
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceResistance and phylogeny guided discovery reveals structural novelty of tetracycline antibiotics†
Ling Yu
Li
a,
Yi Ling
Hu
a,
Jia Lin
Sun
a,
Long Bo
Yu
a,
Jing
Shi
a,
Zi Ru
Wang
a,
Zhi Kai
Guo
b,
Bo
Zhang
a,
Wen Jie
Guo
a,
Ren Xiang
Tan
 *a and
Hui Ming
Ge
*a
*a and
Hui Ming
Ge
*a
aState Key Laboratory of Pharmaceutical Biotechnology, School of Life Sciences, Chemistry and Biomedicine Innovation Center (ChemBIC), Nanjing University, Nanjing 210023, China. E-mail: rxtan@nju.edu.cn; hmge@nju.edu.cn
bKey Laboratory of Biology and Genetic Resources of Tropical Crops, Ministry of Agriculture, Institute of Tropical Bioscience and Bio-technology, Chinese Academy of Tropical Agricultural Sciences, Haikou 571101, China
First published on 17th October 2022
Abstract
Tetracyclines are a class of antibiotics that exhibited potent activity against a wide range of Gram-positive and Gram-negative bacteria, yet only five members were isolated from actinobacteria, with two of them approved as clinical drugs. In this work, we developed a genome mining strategy using a TetR/MarR-transporter, a pair of common resistance enzymes in tetracycline biosynthesis, as probes to find the potential tetracycline gene clusters in the actinobacteria genome database. Further refinement using the phylogenetic analysis of chain length factors resulted in the discovery of 25 distinct tetracycline gene clusters, which finally resulted in the isolation and characterization of a novel tetracycline, hainancycline (1). Through genetic and biochemical studies, we elucidated the biosynthetic pathway of 1, which involves a complex glycosylation process. Our work discloses nature's huge capacity to generate diverse tetracyclines and expands the chemical diversity of tetracyclines.
Introduction
Tetracyclines are a class of bacterial aromatic polyketides featuring linearly fused tetracyclic structures and an oxidized 2-naphthacenecarboxamide framework.1 Tetracyclines target the 30S prokaryotic ribosomal subunit, thus preventing the interaction of aminoacyl-tRNA with the ribosome resulting in the inhibition of translation.2 This family of antibiotics has been widely used clinically as broad-spectrum antibiotics against Gram-positive and Gram-negative pathogens.3 As a result, the widespread use of tetracyclines has been hindered by the emergence of resistance.3–5 Aiming to combat resistant strains, first-generation natural tetracycline antibiotics (chlorotetracycline and oxytetracycline) have been synthetically evolved into second and third-generation tetracyclines.6,7 Notably, tigecycline, a third-generation tetracycline obtained after glycine modification of the C9 position of the tetracyclic core, can effectively escape from efflux pumps and the ribosome resistance protection mechanism and has become a last line of defence against multi-drug resistant bacteria.8However, since the first discovery of chlorotetracycline over 70 years ago,9 only five natural tetracyclines have been reported as exemplified by oxytetracycline,10 SF2575,11 chelocardin,12 and dactylocycline13 (Scheme 1A). It is remarkable that among the five tetracyclines known to date, two (chlorotetracycline and oxytetracycline) have been developed into marked drugs, representing an astonishing 40% success rate. Considering the rarity of tetracycline-type compounds, their fascinating mode of action, and the promise of tetracyclines as novel drug leads, we set out to search for novel members of tetracyclines by exploring nature's biosynthetic repertoire. We reported here the use of a genome mining approach to target tetracycline-type natural products. From the genomic database, we discovered 25 distinct biosynthetic gene clusters (BGCs) that have the potential to produce different tetracyclines. As a proof of concept, we have successfully isolated and characterized a highly glycosylated tetracycline, hainancycline, and elucidated its biosynthetic pathway. This study indicates the huge potential of tetracycline BGCs encoded in actinobacteria.
