
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConfinement and passivation of perovskite quantum dots in porous natural palygorskite toward an efficient and ultrastable light-harvesting system in water†
Genping
Meng‡
 a,
Xijiao
Mu‡
a,
Liping
Zhen
a,
Jun
Hai
a,
Zefan
Zhang
a,
Tianzhi
Hao
a,
Siyu
Lu
*c,
Aiqin
Wang
*b and
Baodui
Wang
*a
a,
Xijiao
Mu‡
a,
Liping
Zhen
a,
Jun
Hai
a,
Zefan
Zhang
a,
Tianzhi
Hao
a,
Siyu
Lu
*c,
Aiqin
Wang
*b and
Baodui
Wang
*a
aState Key Laboratory of Applied Organic Chemistry, Key Laboratory of Nonferrous Metal Chemistry and Resources Utilization of Gansu Province, Lanzhou University, Gansu, Lanzhou 730000, China. E-mail: wangbd@lzu.edu.cn
bKey Laboratory of Clay Mineral Applied Research of Gansu Province, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou, Gansu 730000, China. E-mail: aqwang@licp.cas.cn
cCollege of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou 450001, China. E-mail: sylu2013@zzu.edu.cn
First published on 17th November 2022
Abstract
Perovskite quantum dots (QDs) are promising as representative candidates to construct next-generation superior artificial light-harvesting systems (ALHSs). However, their high sensitivity to external environments, especially to water, imposes a stringent limitation for their actual implementation. Herein, by interface engineering and encapsulation with natural palygorskite (PAL), a water-resistant light-harvesting CsPbBr3@PAL antenna was prepared. Molecular dynamics simulations further confirm a significant shielding protection of the PAL matrix to CsPbBr3, facilitating exceptional stability of the CsPbBr3@PAL antenna when exposed to air for 10 months, to 150 °C thermal stress, and even to water for more than 30 days, respectively. Furthermore, as a result of in situ encapsulation of the PAL matrix and defect passivation caused by H-bonding and coordination-bonding interaction, the CsPbBr3@PAL antenna in water shows a substantially enhanced photoluminescence quantum yield (36.2%) and longer lifetime. After sequentially assembling Eosin Y and Rose Bengal in the pores of the PAL matrix, RB-ESY-CsPbBr3@PAL with a sequential two-step efficient Förster resonance energy transfer process exhibited extremely enhanced photocatalytic activity toward Friedel–Crafts alkylation reactions in aqueous solution, 2.5-fold higher than that of corresponding ESY/RB. Our work provides a feasible strategy for the exploitation of ultra-stable halide perovskite-based ALHSs in aqueous media for solar-energy conversion.
1. Introduction
In nature, photosynthesis is the basic element for plants and other organisms to obtain life-sustaining energy, while helping to keep the terrestrial environment suitable for the survival of other living things.1–3 Inspired by nature, many scientists have begun to pay attention to the development of artificial light-harvesting systems (ALHSs),4–7 which have promoted the remarkable development in optoelectronic and photonic materials and devices such as photocatalysis,7–10 solar cells,11–14 optical sensors,15–18 and luminescent materials.19–22 However, most of the reported sophisticated ALHSs are manufactured in organic solvents rather than in aqueous media as in nature, which hinders their actual applications. Due to the limitations of the solubility of hydrophobic chromophores in water and the quenching effect caused by undesirable aggregation, ALHSs constructed in an aqueous environment usually show unsatisfactory performance.8,23 In addition, only a few studies focused on the natural sequential Förster resonance energy transfer (FRET) process,8 which could achieve larger Stokes shifts and more efficient FRET even without any spectral overlap and/or beyond the Förster radius. Therefore, it is still a challenge to design and fabricate highly efficient ALHSs with the sequential FRET process in an aqueous environment.Perovskite quantum dots (QDs) have attracted tremendous attention as promising candidates for photovoltaic and optoelectronic device applications,24–27 and they possess outstanding optoelectronic properties, such as a high quantum yield (QY), tunable electronic bandgaps and emission spectra in the visible range, narrow emission line widths, and a high molar extinction coefficient.28–30 These advantages make perovskite QDs an attractive alternative to highly efficient light-harvesting antennas for collecting solar energy in ALHSs.31–34 Despite enormous efforts, due to phase transition in water vapor or the aggravation of lattice vibration at high temperature,35,36 the reported perovskite-based ALHSs were only employed in organic solvents and under mild conditions. Therefore, it is a daunting task to realize perovskite-based ALHSs with the sequential FRET regime in aqueous environments, and it has not yet been basically fulfilled. To overcome these obstacles, numerous porous materials such as crosslinked polystyrene beads, zeolites, amorphous alumina and metal–organic frameworks have demonstrated promise for shielding protection of perovskite QDs,37–40 which significantly improved the water and thermal stabilities of perovskite QDs. However, so far, these strategies have not been used to construct ultrastable and efficient perovskite-based ALHSs with the sequential FRET process in aqueous environments.
Palygorskite (PAL)41–44 is a category of natural earth-abundant magnesium aluminum silicate crystalline minerals possessing moderate cation-exchange capacity and regular 3D zeolite-like channels. In such circumstances, porous PAL has provided better confinement and encapsulation for potentially luminescent dye molecules and perovskite QDs owing to their more tunable pores and specific sites though post-processing.5,45–47 Typically, magnesium aluminosilicate layers contain exchangeable cations such as Mg2+, K+ and Na+. These native species can be ion-exchanged for other metal ions,48–51 so that lead ions can be encapsulated through the ion-exchange process. The surface of the PAL channel contains plentiful functional groups, including Si–OH, Mg–OH, and Al–OH, all of which can form H-bonds and coordination bonds with the halide anions and lead ions of the perovskite QDs, respectively. Moreover, PAL could serve as a host matrix to guide the restricted growth of CsPbBr3 QDs and isolate them from each other to diminish the aggregation-caused quenching (ACQ) effect, which enables significantly improved stability and luminescence performance of the perovskite. Motivated by the above inspirations, we demonstrated in situ growth of CsPbBr3 QDs in a porous PAL matrix to construct an efficient water-resistant light-harvesting antenna (denote as CsPbBr3@PAL). The resulting CsPbBr3@PAL composite exhibits ultrastable emission in water for more than 30 days, and it can be further used to encapsulate fluorescence dye Eosin Y (ESY) and Rose Bengal (RB) (Fig. S1†) to build an efficient and ultrastable ALHS (RB-ESY-CsPbBr3@PAL), which could implement an efficient sequential two-step energy-transfer regime in aqueous solution (Fig. 1). Significantly, the harvested solar energy of the RB-ESY-CsPbBr3@PAL system could efficiently promote the photocatalyzed Friedel–Crafts alkylation reactions in aqueous medium due to the in situ encapsulation- and defect passivation-enhanced Förster resonance energy transfer (FRET) process. To the best of our knowledge, it is the first time to fabricate an efficient and water-resistant light-harvesting system using perovskite QDs enabled by chemical interface engineering and encapsulation with natural palygorskite. Therefore, our work is an interesting concept that advances the use of perovskites as a light-harvesting antenna in water.
