
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCutting-edge molecular modelling to unveil new microscopic insights into the guest-controlled flexibility of metal–organic frameworks†
Hengli
Zhao‡
ab,
Camille
Pelgrin-Morvan‡
a,
Guillaume
Maurin‡
 b and
Aziz
Ghoufi‡
*a
b and
Aziz
Ghoufi‡
*a
aInstitut de Physique de Rennes, IPR, CNRS-Université de Rennes 1, UMR CNRS 6251, 35042 Rennes, France. E-mail: aziz.ghoufi@univ-rennes1.fr
bICGM, Univ. Montpellier, CNRS, ENSCM, Montpellier 34293, France
First published on 15th November 2022
Abstract
Metal–organic frameworks are a class of porous solids that exhibit intriguing flexibility under stimuli, leading often to reversible giant structural changes upon guest adsorption. DUT-49(Cu) and MIL-53(Cr) are fascinating flexible MOFs owing to their guest-induced breathing and negative gas adsorption behaviors respectively. Molecular simulation is one of the most relevant tools to examine these phenomena at the atomistic scale and gain a unique understanding of the physics behind them. Although molecular dynamics and Monte Carlo simulations are widely used in the field of porous materials, these methods hardly consider the structural deformation of a soft material upon guest adsorption. In this work, a cutting-edge osmotic molecular dynamics approach is developed to consider simultaneously the fluid adsorption process and material flexibility. We demonstrate that this newly developed computational strategy offers a unique opportunity to gain unprecedented molecular insights into the flexibility of this class of materials.
1 Introduction
One fascinating property of many porous metal–organic frameworks (MOFs) is the diversity of their architectural flexibility, a unique feature in the field of nanoporous materials with respect to other reference materials such as carbons and zeolites.1–5 Guest-induced pore size/shape modulation of this class of MOF framework (breathing, ligand flip, pore gating, etc.) has revealed uncommon adsorption/separation phenomena pointing towards new opportunities for adsorption-based technologies.1–11 Breathing MOFs belong to the most spectacular family of flexible porous solids. Amongst them, prototypical channel-like MIL-53(Cr) (MIL stands for Materials of Institut Lavoisier) has received much attention with the evidence of a guest assisted reversible structural transition between an open pore (op) and a narrow pore (np) form.1,12,13 This intriguing behavior was observed for a series of guest molecules implying a reversible expansion/contraction of the MIL-53(Cr) pore dimensions resulting into about 40% fluctuation of the unit cell volume. Besides its fundamental interest, this phenomenon has aroused great interest not only for the selective capture of gas molecules via size exclusion14 but also for the progressive release of drugs3,15 and for their uses as solvent-stimulated actuators among other applications.16 Remarkably, Cu2-paddle-wheel based-DUT-49(Cu) is undoubtedly the most intriguing breathing MOF owing to its counterintuitive negative gas adsorption (NGA) behavior upon exposure to CH4. This abnormal phenomenon corresponds to a spontaneous guest desorption associated with a spectacular contraction of the framework corresponding to a unit cell variation of more than 50%.17–19Over the last two decades, numerous studies encompassing advanced in situ characterization techniques and simulation tools6,7,20–22 have been reported to shed light on the microscopic mechanisms of these structural transitions. Typically, although X-ray diffraction methods have been able to accurately characterize the extreme steadier phases i.e. the np and op forms and the stable intermediate phases (ip), the structural transition has never been captured so far since it occurs too quickly (10–100 pico-seconds) to be experimentally detectable.6,7,20–22 Similarly, theory using thermodynamics models23–27 based on free energy calculations and macroscopic formulation has been successfully implemented to predict the thermodynamic stability of these different phases depending on the gas pressure and temperature; however, their inherent limitations prevented an understanding of the physics at the origin of these phase transformations. Force field molecular modelling offers an alternative route to deliver a full atomistic picture of such a breathing phenomenon. Indeed, Molecular Dynamics (MD) simulations integrating a reliable flexible force field for the host framework have been able to capture the structural transitions of breathing MOFs for a fixed guest loading.6,7,20–22 Monte Carlo (MC) simulations implemented in the conventional grand canonical ensemble (GCMC) has only been capable of modelling the adsorption isotherms for the rigid phases.28,29 To account for both the gas adsorption process and the resulting guest-triggered flexibility of the MOF, hybrid approaches coupling MC and MD techniques have been developed.30,31 In particular, we earlier applied a hybrid osmotic monte Carlo (HOMC) strategy to gain insight into the CO2-triggered structural switching of MIL-53(Cr) along the gas pressure range.30 In this scenario as depicted in Fig. 1a GCMC and MD simulations are combined in the osmotic statistical ensemble in such a way that the MD run relates to a MD trial move with a frequency of 1/N (N = atom number) similar to a volume change trial move in a standard MC scheme. Recently, R. Snurr et al. used a MC scheme with an MD move and specific trial MC move (HOMC method) to investigate negative thermal expansion in isoreticular MOFs.32 At the same time, J. Jiang et al. examined sorption-induced structural transition of zeolitic imidazolate framework-8 by alternating independent MC and MD simulations involving a breaking of the ergodicity.33 Similarly, D. S. Sholl et al. studied the adsorption and diffusion of small alcohols in zeolitic imidazolate frameworks ZIF-8 and ZIF-90 implementing a MD trial move in a GCMC scheme (similar that HOMC).34 In a recent review35 on sorption-induced polymer rearrangement two strategies have been discussed (i) the concept of iterative pressure MD (IP-MD) simulations to prepare a system with a predefined number of solute molecules and then determine the corresponding loading pressure. This method is based on the calculation of the excess chemical potential from Widom insertion. This method cannot capture stimuli-triggered complex behavior such as NGA since the adsorbed amount is kept fixed.