 | ||
Scheme 1 Genome mining of novel tetracycline gene clusters. (A) Structures of known tetracyclines, (B) the BGCs of known tetracyclines where only resistant genes and KS and CLFs are highlighted in colors, and (C) workflow of the genome mining in this work. 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 393 actinobacteria genome annotation files from the NCBI database; the first round of search probes is based on adjacent regulators and transporters. Within the range of 30 genes near the regulator, the adjacent KS and CLF were used as the probe for the second-round search. Eligible 1744 gene clusters were screened for further analysis. (D) The phylogenetic tree of 1744 CLFs and (E) the genome neighboring network of 30 tetracycline gene clusters (E = 10−10). 393 actinobacteria genome annotation files from the NCBI database; the first round of search probes is based on adjacent regulators and transporters. Within the range of 30 genes near the regulator, the adjacent KS and CLF were used as the probe for the second-round search. Eligible 1744 gene clusters were screened for further analysis. (D) The phylogenetic tree of 1744 CLFs and (E) the genome neighboring network of 30 tetracycline gene clusters (E = 10−10). | ||
Results and discussion
Genome mining of tetracycline biosynthetic gene clusters
In order to find a suitable probe for genome mining, we collected and analysed all known BGCs for tetracyclines (Scheme 1B). Besides the typical type II PKS system that is responsible for synthesizing the tetracyclic skeleton,1 we found all tetracycline BGCs contain a pair of resistant genes, including a transcriptional regulator tetR or marR and a drug resistance transporter like tetA, which are in adjacent positions (Scheme 1B).14–16 The resistance transporter is a tetracycline/metal–proton antiporter located in the membrane, while the regulatory protein TetR is a tetracycline inducible repressor. Once the tetracycline and metal ion complex binds to the TetR repressor and triggers a conformational change, TetR will no longer bind to the tet operator, which leads to the transcription of the transporter. Thus, the transporter pump selectively transports tetracycline antibiotics in time to avoid suicide.17–19Similarly, MarR controls the expression of a multidrug efflux pump.20 As all known tetracycline BGCs share this resistant mechanism, we reasoned that this pair of genes could be a good indicator for mining tetracycline-type natural products.
We thus collected all assembled genome annotation files from the NCBI database which included over 20000 genomes (June 2021). Surprisingly, using tetR/marR and the adjacent transporter gene as probes, we found 351172 BGCs. These BGCs are not all tetracycline biosynthesis-related, because many strains acquired the tetracycline-resistant genes due to the prolonged and extensive use of tetracyclines in the world.21,22 We reasoned that the minimal PKS genes, which are responsible for the assembly of the tetracycline carbon skeleton, should also be colocalized with the resistance genes in the potential tetracycline gene cluster (Scheme 1B). Thus, we took the conserved ketosynthase (KS) and chain length factor (CLF) into consideration. Based on this, an algorithm was developed that can identify various BGCs in which the tetR/marR and transporter genes are colocalized within a 30 gene distance with KS and CLF genes. This refinement resulted in 1744 BGCs, among which all known tetracycline BGCs were obtained, confirming the reliability of this approach. In contrast to the widespread tetracycline-resistant strains and a large number of type II PKS BGCs in actinobacteria, our first genome analysis using a resistance gene and type II PKS gene as probes resulted in only ∼1700 BGCs from over 20000 genomes in the public database, encouraging us to perform further analysis.
We noticed that the identified BGCs included many known BGCs for type II polyketide biosyntheses such as enterocin,23 landomycin,24 and tetracenomycin,25 indicating that they also share a similar resistance mechanism (Fig. S1†). To directly target the tetracycline BGC, we attempted to analyze the CLFs, whose phylogeny highly correlates with the polyketide condensation number and cyclization pattern.26,27 To test if the tetracycline BGCs are in a separate clade, we first generated a phylogenetic tree using CLFs from over 160 characterized BGCs for type II PKS. Indeed, we found that the compounds with different extension lengths and cyclization patterns can be well distinguished (Fig. S2†). Gratifyingly, we observed that five known CLFs from tetracycline BGCs are grouped together, although they are closely related to the BGC from cervimycin, supporting the feasibility of the CLF phylogeny in reflecting the product structure. Thus, the CLF sequences from 1744 BGCs were extracted and a phylogenetic tree was then constructed (Scheme 1D). The most abundant BGCs are angucyclines. Meanwhile spore pigment, naphthoquinone, pentangular polyphenol, and anthracycline BGCs were also widely distributed, indicating that the TetR/MarR-transporter resistance mechanism is widespread in the type II PKS biosynthetic pathway. Notably, we observed a small group that contained 103 CLFs including 5 known tetracycline CLFs clustered together. Among them, 72.8% are from Streptomyces, 14.5% are from Kitasatospora, 4.8% are from Amycolatopsis, and the others are from rare actinomycetes like Spongiactinospora and Micromonospora, indicating a wide spread of tetracycline BGCs in various strains (Fig. S3†). After dereplication, we obtained 25 BGCs that are distinct from all characterized tetracycline BGCs in the literature (Fig. S4 and Tables S3–S27†).