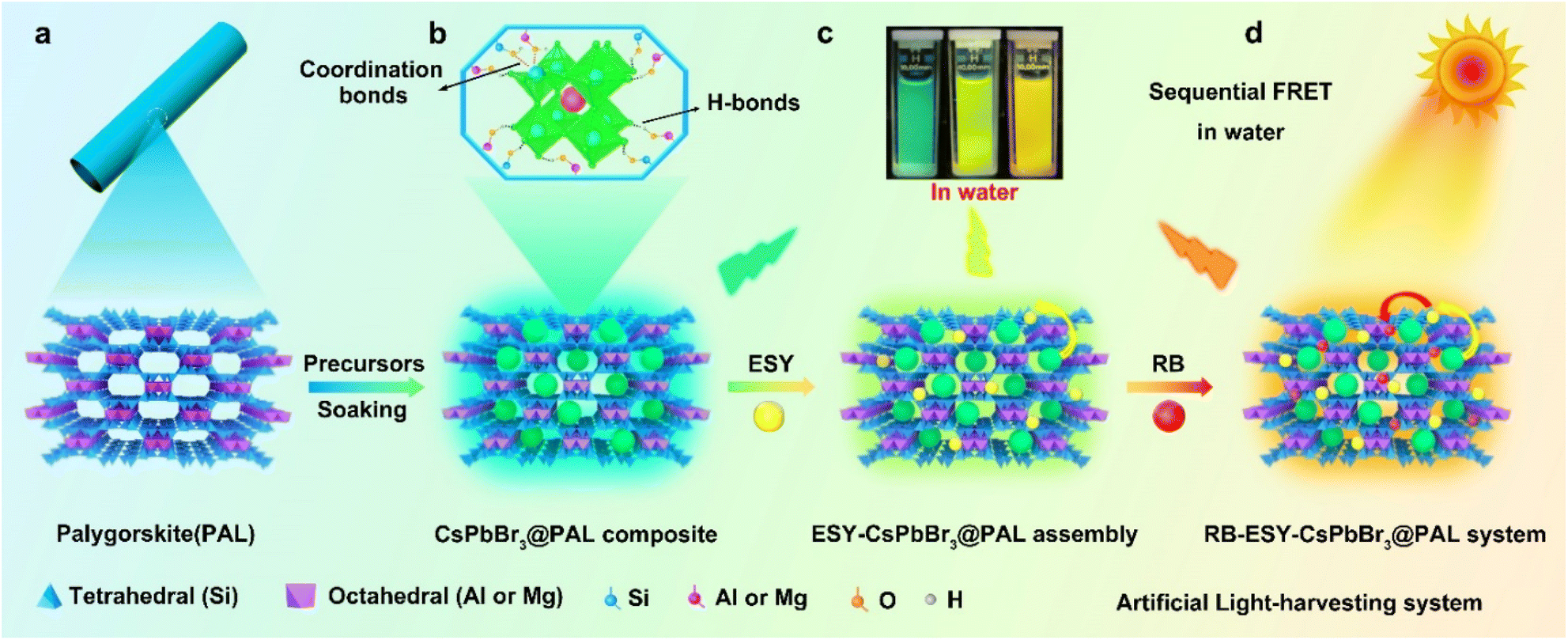 | ||
| Fig. 1 Schematics of the construction of the perovskite–palygorskite composite-based ALHS and the light-harvesting process. (a) The projection of the PAL structure along the (001) direction illustrates its porous nature. (b) Preparation of the water-resistant light-harvesting CsPbBr3@PAL antenna. Illustration of the CsPbBr3 QDs confined in the PAL matrix, showing H-bonding and coordination-bonding interactions between the PAL matrix and perovskite QDs. The Cs+ ion is shown as one red sphere and the PbBr64− octahedra are shown in green. (c) and (d) Construction of the ALHS with sequential two-step energy transfer in water. | ||
2. Results and discussion
2.1 Preparation and characterization of the perovskite–palygorskite antenna
As a proof-of-concept experiment, we rationally designed CsPbX3@PAL (X = Cl, Br or I) composite light-harvesting antennas. In a typical procedure, Fig. 1 shows a schematic illustration of the preparation of the perovskite–palygorskite composite antenna and its sequential two-step energy transfer system (vide infra). The PAL matrix was first treated with hydrochloric acid according to literature protocols to form considerable numbers of silanol groups (Si–OH) and eliminate carbonate impurities of intracrystalline tunnels (Fig. S2†).52–54 The transmission electron microscopy (TEM) images (Fig. S3†) and powder X-ray diffraction (PXRD) patterns (Fig. S4†) showed that the PAL maintained the fibrous morphology and strong (110) crystal plane during the treating process. However, its pore size has been expanded to 3–6 nm (Fig. S5†). Then, the as-treated PAL was immersed in precursor solution of PbBr2 at 80 °C for a week.55,56 Elemental composition analyses (Fig. S6 and Table S1†) showed that the content of Mg2+, Na+ and K+ decreased and the content of Pb2+ and Br− increased after ion exchange, which further confirmed that the ion exchange process occurs. Subsequently, the free PbBr2 was washed with DMF/ethanol by centrifugation. The obtained PbBr2@PAL was dispersed in DMF solution containing Cs-Oleate and subjected to an ultrasonic reaction. Finally, the reaction mixture was precipitated in toluene at room temperature57 to obtain a PAL-encapsulated perovskite composite (denoted as CsPbBr3@PAL composite), where CsPbBr3 QDs in situ nucleated and crystallized inside the PAL matrix (for a full description of the methods, see the ESI†). Such a PAL protection strategy could diminish the ACQ effect and environmental sensitivity, because of the in situ encapsulation and the defect passivation of the PAL matrix.The framework and topological structure of the PAL matrix could guide the restricted growth of CsPbBr3 QDs and keep them isolated from each other, where strong H-bonding and coordination bonding were formed between the hydroxyl group of the PAL matrix and the halide anions and lead ions of the CsPbBr3 QDs, respectively. Such in situ encapsulation and defect passivation enable the improvement of luminescence performance and stability, suggesting that the CsPbBr3@PAL composite could be an attractive alternative to serve as a light-harvesting antenna. Firstly, the structure of the CsPbBr3@PAL antenna was synthetically confirmed. The SEM image (Fig. S7a†) shows that the CsPbBr3@PAL antenna has a fibrous morphology. No free CsPbBr3 QDs were observed, indicating that CsPbBr3 QDs were mainly encapsulated in the PAL matrix. Transmission electron microscopy (TEM) studies revealed that CsPbBr3 QDs were dispersed in the pores of the PAL matrix with an average diameter of 4.4 nm (Fig. 2a and S7b†), and the size of CsPbBr3 QDs matches well with the mesoporous pores of the PAL matrix (Fig. S8†), preliminarily suggesting that the growth of perovskite QDs can be well encapsulated by the cavities of the natural PAL matrix. This phenomenon has also been observed in a previous perovskite-zeolite composite.39Fig. 2b clearly shows the isolated CsPbBr3 QDs with high crystallinity in the CsPbBr3@PAL composite. The representative high-resolution TEM (HRTEM) image shows a well-defined lattice spacing from fringes of about 2.92 Å (inset in Fig. 2b), which matches well with the (200) crystal faces of the cubic phase structure of CsPbBr3 with the Pm![[3 with combining macron]](https://www.rsc.org/images/entities/i_char_0033_0304.gif) m space group, further suggesting that CsPbBr3 QDs were formed in the mesoporous cages of the PAL matrix. The electron diffraction pattern (Fig. 2c) confirms the high crystallinity of CsPbBr3 QDs in the CsPbBr3@PAL composite.58 To better characterize the structure of CsPbBr3 QDs, a HRTEM was used to obtain the atomic image. As presented in Fig. 2d, the HRTEM image can individually resolve the atomic regular distribution of Pb from the <111> projection. To further verify the successful preparation of the composite material, the scanning transmission electron microscopy high-angle annular dark-field (STEM-HAADF) image of the CsPbBr3@PAL antenna clearly shows the CsPbBr3 QDs in the PAL matrix (Fig. 2e), and the corresponding area-selective energy-dispersive X-ray spectroscopy (EDX) mappings (Fig. 2f, g and S9†) further reveal the presence and dispersion of the CsPbBr3 QDs in the PAL matrix. Furthermore, we carried out PXRD measurements for evaluating the crystallinity of the CsPbBr3@PAL antenna (Fig. 2h). The experimental results show characteristic structural peaks at 21.55, 26.48, 30.65 and 37.77°, which are well indexed to the (110), (111), (200) and (211) planes of the CsPbBr3 bulky sample (Pmm, space group No. 221, PDF No. 00-054-0752), respectively. But the peaks of CsPbBr3 QDs are weaker, indicating the formation of CsPbBr3 QDs within the PAL matrix. Brunauer–Emmet–Teller (BET) studies (Fig. 2i, S8 and Table S2†) further support the encapsulation of CsPbBr3 QDs in the PAL matrix. The pore volumes and specific surface area are calculated to be 0.38 m3 g−1 and 110.1 m2 g−1 for the CsPbBr3@PAL antenna, respectively, and 0.47 m3 g−1 and 264.4 m2 g−1 for the PAL matrix, respectively. The pore volume and N2 uptake capacity at saturation are decreased by 19.1% and 58.4% after the encapsulation of CsPbBr3 QDs, respectively, demonstrating the in situ growth of CsPbBr3 QDs into the internal pores of the PAL matrix.39,58 The above results prove that the CsPbBr3@PAL antenna has been successfully built through a simple and rational design. The CsPbBr3 QDs are encapsulated in the pores of the PAL matrix through absorption and cation-exchange processes, in which strong H bonds and coordination bonds can be formed between the plentiful dangling functional groups of the PAL matrix including Si–OH, Al–OH, and Mg–OH and the halide anions and lead ions of CsPbBr3 QDs, respectively. From the Fourier Transform Infrared (FTIR) spectra in Fig. S10,† compared to the PAL matrix, the CsPbBr3@PAL antenna exhibits a wider –OH vibrational peak with a slight shift to a lower wavenumber, which reveals the presence of H bonds between the bromide anion of the CsPbBr3 QDs and the hydroxyl groups on the PAL matrix. Zhao et al. also reported this H-bonding interaction previously between MAPbBr3 QDs and the Al–OH, P–OH, and P
m space group, further suggesting that CsPbBr3 QDs were formed in the mesoporous cages of the PAL matrix. The electron diffraction pattern (Fig. 2c) confirms the high crystallinity of CsPbBr3 QDs in the CsPbBr3@PAL composite.58 To better characterize the structure of CsPbBr3 QDs, a HRTEM was used to obtain the atomic image. As presented in Fig. 2d, the HRTEM image can individually resolve the atomic regular distribution of Pb from the <111> projection. To further verify the successful preparation of the composite material, the scanning transmission electron microscopy high-angle annular dark-field (STEM-HAADF) image of the CsPbBr3@PAL antenna clearly shows the CsPbBr3 QDs in the PAL matrix (Fig. 2e), and the corresponding area-selective energy-dispersive X-ray spectroscopy (EDX) mappings (Fig. 2f, g and S9†) further reveal the presence and dispersion of the CsPbBr3 QDs in the PAL matrix. Furthermore, we carried out PXRD measurements for evaluating the crystallinity of the CsPbBr3@PAL antenna (Fig. 2h). The experimental results show characteristic structural peaks at 21.55, 26.48, 30.65 and 37.77°, which are well indexed to the (110), (111), (200) and (211) planes of the CsPbBr3 bulky sample (Pmm, space group No. 221, PDF No. 00-054-0752), respectively. But the peaks of CsPbBr3 QDs are weaker, indicating the formation of CsPbBr3 QDs within the PAL matrix. Brunauer–Emmet–Teller (BET) studies (Fig. 2i, S8 and Table S2†) further support the encapsulation of CsPbBr3 QDs in the PAL matrix. The pore volumes and specific surface area are calculated to be 0.38 m3 g−1 and 110.1 m2 g−1 for the CsPbBr3@PAL antenna, respectively, and 0.47 m3 g−1 and 264.4 m2 g−1 for the PAL matrix, respectively. The pore volume and N2 uptake capacity at saturation are decreased by 19.1% and 58.4% after the encapsulation of CsPbBr3 QDs, respectively, demonstrating the in situ growth of CsPbBr3 QDs into the internal pores of the PAL matrix.39,58 The above results prove that the CsPbBr3@PAL antenna has been successfully built through a simple and rational design. The CsPbBr3 QDs are encapsulated in the pores of the PAL matrix through absorption and cation-exchange processes, in which strong H bonds and coordination bonds can be formed between the plentiful dangling functional groups of the PAL matrix including Si–OH, Al–OH, and Mg–OH and the halide anions and lead ions of CsPbBr3 QDs, respectively. From the Fourier Transform Infrared (FTIR) spectra in Fig. S10,† compared to the PAL matrix, the CsPbBr3@PAL antenna exhibits a wider –OH vibrational peak with a slight shift to a lower wavenumber, which reveals the presence of H bonds between the bromide anion of the CsPbBr3 QDs and the hydroxyl groups on the PAL matrix. Zhao et al. also reported this H-bonding interaction previously between MAPbBr3 QDs and the Al–OH, P–OH, and P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of AlPO-5 zeolite.39,59 The X-ray photoelectron spectra (XPS) exhibit the characteristic peaks of Cs, Pb, Br, C, Si, Al and O, which are consistent with the elements in the CsPbBr3@PAL antenna (Fig. S11a†). The binding energy related to the oxygen signal shifts from 527.0 eV for the PAL matrix to 527.5 eV for the CsPbBr3@PAL antenna (Fig. S11b and c†). This phenomenon implies that the formation of O–Pb coordination bonds leads to a decrease in the electron density of O atoms.60 Therefore, the cation-exchange properties of the PAL matrix could realize the confined growth of perovskite QDs, which enables the in situ encapsulation of CsPbBr3 QDs. Strong H-bonding and coordination-bonding interactions not only improve the cohesion of CsPbBr3 QDs with the PAL matrix, but also passivate the surface defects of CsPbBr3 QDs, which enhances the luminescence performance and stability of perovskite.
