
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtein charge parameters that influence stability and cellular internalization of polyelectrolyte complex micelles†
Rachel A.
Kapelner‡
a,
Rachel S.
Fisher‡
 ab,
Shana
Elbaum-Garfinkle
bc and
Allie C.
Obermeyer
*a
ab,
Shana
Elbaum-Garfinkle
bc and
Allie C.
Obermeyer
*a
aDepartment of Chemical Engineering, Columbia University, New York, NY 10027, USA. E-mail: aco2134@columbia.edu; Tel: +1-212-853-1215
bStructural Biology Initiative, CUNY Advanced Science Research Center, New York, NY, USA
cPhD Programs in Biochemistry and Biology at the Graduate Center, City University of New York, NY, USA
First published on 29th November 2022
Abstract
Proteins are an important class of biologics, but there are several recurring challenges to address when designing protein-based therapeutics. These challenges include: the propensity of proteins to aggregate during formulation, relatively low loading in traditional hydrophobic delivery vehicles, and inefficient cellular uptake. This last criterion is particularly challenging for anionic proteins as they cannot cross the anionic plasma membrane. Here we investigated the complex coacervation of anionic proteins with a block copolymer of opposite charge to form polyelectrolyte complex (PEC) micelles for use as a protein delivery vehicle. Using genetically modified variants of the model protein green fluorescent protein (GFP), we evaluated the role of protein charge and charge localization in the formation and stability of PEC micelles. A neutral-cationic block copolymer, poly(oligoethylene glycol methacrylate-block-quaternized 4-vinylpyridine), POEGMA79-b-qP4VP175, was prepared via RAFT polymerization for complexation and microphase separation with the panel of engineered anionic GFPs. We found that isotropically supercharged proteins formed micelles at higher ionic strength relative to protein variants with charge localized to a polypeptide tag. We then studied GFP delivery by PEC micelles and found that they effectively delivered the protein cargo to mammalian cells. However, cellular delivery varied as a function of protein charge and charge distribution and we found an inverse relationship between the PEC micelle critical salt concentration and delivery efficiency. This model system has highlighted the potential of polyelectrolyte complexes to deliver anionic proteins intracellularly. Using this model system, we have identified requirements for the formation of PEC micelles that are stable at physiological ionic strength and that smaller protein–polyelectrolyte complexes effectively deliver proteins to Jurkat cells.
Introduction
Advances in genetic engineering and biotechnology have provided the ability to design, develop, and produce proteins as therapeutic treatments. To date, hundreds of protein therapeutics have been developed and approved by the FDA. However, currently approved protein therapies act primarily on extracellular or membrane-bound targets.1 This is largely due to the inability of proteins to cross the cell membrane and access intracellular targets.1–3 To fully realize the promise of protein therapeutics, efficient intracellular protein delivery is required. Intracellular delivery of a protein molecule, as opposed to the cDNA or mRNA for the cell to produce the protein itself, ensures the best control over dosage.4 Current methods of protein delivery have a few key shortcomings including inefficient cellular uptake and poor endosomal escape. Therefore, to capitalize on the potential of genetic and protein engineering to treat disease, new methods for intracellular protein delivery are needed.3,4Two major strategies have been studied to improve intracellular protein delivery: increasing the hydrophobicity of the protein and increasing the cationic charge on the protein.5 It has previously been demonstrated that hydrophobic proteins can translocate across the cell membrane, most likely due to favorable hydrophobic interactions with the lipid bilayer. Mix et al. were able to deliver an esterified green fluorescent protein (GFP) directly to the cytosol.6 Esterification of the carboxyl groups with a hydrophobic diazo compound was sufficient for the protein to directly cross the cellular membrane. Additionally, hydrophobic cell-penetrating peptides (CPPs) have been shown to deliver GFP to HeLa cells.7 Cationic CPPs, particularly peptides with a high fraction of arginine residues, have been shown to similarly facilitate intracellular protein delivery.8–11 Similarly, proteins with significant cationic charge, native or engineered, can also be internalized efficiently by cells.12,13
For anionic proteins to cross the cellular membrane they must overcome the coulombic repulsion from the negatively charged extracellular membrane14 and the hydrophobic nature of the lipid bilayer.6 It is for this reason that anionic proteins remain a particular challenge to deliver intracellularly. Yet at the same time, the delivery of highly anionic nucleic acids has been successfully demonstrated and implemented. Several approaches to deliver anionic proteins have taken inspiration from DNA delivery approaches. These include the formulation of protein nanoparticles,15–18 polymer nanoemulsions,19,20 and liposomes.21–23 While all have demonstrated successes, these techniques tend to be better suited for nucleic acid delivery due to the high charge density of nucleic acids.
An alternative approach for protein delivery uses polyelectrolyte complex (PEC) micelles formed between a protein and diblock copolymer. This approach still looks to DNA delivery for inspiration, as PEC micelles were initially developed by Kataoka et al. for nucleic acid delivery in the late 1990's.24–27 These PEC micelles are formed from the electrostatic attraction between a diblock copolymer with a charged block and a hydrophilic charge-neutral block and a protein of opposite charge.28 Encapsulating supernegatively charged proteins in the core of a PEC micelle could potentially serve to shield the protein from enzymatic degradation prior to delivery, promote intracellular delivery of the protein, and even facilitate endosomal escape.29 This endosomal escape could be enhanced through the proton sponge effect or if that is insufficient through the inclusion of redox sensitive elements.30–34
PEC micelles have been demonstrated to successfully deliver nucleic acids, but because proteins have a lower charge density, initial studies have shown that at physiological ionic strength, protein-based PEC micelles dissociate.35,36 To overcome this challenge, previous work has explored different ways to increase the charge density of the PEC micelle core. One strategy is to introduce a third polyelectrolyte, which would be co-encapsulated in the micelle core with the like-charged protein. While this strategy has been shown to increase the salt stability of the micelle, the protein component partitioned out of the micelle below the micelle critical salt concentration, rendering this approach infeasible for applications at physiological conditions.37,38 Another strategy is to increase the net charge of the protein itself, which can be accomplished through either chemical or genetic modification. Increasing the net negative charge of four model proteins, α-chymotrypsinogen, lysozyme, myoglobin, and RNase A, by chemical supercharging was shown to increase the salt stability of PEC micelles.39 Similarly, supercharging equine heart cytochrome c (CytC) and immunoglobulin G (IgG) with citraconic anhydride or cis-aconitic anhydride resulted in the formation of PEC micelles that remained stable at physiological ionic strength.29,40,41 The reversible nature of the chemical supercharging of proteins in this approach had the added benefit of promoting pH responsive PEC micelle dissolution and endosomal escape in model mammalian cell lines.