 | ||
| Fig. 1 (a) Illustration of the guest-induced breathing phenomenon in MOFs. The adsorbed amount (solid line and left axis) and unit cell volume of the MOF framework (dashed line and right axis) as a function of the gas pressure. Hatched regions correspond to open (op) and close (cp) pore forms while the colored zone are connected to the intermediate pore (ip) and/or phases mixture between both forms. (b) Chart illustrating the three computational approaches allowing us to take into account both the guest adsorption as well as the flexibility of the MOF. μ is the chemical potential, Vi and Ni are the volume and the adsorbed amount at step i, σ is the constraint, p is the pressure, T is the temperature and Nm corresponds to the final adsorbed amount in the case of the hybrid MC/MD method in the canonical ensemble. (c) Overview of the OMD strategy for the prediction of the adsorption isotherm for a breathing material. (d) OMD simulated and experimental CO2 adsorption isotherms at 298 K for MIL-53(Cr) plotted as a semi-logarithmic scale. The OMD simulated unit cell (u.c.) volume changes alongside CO2 adsorption are also provided. | ||
As highlighted by Rogge et al., the applicability of the HOMC approach can be hindered by (i) a premature phase transition as a consequence of a very small free energy barrier between 4 and 5 kJ mol−1 and/or (ii) suppressed phase transitions as a result of a possible high energy barrier of 22–24 kJ mol−1 between metastable states.31,36 To overcome these impediments, these authors proposed a novel hybrid MC/MD scheme to simulate the adsorption isotherms of flexible hosts with their cell volumes maintained fixed.31,37 This procedure, schematized in Fig. 1b, is based on a combination of MD steps in the canonical ensemble and MC simulations in the grand canonical ensemble and requires the construction of different intermediate structures. These structures are geometry optimized and further used as rigid models for GCMC simulations. The series of calculated adsorption isotherms are compared with the experimental data to identify the intermediate structures assigned to each step-change in the experimental profile. However, contrary to the HOMC approach, this method cannot capture the microscopic mechanisms of the adsorption-induced structural transition.
Thus, to unravel the microscopic interplay of guest adsorption and host structural transition, there is a need to bypass both the hurdles mentioned above. To overcome a premature transition as a result of the small free energy barrier, the difference in the adsorbed amounts between the two MD moves has to be very small allowing the minimization of fluctuations of energy and thus avoiding the surpassing of the free energy barrier. The so-suppressed phase transitions due to the potentially high energy barrier between metastable structures can be addressed by promoting the mechanical and thermal fluctuations related to the insertion/deletion of molecules. In the HOMC scheme, a MD move was classically considered as a new simulation of 10–100 ps (ref. 6, 7, 11 and 38) with a relaxation phase preventing thermal, mechanical and energetical fluctuations. Moreover, the principle of dynamic trajectory does not exist anymore in such a HOMC scheme although it is pivotal to capture the reversible structural transition. Herein, we propose to address these two key drawbacks by implementing a novel Osmotic Molecular Dynamics (OMD) scheme applied for the first time to guest-induced breathing MOFs that enables to focus on the dynamic trajectory in which insertion and deletion of molecules are steadily attempted. An osmotic ensemble was selected since the MOF composition is kept constant contrary to the gas concentration that fluctuates. The related statistical ensemble is then NMOFμσT where μ is the chemical potential, σ the applied constraint, T the temperature and NMOF is the number of atoms of the MOF. Fig. 1b summarizes the flowchart of the newly developed OMD approach and its key advantages as compared to the HOMC method: (i) the production of a full dynamical and ergodic trajectory and (ii) the absence of homogenization between MC and MD partition functions to ensure a correct detailed balance.30 Indeed, the reversibility of the Markov chain during the transition between MC and MD parts can be only satisfied if the MC and MD partition functions are similar. The implementation of this OMD strategy is first validated on MIL-53(Cr) a MOF that has already been intensively studied in the past using MD,29,39,40 HOMC,30,31 MC20,41 and theoretical models.8,25,38 As a further stage, the OMD scheme is trained to capture and understand in-depth the NGA phenomenon exhibited by DUT-49(Cu), by far the most complex breathing phenomenon exhibited by a MOF. The microscopic picture of the structural transition occurring during the NGA process is unprecedented and emphasizes the strength of the newly developed OMD approach to tackle the most challenging structural transition in solid state materials.
2 Model and computational procedure
The simulation box consisted of 32 unit cells of MIL-53(Cr)-op deployed according to the x, y and z directions and was built from the crystallographic coordinates previously reported by the X-ray powder diffraction (XRPD) study.12 Thus the box lengths were Lx = 33.2 Å, Ly = 52.8 Å and Lz = 26.8 Å. TThe MIL-53(Cr) material was modelled from the previously developed flexible force field.10 CO2 molecules were simulated by means of the model developed by Harris and Yung.42 The experimental DUT-49(Cu)-op structure18 (Lx = Ly = Lz = 46.427 Å) was considered as the initial configuration. After 100 ps (details of MD simulations are provided below) the transition DUT-49(Cu)-op → DUT-49(Cu)-cp was achieved. The calculated unit cell volume of the empty DUT-49(Cu)-cp was found to be 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 789 ± 98 Å3 corresponding to a lattice parameter of 35.76 ± 0.2 Å that is in fair agreement with the experiment18 (47284.8 Å3 and 36.61 Å). Methane was considered as a single uncharged particle.43 Crossed CO2/MIL-53(Cr) and CH4/DUT-49(Cu) interactions were considered by means of the Lorentz–Berthelot combining rules. The equations of motion were integrated using the velocity Verlet scheme. The Ewald summation was used for calculating the electrostatic interactions and the short range interactions were truncated at 12 Å.
789 ± 98 Å3 corresponding to a lattice parameter of 35.76 ± 0.2 Å that is in fair agreement with the experiment18 (47284.8 Å3 and 36.61 Å). Methane was considered as a single uncharged particle.43 Crossed CO2/MIL-53(Cr) and CH4/DUT-49(Cu) interactions were considered by means of the Lorentz–Berthelot combining rules. The equations of motion were integrated using the velocity Verlet scheme. The Ewald summation was used for calculating the electrostatic interactions and the short range interactions were truncated at 12 Å.
3 Results and discussion
3.1 Development of the OMD method and validation with the description of the CO2 adsorption on breathing MIL-53(Cr)
The developed OMD method relies on the implementation of a trial MC move for the insertion/deletion of a guest molecule during the MD trajectory in the osmotic ensemble. Both insertion and orientation bias are considered to improve the statistical sampling during the insertion/deletion move. The insertion/deletion frequency can be adjusted with respect to the duration of the structural transition and/or the relaxation time of the system once a molecule is inserted/deleted. A compromise has to be found in terms of time and step intervals (tins/del/Nins/del). Four different tins/del pairs were tested, tins/del = 0.0005 (Nins/del = 1), 0.005 (Nins/del = 10), 0.5 (Nins/del = 1000) and 5 ps (Nins/del = 10000) for a timestep of 0.5 × 10−3 ps in the case of the adsorption of CO2 in MIL-53(Cr) for a gas pressure of 0.5 bar (op → np transition). Nins/del = 1 and Nins/del = 10000 correspond to the extreme cases. Beyond Nins/del = 10000, the computational time will be too high to reach the convergence of the adsorbed amount. Fig. S1a and b† show that if tins/del is too long the adsorbed molecules do not equilibrate properly and if tins/del is too short (Nins/del = 1) structural instabilities are observed leading to a premature re-opening of the structure (np → op). However, to set up a more general approach, i.e. system-independent, the frequency of the insertion/deletion move is randomly chosen between 10 and 10000 MD steps. Furthermore, in this OMD scheme only one guest molecule is inserted/deleted while a small time-step of 0.5 × 10−4 ps is used to minimize the perturbation of the dynamics of the system.
