
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganocatalytic asymmetric azidation of sulfoxonium ylides: mild synthesis of enantioenriched α-azido ketones bearing a labile tertiary stereocenter†
Wengang
Guo
 *a,
Feng
Jiang
a,
Shijia
Li
bc and
Jianwei
Sun
*abc
*a,
Feng
Jiang
a,
Shijia
Li
bc and
Jianwei
Sun
*abc
aJiangsu Key Laboratory of Advanced Catalytic Materials & Technology, School of Petrochemical Engineering, Changzhou University, Changzhou, China. E-mail: wgguo@cczu.edu.cn
bDepartment of Chemistry and the Hong Kong Branch of Chinese National Engineering Research Centre for Tissue Restoration & Reconstruction, The Hong Kong University of Science and Technology (HKUST), Clear Water Bay, Kowloon, Hong Kong SAR, China. E-mail: sunjw@ust.hk
cShenzhen Research Institute, HKUST, No. 9 Yuexing 1st Rd, Shenzhen 518057, China
First published on 15th September 2022
Abstract
Disclosed here is a catalytic asymmetric azidation reaction for the efficient synthesis of α-azido ketones bearing a labile tertiary stereocenter. With a superb chiral squaramide catalyst, a mild asymmetric formal H–N3 insertion of α-carbonyl sulfoxonium ylides proceeded with excellent efficiency and enantioselectivity. This organocatalytic process not only complements the previous α-azidation approaches for the formation of quaternary stereocenters and mostly for 1,3-dicarbonyl compounds, but also has advantages over the well-known metal-catalyzed asymmetric carbene insertion chemistry using α-diazocarbonyl compounds. Detailed mechanistic studies via control reactions and NMR studies provided important insights into the reaction pathway, which features reversible protonation and dynamic kinetic resolution. The curiosity in mechanism also led to the development of a simplified alternative protocol with a cheaper HN3 source.
Introduction
Organic azides are valuable molecules with broad applications in different disciplines, including chemical biology, material science, and drug discovery.1,2 They are also important building blocks for the synthesis of other useful compounds, such as amines, nitrenes, and iminophosphoranes.1,3 Specifically, α-azido carbonyl compounds represent a special class of azides with unique applications.4 The proximal azide and carbonyl functionalities enable them to serve as useful precursors toward biologically important molecules, heterocycles, and chiral ligands (e.g., I–III, Scheme 1a).4,5 As a result, a wide range of racemic or achiral methods have been developed for their synthesis.1,6–12 However, their asymmetric synthesis remains a challenge. Among the few methods, catalytic asymmetric α-azidation of carbonyl compounds represents an attractive one, but it was implemented with only limited success (Scheme 1b). These include electrophilic azidation,7 oxidative nucleophilic azidation,8 enantioconvergent halogenophilic nucleophilic substitution (SN2X),9 and radical addition.10 All these methods were uniformly limited to the formation of quaternary stereogenic centers and the majority were even limited to certain 1,3-dicarbonyl molecules. To the best of our knowledge, catalytic asymmetric α-azidation of carbonyl compounds to generate a tertiary stereocenter remains unknown,11 with the exception of a few multi-step approaches from enantioenriched precursors.12 Adjacent to both carbonyl and azide functionalities, this type of doubly activated tertiary stereocenters are very labile (easy to racemize) and vulnerable to many reaction conditions. Moreover, chemoselectivity issues resulting from this tertiary center (e.g., over-azidation) cause additional challenges.13 Herein we address this unmet challenge by a new approach, leading to the first asymmetric azidation toward α-azido ketones bearing a tertiary stereocenter. | ||
| Scheme 1 Introduction to asymmetric α-azidation of carbonyl compounds. | ||
Catalytic asymmetric H–X bond insertion of metal carbenes generated in situ from α-diazocarbonyls represents one of the most powerful approaches to access chiral α-functionalized carbonyl compounds bearing a tertiary stereocenter (Scheme 1c).14 However, despite the great success of this chemistry for the installation of a range of functionalities to the carbonyl α-position, the insertion to a “H–N3” bond remains unknown. This is likely due to the potentially facile decomposition of either the azide reagents or the azido carbonyl products triggered by the same metal catalysts employed for carbene insertion (similar to azide reactivity toward nitrene).
Recently, α-carbonyl sulfur ylides have been developed as powerful surrogates to α-diazocarbonyl compounds, participating in a range of asymmetric cycloaddition and H–X bond insertion reactions, with the latter still in its infancy.15–17 The amenability of this chemistry to mild metal-free organocatalytic conditions, combined with our continued interests in this topic,17 led us to envision that an asymmetric formal H–N3 insertion of α-carbonyl sulfoxonium ylides could be possibly achieved.18 If successful, it would not only eliminate the drawback of the already powerful diazo-based carbene insertion chemistry, but also serve as an important alternative solution to the above challenge in the synthesis of enantioenriched α-azido carbonyl compounds (Scheme 1d).
Results and discussion
Reaction development
We began our study by employing sulfoxonium ylide 1a as the model substrate. The combination of stoichiometric TMSN3 and PhCO2H was used for in situ generation of hydrazoic acid (HN3) in a safe and user-friendly manner.19 In hope of inducing enantiocontrol under mild metal-free conditions, we evaluated a diverse range of chiral hydrogen-bonding organocatalysts.20 Chiral phosphoric acids CPA1 and CPA2 were initially tested, as they were the catalysts of choice in our previous amine N–H insertions.17 The expected azido ketone 2a was successfully generated in almost quantitative yield, but in racemic form (Scheme 2). We also evaluated a wide range of other chiral phosphoric acids (details in the ESI†), but uniformly without asymmetric induction, suggesting that this azidation process is drastically distinct from amination. We then screened other hydrogen bonding catalysts, such as chiral (thio)ureas TU1–4 bearing different chiral motifs (details in the ESI†). However, they also led to no asymmetric induction, although the chemical efficiency could be good. At this point, we realized that PhCO2H or the in situ generated HN3 might be sufficient to protonate sulfoxonium ylides and promote the background reaction. Indeed, without catalyst, the reaction did afford the desired product, thus causing an additional challenge in developing the asymmetric variant. The solutions would be either to inhibit the background reaction or to accelerate the asymmetric pathway, with the latter likely being more feasible. With the notion that chiral hydrogen-bond donors (HBDs) can accelerate Brønsted acid-promoted reactions,21,22 we continued to evaluate another family of HBDs, squaramides, as they feature different acidities and distance between the two HBD motifs from thioureas.20d To our delight, when cinchona alkaloid-derived squaramide SQ1 was used, we started to observe enantiomeric induction (14% ee), albeit low. Subsequent screening indicated that chiral amino acid-derived squaramide SQ2 was able to substantially improve the enantioselectivity (66% ee). Further considerable screening efforts led to the identification of the best catalyst SQ3, giving 72% ee.23 | ||
| Scheme 2 Preliminary results and catalyst screening. Reaction scale: 1a (0.1 mmol), TMSN3 (0.2 mmol), PhCO2H (0.11 mmol), SQ3 (0.01 mmol), solvent (0.5 mL), 24 h. Yield was determined by 1H NMR spectra of the crude mixture using CH2Br2 as an internal standard. Ee was determined by HPLC on a chiral stationary phase. aThe reaction gave a messy mixture, likely due to the incompatibility of the reaction with the basic cinchona alkaloid functionality. | ||
We further examined other reaction parameters with SQ3 as catalyst (Table 1). Less acidic proton sources, such as water, methanol, hexafluoroisopropanol (HFIP), resulted in lower yields and no change in enantioselectivity (entries 1–3). Other carboxylic acids, including AcOH and substituted benzoic acids, gave same results as benzoic acid (entries 4–6). Chiral acids, such as N-Boc-L-proline and N-Boc-D-proline, did not influence the enantioselectivity either (entries 7–8). These results indicated that these acids only served as a proton source to generate HN3, but did not directly function in the enantiodetermining transition state. It was later found that the yield and enantioselectivity were sensitive to solvents. DCM or DCE resulted in a much lower enantioselectivity (entries 9–10). Toluene and THF led to both low conversion/yield and low enantioselectivity (entries 11–12). Drastically, no reaction was observed in EtOAc and MTBE (entries 13–14). It seemed that coordinating solvents were detrimental to this reaction, presumably due to competing hydrogen bonding interactions. An obvious increase in enantioselectivity was observed at a decreased temperature (−10 °C), though at the expense of reaction time (84% ee, entry 15). Variation of the reaction concentration did not affect the outcome (entries 16–17). The presence of background reaction then prompted us to examine an increased catalyst loading, in hope of enhancing enantiocontrol by accelerating the asymmetric path relative to the background reaction. Gratifyingly, the enantioselectivity was further improved to 89% ee with 20 mol% of catalyst (entry 18). Finally, both excellent yield and enantioselectivity were achieved at −15 °C (90% ee, entry 19). Notably, this is a remarkable result in view of the many unfavorable factors, such as labile tertiary stereogenic center, challenging enantiocontrol (no asymmetric induction with most catalysts) as well as complication by the background reaction.
|
|
||||
|---|---|---|---|---|
| Entry | Proton source | Solvent | Conv. (%) | ee (%) |
| a Reaction scale: 1a (0.1 mmol), TMSN3 (0.2 mmol), proton source (0.11 mmol), SQ3 (0.01 mmol), solvent (0.5 mL), 24 h. Yield was determined by 1H NMR spectra of the crude mixture using CH2Br2 as an internal standard. Yield in parentheses refers to isolated yield. Ee was determined by HPLC on a chiral stationary phase. b Run at −10 °C. c 96 h. d c = 0.4 M, 72 h. e c = 0.1 M, 120 h. f Run with 20 mol% of catalyst, 96 h. g Run with 20 mol% of catalyst at −15 °C, 120 h. | ||||
| 1 | H2O | CHCl3 | 30 | 71 |
| 2 | MeOH | CHCl3 | 72 | 71 |
| 3 | HFIP | CHCl3 | 80 | 72 |
| 4 | AcOH | CHCl3 | >95 | 71 |
| 5 | (p-OH)C6H4CO2H | CHCl3 | >95 | 70 |
| 6 | (p-Cl)C6H4CO2H | CHCl3 | >95 | 70 |
| 7 | N-Boc-L-proline | CHCl3 | >95 | 71 |
| 8 | N-Boc-D-proline | CHCl3 | >95 | 71 |
| 9 | PhCO2H | DCM | >95 | 55 |
| 10 | PhCO2H | DCE | >95 | 41 |
| 11 | PhCO2H | Toluene | 45 | 41 |
| 12 | PhCO2H | THF | 25 | 18 |
| 13c | PhCO2H | EtOAc | Trace | — |
| 14 | PhCO2H | MTBE | Trace | — |
| 15b,c | PhCO2H | CHCl3 | >95 | 84 |
| 16b,d | PhCO2H | CHCl3 | >95 | 83 |
| 17b,e | PhCO2H | CHCl3 | >95 | 84 |
| 18b,f | PhCO2H | CHCl3 | >95 | 89 |
| 19g | PhCO2H | CHCl3 | >95 (83) | 90 |
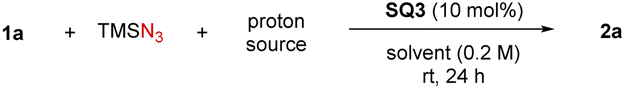
We next investigated the substrate scope of this formal asymmetric H–N3 insertion process (Scheme 3). Sulfoxonium ylides with different substituents (see the ESI for details†), both electron-donating and electron-withdrawing ones, at different positions of the aryl rings reacted smoothly under the optimized conditions, leading to a range of enantioenriched α-azido ketones with good to excellent yields and enantioselectivities.24 It is worth noting that electron-poor arenes on the ylide motif typically reacted at lower reaction rates, but with enhanced enantioselectivity. This might be attributed to less contribution by the background reaction in these cases. Moreover, heterocycles, such as furan and thiophene, could also be incorporated into the azido ketone products with equally good enantioselectivity. These are valuable synthetic precursors to the analogues of monoamine oxidase inhibitors.25,26a
 | ||
| Scheme 3 Reaction scope. Conditions: sulfoxonium ylide 1 (0.20 mmol), BzOH (0.22 mmol), TMSN3 (0.40 mmol), SQ3 (0.04 mmol), CHCl3 (1.0 mL). Isolated yield. ee was determined by HPLC on a chiral stationary phase. | ||
Synthetic application
To demonstrate the practical utility of our process, we carried out a 2 mmol-scale reaction of 1b using the opposite enantiomer of SQ3 as catalyst (Scheme 4). Under the standard conditions, product (R)-2b was obtained with equally good efficiency and enantioselectivity. Notably, the catalyst could be recovered in 90% yield and reused with equal catalytic activity. The enantiopurity of 2b could be enhanced by crystallization. The azide functionality in 2b could be selectively reduced by Pd/C-catalyzed hydrogenation followed by Boc-protection to form α-amino ketone 8, whose absolute configuration was confirmed by X-ray crystallography. Further diastereoselective reduction of 8 furnished amino alcohol 9, representing a family of useful chiral ligands and bioactive molecules.5,25 Finally, a simple 3-step sequence could convert 2b to enantioenriched triazole 10 with good overall efficiency.26b Notably, in all these transformations, the good enantiopurity remained essentially intact. | ||
Scheme 4 Large-scale synthesis of 2b and product transformations. (a) H2, Pd/C, Boc2O, EtOAc, 0 °C, 1 h. (b) NaBH4, MeOH, −20 °C, 5 h. (c) NaBH4; CuSO4·5H2O, PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH; DMP. CH; DMP. | ||
Mechanistic studiFes
Next, we carried out some control experiments to gain insights into the reaction mechanism. First of all, the N-methylated squaramide SQ3-Me2 did not show any catalytic activity (Scheme 5a). Neither rate acceleration (vs. background reaction) nor asymmetric induction was observed, indicating the essential role of the two N–H motifs in the optimal catalyst SQ3, likely serving as hydrogen bond donors. Next, 1H NMR study was used to probe which reactant is activated by the catalyst (Fig. 1). When a solution of 1a in CDCl3 was treated with SQ3, a dramatic high-field shift of the signal at ∼3.6 ppm (the two methyl groups in 1a) was observed. Increasing the loading of SQ3 caused a more pronounced shift, indicating strong but reversible interaction. Such a shift was also observed in the standard reaction mixture. In contrast, mixing SQ3 with HN3 (generated from HCl and NaN3) did not lead to an obvious change in its signal. These observations suggested that the catalyst mainly activates the sulfoxonium ylide partner. | ||
| Scheme 5 Mechanistic studies. (a) Catalytic activity of SQ3-Me2. (b) KIE experiment. (c) Correlation between product er value and catalyst loading. (d) Linear correlation between product and catalyst ee. (e) Proposed mechanism. (f) Computed geometries of the transition states for azide substitution by employing a simplified SQ3 catalyst. | ||
 | ||
| Fig. 1 NMR study of the reaction. | ||
We also carried out a one-pot competition experiment between equal amounts of PhCO2H and PhCO2D.26c With substoichiometric substrate 1e, the reaction did not exhibit a primary kinetic isotope effect. Since the product enantiopurity was stable in the presence of PhCO2H, indicating H/D exchange after product formation was not responsible for this result. Thus, protonation may not be the rate-determining step, which is distinct from the previous S–H insertion (Scheme 5b).16a We believe that the acidity and nucleophilicity of HN3 may have an impact to this observation. Furthermore, an obvious correlation of product enantiomeric excess with catalyst loading was observed, which is consistent with the presence of background reaction (Scheme 5c). This result may also suggest that the catalyst functions prior to the enantiodetermining substitution step (dynamic interconversion).27 Finally, the product ee value showed a linear relation to the catalyst ee, suggesting only one catalyst molecule operates in the enantiodetermining transition state (Scheme 5d).26d
Based on the above results and the typical HBD activation modes as well as DFT computations (Scheme 5e and Fig. S1 in the ESI for details†),16a,20,21,28 we proposed a mechanism for the reaction. The squaramide catalyst initially activates the sulfoxonium ylide with the double N–H motifs. Further reversible protonation of the resulting intermediate A by the in situ generated HN3 leads to ion pair B as a diastereomeric mixture. The azide anion also has hydrogen bond interactions with the squaramide catalyst (Scheme S1†).29 The reversible feature of this step allows (R)-B and (S)-B to rapidly equilibrate with each other, thus epimerizing the α-stereogenic center. Next, the azide substitution takes place to deliver the enantioenriched product through dynamic kinetic resolution. This step is both rate-determining and enantiodetermining. The preference for transition state S-TS2 can be attributed to the hydrogen-bond interactions between the N–H motifs of SQ3 and azide (Scheme 5f, N⋯H1 = 1.91 Å, N⋯H2 = 1.92 Å), while there is only one N–H⋯N interaction in R-TS2 (N⋯H = 1.89 Å). On the other hand, the steric repulsion between azide and the carbonyl moiety of the substrate in R-TS2 leads to higher activation energy. Finally, the catalyst is regenerated, which rests in the DMSO·SQ3 adduct form as indicated by the doublet signal at ∼2.4 ppm (Fig. 1 and also the ESI† for details). This mechanistic rationale agrees with all the experimental results.
We were also curious about whether we could mimic the last step using a preformed sulfoxonium chloride salt. Indeed, mixing substrate 1a with a stoichiometric amount of HCl (aq.) resulted in rapid and clean formation of sulfoxonium salt C within 5 min, which was characterized by 1H and 13C NMR (Fig. 2a, more details in the ESI†). Further addition of ent-SQ3 and NaN3 at −15 °C led to the desired product 2a in 70% yield and 89% ee.29 Indeed, this reaction worked equally well if all the materials (HCl, NaN3, ent-SQ3, and 1a) were added in one portion, regardless of the order of addition. Some selected examples were performed for comparison with the standard protocol, which gave almost the same results (Fig. 2b). The high enantioselectivity achieved here was remarkable, considering that the strong acid HCl could potentially compete with the catalyst to facilitate the racemic background reaction. This new protocol clearly provided a simplified alternative to the standard reaction with a cheaper azide and proton source. Next, we monitored this reaction progress to check whether it proceeded as expected by direct ion-exchange through phase-transfer. To our surprise, salt C immediately reversed to 1a upon addition of NaN3 based on in situ1H NMR study (Fig. 2c). Subsequent addition of SQ3 simply permitted the reaction to proceed slowly to the product, similar to the standard protocol. This result further confirmed that protonation of the sulfoxonium ylide is reversible and salt C is just a combined source of the sulfoxonium ylide and proton. The comparable enantioselectivity between these two protocols also suggested that PhCO2H and TMSN3 do not play any additional roles (e.g., substrate activation or catalyst binding for asymmetric induction) other than the HN3 source, consistent with the observations during condition optimization where different acids did not influence enantioselectivity.
 | ||
| Fig. 2 The use of HCl and NaN3 for the asymmetric azidation reaction. (a) In situ formation of the sulfoxonium chloride salt and their azidation reaction. (b) Selected scope of the azidation protocol with HCl and NaN3. (c) NMR monitoring the formation and azidation reaction of the sulfoxonium chloride salt. | ||
Conclusions
In summary, catalytic asymmetric azidation provides efficient access to enantioenriched α-azido carbonyl compounds. However, known approaches are limitedly available and developed with drawbacks (e.g., all limited to quaternary stereocenters, mostly for 1,3-dicarbonyls). Here we have developed the first catalytic asymmetric azidation for the efficient synthesis of α-azido ketones bearing a labile tertiary stereocenter by formal asymmetric H–N3 insertion of the versatile α-carbonyl sulfoxonium ylides. This mild, base- and metal-free organocatalytic approach represents an extraordinary complement to the powerful metal-catalyzed asymmetric H–X insertion of diazocarbonyl compounds, which remains incapable of H–N3 insertion. The proper choice of a superb chiral squaramide catalyst, together with careful optimization of other parameters, overcame many disfavored factors (e.g., background reaction, product labile chirality) and resulted in excellent efficiency and enantiocontrol. Control experiments and in situ NMR studies suggested that the reaction proceeds via reversible activation and protonation of the sulfoxonium ylide followed by rate- and enantiodetermining azide substitution via dynamic kinetic resolution. Our mechanistic curiosity related to the use of a preformed sulfoxonium salt also led to the discovery of a simplified protocol with a cheaper HN3 source.Data availability
All experimental data is available within the article and the ESI.†Author contributions
W. Guo: conceptualization, methodology, investigation, data curation, and wring-original draft. S. Li: methodology, investigation, data curation, and wring-original draft. F. Jiang: resources. J. Sun: conceptualization, supervision, writing-review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
inancial support was provided by National Natural Science Foundation of China (91956114), Hong Kong Research Grants Council (16303420, 6309321), Innovation and Technology Commission (ITC-CNERC14SC01), Jiangsu Key Laboratory of Advanced Catalytic Materials and Technology (BM2012110), Jiangsu specially appointed professors program, Shenzhen Science and Technology Innovation Committee (JCYJ20200109141408054), and the start-up funding to W. G. by Changzhou University (ZMF21020030). We thank Prof. Lijuan Song for helpful discussion on DFT results. We also thank the Analysis and Testing Center, NERC Biomass of Changzhou University for the assistance in NMR analysis.Notes and references
- Reviews on the utility of organic azides:
(a) S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef PubMed
; (b) S. Bräse and K. Banert, Organic Azides: Synthesis and Applications, Wiley-VCH, Weinheim, 2009 CrossRef
- Application in chemical biology, drug discovery, and material science:
(a) H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS PubMed
- Application in organic synthesis:
(a) F. Palacios, C. Alonso, D. Aparicio, G. Rubiales and J. M. de los Santos, Tetrahedron, 2007, 63, 523–575 CrossRef CAS
- Reviews on α-azido carbonyls:
(a) T. Patonay, K. Kónya and É. Juhász-Tóth, Chem. Soc. Rev., 2011, 40, 2797–2847 RSC
- Selected utility of α-azido ketones:
(a) J. Vantikommu, S. Palle, P. S. Reddy, V. Ramanatham, M. Khagga and V. R. Pallapothula, Eur. J. Med. Chem., 2010, 45, 5044–5050 CrossRef CAS PubMed
- Recent reviews on the synthesis of azides:
(a) P.-G. Ding, X.-S. Hu, F. Zhou and J. Zhou, Org. Chem. Front., 2018, 5, 1542–1559 RSC
- Asymmetric electrophilic azidations:
(a) Q.-H. Deng, T. Bleith, H. Wadepohl and L. H. Gade, J. Am. Chem. Soc., 2013, 135, 5356–5359 CrossRef CAS PubMed
- Asymmetric oxidative nucleophilic azidations:
(a) M. Tiffner, L. Stockhammer, J. Schörgenhumer, K. Röser and M. Waser, Molecules, 2018, 23, 1142–1150 CrossRef PubMed
- Asymmetric SN2X approaches:
(a) X. Zhang, J. Ren, S. M. Tan, D. Tan, R. Lee and C.-H. Tan, Science, 2019, 363, 400–404 CrossRef PubMed
- Asymmetric radical approaches:
(a) L. Wu, Z. Zhang, D. Wu, F. Wang, P. Chen, Z. Lin and G. Liu, Angew. Chem., Int. Ed., 2021, 60, 6997–7001 CrossRef CAS PubMed
- For selected examples of non-azidation approaches (without carbon-azide bond formation in the enantiodetermining step):
(a) N. S. Chowdari, M. Ahmad, K. Albertshofer, F. Tanaka and C. F. Barbas, Org. Lett., 2006, 8, 2839–2842 CrossRef CAS PubMed
- For selected examples from enantioenriched precursors, see:
(a) K. Shibatomi, Y. Soga, A. Narayama, I. Fujisawa and S. Iwasa, J. Am. Chem. Soc., 2012, 134, 9836–9839 CrossRef CAS
- Potential instability and over-azidation issues:
(a) J. H. Boyer and F. C. Canter, Chem. Rev., 1954, 54, 1–57 CrossRef CAS
-
(a) S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365–1377 CrossRef CAS PubMed
- Reviews of sulfur ylides:
(a) A. C. B. Burtoloso, R. M. P. Dias and I. A. Leonarczyk, Eur. J. Org. Chem., 2013, 2013, 5005–5016 CrossRef CAS
- Pioneering studies:
(a) P. B. Momo, A. N. Leveille, E. H. E. Farrar, M. N. Grayson, A. E. Mattson and A. C. B. Burtoloso, Angew. Chem., Int. Ed., 2020, 59, 15554–15559 CrossRef CAS
- For our efforts, see:
(a) W. Guo, Y. Luo, H. H. Y. Sung, I. D. Williams, P. Li and J. Sun, J. Am. Chem. Soc., 2020, 142, 14384–14390 CrossRef CAS PubMed
- For synthesis of racemic α-carbonyl azides through a one pot fluorination/azidation sequence of ketosulfoxonium ylide: D. P. Day, J. A. M. Vargas and A. C. B. Burtoloso, J. Org. Chem., 2021, 86, 12427–12435 CrossRef CAS
- For In situ generation of HN3: D. J. Guerin and S. J. Miller, J. Am. Chem. Soc., 2002, 124, 2134–2136 CrossRef CAS
- Selected reviews of hydrogen bonding catalysis:
(a) Y. Takemoto, Org. Biomol. Chem., 2005, 3, 4299–4306 RSC
-
(a)
L. G. Mancheño, Anion-Binding Catalysis, Wiley-VCH, 5th edn, 2022 CrossRef
- For a selected review and leading examples of enhancing Brønsted acid reactivity by hydrogen bond donors:
(a) H. Yamamoto and K. Futatsugi, Angew. Chem., Int. Ed., 2005, 44, 1924–1942 CrossRef CAS PubMed
-
SQ3 was first developed by Jacobsen and co-workers: H. Zhang, S. Lin and E. N. Jacobsen, J. Am. Chem. Soc., 2014, 136, 16485–16488 CrossRef CAS PubMed
- For unsuccessful examples in scope study, see ESI† for details.
-
(a) D. J. Ager, I. Prakash and D. R. Schaad, Chem. Rev., 1996, 96, 835–875 CrossRef CAS PubMed
- (a) We also attempted to examine aliphatic keto sulfoxonium ylides, but these sulfoxonium ylides were either unreactive or difficult to prepare.; (b) We also tried the direct click reaction, but it led to erosion in enantiopurity.; (c) The enantiopurity of 2a was stable in the presence of PhCO2H, indicating H/D exchange after product formation was not responsible for this result.; (d) Notably, the product enantiopurity decreased in the presence of some acids and bases, such as CPA1, SQ1 (cinchona alkaloid) and Et3N. This also explained the difficulty in search for a suitable catalyst for this process..
- N. M. Rezayee, V. J. Enemærke, S. T. Linde, J. N. Lamhauge, G. J. Reyes-Rodríguez, K. A. Jørgensen, C. Lu and K. N. Houk, J. Am. Chem. Soc., 2021, 143, 7509–7520 CrossRef CAS
- In order to reduce the computational effort, a simplified SQ3 was employed in which a phenyl group was used in place of 1-pyrenyl group.
- For a recent elegant example about catalytic asymmetric azidation under hydrogen bonding phase-transfer catalysis: J. Wang, M. A. Horwitz, A. B. Dürr, F. Ibba, G. Pupo, Y. Gao, P. Ricci, K. E. Christensen, T. P. Pathak, T. D. W. Claridge, G. C. Lloyd-Jones, R. S. Paton and V. Gouverneur, J. Am. Chem. Soc., 2022, 144, 4572–4584 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2149693. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03552a |
| This journal is © The Royal Society of Chemistry 2022 |