While chemical supercharging a protein has produced salt stable PEC micelles for intracellular delivery, there are a few drawbacks to this strategy. Chemical supercharging results in a distribution of supercharged proteins, with polydisperse charge densities; the behavior of the resultant proteins is dependent on reaction conditions and degrees of supercharging, with high degrees of supercharging frequently required for improved delivery efficiency.41 Additionally, many proteins are not stable or catalytically active at the degree of modification required for salt-stable micelle formation. Genetic engineering enables the selection of the location of each charged residue on the protein surface and a monodisperse distribution of net charge. Based on previous work on complex coacervation with homopolymers, protein charge distribution can also impact the conditions where phase separation is observed.42–47 To date, both the effect of the degree of protein supercharging and charge distribution on PEC micelle formation and stability has yet to be established. In this work, genetic engineering was used to precisely control the number and location of charges on a model protein, GFP. A relationship between protein net charge and charge distribution on micelle stability was established and, for isotropically charged proteins, this relationship was consistent with the behavior of analogous bulk-phase coacervates. In addition, the effect of PEC micelle stability on intracellular delivery efficiency was assessed.
Results and discussion
Design of protein–polyelectrolyte delivery system
Given the inability of anionic proteins to enter cells, we designed a polymer delivery vehicle to improve intracellular delivery (Scheme 1a). This approach relies on complexation and microphase separation of anionic proteins with a cationic block copolymer. Mixing GFP with this block copolymer can result in the formation of spherical polyelectrolyte complex (PEC) micelles with a GFP-rich core, depending on the mixing ratio and solution ionic strength. However, in order to effectively deliver proteins using this approach, an understanding of how protein properties impact the formation and stability of PEC micelles is needed. To assess the effect of protein charge on PEC micelle formation and stability, we generated a library of negatively charged superfolder GFP (sfGFP) mutants (Scheme 1b). We have previously demonstrated the relationship between protein charge and both the nature of phase separation (solid or liquid) and the phase behavior at increased ionic strength.42 Based on these prior findings and the requirement for phase separation at physiological ionic strength, only GFP variants with higher net charge were investigated. A panel of GFP mutants with an expected charge of −18, −24, and −30, at pH = 7.4, were evaluated. For each of these variants, the charge was distributed isotropically across the protein surface. | ||
| Scheme 1 (a) Schematic of polyelectrolyte core (PEC) micelle formation with supercharged protein variants. (b) Electrostatic surface potential representation of the solvent accessible surface area at pH 7.4 for the GFP(−7) (superfolder) and supercharged GFP variants studied here. These were prepared using the Adaptive Poisson–Boltzmann Solver (APBS) plugin in pymol (±5 kBT/e with red for negative and blue for positive) (c) synthesis of the diblock copolymer, POEGMA79-b-qP4VP175, via RAFT polymerization. The chain transfer agent (CTA) used was 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid, the indicator used was azobisisobutyronitrile (AIBN), and the OEGMA monomers used had a Mn = 300 g mol−1 (n ∼ 4 and 5). | ||
Variants with equivalent charge localized to a C-terminal polypeptide tag were also generated. The addition of glutamate and aspartate residues at the C-terminus of GFP generated variants with a polyionic tag that provided a localized charge patch for polyelectrolyte complexation. The tagged variants with net charge of −18 and −24 were successfully characterized, but insufficient quantity of pure tag-GFP(−30), with 24 charged residues localized to the C-terminus, was produced for characterization (ESI Fig. S1†). Finally, the unmodified sfGFP protein was also characterized for comparison to the globular domain of the tagged variants. The hydrodynamic radius of purified proteins was characterized by DLS. As expected, all characterized proteins had a hydrodynamic radius of approximately 2 nm, with the exception of GFP(−18) (ESI Fig. S1†). GFP(−18) had a monomeric population with an average radius of 2 nm, but also had a population with a larger radius (∼11 nm). This larger population has been interpreted as protein multimers, which is consistent with the ability of sfGFP to form a weak dimer.48
We then proceeded to design a block copolymer that could complex these anionic proteins and form PEC micelles (Scheme 1c). A neutral-cationic diblock polymer was designed with a poly(oligo(ethylene glycol) methyl ether methacrylate) (POEGMA) charge neutral block and a quaternized poly(4-vinyl-N-methylpyridinium) (qP4VP) cationic block. POEGMA was chosen for the neutral corona block, due to demonstrated cytocompatibility and its comb-like architecture, which has been shown to increase cellular uptake over linear poly(ethylene glycol).49,50 The strong polycation, qP4VP, was selected for the cationic block because previous work had carefully characterized the phase behavior of these anionic GFPs with a qP4VP homopolymer.42 To prepare the block copolymer, the POEGMA block was first synthesized by reversible addition–fragmentation chain-transfer (RAFT) polymerization. The molecular weight of the POEGMA block, Mn = 23.7 kg mol−1, was determined by gel permeation chromatography (ESI Fig. S2†). Subsequently, the poly(4-vinylpyridine) (P4VP) block was synthesized via RAFT polymerization, using the POEGMA block as a macromolecular chain transfer agent. The resulting block copolymer had a block ratio with 2.2 4VP monomers to 1 OEGMA monomer, as determined by 1H NMR (ESI Fig. S3†). The P4VP block was quaternized (∼95%) with an excess of methyl iodide, resulting in the final block copolymer, POEGMA79-b-qP4VP175 (ESI Fig. S3†).