Regarding the thermostat and the barostat we opted for the Martyna–Tuckerman–Klein Barostat44 (MTK) algorithm previously proven to allow sufficient pressure and thermal fluctuations.39 Furthermore the MTK integrator guarantees ergodic sampling. In this algorithm, the barostat relaxation time (τp) controls the magnitude of the internal pressure and simulation box volume fluctuations. As shown in Fig. S2a,† for a system without a structural transition (CO2 gas pressure of 0.1 bar in the MIL-53(Cr)) τp between 0.1 and 5 ps slightly affects the unit cell volume. However with τp = 10 ps a structural transition (op → np) is observed as a result of the increase in pressure fluctuations (Fig. S2b†). Similar results have been found from the op → np structural transition (CO2 gas pressure of 0.5 bar in MIL-53(Cr)). Three relaxation times were tested, 0.5, 1.0 and 5.0 ps to ensure that the OMD simulations do not depend on the relaxation time. As shown in Fig. S2c,† although the structural transition is well recovered, τp, the transition time, sharply differs. This is the result of the pressure fluctuations that increase as τp increases leading to premature transition. Therefore, we opted for τp = 0.5 ps as a compromise to obtain a relaxed system and to avoid a premature transition. Let us mention that for a high value of τp (10 ps) a metastable structure was obtained with chaotic trajectory. Contrary to the HOMC method where MD and MC are decoupled, in the OMD approach there is no need to apply an equilibration step after the insertion/deletion move that might break the ergodicity since only one molecule is inserted or deleted that avoids high fluctuations of energy along the MD trajectory. The OMD trajectory is continuous and the ergodicity is maintained in such a way that the statistical averages can be assimilated to time averages because there is no break in the trajectory contrary to hybrid MC/MD simulations alternating MD and MC runs. The calculation of the adsorption isotherm requires OMD simulations for each gas pressure such that the final configuration obtained for gas pressure pi needs to be the same as the initial configuration at pi+1 (see Fig. 1c). Indeed the molecular mechanisms controlling the guest-induced structural transitions depend on the history of the systems, i.e. retaining a thermal, mechanical and adsorption memory.
Validation of OMD simulations was carried out to examine the CO2 induced structural transition of MIL-53(Cr). Fig. 1d reveals that the OMD simulated CO2 adsorption isotherm at 298 K and σ = 1 bar in MIL-53(Cr) is in fair agreement with the experimental data13 in terms of step change positions associated with both op → np and np → op structural transitions at 0.5 bar and at 5.0 bar, respectively as well as in terms of adsorbed amount. This result demonstrates that the OMD approach implementing our previously developed flexible forcefield to describe the host framework is reliable to account for both the guest adsorption and the concurrent flexibility of the host. Details of the force field are provided in ESI† FIELD-MIL53Cr.txt. As shown in Fig. 1d the step changes are predicted to be sudden contrary to the more gradual evolution as observed experimentally due to the op/np phases mixture.30 Moreover, Fig. S3a and b† report the CO2 adsorbed amount and the unit cell volume (u.c.) change of MIL-53(Cr) as a function of time. These plots reveal that both op → np and np → op transitions are extremely fast (30–50 ps). Meanwhile the CO2 adsorbed amount is quasi-constant during op → np structural switching.
3.2 Transferability of the OMD method to the description of the CH4 triggered NGA in DUT-49(Cu)
Fig. 2a reports the OMD simulated CH4 adsorption isotherms at 111 K and at σ = 1 bar for DUT-49(Cu) showing the presence of two step-changes at 6.0 bar and 120 kPa associated with two subsequent structural switchings from op (100859.9 Å3) to cp (45865.08 Å3) and cp (48170.87 Å3) to op (104173 Å3) respectively. Furthermore, the calculated unit cell volume of the empty DUT-49(Cu)-op was found to be 101023 ± 134 Å3 corresponding to a lattice parameter of 46.37 ± 0.3 Å in fair agreement with the corresponding experimental data18 (100071.8 Å3 and 46.427 Å). This observation emphasizes that the flexible force field used to describe the DUT-49(Cu) framework, a hybrid version of the generic force field DREIDING45 and UFF46 in combination with the united atom Trappe model for CH4 (ref. 47) is accurate enough to capture the NGA phenomenon as highlighted by Fig. 2b with a simulated negative adsorption step (6 kPa) and released amount of CH4 (3.1 mmol g−1) concurrent with the contraction of the structure in line with the corresponding experimental values (10 kPa and 5 mmol g−1 respectively). Details of the force field are provided in ESI† FIELD-DUT49Cu.txt. Furthermore, in the case of an explicit charged description of methane using the OPLS model,48 Fig. S4† shows that the NGA step is well captured. Although the pressure position and the amplitude of NGA are slightly shifted the negative adsorption and its physics are well captured. The re-opening of the structure is predicted to occur at a higher pressure than the experimental value (500 kPa vs. 40 kPa). Besides the choice of the flexible force field, this deviation is most probably associated with slow kinetics of re-opening. Therefore the cp → op transition might be observed at a lower pressure than 500 kPa but using a much longer simulation time. Indeed, a very long-time simulation of 0.75 μs was conducted for a gas pressure of 50 kPa and no transition was observed. This problem is observed in molecular simulations in terms of relaxation time. Typically the capture of the physical properties of some complex systems requires the increase of temperature,49 pressure50 or electrical field.51 Remarkably, this is the first time that a molecular simulation can capture the counterintuitive spectacular NGA behavior of DUT-49(Cu).