To analyze the BGCs, we carried out genome neighboring network (GNN) analysis (Scheme 1E), which has recently become a powerful and visualized bioinformatics tool to predict enzyme functions based on their genomic context.28,29 A GNN consisting of 1021 proteins from 30 BGCs (including five known BGCs) was generated to annotate these tetracycline BGCs. The GNN analysis showed that all of them contain the core genes for tetracyclic skeleton biosynthesis. Besides this, the newly discovered BGCs are diverse, featuring many new enzyme functions that are different from those in known BGCs. Notably, we noticed that the cluster in Streptomyces sp. NAK774 encoded multiple post-modification enzymes including six glycosyltransferases (GTs), three KAS III enzymes, and a FAD-dependent halogenase, suggesting that this strain may produce a tetracycline with interesting peripheral groups (Scheme 2A).
 | ||
| Scheme 2 Biosynthesis of hainancycline (1). (A) The biosynthetic gene cluster for 1 and (B) the proposed biosynthetic pathway for 1. | ||
Isolation and structural characterization of hainancycline
In order to obtain this new tetracycline from the S. sp. NAK774 strain, we first inactivated KS genes to afford the corresponding ΔhaiAB strain (Fig. S5†). After screening various fermentation conditions, we found that a product (1) ([M − H]− ion at m/z 1400.5110), which has a characteristic UV absorption for the tetracyclic core (Fig. S6†),30 is detected in the S. sp. NAK774 wild-type strain, but not in the ΔhaiAB mutant strain (Fig. 1). We hypothesize that 1 might be related to the hai gene cluster. After a large-scale fermentation, this compound was isolated and characterized by extensive analysis of HRESIMS, 1D, and 2D NMR spectroscopy. From HRESIMS, 1 displayed a 3:1 [M − H]− ion signal at m/z 1400.5110 and 1402.5143, revealing that its molecular formula is C68H88ClNO28. The 1D and 2D NMR analysis showed 4 aromatic protons, 9 methyl groups, 6 sugar units and a typical tetracycline skeleton (Table S29†). However, severe overlapped NMR signals hindered structure determination for 1.
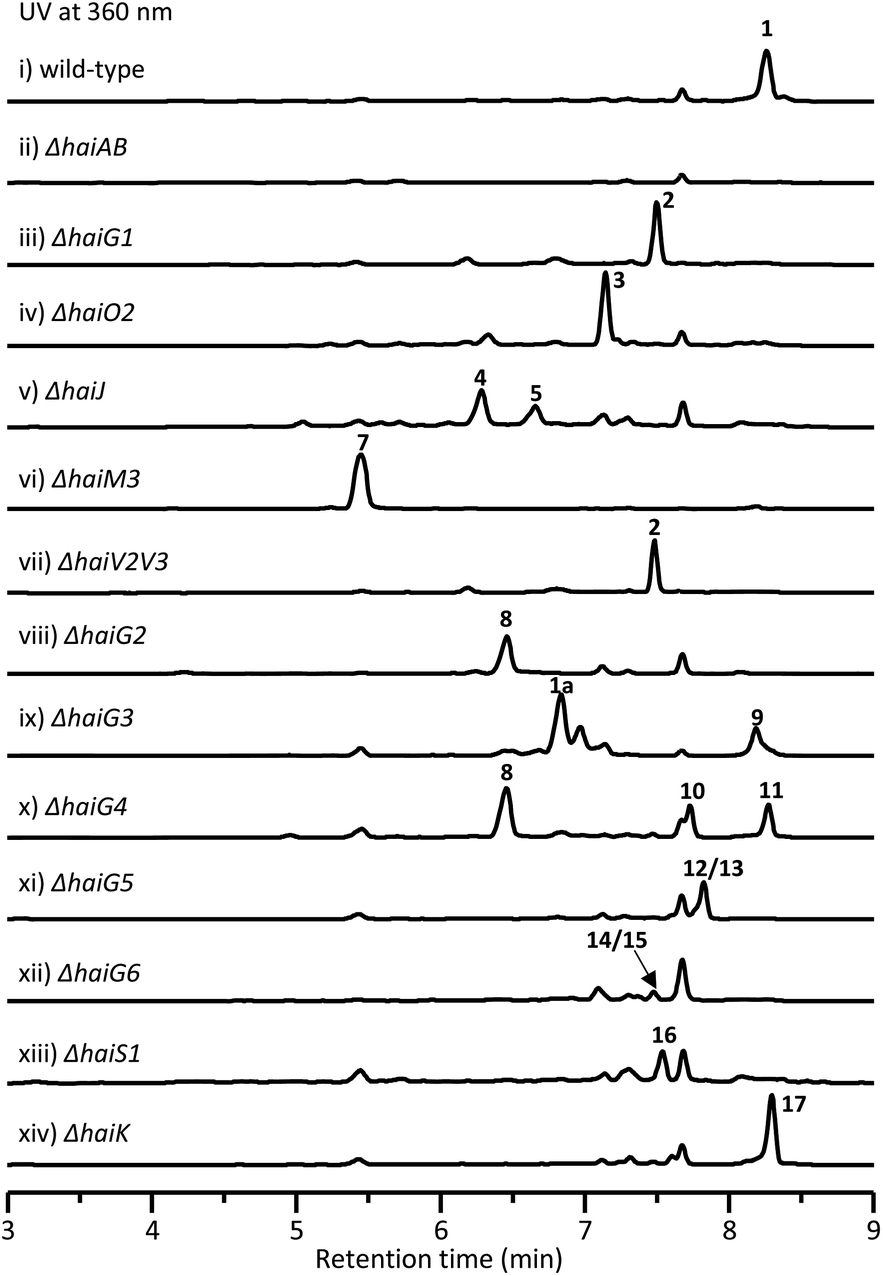 | ||
| Fig. 1 HPLC analysis of metabolites from wild-type strains and mutants. | ||
To simplify the structure elucidation procedure, we attempted to dissect the sugar linkage in 1 through acid hydrolysis. The treatment of 1 with trifluoroacetic acid/MeOH/H2O (1:1:8) at 60 °C for 1 hour led to the complete hydrolysis of 1 and afford three major fragments 1a (m/z 658.2501 [M − H]−), 1b (m/z 329.0794 [M − H]−) and 1c (m/z 483.1390 [M + Na]+). Compound 1a was elucidated to have the same tetracyclic aglycon as observed in SF2575 (Table S30†).16,30 Meanwhile the 1H–1H COSY spectrum showed that the sugars in 1a were different to that in SF2575. The diagnostic HMBC correlations between H8/C1A, H1A/C8, H1A/C9, and H1B/C13 indicated that two sugar moieties were anchored at C9 and N1 positions, respectively. The relative configuration of sugar moieties was determined by the interpretation of the NOESY data and coupling constants. The large diaxial coupling constants, JH1A = 11.1 Hz, and NOE correlations of H1A with H5A, and H2Aβ with H4A indicated that H1A, H4A, and H5A all possessed axial orientations. Thus, sugar A was determined to be amicetose. Similarly, based on NMR analysis, sugar B was also determined to be amicetose. Fragment 1b was elucidated to have two subunits including 5-chloro-6-methylsalicylic acid and a methylated oliose, the stereochemistry of which was determined by NOE analysis (Table S31†). The HMBC correlation of H4F with C1′ suggested the oliose was substituted at the C1′ position. Compared to 1b, fragment 1c has an additional oliose moiety at the C1F position as determined by the HMBC correlation of H3E/C1F and H4F/C1′ (Table S32†). After further scrutiny of the NMR data of 1, two additional sugar moieties, sugar C and sugar D, were identified. The diagnostic HMBC correlations H1B/C13, H1C/C4B, H4C/C1′′, H2′′/C1′′, H1D/C12a, H8/C1A, H4A/C1E, H3E/C1F, H4F/C1′, and 1H–1H COSY of H1B with NH, linked all units, established the complete structure of 1 and designated as hainancycline (Scheme 2B and Table S29†). To the best of our knowledge, 1 represents the most modified tetracycline discovered so far.
Biosynthesis of the aglycon of hainancycline
To investigate the biosynthetic pathway for 1, we analyzed the hai BGC in detail. The hai BGC (GenBank accession number ON755207) spans a ∼55 kb contiguous DNA region consisting of 43 genes responsible for biosynthesis, regulation, and resistance (Scheme 2A). As the aglycon of 1 was identical to SF2575, we carefully compared these two BGCs. The hai BGC encoded all homologous proteins required for aglycon biosynthesis in SF2575 with moderate to high sequence identities (50–80%) (Table S3 and Fig. S7†).30 Thus, we proposed that the tetracycline core in 1 was biosynthesized in a similar manner to that in the SF2575 pathway.To verify this hypothesis, we inactivated haiG1, whose product showed 57% sequence identity to SsfS6 (glycosyltransferase) in SF2575 biosynthesis.30 The resulting ΔhaiG1 mutant abolished the production of 1, but clearly accumulated 2 (Fig. 1, iii and Table S33†), a product that is also isolated from the ΔssfS6/ΔssfM1 double mutant,16 confirming that the function of HaiG1 is the same as SsfS6, which can transfer an amicetose at the C9 position. Based on the same biosynthetic origin, we proposed that 1 possessed the same stereochemistry at C4 and C12a as that in 2. In addition, the NOE correlations between H4a and H5a, H5a and 6-OCH3, and H4a and H1D determined the stereochemistry of C5a and C6 in 1. HaiO2, HaiJ, and HaiM3 were found to be homologous to SsfO1 (oxygenase), SsfP (dehydrogenase), and SsfM2 (O-methyltransferase) (Table S3†),16,30 respectively, which are involved in the successive decoration of the tetracyclic core to form a similar biosynthetic intermediate 8 in SF2575 biosynthesis (Fig. S7†). Indeed, the inactivation of haiO2 led to the disappearance of 1 and the accumulation of 3 (Fig. 1, iv and Table S34†). The inactivation of haiJ, which encoded an NADPH-dependent reductase with 60.4% sequence identity to SsfP, led to the accumulation of 4 and 5 (Fig. 1, v). NMR analysis indicated that 4 is a C-ring rearrange shunt product, and 5 is a C6-methoxyl derivative of 3 (Tables S35 and S36 and Fig. S8†). The structure of 4 is intriguing and has never been isolated from the SF2575 biosynthetic pathway. To test if 4 was derived from the proposed intermediate 6 (Scheme 2B), we overexpressed the HaiO2 protein in E. coli BL21(DE3). The purified HaiO2 protein showed a yellow color and was confirmed to contain FAD as a cofactor (Fig. S9†). When HaiO2 was incubated with 3 in the presence of NADPH, we detected the formation of 4 as a major product, together with a product (m/z 528.1510 (C26H26NO11 [M − H]−)), which matched the formula of 6 (Fig. 2, iv). In contrast, HaiO2 cannot recognize 2 as the substrate that lacks the sugar unit at C9 (Fig. 2, vi). Taking the structural features of 4 and 5 into account, we concluded that HaiO2 is responsible for the hydroxylation of 3 to give 6 (Scheme 2). In the absence of downstream enzyme, 6 can be further hydroxylated to 6a, which can undergo a spontaneous Michael-type addition, followed by a retro-Claisen condensation to give 4 (Fig. S8†). We speculated that the unexpected methoxyl group in 5 could be installed by HaiO2 and HaiM3 or other unknown enzymes on the biosynthetic intermediate 3. In addition, the ΔhaiM3 mutant strain accumulated 7 (Fig. 1, vi, Table S37†). Therefore, the HaiO2 functions immediately after HaiG1, and the roles for HaiO2, HaiJ, and HaiM3 are C6 hydroxylase, C5a–C11a-ene reductase, and C6–OH methyltransferase, respectively.
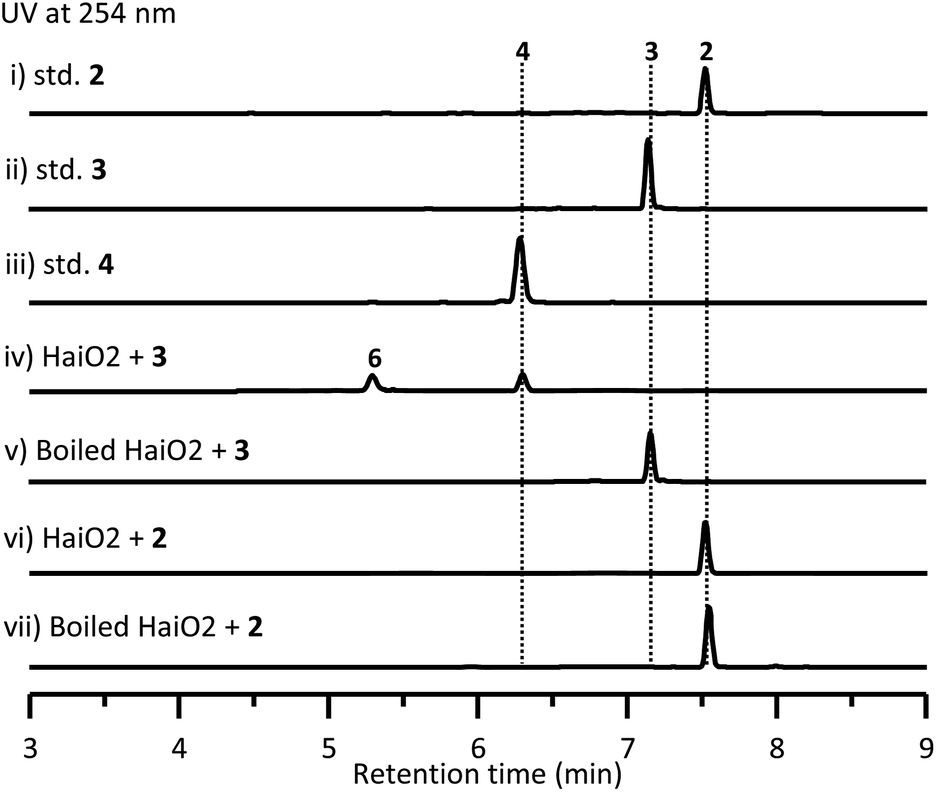 | ||
| Fig. 2 HPLC analysis of HaiO2-catalyzed reactions. (i) authentic 2, (ii) authentic 3, (iii) authentic 4, (iv) HaiO2 incubated with 3 and NADPH, (v) boiled HaiO2 incubated with 3 and NADPH, (vi) HaiO2 incubated with 2 and NADPH, and (vii) boiled HaiO2 incubated with 2 and NADPH. | ||
In vivo characterization of five glycosyltransferases
After the construction of the tetracyclic core, we set out to investigate the glycosylation step in 1 biosynthesis. The deoxysugar units, oliose and amicetose, are also found in mithramycin and tetracenomycin, respectively.31,32 Putative enzymes, HaiV1–HaiV5, encoded in the hai cluster showed high sequence identity to enzymes in the NDP-D-oliose biosynthetic pathway (Fig. S10†).31,33 Additional enzymes, HaiV6–HaiV8, could further convert the NDP-D-deoxysugar intermediate to NDP-amicetose (Fig. S10†).32,33 HaiV7 is annotated as a NDP-hexose 3,5-epimerase showing 42.7% sequence identity to UrdZ1 that epimerizes NDP-D-cinerulose to NDP-L-cinerulose in the biosynthesis of L-rhodinose.33 Thus, we proposed that the configuration of two deoxysugar units in hainancycline is L-amicetose and D-oliose, respectively (Fig. S10†). When we inactivated haiV2 and haiV3 gene, aglycone 2 was detected, confirming that the genes are involved in deoxysugar biosynthesis (Fig. 1, vii).We noticed that in the hai BGC there are five GTs (HaiG2–HaiG6) left in the cluster, which are consistent with the remaining five deoxysugar units in 1 (Table S3†).34,35 To assign their functions, we individually inactivated these GT encoding genes (Fig. S5†). HPLC analysis of metabolic extracts indicated that each mutant showed a metabolic profile distinct from others and a wild-type strain. The subsequent large-scale fermentation led to the isolation of 9 products (8–15 and 1a) from these mutant strains (Fig. 1). All these structures were elucidated by extensive analysis of HRESIMS and NMR data (Tables S38–S45†). Compound 8 isolated from the ΔhaiG2 mutant is a C-glycoside with an intact tetracycline core, suggesting that HaiG2 is a second GT responsible for appending the sugar unit on 8 (Table S38†). Compounds 1a and 9 from the ΔhaiG3 mutant lack the sugar C unit, suggesting that HaiG3 is a terminal GT transferring L-amicetose to sugar B (Tables S29 and S39†). 8, 10 and 11 accumulated in the ΔhaiG4 mutant contain no sugar D unit at the C12a position (Tables S38, S40 and S41†). In addition, 12 and 13 which are produced in the ΔhaiG5 mutant lack sugar E and F units, whereas 14 and 15 from the ΔhaiG6 mutant only lack sugar F units (Tables S42–S45†). These data revealed that HaiG4, HaiG5, and HaiG6 account for transferring sugar D, E, and F units, respectively, which in turn indicated that the role of HaiG2 is transferring sugar B to the amide group of the tetracyclic core (Scheme 2).
Biosynthesis of the salicylic acid
The salicylate substitution at the sugar unit is rare among aromatic polyketides. One similar example is that of SF2575, where the salicylate moiety is directly substituted at the C4 position of the tetracyclic core.16 Moreover, the salicylic acid in SF2575 is biosynthesized from the shikimate pathway that is also observed in the biosynthesis of yersiniabactin36 and mycobactin.37 However, we could not find any homologous enzymes from the shikimate pathway in the hai cluster. Instead, five enzymes, HaiS1–HaiS5, were found to be homologous to ChlB1–ChlB4, and ChlB6 from the chlorothricin biosynthetic pathway, respectively, which collaboratively catalyze the formation of 5-chloro-6-methylsalicylic acid (Fig. S11†).38 Thus, the salicylate substitution in 1 is presumably synthesized via a dedicated iterative type I PKS. When the haiS1 gene was knocked out, a new product 16 was accumulated (Fig. 1, xiii). NMR analysis showed that 16 lacks a salicylate group (Table S46†), thereby confirming our hypothesis.In vivo characterization of HaiK as a malonyl unit transferase
Another interesting structural feature of 1 is the malonate group at the sugar C unit, which is also very rare as a substitution group among natural products. The only remaining unassigned gene in the hai BGC is haiK, whose product is annotated as a KAS III enzyme with 46.6% homology to CerJ.39 The KAS III enzyme in type II PKS biosynthesis usually plays a role in the non-acetate starter unit recognition and polyketide chain initiation.40 However, the inactivation of the haiK gene led to the complete abolishment of 1 along with the emergence of a new product 17 that does not have a malonate group in comparison to the structure of 1, revealing the unique role of HaiK as a malonyl unit transferase (Fig. 1, xiv and Table S47†). Thus, the complete biosynthetic pathway for 1 was disclosed (Scheme 2B). It is worth noting that the late-stage transferases including HaiG4, HaiG5, HaiG6, HaiS5 and HaiK exhibit broad substrate promiscuity, which might be very useful for further combinatorial biosynthesis.The biological activity of isolated compounds
Finally, we performed antibacterial and cytotoxic assays for all isolated compounds. However, all these compounds showed no obvious activity against the tested bacteria. Compounds 1, 2, 9, 11, and 17 showed moderate activities with IC50 values ranging from 8.4 to 28.8 μM against HT29 and HCT116 tumor cell lines, whereas other compounds showed weak activities with IC50 > 30 μM (Table 1). Notably, all active compounds except 2 contain a 5-chloro-6-methylsalicylic acid group, suggesting that this group might play an important role in their activities.| 1 | 2 | 9 | 11 | 17 | |
|---|---|---|---|---|---|
| HCT116 | 12.7 | 8.4 | 15.4 | 15.3 | 28.8 |
| HT29 | 24.8 | 10.1 | 28.5 | 23.5 | >30 |
Conclusions
In summary, our bioinformatic study on known tetracycline gene clusters disclosed that all of them share a similar resistance mechanism. Based on this, we developed a genome mining approach targeting both resistant genes and type II PKS genes, resulting in the discovery of 25 new tetracycline gene clusters. We successfully activated one identified gene cluster and obtained a novel tetracycline named hainancycline, which is the most decorated tetracycline known to date. In vivo gene mutation experiments revealed the biosynthetic pathway of 1 and demonstrated that GTs have relaxed substrate flexibility. Our work demonstrated the feasibility of the discovery of new natural products from a large strain collection. The identification of hai and other putative tetracycline gene clusters sets the stage for future bioengineering of new tetracycline derivatives.Data availability
All experimental data has been uploaded.Author contributions
R. X. T. and H. M. G. conceived and designed the project. L. Y. L., Y. L. H., J. L. S., L. B. Y., J. S., Z. R. W., W. J. G., Z. K. G. and B. Z. performed the experiments and analysed the data. Z. K. G., H. M. G. and L. Y. L. wrote the manuscript. Y. L. H. performed bioinformatics analysis. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by NSFC (22193071, 81925033, 22107048, 81991522, and 81991524), MOST (2018YFA0902000), the Fundamental Research Funds for the Central Universities (14380159) and the GESP program (14915211).Notes and references
- C. Hertweck, A. Luzhetskyy, Y. Rebets and A. Bechthold, Nat. Prod. Rep., 2007, 24, 162–190 RSC
.
- D. E. Brodersen, W. M. Clemons, A. P. Carter, R. J. Morgan-Warren, B. T. Wimberly and V. Ramakrishnan, Cell, 2000, 103, 1143–1154 CrossRef CAS PubMed
- I. Chopra and M. Roberts, Microbiol. Mol. Biol. Rev., 2001, 65, 232–260 CrossRef CAS PubMed
- M. L. Nelson and S. B. Levy, Ann. N. Y. Acad. Sci., 2011, 1241, 17–32 CrossRef CAS PubMed
- M. Thaker, P. Spanogiannopoulos and G. D. Wright, Cell. Mol. Life Sci., 2010, 67, 419–431 CrossRef CAS PubMed
- C. Hobson, A. N. Chan and G. D. Wright, Chem. Rev., 2021, 121, 3464–3494 CrossRef CAS PubMed
- D. Fuoco, Antibiotics, 2012, 1, 1–13 CrossRef CAS PubMed
- G. G. Zhanel, K. Homenuik, K. Nichol, A. Noreddin, L. Vercaigne, J. Embil, A. Gin, J. A. Karlowsky and D. J. Hoban, Drugs, 2004, 64, 63–88 CrossRef CAS PubMed
- B. M. Duggar, Ann. N. Y. Acad. Sci., 1948, 51, 177–181 CrossRef CAS PubMed
- E. Q. King, C. N. Lewis, H. Welch, E. A. Clark, J. B. Johnson, J. B. Lyons, R. B. Scott and P. B. Cornely, J. Am. Med. Assoc., 1950, 143, 1–4 CrossRef CAS PubMed
- M. Hatsu, T. Sasaki, H. Watabe, S. Miyadoh, M. Nagasawa, T. Shomura, M. Sezaki, S. Inouye and S. Kondo, J. Antibiot., 1992, 45, 320–324 CrossRef CAS PubMed
- L. A. Mitscher, J. V. Juvarkar, W. Rosenbrook, W. W. Andres, J. Schenck and R. S. Egan, J. Am. Chem. Soc., 1970, 92, 6070–6071 CrossRef CAS PubMed
- A. A. Tymiak, H. A. Ax, M. S. Bolgar, A. D. Kahle, M. A. Porubcan and N. H. Andersen, J. Antibiot., 1992, 45, 1899–1906 CrossRef CAS PubMed
- W. J. Zhang, B. D. Ames, S. C. Tsai and Y. Tang, Appl. Environ. Microbiol., 2006, 72, 2573–2580 CrossRef CAS PubMed
- T. Lukežič, U. Lešnik, A. Podgoršek, J. Horvat, T. Polak, M. Šala, B. Jenko, P. Raspor, P. R. Herron, I. S. Hunter and H. Petković, Microbiology, 2013, 159, 2524–2532 CrossRef PubMed
- P. Wang, W. Kim, L. B. Pickens, X. Gao and Y. Tang, Angew. Chem., Int. Ed., 2012, 51, 11136–11140 CrossRef CAS PubMed
- T. S. B. Møller, M. Overgaard, S. S. Nielsen, V. Bortolaia, M. O. A. Sommer, L. Guardabassi and J. E. Olsen, BMC Microbiol., 2016, 16, 39 CrossRef PubMed
- J. L. Ramos, M. Martínez-Bueno, A. J. Molina-Henares, W. Terán, K. Watanabe, X. D. Zhang, M. T. Gallegos, R. Brennan and R. Tobes, Microbiol. Mol. Biol. Rev., 2005, 69, 326–356 CrossRef CAS PubMed
- S. Mak, Y. Xu and J. R. Nodwell, Mol. Microbiol., 2014, 93, 391–402 CrossRef CAS PubMed
- I. C. Perera and A. Grove, J. Mol. Cell Biol., 2010, 2, 243–254 CrossRef CAS PubMed
- T. L. Nikolakopoulou, S. Egan, L. S. van Overbeek, G. Guillaume, H. Heuer, E. M. H. Wellington, J. D. van Elsas, J. M. Collard, K. Smalla and A. D. Karagouni, Curr. Microbiol., 2005, 51, 211–216 CrossRef CAS PubMed
- J. M. A. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. V. Piddock, Nat. Rev. Microbiol., 2015, 13, 42–51 CrossRef CAS PubMed
- J. Piel, C. Hertweck, P. R. Shipley, D. M. Hunt, M. S. Newman and B. S. Moore, Chem. Biol., 2000, 7, 943–955 CrossRef CAS PubMed
- O. Yushchuk, M. Kharel, I. Ostash and B. Ostash, Appl. Microbiol. Biotechnol., 2019, 103, 1659–1665 CrossRef CAS PubMed
- P. G. Guilfoile and C. R. Hutchinson, J. Bacteriol., 1992, 174, 3659–3666 CrossRef CAS PubMed
- Z. Feng, D. Kallifidas and S. F. Brady, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 12629–12634 CrossRef CAS PubMed
- S. C. Chen, C. Zhang and L. H. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202202286 CAS
- J. D. Rudolf, X. Yan and B. Shen, J. Ind. Microbiol. Biotechnol., 2016, 43, 261–276 CrossRef CAS PubMed
- J. Shi, C. L. Liu, B. Zhang, W. J. Guo, J. P. Zhu, C. Y. Chang, E. J. Zhao, R. H. Jiao, R. X. Tan and H. M. Ge, Chem. Sci., 2019, 10, 4839–4846 RSC
- L. B. Pickens, W. Kim, P. Wang, H. Zhou, K. Watanabe, S. Gomi and Y. Tang, J. Am. Chem. Soc., 2009, 131(48), 17677–17689 CrossRef CAS PubMed
- A. González, L. L. Remsing, F. Lombó, M. J. Fernández, L. Prado, A. F. Braña, E. Künzel, J. Rohr, C. Méndez and J. A. Salas, Mol. Gen. Genet., 2001, 264, 827–835 CrossRef PubMed
- M. Pérez, F. Lombó, L. Zhu, M. Gibson, A. F. Braña, J. Rohr, J. A. Salas and C. Méndez, Chem. Commun., 2005, 12, 1604–1606 RSC
- D. Hoffmeister, K. Ichinose, S. Domann, B. Faust, A. Trefzer, G. Drage, A. Kirschning, C. Fischer, E. Kunzel, D. W. Bearden, J. Rohr and A. Bechthold, Chem. Biol., 2000, 7, 821–831 CrossRef CAS PubMed
- L. J. Stevenson, J. Bracegirdle, L. Liu, A. V. Sharrock, D. F. Ackerley, R. A. Keyzers and J. G. Owen, RSC Chem. Biol., 2021, 2, 556–567 RSC
- M. Claesson, V. Siitonen, D. Dobritzsch, M. Metsä-Ketelä and G. Schneider, FEBS J., 2012, 279, 3251–3263 CrossRef CAS PubMed
- O. Kerbarh, A. Ciulli, N. I. Howard and C. Abell, J. Bacteriol., 2005, 187, 5061–5066 CrossRef CAS PubMed
- J. Zwahlen, S. Kolappan, R. Zhou, C. Kisker and P. J. Tonge, Biochemistry, 2007, 46, 954–964 CrossRef CAS PubMed
- X. Y. Jia, Z. H. Tian, L. Shao, X. D. Qu, Q. F. Zhao, J. Tang, G. L. Tang and W. Liu, Chem. Biol., 2006, 13, 575–585 CrossRef CAS PubMed
- T. Bretschneider, G. Zocher, M. Unger, K. Scherlach, T. Stehle and C. Hertweck, Nat. Chem. Biol., 2012, 8, 154–161 CrossRef CAS PubMed
- R. Nofiani, B. Philmus, Y. Nindita and T. Mahmud, Med. Chem. Commun., 2019, 10, 1517–1530 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc03965f |
| This journal is © The Royal Society of Chemistry 2022 |