Polyelectrolyte complex (PEC) micelle formation
Micelle formation was initially studied by evaluating several protein and block copolymer mixing ratios using dynamic light scattering (DLS) in the absence of salt (Fig. 1a, ESI Fig. S4 and S5†). For these experiments, the overall macromolecule concentration was kept constant at 0.2 mg mL−1. All of the mutants formed micelles at several mixing ratios. PEC micelle formation was examined as a function of positive charge fraction, f+, which is the ratio of positive charge (M+) to total charge (M+ + M−). M+ and M− are the charge per mass of the positive and negative species respectively. The charge per mass for the proteins, M− was calculated using the expected charge, at pH 7.4, determined by the Henderson–Hasselbalch equation and the isolated side chain pKa values. The charge per mass of the polymer, M+, was calculated using the expected charge based on the degree of quaternization and the estimated molecular weight based on the degree of polymerization. Consistent with prior reports, the hydrodynamic radius of the particles was largely independent of the mixing ratio (ESI Fig. S4a†).39,51 Therefore, the preferred micellar compositions were determined by monitoring the derived count rates, which showed maxima at mixing ratios with excess polycation. We observed that PEC micelles form at f+ greater than 0.5. Because polyelectrolyte complex micelle formation is driven by polyelectrolyte complexation, we would expect micelle formation to be most favorable at charge neutrality or f+ = 0.5. We hypothesize that this shift to higher charge fractions, which indicates that phase separation is favored in the presence of excess positive charge, is largely due to the ionizable nature of proteins. Upon complexation with the polycation, ionizable side chains (His, Lys, Arg) can alter protonation state, making the protein more net negatively charged. Upon charge regulation, fewer proteins are required per polymer chain to achieve charge neutrality. This shift away from charge neutrality has previously been observed in many protein-based complex coacervates, particularly those formed with strong polycations.39,42,51–53 Indeed, the macrophase separation of these supercharged GFPs with a qP4VP homopolymer showed similar shifts away from the expected charge neutral mixing ratio.42 Interestingly, for the isotropic supercharged variants, the charge fraction that resulted in the maximum derived count rate for each mutant decreased with decreasing protein net charge. The derived count rate is correlated with the average scattering intensity measured by DLS and is indicative of higher concentration or larger particles. While potentially counterintuitive, as this implies that fewer of the less negatively charged mutants are required to neutralize the polymer, this is also consistent with previous observations of anionic GFP macrophase separation.42 We hypothesize that for less negatively charged proteins, there are more opportunities for induced charging in the presence of the polycation, resulting in a larger deviation from the hypothetical charge neutral mixing ratio. We also note that while this shift away from f+ = 0.5 can largely be explained by charge regulation of the protein component, using the maximum theoretical charge for each protein still results in a deviation from the expected charge neutral state (f+maxcharge = 0.54–0.67). It is therefore likely that there is some excess polycation, as the protein–polyelectrolyte complexes are unlikely to completely saturate the charges on the strong polycation due to conformation constraints of the protein. In addition to the optimal mixing ratio depending on protein charge, the charge distribution also impacted micelle formation as a function of mixing ratio. For example, tag-GFP(−24) formed PEC micelles over fewer mixing ratios than GFP(−24) (ESI Fig. S4†). This is also consistent with previous findings, where phase separation of proteins with polyionic tags was observed for fewer mixing ratios than their isotropically charged analogs. We hypothesize this behavior can be attributed to either differences in protein geometry or the comparatively lower average distance between like charges for the tagged variants. | ||
| Fig. 1 Effect of protein charge on the formation of polyelectrolyte complex micelles (PECMs). (a) Representative DLS micelle intensity traces for samples prepared with 0.1 mg mL−1 protein and 0.1 mg mL−1 polymer in 10 mM tris, pH 7.4. Traces are an average of 3 samples measured 1 h after mixing. (b) Micelle radius as measured by TEM, n = 50. The black bar indicated the mean. (c) TEM micrographs of PECMs prepared with 0.05 mg mL−1 protein and 0.05 mg mL−1 polymer, with the exception of GFP(−7) and tag-GFP(−24) which were prepared with 0.1 mg mL−1 protein and 0.1 mg mL−1 polymer. Scale bar = 200 nm. | ||
The formation of particles was also probed by transmission electron microscopy (TEM, Fig. 1b and c, ESI Table S4†) for most of the GFP variants. These GFP variants formed spherical assemblies in 10 mM tris, pH 7.4 with 0 mM NaCl. Particles formed from tag-GFP(−18) could not be visualized by TEM, likely due to low stability. Clusters of several spherical particles were observed for some of the isotropic variants and were excluded from subsequent particle size analysis. The radius determined by TEM was smaller than that determined by DLS. We hypothesize this difference is a result of only the core of the micelle having sufficient electron density to generate contrast in TEM.54,55 For the isotropic supercharged variants, the least negatively charged protein, GFP(−18), had the largest radius both by TEM and DLS. One hypothesis for this observation is that PEC micelles formed with the less negatively charged GFP mutant would require more protein molecules in the core to achieve charge neutrality, resulting in a larger core. This is consistent with previous research with lipase, where a positive correlation between the number of lipase molecules in the core of the micelle and the size of the PEC micelle core was observed.38 The other two isotropic variants formed particles with similar radii, as demonstrated by both measurements. In contrast, for the tagged GFP variant, tag-GFP(−24), the radii measured by DLS and TEM were nearly identical. We hypothesize this was due to the swelling of these assemblies upon dilution for TEM analysis. This swelling in the absence of salt was only observed for tag-GFP(−24), most likely due to the lower salt stability of this variant, as described below. It should be noted, that for all of these samples there could be some artifacts due to the drying process required for the TEM analysis.
PEC micelle stability
In the presence of small amounts of NaCl (25 mM), the hydrodynamic radius of PEC micelles formed with isotropically supercharged GFPs decreased (Fig. 2, Table 1, ESI Table S5†). By DLS, particles formed with GFP(−18) decreased by ∼30% and those formed by GFP(−24) and GFP(−30) deceased by ∼25% in the presence of 25 mM NaCl. This behavior, namely a decrease in the hydrodynamic radius in the presence of 25 mM NaCl was also observed by fluorescence correlation spectroscopy (FCS) for micelles formed with GFP(−24) (ESI Fig. S6, ESI Table S2†). In addition to corroborating this size decrease in the presence of small quantities of salt, the FCS measurements of GFP(−24) micelle radius were consistent with those measured by DLS (Table 1, ESI Table S2†). For example, in the absence of salt, the hydrodynamic radius by DLS was 101 ± 12 and by FCS was 97 ± 5. Increasing the system ionic strength would make the core either more liquid-like, if it was solid or gel-like at 0 M NaCl, or decrease the viscosity of a liquid core, resulting in a PEC micelle population with fewer kinetically trapped aggregates and an overall narrower particle size distribution. This was reflected in the decrease in polydispersity index (PDI) as measured by DLS (ESI Tables S4 and S5†). The PDI of the PEC micelles formed in 25 mM NaCl was half that of the PEC micelle solutions with no salt. The decrease in the standard deviation of the intensity peaks of independent samples further reflect that the addition of 25 mM NaCl results in a more homogeneous particle population. | ||
| Fig. 2 PECM stability as a function of protein charge. (a) Temporal stability of micelles was evaluated using the hydrodynamic radius of the DLS intensity of peak 1 over time. Samples were prepared with 0.1 mg mL−1 protein and 0.1 mg mL−1 polymer in 10 mM tris, pH 7.4 with 25 mM NaCl. (b) Salt stability as measured by the hydrodynamic radius determined from the DLS intensity of peak 1. The shaded area corresponds to the standard deviation of three replicates. Samples were prepared with 0.1 mg mL−1 protein and 0.1 mg mL−1 polymer in 10 mM tris, pH 7.4 with varying concentrations of NaCl. Data up to each critical salt concentration is shown. (c) Temporal stability of the PECMs was also evaluated at physiological ionic strength (150 mM NaCl) as in (a). (d) TEM micrographs of GFP(−24) and GFP(−30) based PECMs prepared at different salt concentrations. Samples were prepared with 0.05 mg mL−1 protein and 0.05 mg mL−1 polymer at 25, 125, and 150 mM NaCl. Scale bar = 200 nm. | ||
| Protein | Protein mass fraction | f+ | r H, nm, DLS | r, nm, TEM | r H, nm, DLS | r, nm, TEM | Critical salt concentration, mM |
|---|---|---|---|---|---|---|---|
| 0 mM NaCl | 25 mM NaCl | ||||||
| sfGFP(−7) | 0.5 | 0.91 | 67 ± 3 | 37 ± 9 | 98 ± 3 | — | 25 |
| GFP(−18) | 0.4 | 0.85 | 90 ± 4 | 68 ± 2 | 59 ± 15 | 125 | |
| 0.5 | 0.79 | 117 ± 12 | 57 ± 13 | 83 ± 1 | 59 ± 12 | 125 | |
| GFP(−24) | 0.5 | 0.73 | 101 ± 12 | 36 ± 12 | 72 ± 4 | 45 ± 11 | 175 |
| GFP(−30) | 0.5 | 0.69 | 92 ± 14 | 40 ± 8 | 65 ± 5 | 59 ± 13 | 225 |
| tag-GFP(−18) | 0.5 | 0.79 | 119 ± 7 | — | 105 ± 6 | — | 75 |
| tag-GFP(−24) | 0.5 | 0.74 | 60 ± 1 | 59 ± 13 | 70 ± 2 | — | 100 |
In contrast, the hydrodynamic radius of micelles formed with a tagged GFP variant, tag-GFP(−24), increased by ∼15% under the same conditions and the PDI did not decrease with the addition of a small amount of salt. We hypothesize that the PEC micelles formed with this tagged variant resulted in a liquid-like core with a lower viscosity than PEC micelles formed with isotropic variants. This is based on previous findings that demonstrated that tagged GFP variants were more likely to form liquid complex coacervates with a qP4VP homopolymer.42 Isotropic GFP variants formed solid-like precipitates with the same qP4VP homopolymer. We propose that in the absence of salt, the isotropically supercharged proteins form kinetically trapped PECs, but that formation of micelles in the presence of small quantities of salt enables more rapid chain rearrangement and exchange enabling equilibration of the assemblies. This results in smaller PEC micelle hydrodynamic radii with the isotropic mutants at low salt concentrations. However, the lower net charge tagged variant, tag-GFP(−18), showed similar behavior to the isotropic variants with both a decrease in micelle size and PDI upon the addition of 25 mM NaCl. This variant had a derived count rate that was nearly an order of magnitude lower than the other variants, indicating limited formation and stability of PEC micelles. We attribute the different behavior of tag-GFP(−18) to the relatively low stability micelles even in the absence of salt.
The colloidal stability of the micelles at low salt was monitored for 7 days by DLS (Fig. 2a). In addition to all GFP variants forming PEC micelles at low salt, all mutants formed micelles that remained colloidally stable for 7 days in the presence of 25 mM NaCl. Under these conditions, minimal micelle swelling was observed in the first 8 h for tag-GFP(−18) (+7 ± 5%), tag-GFP(−24) (+4 ± 3%), GFP(−24) (+4 ± 8%), and GFP(−30) (+6 ± 9%). On the other hand, in 25 mM NaCl GFP(−18) micelles swelled +19 ± 1% in the first 8 h and were +43 ± 7% larger than the original size by 24 h. Following this initial swelling, GFP(−18) micelles remained stable at this size for the remainder of the week. Interestingly, this swollen micelle size for GFP(−18) in 25 mM NaCl was similar to the micelle size in the absence of NaCl (117 ± 12 nm at t = 0 in 0 mM NaCl and 119 ± 8 nm at t = 24 h in 25 mM NaCl). This swelling behavior of GFP(−18) micelles at modest ionic strength was observed at non-equal mass mixing ratios as well (ESI Fig. S7†). In addition to changes in micelle size, the derived count rate for GFP(−18) micelles also indicated changes in temporal stability at 25 mM NaCl (ESI Fig. S8†). Without the addition of salt, the micelles maintained a constant derived count rate over 7 days. But, with the addition of 25 mM NaCl, micelles formed with GFP(−18) saw an initial increase in the derived count rate over the first 24 h, but then had a stable count rate for the next 6 days, again indicative of an increased particle size in solution.
After establishing the colloidal stability at low salt, micelle formation and stability as a function of ionic strength was assessed (Fig. 2b). Mixtures of protein and polymer at varying ionic strengths were prepared by first mixing protein solutions with different quantities of a 5 M NaCl solution, followed by addition of the polymer solution. Shortly after mixing, the particles in solution were monitored by DLS. Critical salt concentrations were established by both the observed swelling of the hydrodynamic radius and a decrease in the derived count rate, indicating a decrease in the average scattering of objects in solution, as a function of salt concentration (Fig. 2b, Table 1, ESI Fig. S11 and S12†). Both of these metrics gave consistent indications of the critical salt concentration within 25–50 mM increments of NaCl. Micelle swelling as a function of NaCl concentration was also monitored for one GFP variant, GFP(−24), by FCS. At physiological ionic strength, just below the critical salt concentration determined by DLS, the particles hydrodynamic radius determined by FCS also increased slightly. These FCS measurements also enabled the estimation of the number of GFP molecules per particle. As expected, nearly all of the GFP was incorporated into the particles, though there was more free GFP in solution as the salt concentration increased. This resulted in approximately 300 GFP per micelle in the absence of salt, which decreased to approximately 100 as the NaCl concentration approached the critical salt concentration.
The isotropically charged mutants demonstrated a clear trend with increasing negative charge improving the micelle salt stability (Table 1). As the magnitude of protein charge increases, so does the number of potential interaction sites with the polycation, resulting in an increase in the overall strength of interaction between the polymer and protein. Therefore, a higher salt concentration was required for microphase separation to no longer be entropically favorable. In comparison, we observed that micelles formed from tagged GFP variants had lower critical micelle salt concentrations. We hypothesize that this is due to differences between the interactions of the globular domain with the qP4VP block and the polyionic tag with the qP4VP block. It has previously been demonstrated that all of these GFP mutants remain phase separated with a qP4VP homopolymer at physiological ionic strength, albeit at a 5-fold higher macromolecule concentration.42 The salt stability data for macrophase separation indicates that the tag interacts more strongly with qP4VP than the isotropic equivalent. This resulted in a comparatively higher critical salt concentrations for tagged GFPs with the qP4VP homopolymer than the isotropically charged counterparts (Fig. 3). In contrast, for PEC micelles the isotropically supercharged GFP micelles remain assembled at higher salt concentrations than the tagged GFP variants. However, while micelles formed with a tagged variant, tag-GFP(−24), swelled and then disassembled above 100 mM NaCl, particles with a hydrodynamic radius slightly larger than the free protein (∼7 nm particles; ∼2 nm protein) were observed via the DLS number distribution at higher salt concentrations (ESI Fig. S13†). This behavior was not observed for GFP(−24) PEC micelles. We hypothesize that the increasing salt concentration effectively screens the interactions between the polymer and the protein globular domain, which has an expected charge of −7, but not between the polymer and the charged tag, which has an expected charge of −18. Polymer interactions with both the tag and globular domain would allow more than one polymer chain to interact with each protein within the PEC micelle core. Screening the polymer–globular domain interactions could result in PEC micelle swelling and dissociation. Given that the tagged variants have the least negative globular domain, we would then expect the critical micelle salt concentration of the tagged variants to be the lowest. But concurrently, the tag domain could still facilitate interactions with the polymer above the critical micelle salt concentration but below the critical salt concentration established with the qP4VP homopolymer. This could result in the formation of smaller protein–polymer complexes with one polymer molecule complexed with a few proteins, indicated by the ∼7 nm particle size that is observed by DLS.56,57
 | ||
| Fig. 3 Relationship between critical salt concentration and the ratio of negative to positive residues on the protein. (a) For isotropically supercharged variants, the critical salt concentration for macro- (open symbols, dashed line) and microphase separation (filled symbols, solid line) are nearly identical. (b) For GFP variants that were supercharged via a polyionic peptide tag, the critical salt concentration for microphase separation (filled symbols, solid line) is similar to the critical salt concentration for macrophase separation of the globular domain (GFP(−7), grey open circle). | ||
To better understand the particle populations present during protein delivery applications, the temporal stability of the PEC micelles at physiological ionic strength was monitored (Fig. 2c, ESI Fig. S8 and S10†). GFP(−24) and GFP(−30), which formed PEC micelles with a critical salt concentration greater than 150 mM NaCl, remained colloidally stable for 7 days. The less negatively charged variant, GFP(−18), which had a critical salt concentration near physiological ionic strength, formed micelles that swelled slightly over time and over the course of the week had a slight decrease in the time averaged scattering intensity of the solution, as monitored by the derived count rate. Conversely, the tagged GFP variants rapidly swelled (∼2–8 h) in the presence of 150 mM NaCl and demonstrated a decreased derived count rate (ESI Fig. S8†). To support these DLS results, micelles formed with GFP(−24) and GFP(−30) at increased salt concentrations were characterized by TEM (Fig. 2d, ESI Tables S6 and S7†). Analysis of the TEM images confirms that there was no increase in the hydrodynamic radius as the salt concentration was increased from 25 mM to 125 mM NaCl. As the salt concentration was further increased to 150 mM, swollen PEC micelles were observed for micelles formed with GFP(−24). While this is below the critical salt concentration determined by DLS, the samples were diluted immediately prior to TEM imaging, which may impact the observed micelles. As the critical salt concentration by DLS was even higher for GFP(−30) micelles, a similar increase in size was not observed by TEM at 150 mM NaCl.
GFP delivery with PEC micelles
With an understanding of the PEC micelle size and stability at physiological ionic strength, we then proceeded to study the delivery of anionic GFP mutants to Jurkat T cells. We investigated the delivery of the anionic proteins and their PEC micelle counterparts for the variants that formed micelles at or near physiological ionic strength (GFP(−18), GFP(−24), GFP(−30), and tag-GFP(−24)). Solutions containing PEC micelles were incubated with Jurkat cells at 37 °C for 1 h (final macromolecule concentrations of 0.05 and 0.1 mg mL−1). After washing, the cell fluorescence was quantified by flow cytometry (Fig. 4, ESI Fig. S14–S21†). In addition to the GFP loaded micelles, the delivery of a cationic variant, containing a poly(arginine) tag (GFP(+6)-6R), was evaluated for comparison. It has previously been demonstrated that arginine rich peptide fusions promote cellular internalization of fluorescent proteins.58,59 | ||
| Fig. 4 Effect of protein charge on the delivery of GFP via PECMs. All samples treated with Sytox Aadvanced. Poly and +6-6R samples contain 0.05 mg mL−1 of polymer and protein, respectively. PECM samples contain 0.05 mg mL−1 of polymer and protein each. (a) Ungated fraction of Sytox PECM Jurkat T cells. The mean, standard deviation, and individual data points of 3 biological replicates are shown. (b) Fraction of GFP+ treated Jurkat T cells, gated for T cells. The mean, standard deviation and individual data points of 3 biological replicates are shown. (c) Histograms of GFP intensity of PECM treated Jurkat T cells, gated for T cells. Shown are overlayed histograms for 3 biological replicates. | ||
The flow cytometry data indicated a clear relationship between cytotoxicity and micelle stability. Exposure to the GFP(+6)-6R positive control, as well as the free protein solutions of the anionic GFPs, resulted in a minimal decrease in cell viability (Fig. 4a, ESI Table S8†). Treatment with the free polymer, however, resulted in approximately a 20% decrease in cell viability (Fig. 4a, ESI Table S8†). For the micelles formed with tag-GFP(−24) and GFP(−18), which have a critical salt concentration below 150 mM, cell viability saw a similar 20% decrease upon incubation with micelles. GFP(−24) and GFP(−30) micelles, which were stable at physiological ionic strength, resulted in modest decreases in viability (8% and 5%, respectively, Fig. 4a). We hypothesize that the decrease in cell viability predominantly correlates with the concentration of free polymer in solution, which increases as micelle stability decreases. Even though the mixing ratios for all of the protein variants imply a potential excess of free polycation in solution (f+ > 0.5), the cytotoxicity data indicates that proteins that formed stable micelles at physiological ionic strength were unlikely to have a significant amount of free polymer in solution. Importantly though, the interest in using polycations for nucleic acid delivery has spurred the study of cytocompatible polycations.60 Altering the polycation in this system could, therefore, further reduce the observed cytotoxicity.
GFP delivery efficiency to Jurkat cells was quantified by flow cytometry (Fig. 4b and c ESI Table S8†). Cellular uptake of GFP(+6)-6R was 10-fold greater than uptake of any of the anionic proteins variants, demonstrating the challenge of delivering negatively charged proteins (ESI Fig. S15–S21†). Critically, complexation of the anionic proteins with the cationic block copolymer resulted in successful internalization of the GFP variants. PEC micelles formed with the most anionic protein, GFP(−30), resulted in the least efficient cellular uptake, with only a 5-fold increase over the free protein solution (Fig. 4c). Cellular delivery efficiency further increased for both GFP(−24) and GFP(−18) PEC micelles, with uptake similar to the cationic GFP control (Fig. 4b and c). GFP(−18) delivery by micelles was also modestly dependent on the protein and polymer mixing ratio, with the higher positive charge fraction (f+ = 0.85) showing a 50% increase relative to a mixing ratio with more of the anionic protein (f+ = 0.79) (ESI Fig. S16 and S21†). We hypothesize that these changes in cellular delivery correlate with the DLS derived count rate for the micelles at low salt, which would be indicative of more particles scattering in solution (ESI Fig. S5†) as well as with the presence of a slight excess of cationic charge in the assembled particles, as indicated by the increased positive charge fraction. Tag-GFP(−24) PEC micelles resulted in the most efficient cellular uptake, with a 100-fold increase over anionic proteins alone and a 10-fold increase compared to the cationic GFP(+6)-6R control (Fig. 4c). These combined results reveal a convincing inverse relationship between micelle stability and delivery efficiency. We initially hypothesized that a PEC micelle <200 nm in diameter at physiological ionic strength would result in the most efficient GFP delivery. However, the most salt stable micelles, formed with the GFP(−30) mutant, have a hydrodynamic diameter less than 200 nm at physiological ionic strength but delivered the least efficiently. We therefore propose that smaller protein/polymer complexes are more efficiently delivered than intact micelles. While PEC micelles formed with tag-GFP(−24) were not stable in solution at physiological ionic strength, the anionic protein likely still interacts with the polymer at this ionic strength forming small complexes, as evidenced by DLS (ESI Fig. S13†). These smaller complexes, particularly if they contain excess cationic charge, would more preferentially interact with the cellular membrane and be more effectively endocytosed. While not as drastic for the isotropic GFP variants with intermediate charge, the modest PEC micelle stability near physiological ionic strength suggests that these proteins may similarly have preferential uptake through smaller protein/polymer complexes.
Finally, the uptake of GFP was corroborated by confocal microscopy, which correlated with the flow cytometry analysis (Fig. 5, ESI Fig. S22†). For all GFP variants, cellular internalization was observed after 1 h of incubation with PEC micelles or cationic protein. The cationic poly(arginine) tag protein was observed to co-localize with a dye that stains lysosomes (LysoTracker), consistent with internalization via endocytosis (ESI Fig. S17†).9,61,62 GFP delivered by PEC micelles was observed to be associated with the perimeter of the cell and primarily localized in puncta, the latter also consistent with internalization via endocytosis.63
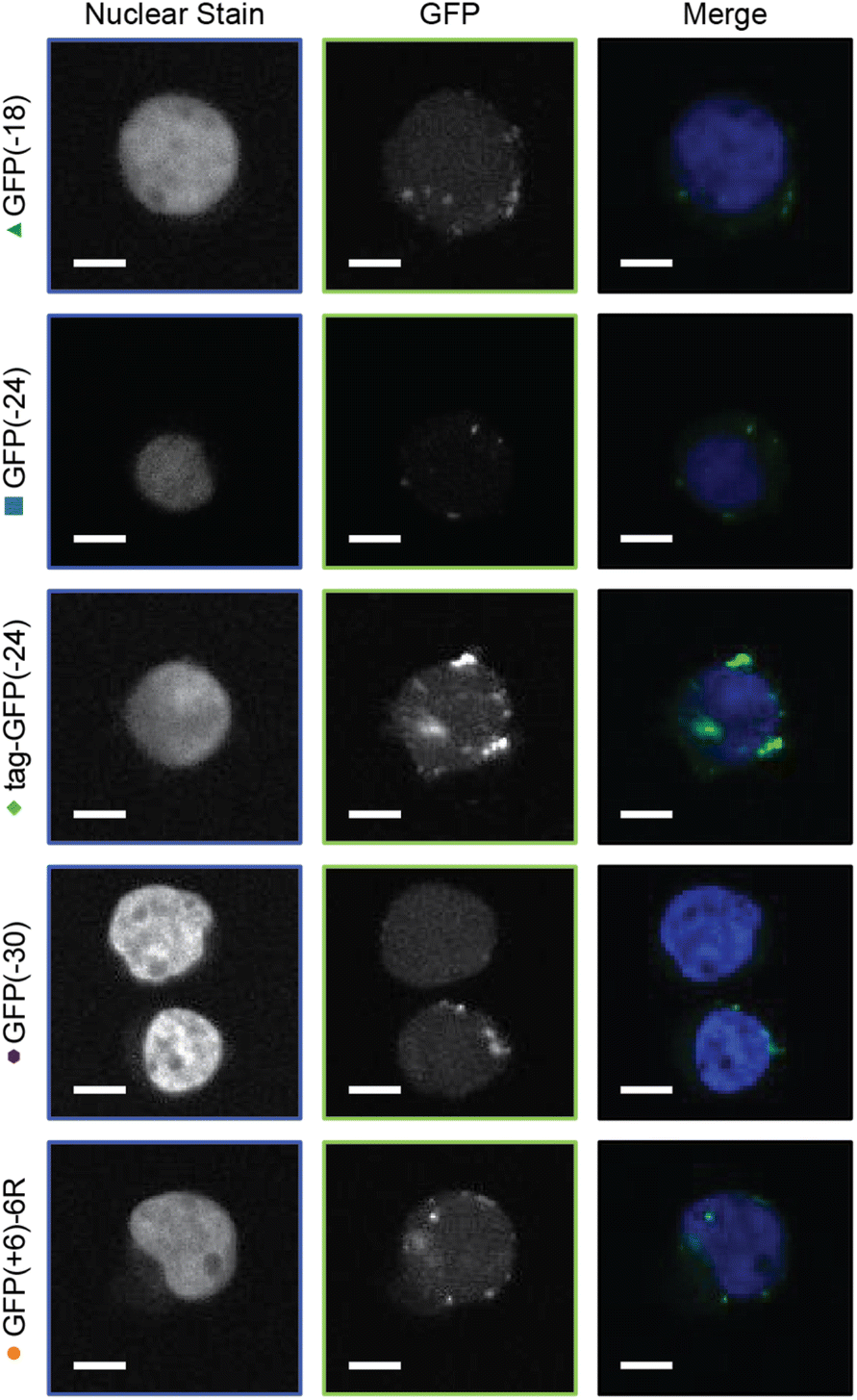 | ||
| Fig. 5 Confocal images of Jurkat T cells. Cells were incubated with GFP containing micelles for 1 h and then washed 3× with DPBS. Cells were fixed with 4% formaldehyde in DPBS and stained with NucBlue for 15 min. Scale bar = 5 μm. | ||
Conclusions
A panel of anionic supercharged GFPs and a cationic diblock copolymer were developed to study the impact of protein charge and charge distribution on PEC micelle formation and stability for the potential application of intracellular protein delivery. All of the proteins formed PEC micelles with the diblock copolymer at several mixing ratios. Supercharging the model protein with a polyionic tag produced PEC micelles with low dispersity, whereas isotropically supercharging GFP resulted in PEC micelles with higher dispersity in the absence of salt. Increasing the solution ionic strength, resulted in PEC micelles with isotropically supercharged GFPs with lower dispersity, which we interpreted to be a result of kinetic trapping of complexes in the absence of salt. All mutants produced PEC micelles that remained colloidally stable for 1 week at low ionic strength. Supercharging GFP with an anionic tag produced PEC micelles with lower critical salt concentrations, suggesting that interaction with the high charge density ionic tag did not sufficiently stabilize micelle formation. The critical salt concentration of the isotropic variants increased with decreasing protein net charge, which is consistent with prior work showing a monotonic increase as a function of protein charge. The intracellular delivery of the PEC micelles was compared to a GFP control with a cell-penetrating poly(arginine) peptide. Complexation of the anionic proteins with the cationic block copolymer resulted in internalization of the GFP variants at least as efficiently as the cationic protein control. An inverse relationship was established between GFP delivery efficiency and PEC micelle salt stability, indicating that smaller protein/polymer complexes are more efficiently delivered to cells. This highlights the importance of characterizing both the solution structure and function of polyelectrolyte complexes as they are not inherently directly correlated. Indeed, future work will probe how to further tune protein and polymer interactions to maximize cytosolic delivery. While the system described herein successfully delivered anionic proteins to cells, it came at the cost of some modest cytotoxicity. Future work will focus on approaches to maintain and improve this delivery efficiency while minimizing cytotoxicity. This can potentially be accomplished through the development of smaller PECs, non-toxic cationic delivery agents, cell-surface targeting ligands, and methods to enhance endosomal escape.Data availability
Data are available from the corresponding author upon request.Author contributions
R. A. K. and A. C. O. are jointly responsible for the conception, design, and performance of the protein engineering and DLS, TEM, and cell delivery studies described herein. R. S. F. and S. E. G. designed, performed, and analyzed the FCS experiments. R. A. K., R. S. F., and A. C. O. were responsible for the analysis, data presentation, and writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the National Institutes of Health under NIH-NIGMS-R35GM138378, the Fu Foundation School of Engineering and Applied Sciences at Columbia University, the Air Force Office of Scientific Research MURI (Grant 12936019), and the Sloan Foundation CUNY JFRASE (G-2018-11286) for funding. These studies used the resources of the Herbert Irving Comprehensive Cancer Center Confocal and Specialized Microscopy Shared Resource funded in part through the Center Grant P30CA013696 as well as the CUNY Advanced Science Research Center Imaging Facility.Notes and references
- D. S. Dimitrov, in Therapeutic Proteins: Methods and Protocols, ed. V. Voynov and J. A. Caravella, Humana Press, Totowa, NJ, 2012, pp. 1–26 Search PubMed.
- D. S. Pisal, M. P. Kosloski and S. V. Balu-Iyer, J. Pharm. Sci., 2010, 99, 2557–2575 CrossRef CAS PubMed.
- A. Fu, R. Tang, J. Hardie, M. E. Farkas and V. M. Rotello, Bioconjug. Chem., 2014, 25, 1602–1608 CrossRef CAS PubMed.
- B. Leader, Q. J. Baca and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7, 21–39 CrossRef CAS PubMed.
- J. M. Horn and A. C. Obermeyer, Biomacromolecules, 2021, 22, 4883–4904 CrossRef CAS PubMed.
- K. A. Mix, J. E. Lomax and R. T. Raines, J. Am. Chem. Soc., 2017, 139, 14396–14398 CrossRef CAS PubMed.
- S. Schmidt, M. J. W. Adjobo-Hermans, R. Kohze, T. Enderle, R. Brock and F. Milletti, Bioconjug. Chem., 2017, 28, 382–389 CrossRef CAS PubMed.
- S. Ramakrishna, A.-B. Kwaku Dad, J. Beloor, R. Gopalappa, S.-K. Lee and H. Kim, Genome Res., 2014, 24, 1020–1027 CrossRef CAS PubMed.
- J. Hu, Y. Lou and F. Wu, J. Phys. Chem. B, 2019, 123, 2636–2644 CrossRef CAS PubMed.
- D. M. Copolovici, K. Langel, E. Eriste and Ü. Langel, ACS Nano, 2014, 8, 1972–1994 CrossRef CAS PubMed.
- J. Ye, E. Liu, Z. Yu, X. Pei, S. Chen, P. Zhang, M.-C. Shin, J. Gong, H. He and V. C. Yang, Int. J. Mol. Sci., 2016, 17, E1892 CrossRef PubMed.
- J. J. Cronican, K. T. Beier, T. N. Davis, J.-C. Tseng, W. Li, D. B. Thompson, A. F. Shih, E. M. May, C. L. Cepko, A. L. Kung, Q. Zhou and D. R. Liu, Chem. Biol., 2011, 18, 833–838 CrossRef CAS PubMed.
- J. J. Cronican, D. B. Thompson, K. T. Beier, B. R. McNaughton, C. L. Cepko and D. R. Liu, ACS Chem. Biol., 2010, 5, 747–752 CrossRef CAS PubMed.
- Y. Ma, K. Poole, J. Goyette and K. Gaus, Front. Immunol., 2017, 8, 1513 CrossRef PubMed.
- F. Scaletti, J. Hardie, Y.-W. Lee, D. C. Luther, M. Ray and V. M. Rotello, Chem. Soc. Rev., 2018, 47, 3421–3432 RSC.
- L. P. Herrera Estrada and J. A. Champion, Biomater. Sci., 2015, 3, 787–799 RSC.
- Z. Gu, A. Biswas, M. Zhao and Y. Tang, Chem. Soc. Rev., 2011, 40, 3638–3655 RSC.
- A. Biswas, K.-I. Joo, J. Liu, M. Zhao, G. Fan, P. Wang, Z. Gu and Y. Tang, ACS Nano, 2011, 5, 1385–1394 CrossRef CAS PubMed.
- J. Lv, E. Tan, Y. Wang, Q. Fan, J. Yu and Y. Cheng, J. Controlled Release, 2020, 320, 412–420 CrossRef CAS PubMed.
- C. Van Bruggen, J. K. Hexum, Z. Tan, R. J. Dalal and T. M. Reineke, Acc. Chem. Res., 2019, 52, 1347–1358 CrossRef CAS PubMed.
- B. Chatin, M. Mével, J. Devallière, L. Dallet, T. Haudebourg, P. Peuziat, T. Colombani, M. Berchel, O. Lambert, A. Edelman and B. Pitard, Mol. Ther.–Nucleic Acids, 2015, 4, e244 CrossRef CAS PubMed.
- A. Wang, T. Yang, W. Fan, Y. Yang, Q. Zhu, S. Guo, C. Zhu, Y. Yuan, T. Zhang and Y. Gan, Adv. Healthcare Mater., 2019, 8, e1801123 CrossRef PubMed.
- J. A. Zuris, D. B. Thompson, Y. Shu, J. P. Guilinger, J. L. Bessen, J. H. Hu, M. L. Maeder, J. K. Joung, Z.-Y. Chen and D. R. Liu, Nat. Biotechnol., 2015, 33, 73–80 CrossRef CAS PubMed.
- A. Harada and K. Kataoka, Macromolecules, 1998, 31, 288–294 CrossRef CAS.
- A. Harada and K. Kataoka, Macromolecules, 1995, 28, 5294–5299 CrossRef CAS.
- A. Harada and K. Kataoka, Langmuir, 1999, 15, 4208–4212 CrossRef CAS.
- M. Jaturanpinyo, A. Harada, X. Yuan and K. Kataoka, Bioconjug. Chem., 2004, 15, 344–348 CrossRef CAS PubMed.
- J. R. Magana, C. C. M. Sproncken and I. K. Voets, Polymers, 2020, 12, 1953 CrossRef CAS PubMed.
- Y. Lee, T. Ishii, H. Cabral, H. J. Kim, J.-H. Seo, N. Nishiyama, H. Oshima, K. Osada and K. Kataoka, Angew. Chem., Int. Ed., 2009, 48, 5309–5312 CrossRef CAS PubMed.
- J.-P. Behr, Chimia, 1997, 51, 34 CrossRef CAS.
- Y. Hu, P. U. Atukorale, J. J. Lu, J. J. Moon, S. H. Um, E. C. Cho, Y. Wang, J. Chen and D. J. Irvine, Biomacromolecules, 2009, 10, 756–765 CrossRef CAS PubMed.
- S. Foster, C. L. Duvall, E. F. Crownover, A. S. Hoffman and P. S. Stayton, Bioconjug. Chem., 2010, 21, 2205–2212 CrossRef CAS PubMed.
- S. Cerritelli, D. Velluto and J. A. Hubbell, Biomacromolecules, 2007, 8, 1966–1972 CrossRef CAS PubMed.
- T. Bus, A. Traeger and U. S. Schubert, J. Mater. Chem. B, 2018, 6, 6904–6918 RSC.
- K. Iwashita, A. Handa and K. Shiraki, Int. J. Biol. Macromol., 2018, 120, 10–18 CrossRef CAS PubMed.
- X. Yuan, Y. Yamasaki, A. Harada and K. Kataoka, Polymer, 2005, 46, 7749–7758 CrossRef CAS.
- S. Lindhoud, R. de Vries, W. Norde and M. A. C. Stuart, Biomacromolecules, 2007, 8, 2219–2227 CrossRef CAS PubMed.
- S. Lindhoud, R. de Vries, R. Schweins, M. A. C. Stuart and W. Norde, Soft Matter, 2008, 5, 242–250 RSC.
- A. C. Obermeyer, C. E. Mills, X.-H. Dong, R. J. Flores and B. D. Olsen, Soft Matter, 2016, 12, 3570–3581 RSC.
- Y. Lee, T. Ishii, H. J. Kim, N. Nishiyama, Y. Hayakawa, K. Itaka and K. Kataoka, Angew. Chem., Int. Ed., 2010, 49, 2552–2555 CrossRef CAS PubMed.
- A. Kim, Y. Miura, T. Ishii, O. F. Mutaf, N. Nishiyama, H. Cabral and K. Kataoka, Biomacromolecules, 2016, 17, 446–453 CrossRef CAS PubMed.
- R. A. Kapelner and A. C. Obermeyer, Chem. Sci., 2019, 10, 2700–2707 RSC.
- W. N. Ainis, A. Boire, V. Solé-Jamault, A. Nicolas, S. Bouhallab and R. Ipsen, Langmuir, 2019, 35, 9923–9933 CrossRef CAS PubMed.
- A. J. Simon, Y. Zhou, V. Ramasubramani, J. Glaser, A. Pothukuchy, J. Gollihar, J. C. Gerberich, J. C. Leggere, B. R. Morrow, C. Jung, S. C. Glotzer, D. W. Taylor and A. D. Ellington, Nat. Chem., 2019, 11, 204–212 CrossRef CAS PubMed.
- S. Kim, H. V. Sureka, A. B. Kayitmazer, G. Wang, J. W. Swan and B. D. Olsen, Biomacromolecules, 2020, 21, 3026–3037 CrossRef CAS PubMed.
- J. J. van Lente, M. M. A. E. Claessens and S. Lindhoud, Biomacromolecules, 2019, 20, 3696–3703 CrossRef CAS.
- W. C. B. McTigue and S. L. Perry, Soft Matter, 2019, 15, 3089–3103 RSC.
- J.-D. Pédelacq, S. Cabantous, T. Tran, T. C. Terwilliger and G. S. Waldo, Nat. Biotechnol., 2006, 24, 79–88 CrossRef.
- J. Teo, J. A. McCarroll, C. Boyer, J. Youkhana, S. M. Sagnella, H. T. T. Duong, J. Liu, G. Sharbeen, D. Goldstein, T. P. Davis, M. Kavallaris and P. A. Phillips, Biomacromolecules, 2016, 17, 2337–2351 CrossRef CAS PubMed.
- I. Ozer, A. Tomak, H. M. Zareie, Y. Baran and V. Bulmus, Biomacromolecules, 2017, 18, 2699–2710 CrossRef CAS PubMed.
- A. Nolles, A. H. Westphal, J. A. de Hoop, R. G. Fokkink, J. M. Kleijn, W. J. H. van Berkel and J. W. Borst, Biomacromolecules, 2015, 16, 1542–1549 CrossRef CAS PubMed.
- W. M. de Vos, F. A. M. Leermakers, A. de Keizer, M. A. Cohen Stuart and J. M. Kleijn, Langmuir, 2010, 26, 249–259 CrossRef CAS PubMed.
- C. S. Cummings and A. C. Obermeyer, Biochemistry, 2018, 57, 314–323 CrossRef CAS PubMed.
- Y. Zheng, Y.-Y. Won, F. S. Bates, H. T. Davis, L. E. Scriven and Y. Talmon, J. Phys. Chem. B, 1999, 103, 10331–10334 CrossRef CAS.
- D. Sprouse, Y. Jiang, J. E. Laaser, T. P. Lodge and T. M. Reineke, Biomacromolecules, 2016, 17, 2849–2859 CrossRef CAS PubMed.
- S. Shah and L. Leon, Curr. Opin. Colloid Interface Sci., 2021, 53, 101424 CrossRef CAS.
- M. Amann, J. S. Diget, J. Lyngsø, J. S. Pedersen, T. Narayanan and R. Lund, Macromolecules, 2019, 52, 8227–8237 CrossRef CAS.
- M. Chang, J.-C. Chou and H.-J. Lee, Plant Cell Physiol., 2005, 46, 482–488 CrossRef CAS PubMed.
- S. M. Fuchs and R. T. Raines, Protein Sci. Publ. Protein Soc., 2005, 14, 1538–1544 CrossRef CAS PubMed.
- R. Kumar, N. Le, Z. Tan, M. E. Brown, S. Jiang and T. M. Reineke, ACS Nano, 2020, 14, 17626–17639 CrossRef CAS PubMed.
- J. K. Allen, D. J. Brock, H. M. Kondow-McConaghy and J.-P. Pellois, Biomolecules, 2018, 8, E50 CrossRef PubMed.
- A. Erazo-Oliveras, N. Muthukrishnan, R. Baker, T.-Y. Wang and J.-P. Pellois, Pharm. Basel Switz., 2012, 5, 1177–1209 CAS.
- D. C. Luther, T. Jeon, R. Goswami, H. Nagaraj, D. Kim, Y.-W. Lee and V. M. Rotello, Bioconjug. Chem., 2021, 32, 891–896 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc00192f |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2022 |