 | ||
| Fig. 2 (a) OMD simulated and experimental CH4 adsorption isotherms at 111 K for DUT-49(Cu) plotted as a semi-logarithmic scale. The OMD simulated unit cell volume changes alongside CH4 adsorption are also provided. (b) OMD simulated and experimental adsorption isotherms at a low pressure to emphasize the NGA (ΔnNGA). | ||
Fig. 3a that represents the time evolution of the unit cell volume of DUT-49(Cu) during both transitions shows that at 500 kPa the re-opening of the structure is extremely long (about 4 ns) as compared to the cp → op one (200–500 ps range). The inset in Fig. 3a shows that the CH4 adsorbed is well correlated with the unit cell evolution of the MOF framework. Thermodynamically Fig. 3c and d show that the energy cost (intramolecular energy) of the op → cp transition due to the increase of the internal constraints is compensated by the interactions between CH4 molecules and the MOF framework. The intramolecular energy corresponds to the bonds, angles, dihedrals and non-bonded potentials of DUT-49(Cu) while the intermolecular one is related to the van der Waals (modeled from Lennard-Jones potential) and electrostatic interactions between methane molecules and DUT-49(Cu) and between molecules themselves. This results from the shrinkage of the unit-cell of DUT-49(Cu) involving a shortening of the intermolecular distances. This energy compensation is not observed during the reopening where both intramolecular and intermolecular interactions decrease in favour of the op form since the internal tension decreases and the adsorbed amount increases. Interestingly, this compensation between the intramolecular and the intermolecular energies during the op → cp transition was also observed in MIL-53(Cr) upon CO2 adsorption as well as the intramolecular behavior during the re-opening.6
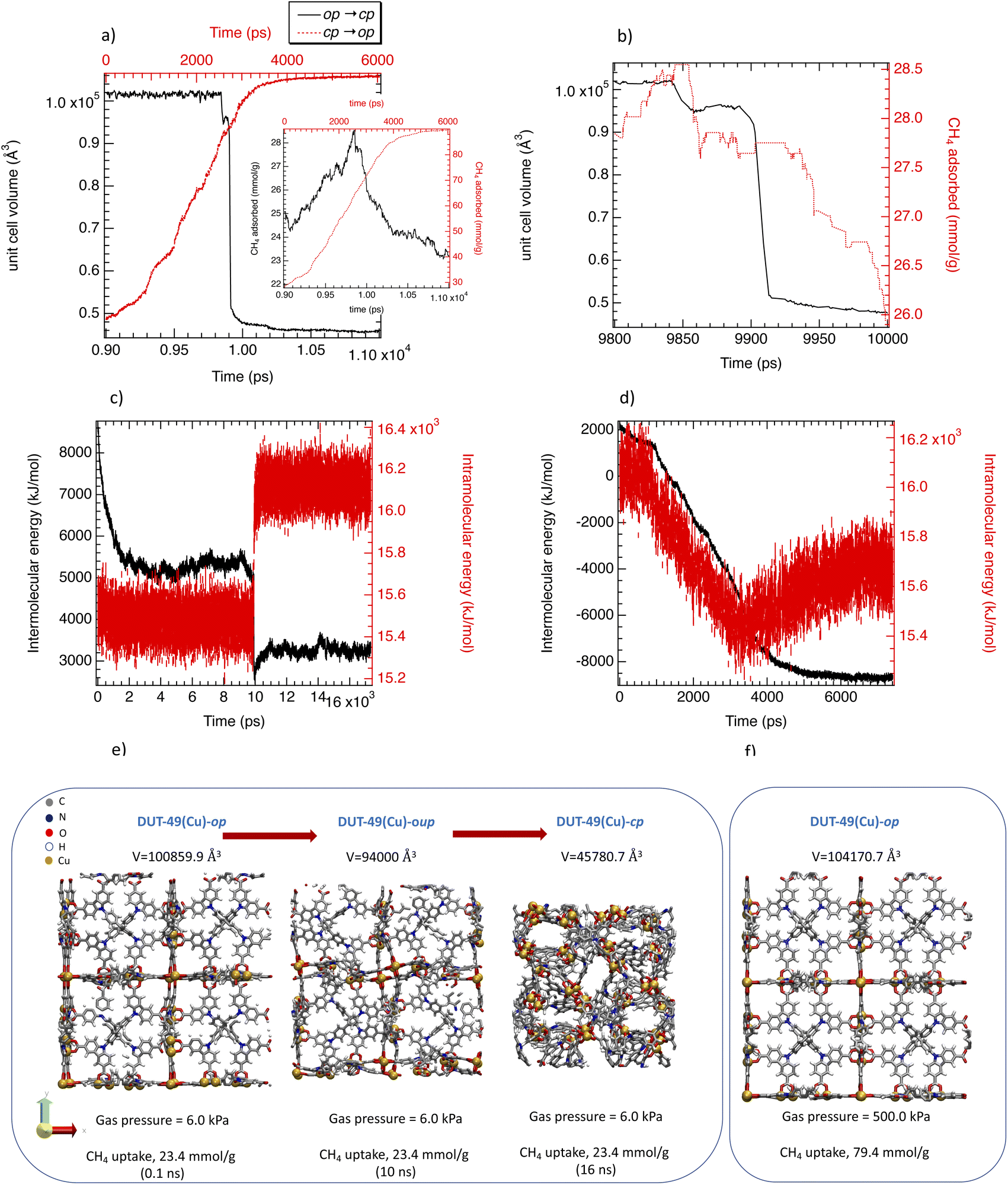 | ||
| Fig. 3 (a) OMD simulated unit cell volume changes alongside the CH4-triggered op–cp and cp–op structural changes. Inset figure represents the corresponding CH4 adsorbed amount with the same color code. (b) Enlargement of part a between 9.8 ns and 10 ns. Intermolecular and intramolecular energy as a function of time during the op → cp (c) and cp → op (d) transitions. (e) Illustrations of the DUT-49(Cu) structure configuration during the structural transitions: op, cp and unstable phases. (f) Illustration of the final configuration of DUT-49(Cu) for a CH4 pressure of 500 kPa. | ||
Interestingly, Fig. 3a and b evidence a small plateau at 10 ns corresponding to a unit cell volume of 94000 Å3 for DUT-49(Cu) suggesting an unstable open pore labelled as DUT-49(Cu)-oup and associated with a short lifetime. Fig. 3d and e illustrate the oup, cp and the op forms obtained by our OMD simulations. Fig. 3e and f show a progressive contraction involving strong deformation of the biphenyl axis schematized in Fig. 4a and quantified in Fig. 4b with the instantaneous angle reported between the nitrogen atoms of the carbazole groups and a carbon atom of the biphenyl group connecting them. This angle evolves from 178° (almost linear shape) to a value between 170° and 150° highlighting different degrees of the curved shape. This deformation is a result of a relatively important stress exerted by the adsorbed methane molecules that induces a bending type mechanical response of the host and its subsequent deformation to minimize the constraint and hamper a structure collapse related to a buckling deformation. This result is in line with the work reported by Evans et al. who studied the stress–strain curves from the deformation of the N–N distance of the empty DUT-49(Cu) by using DFT optimizations of a single isolated ligand.38 For deformations higher than 3 Å, a buckling transition occurs, with a sudden large deflection of the ligand and a non-linear mechanical response related to the NGA structural response. Comparatively, the OMD simulation clearly evidenced the microscopic mechanism of the NGA in the presence of methane. Fig. 5a shows the average number of CH4 molecules with an interacting distance with the atoms of DUT-49(Cu) lower than 4.5 Å for a gas pressure of 5 kPa. This figure reveals that CH4 is relatively homogeneously distributed with however a preferential region near the carbazole groups. We have also reported in Fig. 5b the local three-dimensional free energy profile. This figure highlights the strong force exerted on carbazole groups on the biphenyl axis which probably leads to a tension at the origin of buckling deformation. From an energy point of view, the situation is similar to that observed with MIL-53(Cr).6 Indeed, Fig. S5† shows an increase in intramolecular interactions (mainly dihedral) compensated by the intermolecular contribution during the op → cp transition.
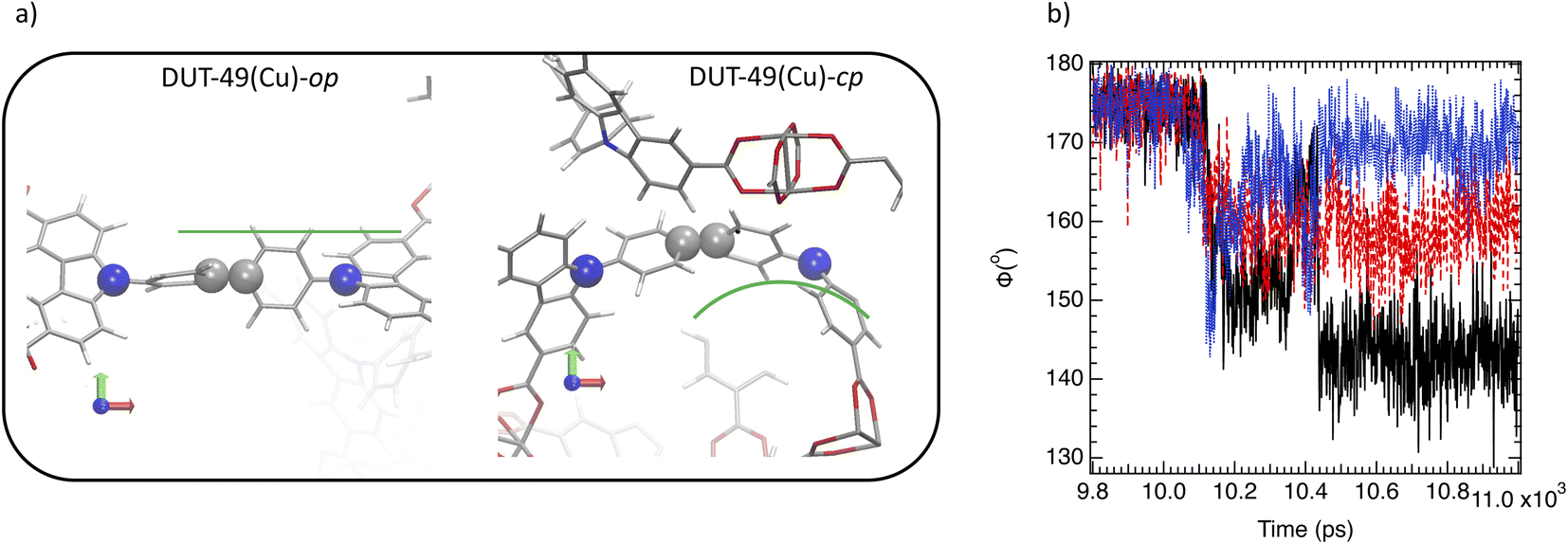 | ||
| Fig. 4 (a) Illustration of the typical angular deflection in both DUT-49(Cu)-op and DUT-49(Cu)-cp. (b) Time evolution of three angular deflections, ϕ1 (black), ϕ1 (red) and ϕ3 (blue) randomly selected during the op → cp transition. | ||
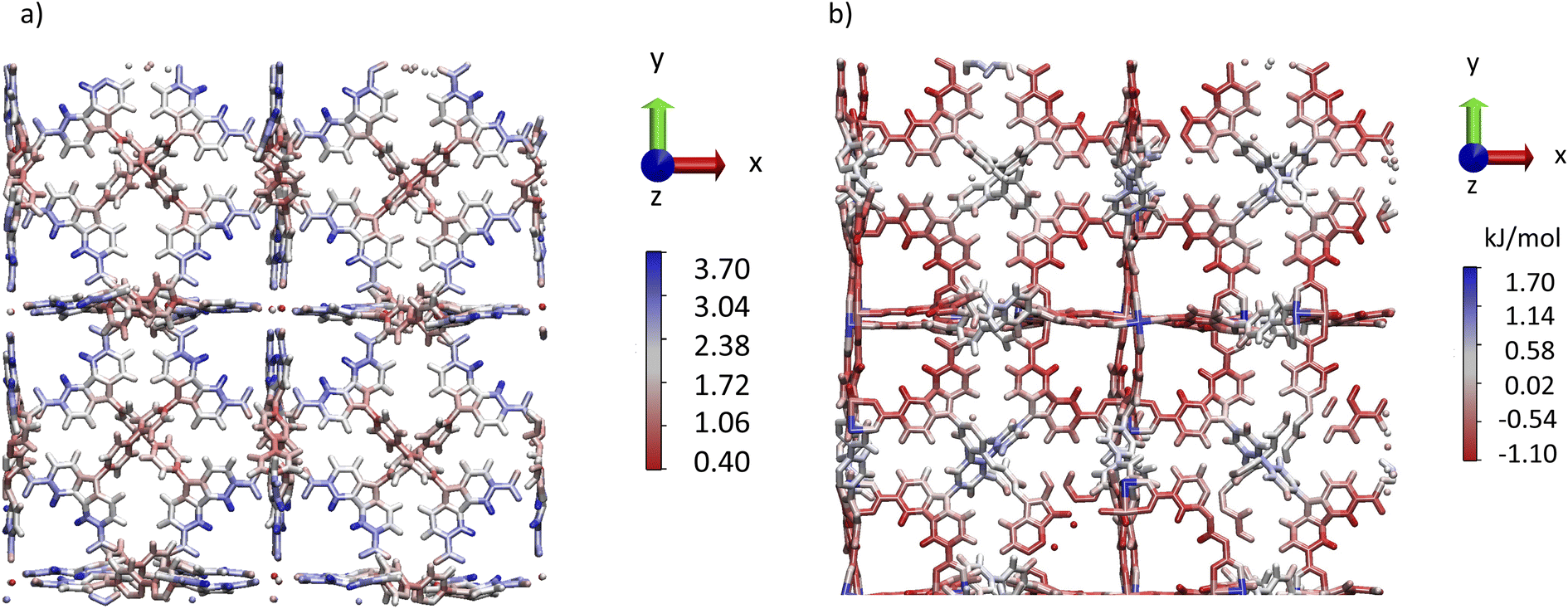 | ||
| Fig. 5 Three dimensional distribution (a) of density (number of molecules) and (b) free energy of the CH4 molecules with close contact with the DUT-49(Cu)-op below 4.5 Å. | ||
Fig. 6 focuses on the time evolution of the unit cell volume of DUT-49(Cu) and the adsorbed amount in the range of the op → cp transition related to the NGA. Five regions can be distinguished in three domains of time; from 9.8 to 1.0 ns (Fig. 6a corresponding to regions (I), (II), (III) and (IV)), from 1.0 to 1.1 ns (Fig. 6b associated with region (IV)) and from 1.1 to 1.6 ns (Fig. 6c related to region (V)); region (I) corresponds to the op → oup transition and ΔnNGA = −0.5 mmol g−1, region (II) corresponds to the oup form where the CH4 adsorbed amount is constant, region (III) related to the oup → cp′ such that cp′ is the form obtained from the sudden transition of the DUT-49(Cu)-oup and ΔnNGA = −2.4 mmol g−1, region (IV) is related to a slow cp′ → cp transition with ΔnNGA = −2.4 mmol g−1 and a region (V) corresponding to the cp form. The op → cp transition begins from a critical CH4 adsorbed value of 28.5 mmol g−1 (6 kPa) higher than the calculated value at 5.9 kPa corresponding to 26.1 mmol g−1 on the op form. Fig. 6 shows that the structural transition between both the op and cp phases is progressive via unstable and transitory forms (oup and cp′) depending on the adsorbed amount that underline the metastable character of DUT-49(Cu).
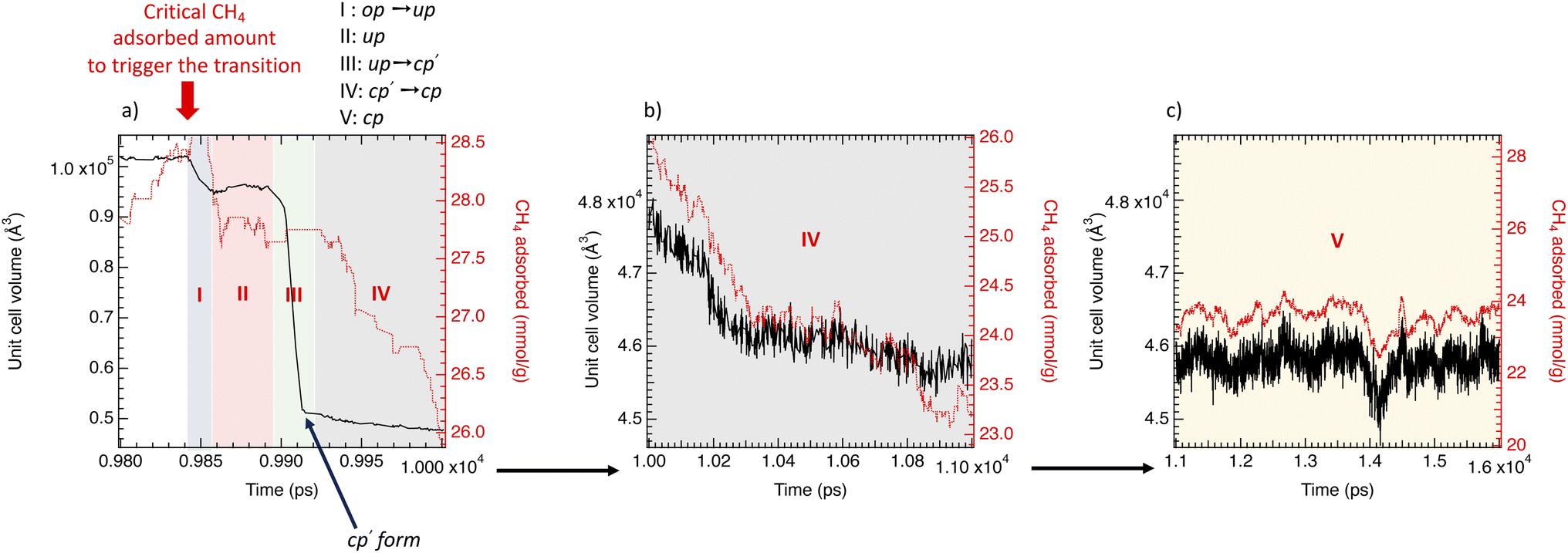 | ||
| Fig. 6 Simultaneous evolution of the unit cell volume (solid black line) and adsorbed amount of CH4 (dotted red line) as a function of time (between 0.98 ns and 1.6 ns) into regions I, II, III and IV (a) IV (b) and V (c) for a gas pressure of 6 kPa. | ||
Contrary to the standard MD and GCMC simulations, the OMD method offers a unique opportunity to capture the structural transition as a function of the CH4 adsorbed and to highlight the existence of a transient and unstable phase reminiscent of the metastable forms observed from desorption experiments.18 We report in Fig. 7a the CH4 desorption isotherm for DUT-49(Cu). Fig. 7a evidences the presence of a hysteresis which is in good qualitative agreement with the experimental trend previously reported. The desorption isotherm shows a progressive decrease in CH4 adsorbed during the op → cp transition contrary to the adsorption isotherm that exhibits a sudden increase during the re-opening of the structure. Fig. 7b sheds light on the fact this progressive desorption is the result of the formation of DUT-49(Cu) with intermediate pore sizes (DUT-49(Cu)-ip) characterized by a progressive pore contraction (see Table S1† where the lattice parameters are provided) which is in line with the experimental observation.18 Furthermore, as shown in Fig. 7b, the OMD simulations are also capable of capturing the absence of re-opening at low gas pressure during the desorption. For a gas pressure of 2 kPa a slightly more contracted form than the cp form is obtained (the contracted pore form (cp)) that is again in fair concordance with the experimental observation. This clearly emphasizes the ability of the OMD method to capture accurately the underlying physics of the DUT-49(Cu) transition.
 | ||
| Fig. 7 (a) OMD simulated CH4 adsorption and desorption isotherms at 111 K on DUT-49(Cu) and (b) unit cell volume of DUT-49(Cu) as a function of the gas pressure in both adsorption and desorption processes. | ||
4 Conclusion
In this work a cutting-edge atomistic simulation method was developed by integrating a GCMC trial move into a MD scheme to take into account both the fluid adsorption process as well as the flexibility of the material in the osmotic statistical ensemble. The corresponding OMD method surpasses classical MD, MC and HOMC simulations since it can capture simultaneously the fluctuations of the number of adsorbed molecules and the associated structural and dynamics consequences on the structure of the host material. This newly developed OMD method was demonstrated to capture accurately the reversible guest-induced structural transition of two prototypal highly breathing MOFs, MIL-53(Cr) and DUT-49(Cu). While the structural transitions exhibited by MIL-53(Cr) between the open and narrow pore forms are sudden, the volume change of DUT-49(Cu) was found to be progressive highlighting the existence of intermediate pore forms between the narrow and open pore structures. Compared to recent theoretical studies on the NGA phenomenon exhibited by DUT-49(Cu) using thermodynamical24,26,27,36,38 and mechanical38 approaches, this OMD method sheds light for the first time on the molecular origin of this abnormal structural behavior under fluid adsorption. We demonstrate that the NGA phenomenon is triggered by a critical amount of CH4 and controlled by the high flexibility of the carbazole–bipheny–carbazole axis inducing a strong bending. Contrary to macroscopic approaches and classical molecular methods, this OMD method is probably the only way to highlight the fine interplay between structural transformations in soft materials and fluid adsorption. The OMD method is therefore well suited to be combined with experiments to obtain a full microscopic picture of the structural transformations triggered from internal and external stimuli. This newly developed approach paves the way towards the study of a more complex scenario with the combination of several stimuli such as adsorption with electric field51 and mechanical pressure10 on these flexible materials.Data availability
Data for this paper, including force fields of DUT-49(Cu) and MIL-53(Cr) are available at https://github.com/aghoufi/DUT-49-Cu--OMD.Author contributions
A. G. and G. M. supervised the project. C. P., H. Z. and A. G. carried out OMD simulations. All authors were involved in the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank ANR MEACOPA (ANR-17-CE29-0003) for funding.Notes and references
- C. Serre and G. Férey, Chem. Soc. Rev., 2009, 38, 1380–1399 RSC.
- S. Horike, S. Shimomora and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed.
- G. Férey, C. Serre, T. Devic, G. Maurin, H. Jobic, P. L. Llewellyn, G. D. Weireld, A. Vimont, M. Daturi and J.-S. Chang, Chem. Soc. Rev., 2011, 40, 550–562 RSC.
- S. Kitawaga, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef PubMed.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R.-A. Fischer, Chem. Soc. Rev., 2014, 16, 6062–6096 RSC.
- F. Salles, A. Ghoufi, G. Maurin, R.-G. Bell, C. Mellot-Draznieks, P.-L. Llewellyn, C. Serre and G. Ferey, Angew. Chem., Int. Ed., 2008, 47, 8487–8491 CrossRef CAS PubMed.
- A. Ghoufi, G. Maurin and G. Férey, J. Phys. Chem. Lett., 2010, 1, 2810–2815 CrossRef CAS.
- A. Boutin, F.-X. Coudert, M.-A. Springuel-Huet, A.-V. Neimark, G. Férey and A.-H. Fuchs, J. Phys. Chem. C, 2010, 114, 22237–22244 CrossRef CAS.
- M. Gaab, N. Trukhan, S. Maurer, R. Gummaraju and U. Müller, Microporous Mesoporous Mater., 2012, 157, 131–136 CrossRef CAS.
- A. Ghoufi, A. Subercaze, Q. Ma, P.-G. Yot, Y. Ke, I. Puente-Orench, T. Devic, V. Guillerm, C. Zhong, C. Serre, G. Ferey and G. Maurin, J. Phys. Chem. C, 2012, 116, 13289–13295 CrossRef CAS.
- Q. Ma, Q. Yang, A. Ghoufi, G. Férey, C. Zhong and G. Maurin, Dalton Trans., 2012, 41, 3915–3919 RSC.
- C. Serre, F. Millange, C. Thouvenot, M. Noguès, G. Marsolier, D. Louer and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519–13526 CrossRef CAS PubMed.
- C. Serre, S. Bourrelly, A. Vimont, N.-A. Ramsahye, G. Maurin, P.-L. Llewellyn, M. Daturi, Y. Filinchuk, O. Leynaud, P. Garnes and G. Férey, Adv. Mater., 2007, 19, 2246–2251 CrossRef CAS.
- N. Chanut, A. Ghoufi, M.-V. Coulet, S. Bourrelly, B. Kuchta, G. Maurin and P. Llewellyn, Nat. Commun., 2020, 11, 1216 CrossRef CAS PubMed.
- P. Horcajada, G.-R. Baati, P.-K. Allan, G. Maurin, P. Couvreur, G. Férey, R.-E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- P. Freund, I. Senkovska, B. Zheng, V. Bon, B. Krause, G. Maurin and S. Kaskel, Chem. Commun., 2020, 56, 7411–7414 RSC.
- V. Bon, N. Kavoosi, I. Senkovska and S. Kaskel, ACS Appl. Mater. Interfaces, 2015, 7, 22292–22300 CrossRef CAS PubMed.
- S. Krause, V. Bon, I. Senkovska, U. Stoeck, D. Wallacher, D.-M. Tobbens, S. Zander, R.-S. Pillai, G. Maurin, F.-X. Coudert and S. Kaskel, Nature, 2016, 532, 348–352 CrossRef CAS PubMed.
- S. Krause, J. Evans, V. Bon, I. Senkovska, P. Lacomi, F. Kolbe, S. Ehrling, E. Troschke, J. Getzschmann, D. M. Tobbens, A. Franz, D. Wallacher, P. Yot, G. Maurin, E. brunner, P. Llewellyn, F. Coudert and S. Kaskel, Nat. Commun., 2019, 10, 3632 CrossRef PubMed.
- S. Krause, J.-D. Evans, V. Bon, I. Senkovska, S. Ehrling, U. Stoeck, P.-G. Yot, P. lacomi, P.-L. Llewellyn, G. Maurin, F. X. Coudert and S. Kaskel, J. Phys. Chem. C, 2018, 122, 19171 CrossRef CAS PubMed.
- J. Heinen and D. Dubbeldam, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, 1363 Search PubMed.
- J.-D. Evans, J.-P. Durholt, S. Kaskel and R. Schmid, J. Mater. Chem. A, 2019, 7, 24019–24026 RSC.
- R. Demuynck, S.-M. J. Rogge, L. Vanduyfhus, J. Wieme, M. Waroquier and V. V. Speybroeck, J. Chem. Theory Comput., 2017, 13, 5861 CrossRef CAS PubMed.
- L. Vanduyfhuys, S. Rogge, W. Wieme, S. Vandenbrande, G. Maurin, M. Waroquier and V. V. Speybroeck, Nat. Commun., 2018, 9, 204 CrossRef CAS PubMed.
- L. Vanduyfhuys and G. Maurin, Adv. Theory Simul., 2019, 1900124 CrossRef CAS.
- J. Evans, S. Krause, S. Kaskel, M. Sweatman and L. Sarisov, Chem. Sci., 2019, 10, 5011–5017 RSC.
- R. Goeminne, S. Krause, S. Kaskel, T. Verstraelen and J. Evans, J. Am. Chem. Soc., 2021, 143, 4143–4147 CrossRef CAS PubMed.
- N. Ramsahye, G. Maurin, S. Bourrelly, P. L. Llewellyn, T. Loiseau and G. Férey, Chem. Commun., 2007, 31, 3261 RSC.
- P. lacomi, B. Zheng, S. Krause, S. Kaskel, G. Maurin and P.-L. Llewellyn, Chem. Mater., 2020, 32, 3489 CrossRef PubMed.
- A. Ghoufi and G. Maurin, J. Phys. Chem. C, 2010, 114, 6496–6502 CrossRef CAS.
- S.-M. Rogge, R. Goeminne, R. Demuynck, J.-J. Gutiérrez-Sevillano, S. Vandenbrande, L. Vanduyfhuys, M. Waroquier, T. Verstraelen and V. Van Speybroeck, Adv. Theory Simul., 2019, 2, 1800177 CrossRef CAS.
- D. Dubbeldam, K. Walton, D. Ellis and R. Snurr, Angew. Chem., Int. Ed., 2007, 46, 4496 CrossRef CAS PubMed.
- L. Zhang, Z. Hu and J. Jiang, J. Am. Chem. Soc., 2013, 135, 3722–3728 CrossRef CAS PubMed.
- J. Gee, J. Chung, S. Sankar and D. Sholl, J. Phys. Chem. C, 2013, 117, 3169–3176 CrossRef CAS.
- D. Anstine and C. Colina, Polym. Int., 2020, 70, 984–989 CrossRef.
- S. Rogge, S. Caroes, R. Demuynck, M. Waroquier, V. V. Speybroeck and A. Ghysels, J. Chem. Theory Comput., 2018, 14, 1186–1197 CrossRef CAS PubMed.
- J. Rogacka, F. Formalik, A.-L. Triguero, L. Firlej, B. Kuchta and S. Calero, Phys. Chem. Chem. Phys., 2019, 21, 3294 RSC.
- J.-D. Evans, L. Bocquet and F.-X. Coudert, Chem, 2016, 1, 874 Search PubMed.
- S. Rogge, L. Vanduyfhuys, A. Ghysels, M. Waroquier, T. Verstraelen, G. Maurin and V. V. Speybroeck, J. Chem. Theory Comput., 2015, 11, 583 CrossRef PubMed.
- P.-G. Yot, L. Vanduyfhus, E. Alvarez, J. Rodriguez, J.-P. Itié, P. Fabry, N. Guillou, T. Devic, I. Beurroies, P.-L. Llewellyn, V. V. Speybroeck, C. Serre and G. Maurin, Chem. Sci., 2016, 7, 446–450 RSC.
- F. Salles, G. Maurin, C. Serre, P. L. Llewellyn, C. Knofel, H.-J. Choi, Y. Filinchuk, L. Oliviero, A. vimon, J.-R. Long and G. Ferey, J. Am. Chem. Soc., 2010, 132, 13782 CrossRef CAS PubMed.
- J. Harris and K. Yung, J. Phys. Chem., 1995, 99, 12021–12024 CrossRef CAS.
- D. Moller, J. Oprzynski, A. Muller and J. Fischer, Mol. Phys., 1992, 75, 363 CrossRef.
- G. Martyna, M. Tuckerman, D. Tobias and M. Klein, Mol. Phys., 1996, 87, 1117 CrossRef CAS.
- S. Mayo, B. Olafson and W. Goddard, J. Phys. Chem., 1990, 94, 8897 CrossRef CAS.
- A. Rappe, C. Casewit, K. Colwell, W. A. Goddard III and W. Skiff, J. Am. Chem. Soc., 1992, 114, 10024 CrossRef CAS.
- M. Martin and J. Siepman, J. Phys. Chem. B, 1998, 102, 2569–2577 CrossRef CAS.
- W. Jorgensen, D. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225 CrossRef CAS.
- A. Khudozhitov, H. Zhao, S. Arzumanov, D. Kolokolov, G. Maurin and A. Stepanov, ACS Appl. Mater. Interfaces, 2021, 13, 33685–33692 CrossRef PubMed.
- M. Ding, A. ghoufi and A. Szymczyk, Desalination, 2014, 343, 48–53 CrossRef CAS.
- A. Ghoufi, K. Benhamed, L. Boukli-Hacene and G. Maurin, ACS Cent. Sci., 2017, 3, 394 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc04174j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |