G. Burdziński and R. Cibulka, Org. Biomol. Chem., 2018, 16, 6809 RSC.
- For similar studies with alkylated flavins, see: (a) A. Niemz, J. Imbriglio and V. M. Rotello, J. Am. Chem. Soc., 1997, 119, 887 CrossRef CAS; (b) S. L. J. Tan and R. D. Webster, J. Am. Chem. Soc., 2012, 134, 5954 CrossRef CAS; (c) L. N. Mataranga-Popa, I. Torje, T. Ghosh, M. J. Leitl, A. Späth, M. L. Novianti, R. D. Webster and B. König, Org. Biomol. Chem., 2015, 13, 10198 RSC; (d) S. L. J. Tan, M. L. Novianti and R. D. Webster, J. Phys. Chem. B, 2015, 119, 14053 CrossRef CAS.
- For E/Z-isomerisation with flavins, see: (a) J. B. Metternich and R. Gilmour, J. Am. Chem. Soc., 2015, 137, 11254 CrossRef CAS; (b) J. B. Metternich, D. G. Artiukhin, M. C. Holland, M. von Bremen-Kühne, J. Neugebauer and R. Gilmour, J. Org. Chem., 2017, 82, 9955 CrossRef CAS.
- (a) A. Studer, Angew. Chem., Int. Ed., 2000, 39, 1108 CrossRef CAS; (b) E. C. Gentry, L. J. Rono, M. E. Hale, R. Matsuura and R. R. Knowles, J. Am. Chem. Soc., 2018, 140, 3394 CrossRef CAS; (c) S. Heindl, M. Riomet, J. Matyasovsky, M. Lemmerer, N. Malzer and N. Maulide, Angew. Chem., Int. Ed., 2021, 60, 19123 CrossRef CAS.
- For examples of reactions with TEMPO-functionalised substrates, see: (a) T. Inokuchi and H. Kawafuchi, Tetrahedron, 2004, 60, 11969 CrossRef CAS; (b) L. Tebben and A. Studer, Angew. Chem., Int. Ed., 2011, 50, 5034 CrossRef CAS PubMed; (c) G. Audran, P. Brémond and S. R. A. Marque, Chem. Commun., 2014, 50, 7921 RSC; (d) Q. Zhu, E. C. Gentry and R. R. Knowles, Angew. Chem., Int. Ed., 2016, 55, 9969 CrossRef CAS; (e) G. Schulz and A. Kirschning, Org. Biomol. Chem., 2021, 19, 273 RSC.
- M. L. Graziano, A. Carotenuto, M. R. Iesce and R. Scarpati, Tetrahedron Lett., 1977, 18, 447 CrossRef.
- Y. Qian, C. Jing, C. Zhai and W.-h. Hu, Adv. Synth. Catal., 2012, 354, 301 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc04341f |
| This journal is © The Royal Society of Chemistry 2022 |