O groups of AlPO-5 zeolite.39,59 The X-ray photoelectron spectra (XPS) exhibit the characteristic peaks of Cs, Pb, Br, C, Si, Al and O, which are consistent with the elements in the CsPbBr3@PAL antenna (Fig. S11a†). The binding energy related to the oxygen signal shifts from 527.0 eV for the PAL matrix to 527.5 eV for the CsPbBr3@PAL antenna (Fig. S11b and c†). This phenomenon implies that the formation of O–Pb coordination bonds leads to a decrease in the electron density of O atoms.60 Therefore, the cation-exchange properties of the PAL matrix could realize the confined growth of perovskite QDs, which enables the in situ encapsulation of CsPbBr3 QDs. Strong H-bonding and coordination-bonding interactions not only improve the cohesion of CsPbBr3 QDs with the PAL matrix, but also passivate the surface defects of CsPbBr3 QDs, which enhances the luminescence performance and stability of perovskite.
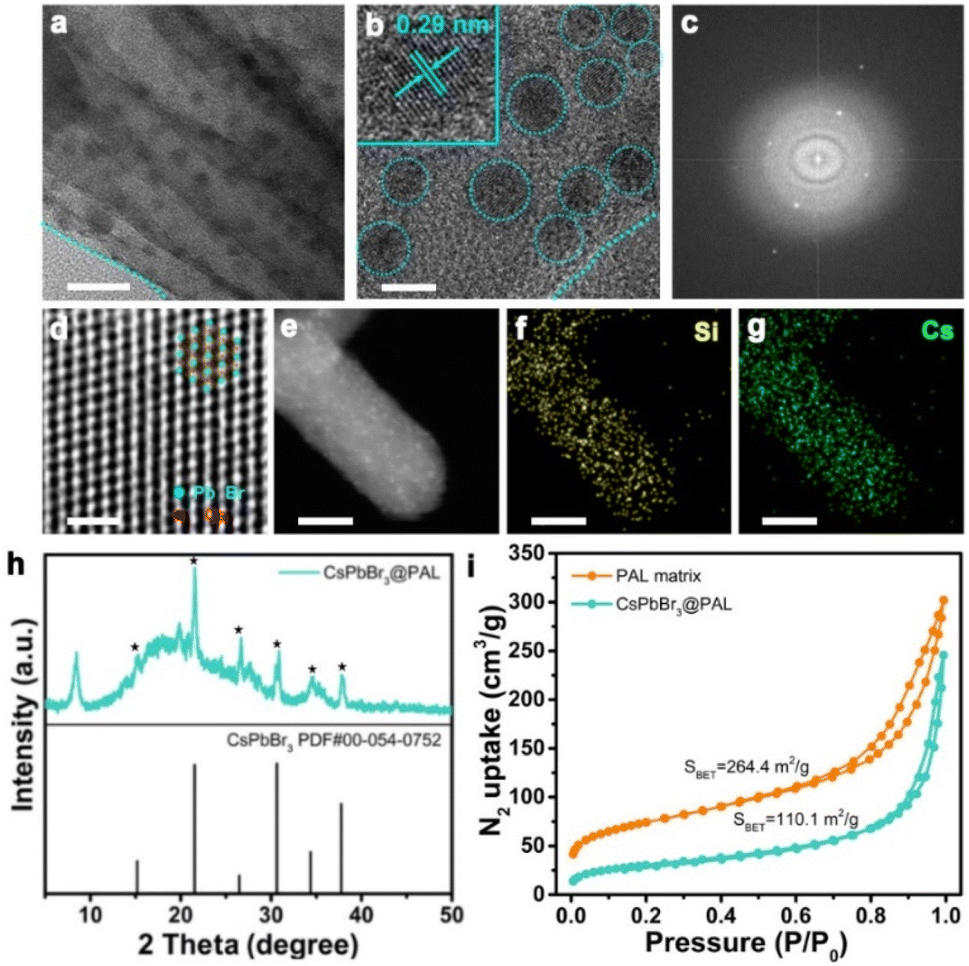 | ||
| Fig. 2 Characterization of the CsPbBr3@PAL antenna. (a) and (b) TEM image of the CsPbBr3@PAL antenna; the black dots are CsPbBr3 QDs. Inset shows the HRTEM image of (b) the lattice spacing of the confined CsPbBr3 QDs. (c) The electron diffraction pattern of CsPbBr3 QDs in the CsPbBr3@PAL composite. The electron diffraction pattern reveals the high crystallinity of the CsPbBr3 QDs. (d) Magnified HRTEM image of CsPbBr3 QDs showing an atomic arrangement from the <111> projection. (e) HAADF image of PAL filled with CsPbBr3 QDs. (f) and (g) The Si and Cs elemental mapping by EDX on the same area. Scale bars: (a) 20 nm; (b) 5 nm; (d) 1 nm; (e), (f), and (g) 30 nm. (h) PXRD patterns of the CsPbBr3@PAL composite. (i) N2 sorption isotherm at 77 K and Brunauer–Emmett–Teller plot (inset) for the CsPbBr3@PAL composite. | ||
The obtained CsPbBr3@PAL antenna shows a yellow powder, indicating the formation of the CsPbBr3 @PAL composite.61 The as-synthesized CsPbBr3@PAL antenna exhibits strong green fluorescence emission under UV irradiation with a wavelength of 365 nm (Fig. S12†). To verify that the strong photoluminescence of the CsPbBr3@PAL antenna is derived from encapsulation and defect passivation, neat CsPbBr3 QDs with an average size of 4.3 nm were synthesized (Fig. S13†). Fig. S12† shows that the CsPbBr3 QDs emit faintly negligible fluorescence under ultraviolet irradiation, caused by the typical non-radiative recombination at the defects of CsPbBr3 QDs.39 Benefiting from the encapsulation and in situ passivation in the PAL matrix, the antenna exhibits good optical properties. UV/Vis diffuse reflectance spectroscopy (DRS) of CsPbBr3@PAL shows a well-resolved band-edge exciton peak at a wavelength of ca. 504 nm (Fig. 3a) and the direct band gap is calculated from Tauc plots to be approximately 2.37 eV (Fig. S14†), which is analogous to previously reported perovskite composites.39,62 Impressively, the photoluminescence (PL) of CsPbBr3 QDs embedded into the PAL matrix exhibits a sharp emission peak at 516 nm (Fig. 3a), blueshifted by ≈21 nm with respect to the CsPbBr3 bulk crystals (BCs) (Fig. S15†). This could be connected with the quantum-confinement effect.63–65 Moreover, for the CsPbBr3@PAL antenna, the emission line width of the PL peak is 19.6 nm. Such a narrow PL peak width of the composite material is due to the encapsulation of CsPbBr3 QDs by the PAL matrix, which cause the reduction of the size distribution range of CsPbBr3 QDs.66,67 More importantly, Fig. 3b shows that the neat CsPbBr3 QDs decomposed easily in water and show negligible fluorescence. However, the CsPbBr3@PAL composite still retains strong fluorescence emission in water. To further confirm defect passivation caused by H-bonding and coordination-bonding interactions, time-resolved fluorescent decay spectroscopy (TRFDS) of traces was performed and the results are shown in Fig. 3c and Table S3.† Two curves, a faster one for the CsPbBr3 QDs and a slower one for the CsPbBr3@PAL antenna, were obtained and the slower decay means a longer lifetime. Meanwhile, the decay curves were fitted, respectively, with a fast decay relevant to the high defect sites on the crystal surface and a slow decay relative to the carrier transport inside the quasi-perfect crystals.39 The short and long lifetimes of the CsPbBr3@PAL antenna are computed to be τ1 = 19.61 ns and τ2 = 121.64 ns, respectively, almost 5-fold and 6-fold that of the CsPbBr3 QDs (Table S3†). This alludes that the CsPbBr3 QDs within the PAL matrix have a much lower defect density. The nonradiative recombination and the shortened lifetimes observed in the CsPbBr3 QDs might originate from the formation of anti-site Pb–Br defects and vacancies, which is due to under-coordinated Pb atoms.39,68,69 Therefore, H-bonding and coordination-bonding interactions between the functional hydroxyl groups of the PAL matrix and the CsPbBr3 QDs play a key role in passivating the surface defects and suppressing ion migrations. The as-obtained CsPbBr3@PAL antenna displays enhanced PL emission, which benefits from the in situ encapsulation and defect passivation of the PAL matrix. Therefore, the absolute QY of the CsPbBr3@PAL antenna reaches 36.2% (Fig. S16†) in water, which is higher than that of CsPbBr3–zeolite-Y, CsPbBr3–SiO2 and CsPbBr3–AlPO-5 composite phosphors.37,39,64 However, the CsPbBr3 QDs have almost no PL (Fig. S17†) in water, which is in agreement with previous reports about the low QY for perovskite QDs without ligand passivation or matrix encapsulation.29,39
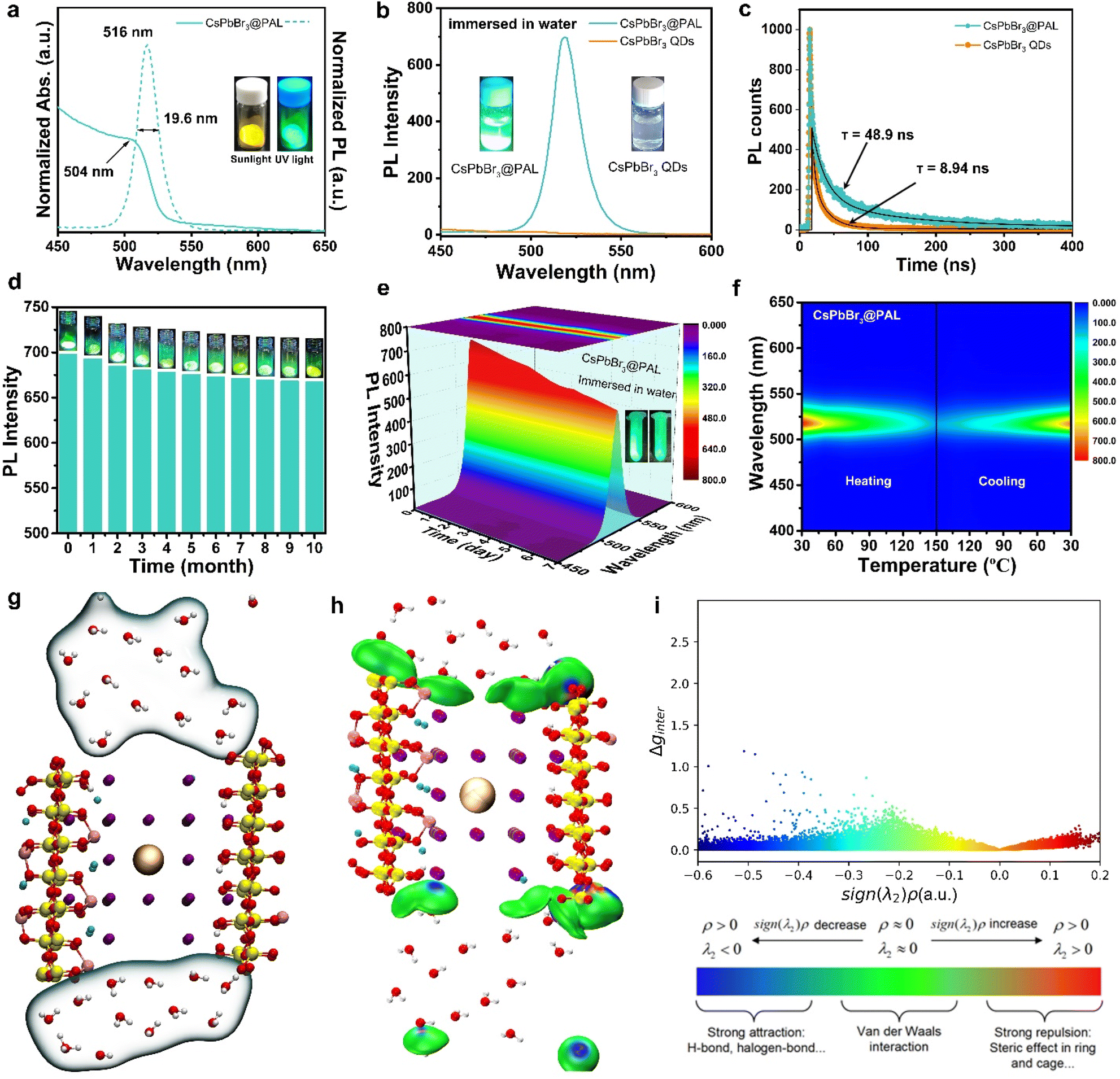 | ||
| Fig. 3 The optical properties and stability of the CsPbBr3@PAL antenna. (a) UV/Vis DRS (solid lines) and PL (dotted lines) spectra of the CsPbBr3@PAL antenna. Inset: digital photograph of the CsPbBr3@PAL antenna taken under a 365 nm xenon lamp. (b) The PL spectra of the CsPbBr3@PAL antenna and neat CsPbBr3 QDs in water. (c) TRFDS spectra of the CsPbBr3@PAL antenna. The black curves are the fitting curves of the two PL decay curves, respectively. (d) PL intensity of the stored CsPbBr3@PAL antenna stored in air environments for 10 months. Inset: corresponding digital photos under ultraviolet irradiation at 365 nm. (e) 3D Colour Map Surface image of the PL spectrum of the CsPbBr3@PAL antenna immersed in water for a week. Inset: digital photos under ultraviolet irradiation at 365 nm before and after immersion in water. (f) Pseudo-colour image of the temperature-dependent PL for the CsPbBr3@PAL antenna, including heating up and cooling down processes. (g) Molecular dynamics equilibrium structure of the composite CsPbBr3@PAL and water. (h) Equivalent surface diagram of the independent gradient model. (i) Scatter plot. | ||
2.2 Evaluation of ambient, water and thermal stability of the CsPbBr3@PAL antenna
To probe the stability toward air, the CsPbBr3@PAL antenna was exposed to ambient conditions with ≈40% humidity for 10 months. The specimen still exhibits remarkable PL performance (Fig. 3d), and the PL intensity of the CsPbBr3@PAL antenna dropped by only 4.4%. Such excellent ambient stability demonstrates that the PAL matrix can effectively diminish the contact of the CsPbBr3 QDs with the external moisture on account of the shielding protection and passivation of the PAL matrix.37,70 Paramount to this feature, the CsPbBr3@PAL antenna exhibits striking water durability. To obtain more direct evidence, the specimen was ultrasonically soaked in water to monitor PL intensity. It can still show 74.1% luminescence under ultraviolet irradiation after a week, as presented in Fig. 3e and S18a.† After 30 days, the sample in water still maintains a certain fluorescence emission (Fig. S18b†). The prominent ambient and water stabilities of the antenna suggest that the PAL matrix can effectively protect CsPbBr3 QDs and suppress the decomposition of CsPbBr3 QDs by water. Additionally, the thermal stability was investigated by monitoring the PL intensity of the CsPbBr3@PAL antenna during a heating–cooling cycle at 30–150 °C. As presented in Fig. 3f and S19.† The PL emission intensity of the CsPbBr3@PAL antenna reduces with the increase of the temperature in the heating phase, which may be ascribed to more non-radiative transitions that stem from the intensified lattice vibration.39 The PL intensity of CsPbBr3@PAL can maintain 47.7% and 17.3% of its initial PL value at 90 °C and 150 °C, respectively. Intriguingly, the PL intensity of CsPbBr3@PAL could progressively recover to 89.5% of its initial PL value after cooling down to 30 °C. These results demonstrate that the PAL matrix can productively suppress the lattice vibration of the perovskite QDs at high temperature.To further understand the stability of CsPbBr3 in water, we employed molecular dynamics simulations to investigate the interaction between water and CsPbBr3@PAL. Fig. 3g shows that water molecules cannot enter the cavity of PAL, thus reducing the quenching of perovskite by water. Further interaction analysis showed the existence of van der Waals forces between water and the cavity (Fig. 3h). The scatter plot also shows that the existence of surface tension of water molecules makes it difficult for water molecules to enter the cavity of PAL (Fig. 3i). These results indicate that the PAL in CsPbBr3@PAL can efficiently prevent the contact between water and CsPbBr3 QDs, thereby greatly improving the water stability of perovskite. Notably, CsPbX3@PAL (X = Cl, Br or I) composites can accurately achieve color tunable emission under the excitation of UV light (Fig. 4a). Fig. 4b clearly shows that the normalized PL spectra can be effectively tuned in the range from 438 to 640 nm by introducing the Cl and I halogen anions, which is conducive to achieving overlap as considerable as possible between the CsPbX3@PAL antenna emission and the dye molecule absorption. Fig. S20† shows more detailed spectral information. All specimens exhibit extremely pure and saturated colors, as presented in the Commission Internationale de l’Eclairage (CIE) chromaticity diagram (Fig. 4c). Compared to the National Television System Committee (NTSC) standard colors (stars), the color-tunable CsPbX3@PAL antennas can cover the upper left part of the CIE diagram. Therefore, the CsPbX3@PAL composites can be used as an extraordinary antenna candidate to construct ALHSs.
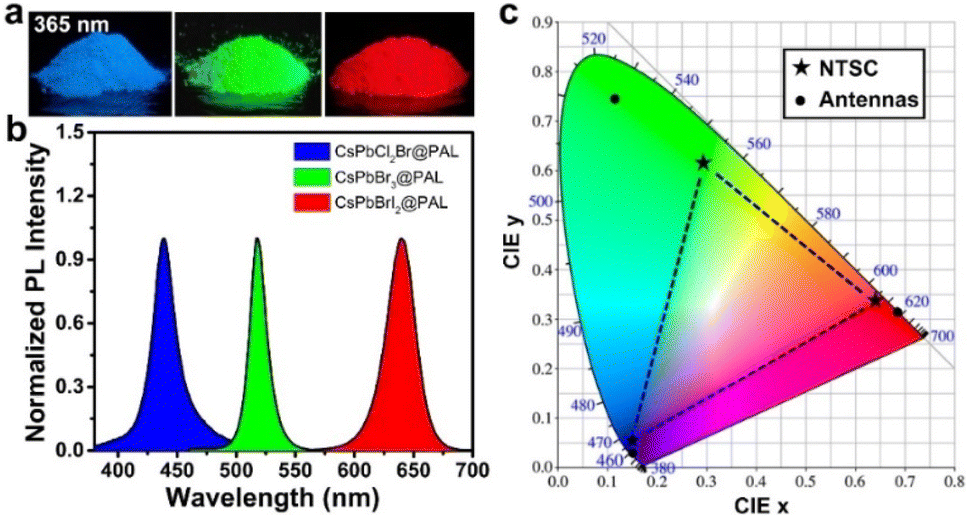 | ||
| Fig. 4 Composition-tunable PL spectra of the CsPbX3@PAL composite. (a) Digital photos of the CsPbX3@PAL composite under ultraviolet irradiation at 365 nm. (b) PL spectra of the CsPbX3@PAL composite. (c) CIE coordinates of the three-color CsPbX3@PAL composite (circular) compared to the NTSC color standards (stars). | ||
2.3 Construction of an artificial light-harvesting system with sequential two-step energy transfer in water
Taking the above considerations into account, it is feasible to construct an efficient ALHS with a sequential two-step FRET regime, in which the effective transfer of solar energy from the CsPbBr3@PAL donor to the dye acceptors can be implemented. ESY dye was selected as the first acceptor, because the fluorescence spectrum of the CsPbBr3@PAL antenna overlapped well with the absorption spectrum of ESY dye (Fig. S21a†). As depicted in Fig. 5a, with the progressive increase of the ratio of the ESY acceptor to the CsPbBr3@PAL antenna, the PL intensity of the ESY acceptor at 541 nm gradually appeared and increased as indicated by emission spectra, while the PL intensity of the CsPbBr3@PAL antenna at 516 nm decreased upon excitation under 365 nm illumination in water. In contrast, the PL intensity of free ESY was negligible upon direct excitation at 365 nm (Fig. S22a†). Moreover, the visual fluorescent color changed from green of the CsPbBr3@PAL antenna to yellow-green of the ESY-CsPbBr3@PAL assembly, obviously implying efficient energy transfer from the CsPbBr3@PAL antenna to the ESY acceptor. In addition, the TRFDS tests showed that the fluorescence lifetimes (τ) declined from the CsPbBr3@PAL antenna (τ1 = 19.61 ns and τ2 = 121.64 ns) to the ESY-CsPbBr3@PAL assembly (τ1 = 2.21 ns and τ2 = 29.00 ns) (Fig. 5b and Table S5†), demonstrating that the collected photons by the CsPbBr3@PAL antenna were successfully transported to the ESY acceptor. Moreover, based on the TRFDS of ESY (Fig. S23 in the ESI†), the average lifetime (τ) of EYB (λex = 516 nm) was 2.9 ns, providing more evidence for the energy transfer from the CsPbBr3@PAL donor to the ESY acceptor. In the ESY-CsPbBr3@PAL assembly, the energy transfer efficiency (ΦET) eventually reached 72.2% at a donor/acceptor ratio of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 in water (Fig. S24a, b and Table S6†).
:7 in water (Fig. S24a, b and Table S6†).
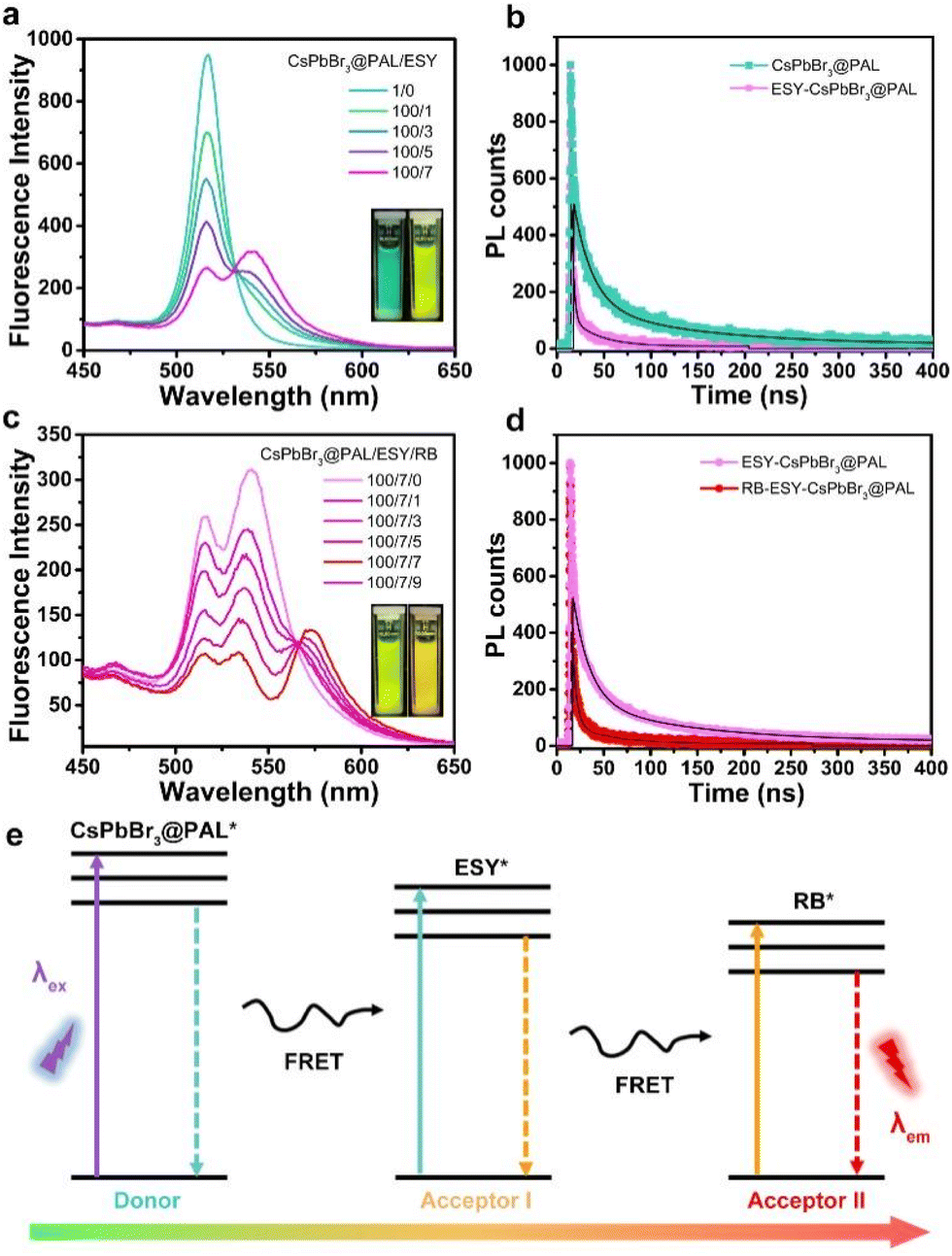 | ||
| Fig. 5 Artificial light-harvesting system with sequential energy transfer. Emission spectra of the CsPbBr3@PAL antenna with different amounts of ESY receptor (a) and ESY-CsPbBr3@PAL (100:7) with different amounts of RB receptor (c) in water (λex = 365 nm). The illustrations are digital photos. The TRFDS spectra of the CsPbBr3@PAL antenna and ESY-CsPbBr3@PAL recorded at λ = 516 nm (b) and ESY-CsPbBr3@PAL and RB-ESY-CsPbBr3@PAL recorded at λ = 541 nm (d) in water (λex = 365 nm). (e) Illustration of the energy level diagram for sequential FRET-associated PL. | ||
Considering that the natural light-harvesting process features sequential multistep FRET, we further constructed a sequential two-step FRET system by using the CsPbBr3@PAL antenna. RB was selected as the second acceptor, because its absorption spectrum and the fluorescence spectrum of the ESY-CsPbBr3@PAL assembly matched well (Fig. S21b†). With the gradual increase of the ratio of the RB acceptor to the ESY-CsPbBr3@PAL assembly, the PL intensity at 570 nm ascribed to RB burgeoned and progressively increased, while the emission peak of ESY at 541 nm decreased upon excitation under 365 nm illumination in water (Fig. 5c), along with visual changes from yellow-green to orange in the fluorescence color. In contrast, the PL intensity of free RB was negligible upon direct excitation at 365 nm (Fig. S22b†). In the RB-ESY-CsPbBr3@PAL system, the second-step energy transfer efficiency was calculated to be 65.3% (Fig. S24c, d and Table S6†) at a CsPbBr3@PAL:ESY:RB ratio of 100:7:9. A decline of the fluorescence lifetime (τ), from the ESY-CsPbBr3@PAL assembly (τ1 = 18.04 ns and τ2 = 121.89 ns) to the RB-ESY-CsPbBr3@PAL system (τ1 = 4.31 ns and τ2 = 41.94 ns) (Fig. 5d and Table S5†), was recorded by TRFDS experiments. Moreover, the TRFDS of RB (ESI,† Fig. S23†) shows that the average lifetime (τ) of RB (λex = 541 nm) was 1.0 ns, providing more evidence for the sequential two-step energy transfer. These results suggest that the CsPbBr3@PAL composite successfully behaves as a donor in ALHS, and sequential two-step FRET instead of charge transfer34 is achieved from the CsPbBr3 QDs to ESY, and then to RB in the relay mode (Fig. 5e). More importantly, as presented in Fig. S25,† the CsPbBr3@PAL antenna remains dispersed for at least 24 h at room temperature, indicating that the hydrophily of the PAL matrix can restrain its agglomeration and improve its dispersity in water.
Furthermore, in order to illustrate that it is the combination of the PAL matrix and perovskite QDs that endows the CsPbBr3@PAL antenna with excellent performance, the CsPbBr3 QDs with an average size of 4.3 nm are used as an antenna to construct an ALHS for comparison. Fig. S17† shows that the CsPbBr3 QDs without the PAL matrix decomposed easily in water and show negligible fluorescence. Therefore, CsPbBr3 QDs without the PAL matrix fail to construct an ALHS in water. When CsPbBr3 QDs and ESY were dispersed in toluene, an inconspicuous FRET process was observed (Fig. S26†). We speculate, on the basis of the above experimental results, that ESY and RB could be absorbed and encapsulated into the pores of the PAL matrix, significantly shortening the distance between CsPbBr3 QDs and ESY/RB, which guaranteed more efficient energy transfer.
It is well known that ESY and RB dyes, as efficient photosensitizers, can be utilized for green chemical synthesis.71–74 In this context, to explore the utilization of the ALHS with sequential two-step FRET as a sustainable energy source, the photocatalytic activity of the RB-ESY-CsPbBr3@PAL system is evaluated by catalyzing the Friedel–Crafts alkylation reactions between indol (1a) and N,N-dimethylaniline (b) in an aqueous solution (Vwater:VMeCN = 1:1, considering the solubility of the substrates) under visible light (Xe lamp as the sunlight simulator) to synthesize the target product 4-(1H-indol-3-ylmethyl)-N,N-dimethylaniline (1c) that is an important structural skeleton in various naturally occurring products and pharmaceuticals (Fig. S27†).75,76 The CsPbBr3@PAL composite can act as an efficient antenna to harvest UV/Vis light and pass photoexcitation to ESY and RB successively through the sequential two-step FRET mechanism, where it is utilized to synthesize value chemicals (Fig. S28†). Hence, the solar energies can be directly transformed. The Friedel–Crafts alkylation reaction was carried out by 1H NMR spectroscopy (Fig. S29†), and the targeted product was isolated and purified by column chromatography on silica gel. As presented in Table 1, when RB-ESY-CsPbBr3@PAL functions as a nanoreactor, the yield of 1c reached 65.5% after 12 h irradiation under optimized conditions. Inversely, only 26.7% and 7.4% yields were collected by using ESY/RB dyes and CsPbBr3@PAL under otherwise identical conditions, respectively. In addition, the amount of organic photocatalyst and photocatalytic reaction time in this catalytic system are much lower than those reported in the literature.73 Moreover, no photocatalytic activity was exhibited when the reaction was performed in the absence of light. The RB-ESY-CsPbBr3@PAL system as a nanoreactor not only conspicuously enhanced the reaction yield but also successfully fulfilled the Friedel–Crafts alkylation reaction in aqueous solution. In addition, the system did not undergo dye photobleaching in the catalytic reaction process owing to the protection of the PAL matrix (Fig. S27†). Furthermore, to evaluate the scope of the RB-ESY-CsPbBr3@PAL system as a nanoreactor, the reactivities of indoles bearing an electron-donating group (methyl) and bearing an electron-withdrawing group (bromine) and N,N-dimethylaniline to afford 3-arylmethyl indole derivatives were investigated, respectively (shown in Fig. S30 and S31†). The light-harvesting RB-ESY-CsPbBr3@PAL system exhibits higher photocatalytic activities compared to dye alone (Table S7†). The excellent photocatalytic activity of the RB-ESY-CsPbBr3@PAL system in aqueous solution results from three possible factors: (i) the PAL matrix as a protection shell endows CsPbBr3 QDs with ultrastability in water. In addition, in situ encapsulation and defect passivation of the PAL matrix make CsPbBr3@PAL show an enhanced quantum yield and a longer fluorescence lifetime compared to CsPbBr3 QDs in water. (ii) The RB-ESY-CsPbBr3@PAL system with sequential two-step FRET could elevate the utilization efficiency of solar energy. (iii) Loading ESY/RB into the channels of the PAL matrix and assembling into the surface of the perovskite QDs can reduce photobleaching, improve energy transfer, and shorten the distance between the donor and the acceptor. These results suggested that the ESY-RB-CsPbBr3@PAL system could make full utilization of the captured solar photons to effectively photocatalyze the chemical reaction in aqueous medium, which presents a versatile approach to enhance the photocatalytic activities of photosensitizers.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Light | Solvent | Isolated yield% |
| a 1.03 × 10−6 mol% catalyst at room temperature for 12 h (this work). b 5 mol% catalyst at room temperature for 24 h.7 | ||||
| 1a | ESY + RB | Yes | H2O:CH3CN = 1:1 |
26.7 |
| 2 | CsPbBr3@PAL | Yes | H2O:CH3CN = 1:1 |
7.4 |
| 3a | ALHS | Yes | H2O:CH3CN = 1:1 |
65.5 |
| 4a | ALHS | No | H2O:CH3CN = 1:1 |
Trace |
| 5b | RB | Yes | CH3CN:H2O = 4:1 |
52 |

3. Conclusions
In summary, a water-resistant perovskite–palygorskite light-harvesting antenna has been successfully synthesized by in situ passivating and encapsulating CsPbBr3 QDs into the pores of a natural PAL matrix. Experimental data and molecular dynamics simulations showed that the PAL in CsPbBr3@PAL could efficiently prevent the contact between water and CsPbBr3 QDs, which greatly improved the structural stability of CsPbBr3 QDs under an external harsh environment, including exposure to ambient air conditions, heat (150 °C), and upon immersion in water for more than 30 days. Meanwhile, the synergy of in situ encapsulation of the PAL matrix and defect passivation caused by H-bonding and coordination-bonding interactions contributes to excellent luminescence for the CsPbBr3 QDs with a QY value of 36.2% in water and a longer exciton lifetime, which is the highest value among the porous matrices emasculated CsPbBr3 QDs. Furthermore, by introducing Cl and I halogen anions, other halide perovskites could successfully load into the PAL matrix, and thus the formed CsPbX3@PAL composites accurately achieve color tunable emission under the excitation of UV light. The above advantages make CsPbX3@PAL a good light-harvesting antenna to sequentially assemble ESY and RB for constructing an efficient light-harvesting system in water. The experimental results showed that RB-ESY-CsPbBr3@PAL exhibited extremely enhanced photocatalytic activity toward Friedel–Crafts alkylation reactions in aqueous solution, 2.5-fold higher than that of corresponding ESY/RB. Our reported interface engineering and encapsulation strategy with natural PAL will provide an alternative route to fabricate stable and efficient perovskite based artificial light-harvesting systems in aqueous media for solar-energy conversion.4. Experimental
4.1 Preparation of acid-activated palygorskite
Natural palygorskite (PAL) was premodified by acid activation according to the protocol described in the previous literature.47,77 Typically, 5 g of natural PAL micro-powder was soaked in 50 mL of HCl aqueous solution with a concentration of 5 M and was stirred vigorously at 80 °C for 1 h under reflux. After this, heating was stopped and the sample was maintained for 4 h in acid solution at room temperature. Subsequently, the acid-activated PAL was washed using deionized water until pH ∼ 6 and dried at 105 °C overnight. The final sample is denoted as PAL. The purpose of acid modified PAL is to eliminate mineral impurities and increase silanol groups (Si–OH).4.2 Preparation of PbBr2@PAL
0.5 mmol of PbBr2 was dissolved in 10 mL of DMF. DMF acted as a good solvent to dissolve inorganic salts. Then, 100 mg of porous resultant PAL matrix materials was soaked in 1.0 mL of the precursor solution at 80 °C for 7 days. Subsequently, DMF/ethanol was utilized to centrifugally wash away free PbBr2 attached to the surface of the PAL matrix. About 110 mg of solid powder was obtained by vacuum drying. The synthesis of PbX2@PAL (X = Cl, I) was similar to that of PbBr2@PAL.4.3 Preparation of the CsPbBr3@PAL antenna
The as-prepared PbBr2@PAL (110 mg) was immersed in 200 μL of Cs-Oleate DMF solution (50 mM) under ultrasound, and then stirred vigorously for 1 h. After that, the CsPbBr3@PAL composite was prepared by putting the above reaction mixture into 10.0 mL dried PhMe. Finally, the CsPbBr3@PAL antenna was obtained after washing with PhMe. About 118 mg of solid powder was obtained by vacuum drying. The synthesis of CsPbX3@PAL (X = Cl, I) was similar to that of CsPbX3@PAL.4.4 Preparation of the ESY-CsPbBr3@PAL assembly
The as-prepared PbBr2@PAL (110 mg) was immersed in 300 μL of DMF solution containing dye ESY (8.2 mg) under vigorous stirring for 1 h. Subsequently, 200 μL of Cs-Oleate DMF solution (50 mM) was added to the above mixture to obtain the ESY-CsPbX3@PAL precursor. Then, the reaction mixture was soaked in 10.0 mL dried PhMe to prepare the ESY-CsPbBr3@PAL assembly.4.5 Preparation of the RB-ESY-CsPbBr3@PAL system
The as-prepared PbBr2@PAL (110 mg) was immersed in 300 μL of DMF solution containing ESY (8.2 mg) and RB (10.6 mg) under vigorous stirring for 1 h. Subsequently, 200 μL of Cs-Oleate DMF solution (50 mM) was added to the above mixture to obtain the RB-ESY-CsPbX3@PAL precursor. Then, the reaction mixture was soaked in 10.0 mL dried PhMe to prepare the RB-ESY-CsPbBr3@PAL system.Data availability
All the data supporting this article have been included in the main text and the ESI.†Author contributions
B. D. W. designed and conceived the whole project. A. Q. W and S. Y. L. conceived the project. G. P. M. carried out the materials synthesis, experimental test and data analysis. M. X. J. conducted molecular dynamics simulations. L. P. Z., G. P. M., J. H. and B. D. W. wrote the paper together. Z. F. Z. and T. Z. H. performed the electron microscopy tests. All the authors contributed to the discussion during the whole project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by the National Natural Science Foundation of China (21977041, 21904052, and 22221001), the Natural Science Foundation of Gansu Province (20JR5RA297), and the Key Laboratory of Clay Mineral Applied Research of Gansu Province (CMAR-2019-1). We would like to acknowledge the assistance extended by the Electron Microscopy Centre of Lanzhou University in performing the microscopy experiments.Notes and references
- C. D. P. Duffy, Science, 2020, 368, 1427–1428 CrossRef PubMed.
- R. Croce and H. van Amerongen, Nat. Chem. Biol., 2014, 10, 492–501 CrossRef.
- N. Nelson and A. Ben-Shem, Nat. Rev. Mol. Cell Biol., 2004, 5, 971–982 CrossRef PubMed.
- Y. Kuramochi, R. Sato, H. Sakuma and A. Satake, Chem. Sci., 2022, 13, 9861–9879 RSC.
- Y. Ishida, T. Shimada, D. Masui, H. Tachibana, H. Inoue and S. Takagi, J. Am. Chem. Soc., 2011, 133, 14280–14286 CrossRef PubMed.
- X. Qian, Y. Zhao, Y. Alsaid, X. Wang, M. Hua, T. Galy, H. Gopalakrishna, Y. Yang, J. Cui, N. Liu, M. Marszewski, L. Pilon, H. Jiang and X. He, Nat. Nanotechnol., 2019, 14, 1048–1055 CrossRef PubMed.
- X.-B. Li, C.-H. Tung and L.-Z. Wu, Nat. Rev. Chem., 2018, 2, 160–173 CrossRef.
- D. Zhang, W. Yu, S. Li, Y. Xia, X. Li, Y. Li and T. Yi, J. Am. Chem. Soc., 2021, 143, 1313–1317 CrossRef PubMed.
- H.-J. Yu, X.-L. Zhou, X. Dai, F.-F. Shen, Q. Zhou, Y.-M. Zhang, X. Xu and Y. Liu, Chem. Sci., 2022, 13, 8187–8192 RSC.
- A. H. Proppe, Y. C. Li, A. Aspuru-Guzik, C. P. Berlinguette, C. J. Chang, R. Cogdell, A. G. Doyle, J. Flick, N. M. Gabor, R. van Grondelle, S. Hammes-Schiffer, S. A. Jaffer, S. O. Kelley, M. Leclerc, K. Leo, T. E. Mallouk, P. Narang, G. S. Schlau-Cohen, G. D. Scholes, A. Vojvodic, V. W.-W. Yam, J. Y. Yang and E. H. Sargent, Nat. Rev. Mater., 2020, 5, 828–846 CrossRef.
- Z. Fan, Y. Yin, B. Cai, Q. Ma, Q. Liu, X. Liu, L. Yinhua and W.-H. Zhang, Chem. Sci., 2022, 13, 10512–10522 RSC.
- A. Starkholm, L. Kloo and P. H. Svensson, J. Am. Chem. Soc., 2020, 142, 18437–18448 CrossRef PubMed.
- X. Zhang, Y. Xu, F. Giordano, M. Schreier, N. Pellet, Y. Hu, C. Yi, N. Robertson, J. Hua, S. M. Zakeeruddin, H. Tian and M. Gratzel, J. Am. Chem. Soc., 2016, 138, 10742–10745 CrossRef PubMed.
- L. Hu, Q. Zhao, S. Huang, J. Zheng, X. Guan, R. Patterson, J. Kim, L. Shi, C. H. Lin, Q. Lei, D. Chu, W. Tao, S. Cheong, R. D. Tilley, A. W. Y. Ho-Baillie, J. M. Luther, J. Yuan and T. Wu, Nat. Commun., 2021, 12, 466 CrossRef PubMed.
- N. Melnychuk, S. Egloff, A. Runser, A. Reisch and A. S. Klymchenko, Angew. Chem., Int. Ed., 2020, 59, 6811–6818 CrossRef.
- Z. Zhang, Z. Zhao, L. Wu, S. Lu, S. Ling, G. Li, L. Xu, L. Ma, Y. Hou, X. Wang, X. Li, G. He, K. Wang, B. Zou and M. Zhang, J. Am. Chem. Soc., 2020, 142, 2592–2600 CrossRef.
- X. Zhang, S. Rehm, M. M. Safont-Sempere and F. Wurthner, Nat. Chem., 2009, 1, 623–629 CrossRef.
- K. Trofymchuk, A. Reisch, P. Didier, F. Fras, P. Gilliot, Y. Mely and A. S. Klymchenko, Nat. Photonics, 2017, 11, 657–663 CrossRef.
- Q. Zhang, M. He, Q. Wan, W. Zheng, M. Liu, C. Zhang, X. Liao, W. Zhan, L. Kong, X. Guo and L. Li, Chem. Sci., 2022, 13, 3719–3727 RSC.
- Y. X. Yuan, J. H. Jia, Y. P. Song, F. Y. Ye, Y. S. Zheng and S. Q. Zang, J. Am. Chem. Soc., 2022, 144, 5389–5399 CrossRef.
- X. M. Chen, Q. Cao, H. K. Bisoyi, M. Wang, H. Yang and Q. Li, Angew. Chem., Int. Ed., 2020, 59, 10493–10497 CrossRef PubMed.
- Y. X. Hu, X. Hao, L. Xu, X. Xie, B. Xiong, Z. Hu, H. Sun, G. Q. Yin, X. Li, H. Peng and H. B. Yang, J. Am. Chem. Soc., 2020, 142, 6285–6294 CrossRef PubMed.
- Q. Song, S. Goia, J. Yang, S. C. L. Hall, M. Staniforth, V. G. Stavros and S. Perrier, J. Am. Chem. Soc., 2021, 143, 382–389 CrossRef.
- Y. Shen, L. P. Cheng, Y. Q. Li, W. Li, J. D. Chen, S. T. Lee and J. X. Tang, Adv. Mater., 2019, 31, e1901517 CrossRef PubMed.
- J. Yuan, X. Zhang, J. Sun, R. Patterson, H. Yao, D. Xue, Y. Wang, K. Ji, L. Hu, S. Huang, D. Chu, T. Wu, J. Hou and J. Yuan, Adv. Funct. Mater., 2021, 31(27), 2101272 CrossRef.
- K. L. Wang, Y. H. Zhou, Y. H. Lou and Z. K. Wang, Chem. Sci., 2021, 12, 11936–11954 RSC.
- K. Chen, W. Jin, Y. Zhang, T. Yang, P. Reiss, Q. Zhong, U. Bach, Q. Li, Y. Wang, H. Zhang, Q. Bao and Y. Liu, J. Am. Chem. Soc., 2020, 142, 3775–3783 CrossRef.
- A. Dey, J. Ye, A. De, E. Debroye, S. K. Ha, E. Bladt, A. S. Kshirsagar, Z. Wang, J. Yin, Y. Wang, L. N. Quan, F. Yan, M. Gao, X. Li, J. Shamsi, T. Debnath, M. Cao, M. A. Scheel, S. Kumar, J. A. Steele, M. Gerhard, L. Chouhan, K. Xu, X.-g. Wu, Y. Li, Y. Zhang, A. Dutta, C. Han, I. Vincon, A. L. Rogach, A. Nag, A. Samanta, B. A. Korgel, C.-J. Shih, D. R. Gamelin, D. H. Son, H. Zeng, H. Zhong, H. Sun, H. V. Demir, I. G. Scheblykin, I. Mora-Seró, J. K. Stolarczyk, J. Z. Zhang, J. Feldmann, J. Hofkens, J. M. Luther, J. Pérez-Prieto, L. Li, L. Manna, M. I. Bodnarchuk, M. V. Kovalenko, M. B. J. Roeffaers, N. Pradhan, O. F. Mohammed, O. M. Bakr, P. Yang, P. Müller-Buschbaum, P. V. Kamat, Q. Bao, Q. Zhang, R. Krahne, R. E. Galian, S. D. Stranks, S. Bals, V. Biju, W. A. Tisdale, Y. Yan, R. L. Z. Hoye and L. Polavarapu, ACS Nano, 2021, 15, 10775–10981 CrossRef PubMed.
- J. De Roo, M. Ibanez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef PubMed.
- Q. A. Akkerman, G. Raino, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394–405 CrossRef.
- X. Han, Y. Lei, Q. Liao and H. Fu, Angew. Chem., Int. Ed., 2021, 60, 3037–3046 CrossRef PubMed.
- H. Zhang, Y. Wu, Q. Liao, Z. Zhang, Y. Liu, Q. Gao, P. Liu, M. Li, J. Yao and H. Fu, Angew. Chem., Int. Ed., 2018, 57, 7748–7752 CrossRef PubMed.
- G. Meng, L. Zhen, S. Sun, J. Hai, Z. Zhang, D. Sun, Q. Liu and B. Wang, J. Mater. Chem. A, 2021, 9, 24365–24373 RSC.
- K. Zhong, S. Lu, W. Guo, J. Su, S. Sun, J. Hai, F. Chen, A. Wang and B. Wang, J. Mater. Chem. A, 2021, 9, 10180–10185 RSC.
- M. V. Kovalenko, L. Protesescu and M. I. Bodnarchuk, Science, 2017, 358, 745–750 CrossRef PubMed.
- Q. Zhang and Y. Yin, ACS Cent. Sci., 2018, 4, 668–679 CrossRef PubMed.
- Y. Wei, X. Deng, Z. Xie, X. Cai, S. Liang, P. a. Ma, Z. Hou, Z. Cheng and J. Lin, Adv. Funct. Mater., 2017, 27, 1703535 CrossRef.
- A. Loiudice, S. Saris, E. Oveisi, D. T. L. Alexander and R. Buonsanti, Angew. Chem., Int. Ed., 2017, 56, 10696–10701 CrossRef PubMed.
- P. Wang, B. Wang, Y. Liu, L. Li, H. Zhao, Y. Chen, J. Li, S. F. Liu and K. Zhao, Angew. Chem., Int. Ed., 2020, 59, 23100–23106 CrossRef PubMed.
- J. Hou, P. Chen, A. Shukla, A. Krajnc, T. Wang, X. Li, R. Doasa, L. H. G. Tizei, B. Chan, D. N. Johnstone, R. Lin, T. U. Schülli, I. Martens, D. Appadoo, M. S. Ari, Z. Wang, T. Wei, S.-C. Lo, M. Lu, S. Li, E. B. Namdas, G. Mali, A. K. Cheetham, S. M. Collins, V. Chen, L. Wang and T. D. Bennett, Science, 2021, 374, 621–625 CrossRef CAS PubMed.
- W. F. Bradley, Am. Mineral., 1940, 25, 405–410 CAS.
- W. L. Haden and A. Schwint, Ind. Eng. Chem., 1967, 59, 58–69 CrossRef CAS.
- W. T. Granquist and R. C. Amero, J. Am. Chem. Soc., 1948, 70, 3265–3270 CrossRef.
- B. Mu, Y. Kang and A. Wang, J. Mater. Chem. A, 2013, 1, 4804–4811 RSC.
- H. Guo, H. Zhang, F. Peng, H. Yang, L. Xiong, C. Huang, C. Wang, X. Chen and L. Ma, Appl. Clay Sci., 2015, 111, 83–89 CrossRef.
- X.-Y. Li, D.-Y. Zhang, X.-Q. Liu, L.-Y. Shi and L.-B. Sun, Chem. Eng. Sci., 2016, 141, 184–194 CrossRef.
- W. Wang, W. Dong, G. Tian, L. Sun, Q. Wang, A. Hui, B. Mu and A. Wang, Powder Technol., 2019, 354, 1–10 CrossRef.
- M. K. Uddin, Chem. Eng. J., 2017, 308, 438–462 CrossRef.
- L. Wang, Y. Shi, D. Yao, H. Pan, H. Hou, J. Chen and J. C. Crittenden, Chem. Eng. J., 2019, 366, 569–576 CrossRef.
- A. Kilincarslan Kaygun, M. Eral and S. Akyil Erenturk, J. Radioanal. Nucl. Chem., 2016, 311, 1459–1464 CrossRef.
- X. Liu, X. Xu, J. Sun, A. Alsaedi, T. Hayat, J. Li and X. Wang, Chem. Eng. J., 2018, 343, 217–224 CrossRef.
- H. Chen, J. Zhao, A. Zhong and Y. Jin, Chem. Eng. J., 2011, 174, 143–150 CrossRef.
- T. Chen, H. Liu, J. Li, D. Chen, D. Chang, D. Kong and R. L. Frost, Chem. Eng. J., 2011, 166, 1017–1021 CrossRef.
- W. Kuang, G. A. Facey and C. Detellier, Clays Clay Miner., 2004, 52, 635–642 CrossRef.
- Y. C. Zou, L. Mogg, N. Clark, C. Bacaksiz, S. Milovanovic, V. Sreepal, G. P. Hao, Y. C. Wang, D. G. Hopkinson, R. Gorbachev, S. Shaw, K. S. Novoselov, R. Raveendran-Nair, F. M. Peeters, M. Lozada-Hidalgo and S. J. Haigh, Nat. Mater., 2021, 20, 1677–1682 CrossRef.
- L. Mogg, G. P. Hao, S. Zhang, C. Bacaksiz, Y. C. Zou, S. J. Haigh, F. M. Peeters, A. K. Geim and M. Lozada-Hidalgo, Nat. Nanotechnol., 2019, 14, 962–966 CrossRef.
- S. Sun, D. Yuan, Y. Xu, A. Wang and Z. Deng, ACS Nano, 2016, 10, 3648–3657 CrossRef PubMed.
- G. Y. Qiao, D. Guan, S. Yuan, H. Rao, X. Chen, J. A. Wang, J. S. Qin, J. J. Xu and J. Yu, J. Am. Chem. Soc., 2021, 143, 14253–14260 CrossRef PubMed.
- X. Li, M. I. Dar, C. Yi, J. Luo, M. Tschumi, S. M. Zakeeruddin, M. K. Nazeeruddin, H. Han and M. Gratzel, Nat. Chem., 2015, 7, 703–711 CrossRef PubMed.
- J. Yang, E. H. Sargent, S. O. Kelley and J. Y. Ying, Nat. Mater., 2009, 8, 683–689 CrossRef.
- X. Yu, L. Wu, D. Yang, M. Cao, X. Fan, H. Lin, Q. Zhong, Y. Xu and Q. Zhang, Angew. Chem., Int. Ed., 2020, 59, 14527–14532 CrossRef.
- Y. Liu, Z. Yang, D. Cui, X. Ren, J. Sun, X. Liu, J. Zhang, Q. Wei, H. Fan, F. Yu, X. Zhang, C. Zhao and S. F. Liu, Adv. Mater., 2015, 27, 5176–5183 CrossRef.
- L. Xie, Z. Hong, J. Zan, Q. Wu, Z. Yang, X. Chen, X. Ou, X. Song, Y. He, J. Li, Q. Chen and H. Yang, Adv. Mater., 2021, 33, e2101852 CrossRef PubMed.
- V. Malgras, S. Tominaka, J. W. Ryan, J. Henzie, T. Takei, K. Ohara and Y. Yamauchi, J. Am. Chem. Soc., 2016, 138, 13874–13881 CrossRef PubMed.
- K. Tanaka, T. Takahashi, T. Ban, T. Kondo, K. Uchida and N. Miura, Solid State Commun., 2003, 127, 619–623 CrossRef.
- X. Peng, J. Wickham and A. P. Alivisatos, J. Am. Chem. Soc., 1998, 120, 5343–5344 CrossRef.
- Y. Yin and A. P. Alivisatos, Nature, 2005, 437, 664–670 CrossRef PubMed.
- V. Adinolfi, O. Ouellette, M. I. Saidaminov, G. Walters, A. L. Abdelhady, O. M. Bakr and E. H. Sargent, Adv. Mater., 2016, 28, 7264–7268 CrossRef.
- T. Niu, J. Lu, R. Munir, J. Li, D. Barrit, X. Zhang, H. Hu, Z. Yang, A. Amassian, K. Zhao and S. F. Liu, Adv. Mater., 2018, 30, e1706576 CrossRef PubMed.
- S. Wu, Z. Li, M. Q. Li, Y. Diao, F. Lin, T. Liu, J. Zhang, P. Tieu, W. Gao, F. Qi, X. Pan, Z. Xu, Z. Zhu and A. K. Jen, Nat. Nanotechnol., 2020, 15, 934–940 CrossRef PubMed.
- X. Z. Fan, J. W. Rong, H. L. Wu, Q. Zhou, H. P. Deng, J. D. Tan, C. W. Xue, L. Z. Wu, H. R. Tao and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 8514–8518 CrossRef.
- D. M. Yan, J. R. Chen and W. J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 378–380 CrossRef.
- A. Srivastava, P. K. Singh, A. Ali, P. P. Singh and V. Srivastava, RSC Adv., 2020, 10, 39495–39508 RSC.
- X.-Q. Dai, W.-X. Xu, Y.-L. Wen, X.-H. Liu and J.-Q. Weng, Tetrahedron Lett., 2018, 59, 2945–2949 CrossRef.
- R. Contractor, I. J. Samudio, Z. Estrov, D. Harris, J. A. McCubrey, S. H. Safe, M. Andreeff and M. Konoplev, Cancer Res., 2005, 65, 2890–2898 CrossRef PubMed.
- M. Lounasmaa and A. Tolvanen, Nat. Prod. Rep., 2000, 17, 175–191 RSC.
- H. Chen, Y. Zhaoa and A. Wang, J. Hazard. Mater., 2007, 149, 346–354 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc05220b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |