
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe mechanism of Fe induced bond stability of uranyl(V)†
Tonya
Vitova
 *a,
Radmila
Faizova
b,
Jorge I.
Amaro-Estrada
c,
Laurent
Maron
*c,
Tim
Pruessmann
a,
Thomas
Neill
a,
Aaron
Beck
a,
Bianca
Schacherl
a,
Farzaneh Fadaei
Tirani
b and
Marinella
Mazzanti
*b
*a,
Radmila
Faizova
b,
Jorge I.
Amaro-Estrada
c,
Laurent
Maron
*c,
Tim
Pruessmann
a,
Thomas
Neill
a,
Aaron
Beck
a,
Bianca
Schacherl
a,
Farzaneh Fadaei
Tirani
b and
Marinella
Mazzanti
*b
aKarlsruhe Institute of Technology (KIT), Institute for Nuclear Waste Disposal (INE), P.O. 3640, D-76021 Karlsruhe, Germany. E-mail: tonya.vitova@kit.edu
bGroup of Coordination Chemistry, Institut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland
cLPCNO, University of Toulouse, INSA Toulouse 135, Avenue de Rangueil, Toulouse Cedex 31077, France
First published on 5th August 2022
Abstract
The stabilization of uranyl(V) (UO21+) by Fe(II) in natural systems remains an open question in uranium chemistry. Stabilization of UVO21+ by Fe(II) against disproportionation was also demonstrated in molecular complexes. However, the relation between the Fe(II) induced stability and the change of the bonding properties have not been elucidated up to date. We demonstrate that U(V) – oaxial bond covalency decreases upon binding to Fe(II) inducing redirection of electron density from the U(V) – oaxial bond towards the U(V) – equatorial bonds thereby increasing bond covalency. Our results indicate that such increased covalent interaction of U(V) with the equatorial ligands resulting from iron binding lead to higher stability of uranyl(V). For the first time a combination of U M4,5 high energy resolution X-ray absorption near edge structure (HR-XANES) and valence band resonant inelastic X-ray scattering (VB-RIXS) and ab initio multireference CASSCF and DFT based computations were applied to establish the electronic structure of iron-bound uranyl(V).
Introduction
A deep understanding of the stability and mobilization behavior of uranium (U) in the environment is of high importance for the implementation of remediation strategies at contaminated sites resulting from uranium mining and for developing safety cases for the geological disposal of nuclear waste.1–3 Understanding bonding interactions for uranium and more generally for actinides is also of upmost relevance for applications in medicine and in nuclear energy production.4,5 The early actinide (An) elements (thorium (Th) – plutonium (Pu)) have complex and fascinating chemical properties that are still not well understood and a forefront research topic in actinide science. Due to the heavy nuclei, relativistic effects significantly influence the electronic structure of the Ans and result in an increased radial extension of the 5f valence orbitals compared to 4f orbitals allowing the 5f electrons to actively participate in chemical bonding of Ans up to plutonium (Pu).6–14 Uranium (U) is the actinide element with the largest economic and environmental importance. Uranium is most stable and abundant in nature in the oxidation states +IV and +VI and uranium redox chemistry is of the upmost interest due to the relatively high environmental solubility and mobility of U(VI) compared to U(IV). The U(V) and U(VI) oxidation states tend to form short covalent axial trans dioxo bonds often referred to as uranyl (UO21+ = U(V)-yl and UO22+ = U(VI)-yl). The electronic structure of the U(VI)-yl moiety has been the subject of intensive study,15–17 but less reports are available on U(V)-yl18–21 because uranyl(V) species are much less stable in aerobic aqueous conditions than uranyl(VI) species and their synthetic preparation is not trivial.22–24 Only one natural uranium mineral, wyartite (CaU(V)(UO2)2(CO3)O4(OH)·7H2O) was so far reported to contain U(V).25,26 However, uranyl(V) species have been identified during the U(VI) reduction by Fe(II)-bearing minerals such as mica27 or magnetite ([Fe2+(Fe3+)2O4])28–32 or in the bacteria-mediated reduction.33 The Vitova group revealed that U(V), when incorporated in magnetite and bound by Fe in the second coordination sphere, is stable also under oxic conditions.31 However, these systems are highly disordered and often transient making detailed structural studies challenging. Only three examples of synthetic heteropolymetallic molecular complexes presenting a UO2+⋯Fe2+ interaction have been reported so far.34–36 Such well-characterised, crystalline compounds are very valuable for investigating how Fe influences the electronic and geometric structure of U(V)-yl and the covalency as well as stability of the U–O axial and U–O/U–N equatorial bonds. Recent studies from the Mazzanti group showed that the presence of Fe2+ bound to the uranyl(V) oxo group leads to increased stability of a UO2+ Schiff base complex with respect to proton induced disproportionation. In addition, previously reported35 cyclic voltammetry studies showed an increased stability of the +V oxidation state compared to +VI and +IV oxidation state for the Fe–U(V)-yl compared to the K–U(V)-yl. Both reduction and oxidation of the uranyl(V) cation are more difficult in the presence of Fe(II). This gives a quantitative measure of the intrinsic stabilization of the U(V)-yl oxidation state by Fe(II) independently of the role of the capping ion in the formation of polynuclear disproportionation intermediates or in proton induced disproportionation.37 The stabilizing effects of Fe2+ binding on U(V)-yl oxo bonding were previously investigated in synthetic studies,33,35 but the origins of such effects were not elucidated and the electronic structure of these complexes was never computed or spectroscopically characterized. These studies are crucial to gain insight on the role that iron binding plays in stabilizing pentavalent uranyl species in the environmental Fe2+-mediated reduction of uranium(VI). Another important factor in the prevalence of uranyl(V) species in the environmental migration of uranium is coordinating ligands which can stabilize U(V). It has been known for more than 40 years that uranyl(V) can be stabilized in water by high carbonate concentrations yielding the uranyl(V) carbonate complex [UO2(CO3)3]4−. However, in spite of the fact that several uranyl(V) compounds stable in organic solvents have been isolated,22,34,37–48 the first example of a well-defined and crystallographically characterized complex of uranyl(V) which is stable in anaerobic water was prepared as recently as 2018, using the pentadentate aminephenolate ligand dpaea (H2dpaea = bis(pyridyl-6-methyl-2-carboxylate)-ethylamine).24 In order to elucidate the parameters leading to the stabilization of uranyl(V) in water by the dpaea ligand, it is important to analyse the electronic effects of supporting ligands on the U(V)-yl oxo bonding.Here, we present a spectroscopic and computational study of the impact of the supporting ligand and of Fe2+ on the bonding properties of the U(V)-yl moiety and of the U(V) – equatorial ligand bonds. Moreover, we correlate changes in bonding to the U(V) stability. For the first time we characterize the bond covalency in uranyl(V) and uranyl(VI) molecular compounds using a combination of U M4,5 edge High energy Resolution X-ray Absorption Near Edge Structure (HR-XANES) and U Valence Band – Resonant Inelastic X-ray Scattering (VB-RIXS) experimental techniques, which probe the U unoccupied and occupied 5f and 6p/7p valence states with high energy resolution and are sensitive to changes of the oxidation states and covalency of the U axial and equatorial chemical bonds.19,49–54
Results and discussion
Our systematic approach includes the variation of the U oxidation state – U(VI), U(V), U(IV) – taking care of minimizing the modification of the coordination sphere. This series is explored for two pentadentate ligands, i.e. Mesaldien2− (H2Mesaldien = N,N′-(2-aminomethyl)diethylenebis (salicyl-imine)) and dpaea2− (H2dpaea = bis(pyridyl-6-methyl-2-carboxylate)-ethylamine) which lead to uranyl(V) complexes stable in organic solvents (Mesaldien2−) or in water (dpaea2−). Moreover, complexes with one or two Fe atoms bound to the axial O atoms of [UVO2(Mesaldien)]− are investigated allowing to detect electronic and geometric changes induced by Fe on the U(V)-yl bond.Molecular structures
The complexes [UVIO2(Mesaldien)] (1), {[UVO2(Mesaldien)]K}n (2),43 [{Fe(TPA)Cl}{UO2(Mesaldien)}{Fe(TPA)Cl}]I, (4),35 [UIV(Mesaldien)I2](5),43 [UVIO2(dpaea)](6), [K(2.2.2.cryptand)][UVO2(dpaea)] (7), [UIV(dpaea)2] (8)24 were prepared in microcrystalline form as previously described. We also prepared and crystallographically characterized the Fe(II)–U(V) complex [Fe(TPA)(Py)UVO2(Mesaldien)]I (3) which was not previously reported. The complex 3 allows to study the effect of Fe binding to the U–O(yl) group on the stability of the uranyl(V) complex. Complex 3 was prepared by reacting complex 2 with 1 equiv. of [Fe(TPA)l2] in pyridine. The schematic structure of complexes 1–8 is given in Fig. 1 and the X-ray determined solid-state molecular structures are shown in Fig. S3.†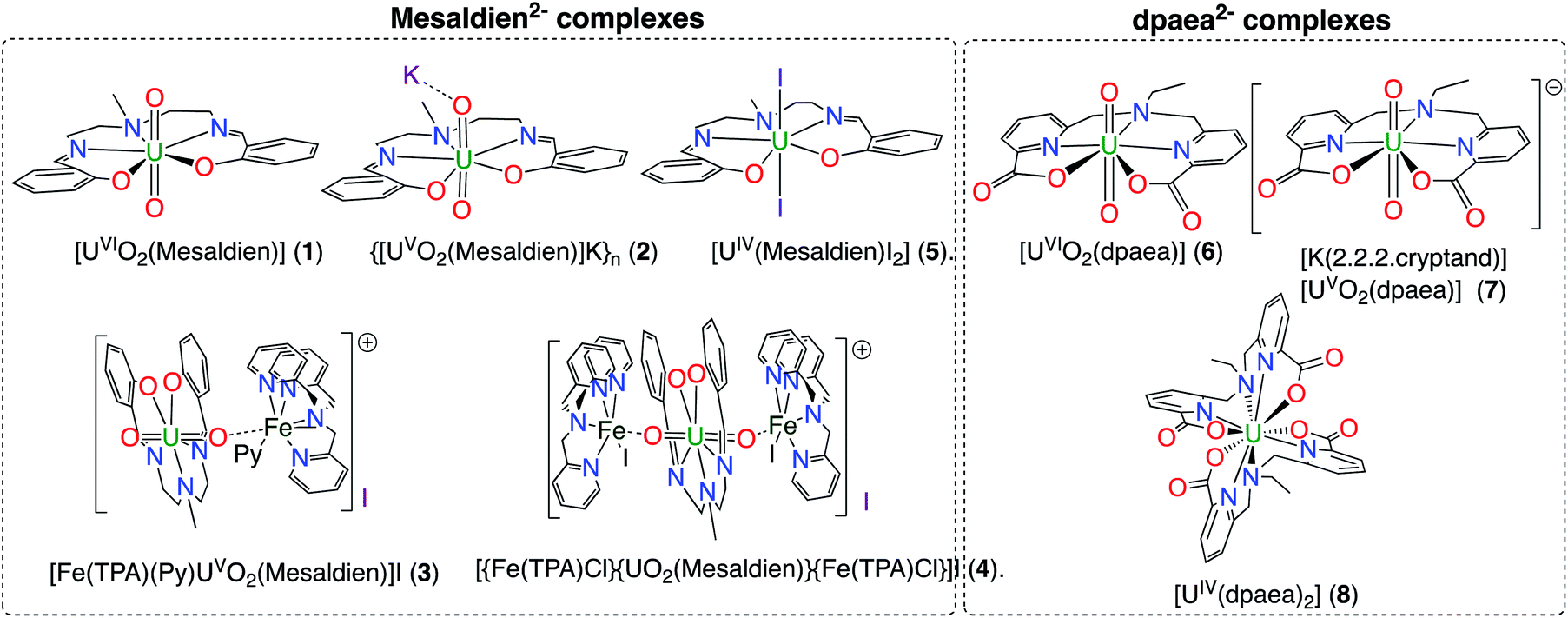 | ||
| Fig. 1 Schematic structures of complexes 1–8. | ||
DFT and CASSCF computations
To gain insights on the bonding and on the electronic structure of the complexes 1, 2, 3, 6 and 7, a computational analysis was carried out at the DFT level (B3PW91) but also at the CASSCF level. The optimized geometries are in good agreement with the experimental ones. Focusing on the U(VI) complexes (1 and 6, singlet spin state), the U–O(yl) bonds are correctly reproduced computationally with a maximum deviation of 0.01 Å. In complex 1, the equatorial bonds (U–O and U–N(imine) are also correctly reproduced with a maximum deviation of 0.05 Å. For complex 6, the U–O bonds are correctly reproduced with a deviation of 0.05 Å. For both complexes, the donor–acceptor interaction with amine or pyridine is slightly less accurate deviation close to 0.08 Å) but remains acceptable. The nature of the U–O(yl) bonds was therefore analyzed using Natural Bonding Orbital Analysis (NBO). In complex 1, NBO indicates the presence of two triple bonds U–O, in line with a classical uranyl complex. These bonds are found to be strongly polarized toward O (79, 79, 78%). The bonding implies that the overlap between a 6d/5f orbital (roughly 50–50) with either pure 2p (π-type orbital) or hybrid sp (σ orbital). A similar situation is found for complex 6, with the presence of two strongly polarized toward O (79, 79, 79%) triple bonds. The covalency in these bonds was thus analyzed using the Wiberg Bond Indexes that relate to the overlap population.The U–O(yl) WBI in complexes 1 and 6 are again quite similar 2.17 and 2.18, respectively, indicating the presence of covalent interactions. The calculations show no evidence of any substantial difference in the covalency of the U–O(yl) bonds in these two complexes. However, in complex 1, the NBO indicates the presence of polarized bonds between U (10%) and the two oxygen and three nitrogen (90%) atoms whereas no bonds could be identified by NBO in complex 6. The stronger interactions found in the equatorial plane for complex 1 may explain why experimental data, discussed below, point to a more covalent U–O(yl) interaction in 6 than in 1.
Like their U(VI) analog, the optimized geometries of complexes 2 and 7 (doublet spin state) compared well to the experimental ones. The U–O(yl) bond in complex 2 is reproduced within 0.04 Å whereas for complex 7, the accuracy is up to 0.02 Å. The increase of the U–O(yl) and equatorial bond lengths by up to 0.1 Å is reproduced. At the NBO level, the U–O(yl) bonds in complex 2 are described as singly polarized σ bonds (80% contribution from O) in line with a reduced covalency with respect to complex 1. The latter is highlighted by the WBI which drops down to 1.70. This value indicates some energy-driven interactions between O (2p) and U (6d/5f) since the overlap is decreasing according to the NBO (single bond found). The description of the U–O(yl) bond is very different in complex 7. Indeed, NBO shows the presence of two triple U–O bonds (two bonds found at the second order to describe the axial U–O bonds). These bonds are strongly polarized toward O (78, 82, 82% for the triple bond and 82, 82% for the double). This is in line with a less covalent complex than complex 6 but also with a stronger covalent interaction in complex 7 than in complex 2. That is related to the difference of covalency in the parent complexes 1 and 6 found by the experiments. The 2.0 value of the WBI U–O(yl) bonds is also in line with this statement. Indeed, the CASSCF calculations show four different states close in energy for the ground state of complex 6, a triplet at 0 eV and three singlets lying at 0.04, 0.05 and 0.06 eV. The previously reported solution 1H NMR data for complex 6 show a classical diamagnetic spectrum in agreement with the presence of a U(VI) complex.15 However, the CASSCF calculations suggest that reduction of the U(VI) complex 6 to its U(V) analogue is easily accessible. This is a striking difference with complex 1. The CASSCF results found for complex 7 indicate that the ground state is a doublet with another doublet excited state (multireference) lying 0.09 eV higher in energy. Therefore, the ground state has a U(V) character and the first excited state brings some U(IV) character. Thus, complexes 2 and 7 might have very different behavior in solution and/or reactivity due to these very different electronic configurations. Notably, reduction of U(VI) to U(V) and U(V) to U(IV) is favored by the electron configuration of the dpaea complexes compared to the Mesaldien complexes.
The optimized geometry of 3 is again in good agreement with the experimental one. The U–O(yl) bonds are correctly reproduced (1.91 vs. 1.93 Å and 1.82 vs. 1.84 Å) as well as the O–Fe (0.04 Å) and all the bonds in the equatorial plane (0.03 Å). At the NBO level the U–O(Fe) bond has no overlap contribution (and is therefore mainly an electrostatic interaction) whereas the U–O(yl) trans is a triple bond strongly polarized toward O (80, 80, 75%), similar to the one found in complex 1. The U–O(Fe) WBI is 1.50 whereas the U–O(yl) one is 2.10. The former is even lower than in complex 2 (1.70), in line with a decrease in covalency, whereas the latter is similar to that found in 1 (2.17) and therefore higher than in 2. However, the decrease of covalency in the U–O(Fe) bond is due to the formation of a covalent Fe–O bond (WBI of 0.5). The decrease in the overlap contribution of uranium in the U–O(Fe) bonds also allows stronger interaction with the ligand in the equatorial plane (U–O WBI of 0.68 in 3vs. 0.54 in 2). The coordination of iron is thus stabilizing the U(V) complex by slightly decreasing the covalency in the U–O(Fe) in order to form a Fe–O bond and allowing better interaction with the equatorial ligand. The NBO analysis indicates U(V) complex and a low spin Fe(II). The latter is further corroborated by the CASSCF calculations where the ground state is a doublet (monoreference). The first excited state, of a U(VI)–Fe(III) character, is 1.3 eV higher in energy. This is different from what is found for complex 2 where the first excited state was more than 4.0 eV higher in energy.
Uranium M4 and M5 edge HR-XANES
The U M4,5 edge High Energy Resolution X-ray Absorption Near Edge Structure (HR-XANES) probes predominantly the unoccupied 5f valence orbitals of U and provides detailed insights into the U bonding properties. The characterisation capabilities of the spectroscopic technique are summarized in Fig. 2a. It is a tool for evaluating the electron density on U and the covalency of the U(V)-Oyl chemical bonds. This spectroscopic technique has been applied previously to study binding differences between uranyl(V) and uranyl(VI) molecular compounds.19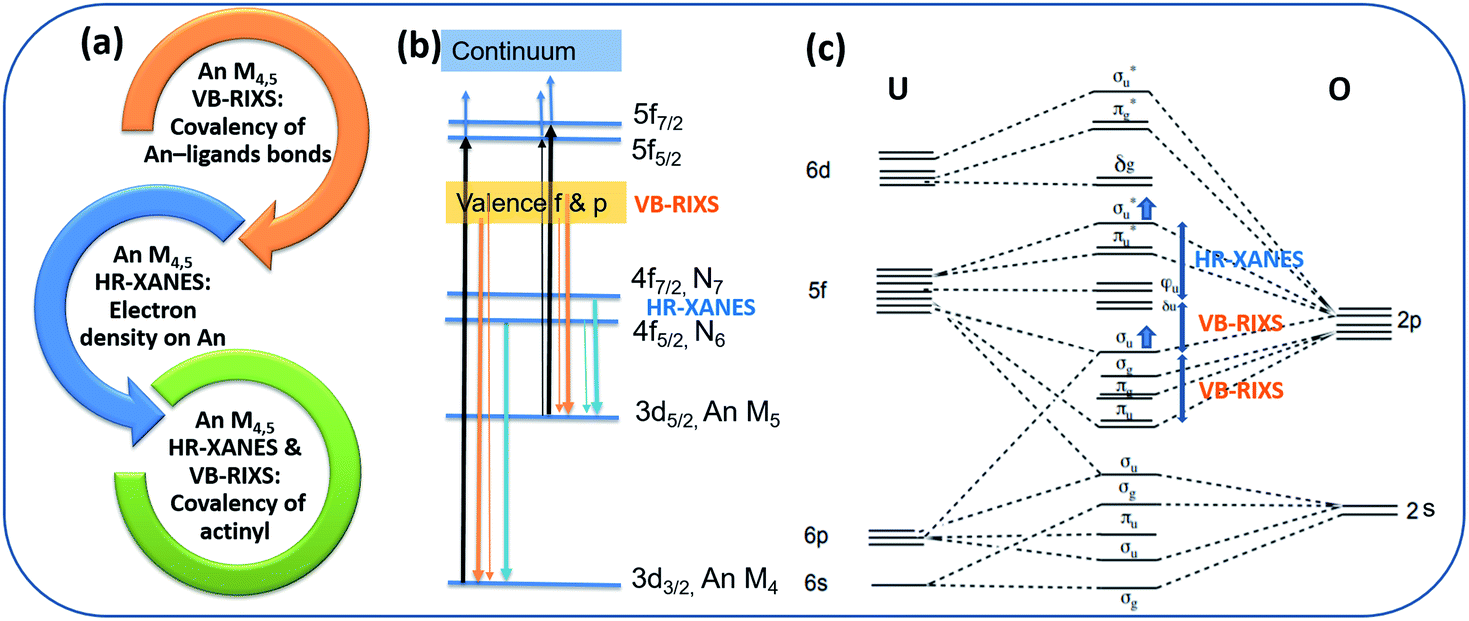 | ||
| Fig. 2 (a) Key characterization capabilities of the An – ligand bond properties of the An M4,5 HR-XANES and VB-RIXS spectroscopy tool. (b) The actinide (An) atomic orbitals and electronic transitions relevant for An M4,5 HR-XANES and VB-RIXS are marked. The electronic transitions follow the selection rule ΔJ = 0, ±1 (Δl = ±1). The probability for the transitions is coded in the thickness of the lines – thick line indicates high probability for transition, thin line indicates low probability for transition. (c) The molecular orbital scheme of uranyl (U(VI)/(V)O22+/1+). The one additional electron in the LUMO is omitted for U(V). The “pushing from below” effect is marked with two blue arrows. The relative energy shifts between specific unoccupied and occupied orbitals measured with U M4,5 edge HR-XANES and VB-RIXS, respectively, are noted with double arrows. | ||
The basic principle of the U M4,5 edge HR-XANES technique includes excitation of U 3d3/2 (M4 edge) or 3d5/2 (M5 edge) electrons to unoccupied orbitals with predominant 5f character (cf.Fig. 2b, black arrows) followed by emission (4f → 3d) (cf.Fig. 2b, blue arrows), both governed by selection rules (ΔJ = 0, ±1). Multiplet effects caused by electron–electron/hole interactions can in principle play a substantial role for the broadening/splitting of spectral peaks for materials with 5fN (N > 0) ground state electronic structure configuration. However, different computational approaches verified that for energy resolution in the order of 1 eV relevant for the An M4,5 edge HR-XANES, the ligand field has the largest influence on the splitting of the 5f orbitals for actinyls and the multiplet effects mainly add broadening to the spectral peaks.51,53,55–57
The linear structure of the uranyl molecule leads to the U valence orbitals being most usefully described in terms of their σ, π, δ and ϕ character with respect to rotation about the axial U(V)/(VI)–O(yl) z axis (D∞h point group symmetry) (cf.Fig. 2c).6,17 The magnitude of the influence of the axial, equatorial ligand fields and the spin–orbit coupling on the splitting of the unoccupied orbitals with 5f character can be ordered as follows: axial field (5fσ*, 5fπ*) > spin–orbit > axial field(5fδ, 5fϕ) + equatorial field.58 The highest occupied σu bonding and the respective unoccupied σu* (σ*) antibonding orbital are a mixture of a U hybrid orbital containing mainly U 5f, 6p and axial oxygen (Oax) 2p orbitals, which are oriented along the O–U–O axis, therefore they are naturally very sensitive to changes of bond lengths and distribution of charge along the U–Oax bond. Due to interaction of the valence σu bonding and the 6p semi-core orbitals, the σu shifts to higher energy and this coincides with an increased mixing of 5f and 2p orbitals in the σu orbital, i.e. with increased covalency of the U–Oax bond. This effect is known as “pushing from below”. At the same time the σ* orbital is “pushed” to higher energies, whereas the largely non-bonding δ/ϕ orbitals remain at similar energies. Note that also for the complexes studied here the U δ/ϕ orbitals remain substantially non-bonding. It is evident from Fig. S32† where no mixing of U f-DOS with U and N s and p DOS is visible in the first 2 eV above the Fermi energy.
For the first time we compare experimental and computed spectra of both U M4 and M5 edge HR-XANES spectra of actinyl(V) compounds and their actinyl(VI) analogues for complexes 1, 2 and 3 (cf.Fig. 3). The U M4 and M5 edge computed and experimental spectra are in a very good agreement. The An M4,5 edge HR-XANES are the only experimental techniques allowing to measure for all actinyls(V)/(VI) the energy shifts between the δ/ϕ and π* (in some cases) and δ/ϕ and σ* orbitals from one spectrum without interference with transitions to other orbitals.17,50,56,59 These orbitals manifest as three identifiable peaks in HR-XANES spectra of actinyls, commonly referred to as A (δ/ϕ), B (π*) and C (σ*) peaks (cf.Fig. 3b). We previously illustrated that the change of this energy shift (between δ/ϕ and σ*, see top blue double arrow in Fig. 2c) can be used as a tool to measure the magnitude of the “pushing from below” effect and thus the variations of the covalency of the U–Oax bond (cf.Fig. 3b).19,56 The energy shift between δ/ϕ and σ* (A–C), decreases in the order U(VI) (1) > U(V) (2) > U(V) Fe (3) in both experimental and theoretical data and is about 1 eV smaller for the U M5 compared to M4 edge HR-XANES spectra (cf.Fig. 3b and 4). This can be explained by the fact that at U M5 edge excitations to higher energy U δ/ϕ orbitals take place (Fig. 3b). The good agreement between computations and experiments corroborates that the U M4 and M5 edge HR-XANES spectra describe well the ground state electronic structure for U(VI)-yl and U(V)-yl. Relativistic multireference ab initio wave function calculations within the restricted active space verified this notion for uranyl(VI) for both ground and excited state.60 It was, however, discussed whether the A–C shift and the mixing of metal with ligand orbitals, i.e. the bond covalency, decreases for neptunyl(VI) and plutonyl(VI) in the excited state. It was suggested that this results from a combined effect of (1) larger effective charge for Pu(VI) and Np(VI) compared to U(VI) making the 6p electrons more core-like upon excitation and (2) repulsion interaction between the electrons in δ/ϕ (for 5fn, n > 0) and the excited electron in the σ* orbital. However, we can systematically compare the A–C energy shifts for the same An element, uranium, since the former effect (1) is comparable. The latter effect (2) is not confirmed from our data since the A–C shift for the compound 3 decreases and the electron density on U also decreases as illustrated by the energy shift of the U M4 and M5 edge spectra of 3 to higher energy compared to 2 (Fig. 4). Considering the above arguments, we will use here the change of the A–C energy shift as a tool for comparison of bond covalency of the U(VI)–O(yl) and U(V)–O(yl). The energy shift between δ/ϕ and σ* (A–C) decreases in the order U(VI) (1) > U(V) (2) > U(V) Fe (3) in both experimental and theoretical data and this shows that the U(V)/(VI)–O(yl) bond covalency decreases in the same order 1 > 2 > 3.
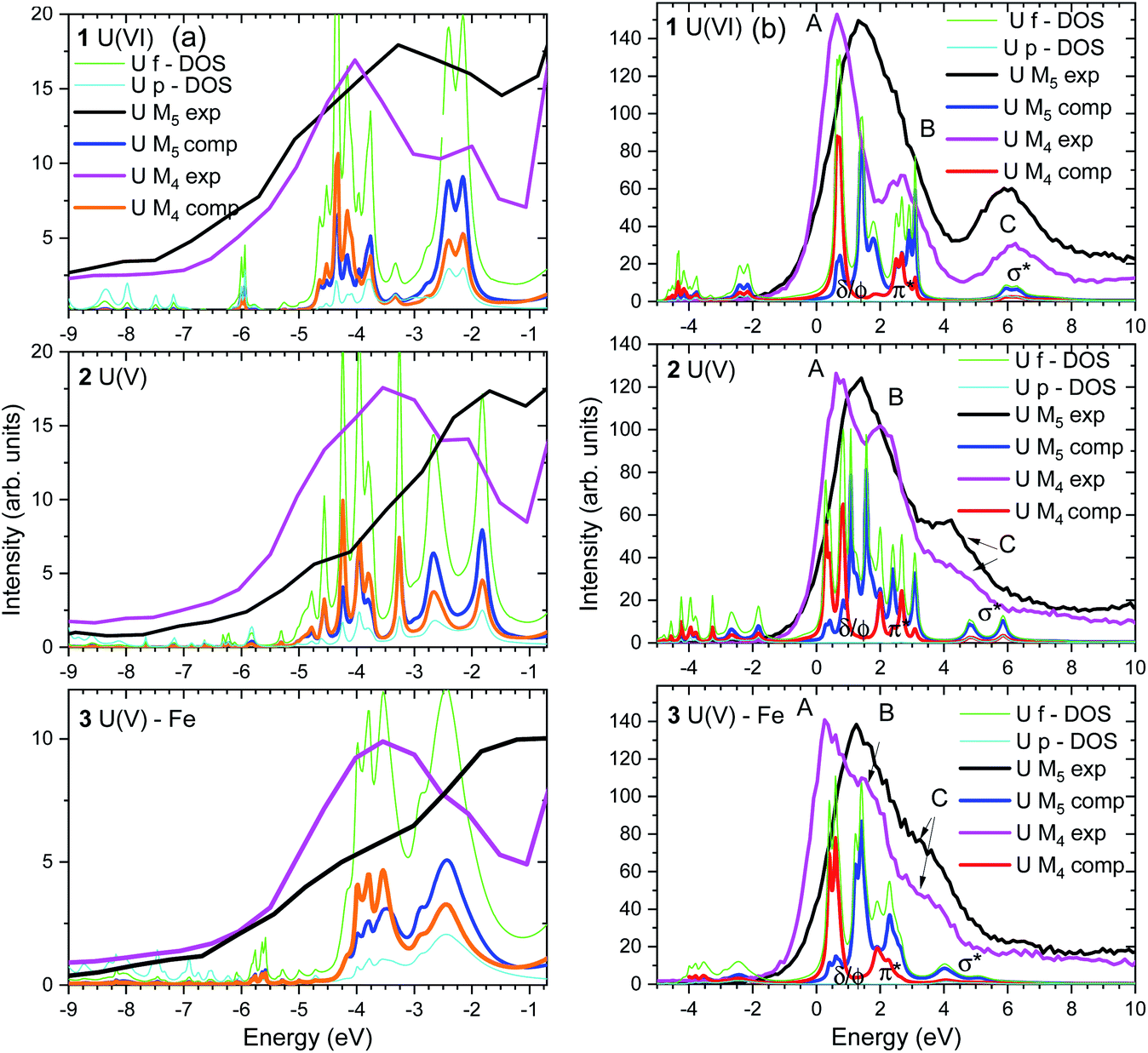 | ||
| Fig. 3 (a) U M4 and M5 edge VB-RIXS and (b) HR-XANES and U f, p – DOS (only computed) spectra of the 1, 2 and 3 compounds experimentally measured and computed with the FDMNES code. The experimental spectra are shifted along the x axis to best match the theoretical spectra. Relative energy shift of 1 eV is applied between the U M4 and M5 edge HR-XANES, and U M4 and M5 edge VB-RIXS spectra. | ||
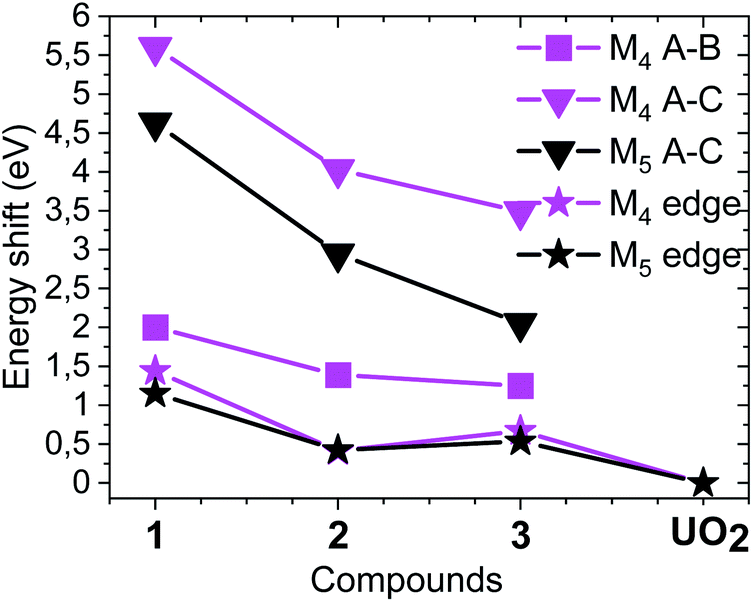 | ||
| Fig. 4 Energy shifts between A, B and A, C spectral peaks as well as the relative energy shifts of the M4 and M5 absorption edges compared to the spectrum of UO2. The spectra of the compounds are shown in Fig. 3b (cf. Fig. S33† including UO2). | ||
Uranium M4,5 edge VB-RIXS
The U M4,5 edge Valence Band Resonant Inelastic X-ray Scattering (VB-RIXS) spectra probe predominantly the U 5f contribution in the occupied valence bonding orbitals. As a result, it is directly sensitive to the bond covalency of all U chemical bonds. Fig. 2a depicts the main capabilities of the spectroscopic technique. VB-RIXS is also a tool for detecting the uranyl(V) and uranyl(VI) axial bond covalency, similarly to U M4,5 edge HR-XANES. We demonstrate the potential of this technique for in-depth studies of binding properties of uranium, and generally of the actinide elements, here for the first time.The U M4 and M5 edge VB-RIXS spectra for U(VI) (1), U(V) (2) and U(V) Fe (3) are depicted in Fig. 3a and 5. The relevant electronic transitions are depicted in Fig. 2b. U 3d3/2 (M4 edge) or 3d5/2 (M5 edge) electrons are excited to unoccupied orbitals with predominant 5f character (cf.Fig. 2b, black arrows) followed by transitions of 5f and 6p electrons in the occupied valence band to the 3d states (cf.Fig. 2b, orange arrows), both processes are governed by selection rules (ΔJ = 0, ±1). The differences in probability for the transition process is coded in the thickness of the arrow lines in Fig. 2b (e.g. more probable transition is marked with a thicker line).
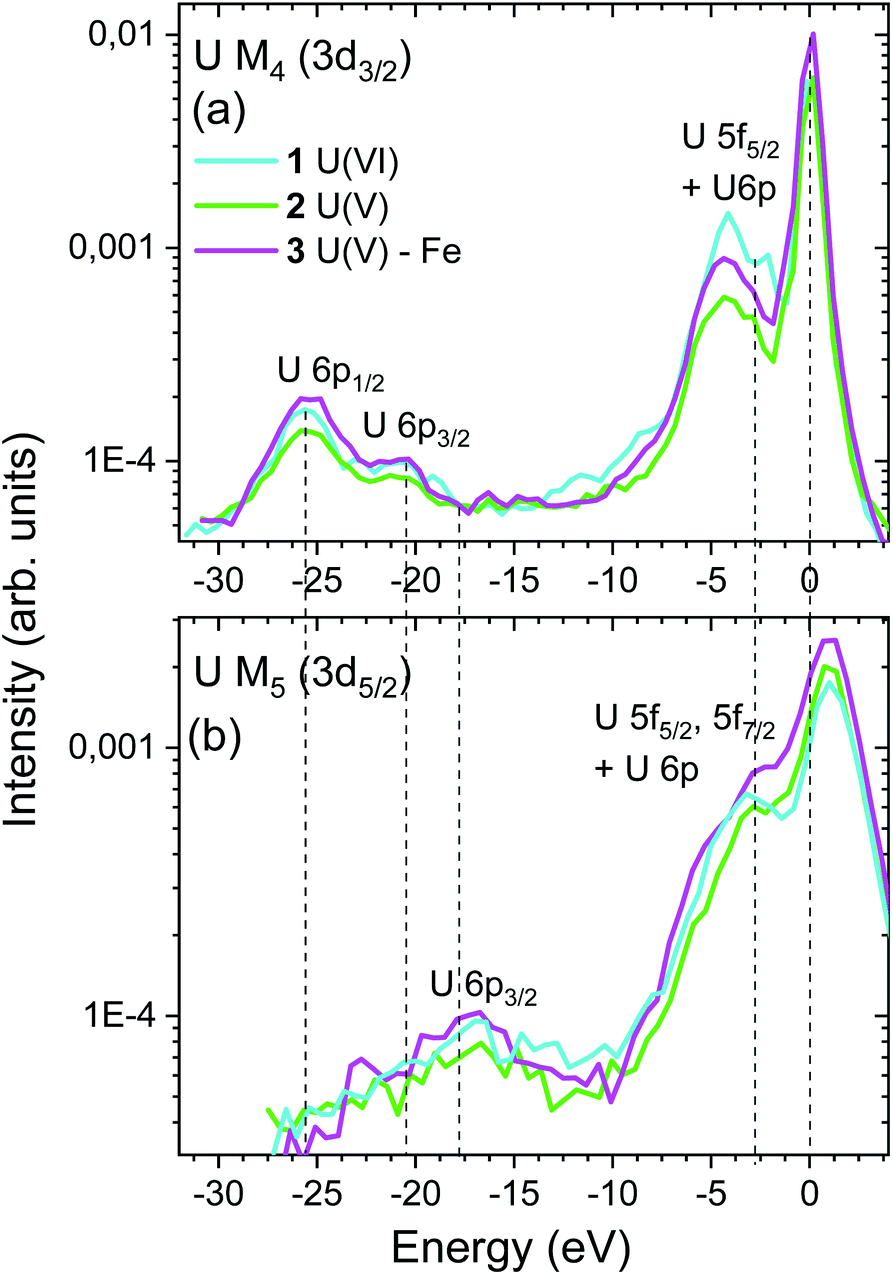 | ||
| Fig. 5 Energy shift of (a) U M4 and (b) M5 VB-RIXS spectra of compounds 1, 2 and 3. The elastic scattering peaks are aligned and shifted to 0 eV and – 1 eV for U M4 and M5 edge VB-RIXS, respectively. The peaks at about – 5 eV measure the U 5f and 6p electron density in the occupied valence band. The peaks at lower energies probe the U 6p electron density in the U semi-core orbitals (P2, P3). | ||
The VB-RIXS spectra provide the following insights into the bonding properties of the U – ligand chemical bond:
(1) The areas of the U M4 and M5 edge VB-RIXS spectra are a relative measure of the U f and p, electron density in the occupied valence band of uranium. As a result, the spectra directly measure the average bond covalency for the chemical bonds between U and all binding atoms in the axial and equatorial plan. In case of ionic bonds, the U f electron density will be transferred to the ligand and no intensity will be visible in the VB-RIXS spectrum.
The integrated intensity of the VB-RIXS spectra follow the trend U(VI) (1) < U(V) Fe (3) < U(V) (2) (cf.Fig. 5); only the peaks below the elastic scattering peak, up to – 10 eV, are considered since they represent the 5f contribution in the occupied valence band; note that the U 6p contribution is small. This result is evidence that the total bond covalency between U and the axial and equatorial ligands is larger for U(V) Fe (3) compared to U(V) (2).
(2) Specifically for the uranyls (valid also for the actinyls), “the pushing from below” effect shifts up the energy of the σu orbital compared to the πu orbital and this shift depends on the degree of covalency of the U(V)/U(VI)–O(yl) bond (cf.Fig. 2a, bottom blue double arrow). The energy shifts between the σu and the πu orbitals can be measured by VB-RIXS, as a result, it is a spectroscopic tool sensitive specifically to the U(V)/U(VI)–O(yl) bond covalency similarly to the A–C shift in the U M4,5 HR-XANES spectra.
The computed U p and f occupied DOS for the 1, 2 and 3 compounds are shown in Fig. 3a and agree well with the experimental data. Note that the U p and f states are completely hybridized. There are two groups of peaks in the computed DOS, well separated for U(VI), corresponding to the πu and σu occupied valence orbitals (peaks at about −4 and −2 eV for U(VI) in Fig. 3a, 5a). Due to selection rules for the electronic transitions leading to different probabilities for transitions (cf.Fig. 2b, thin and thick arrow lines), the VB-RIXS spectra have distribution of intensities. The U M5 edge is more sensitive to the high energy part of the DOS spectrum (σu orbital, Fig. 2c), whereas the U M4 to the part at lower energy (πu orbital, Fig. 2c). The two clearly visible peaks in the U(VI) M4 edge spectrum are barely resolved in the U(V) spectrum and overlap completely in the U(V)–Fe spectrum. This is evidence that the covalency of the U(V)/U(VI)–O(yl) bond decreases in the order U(VI) (1) > U(V) (2) > U(V) Fe (3). The U M5 VB-RIXS spectra are less instructive since they are much broader, as a result of the lower experimental resolution.
(3) The energy shift between the VB-RIXS spectra (peaks up to −10 eV) and the maximum of the elastic scattering peak at 0 eV decreases for U(VI) compared to the two U(V) compounds (cf.Fig. 5). This is a measure of the energy shift between the lowest unoccupied (nonbonding δ/ϕ) and highest occupied orbitals (σu) with U f character and thus a measure of the “pushing from below” effect (cf.Fig. 2c, middle blue double arrow). Considering that the shift is smaller for U(VI) compared to U(V), this is also an indication for higher bond covalency for U(VI) compared to U(V). The small differences between the spectra of the two U(V) compounds do not allow to clearly distinguish between them. Note that U M5 VB-RIXS spectra overlap strongly with the elastic scattering peak preventing reliable data analyses. The energy peaks below – 10 eV in the U M4 and M5 VB-RIXS spectra in Fig. 5 are 6p1/2/6p3/2 → 3d electronic transitions of semi-core 6p electrons (P2, P3), which is a demonstration that the U M4, M5 edge VB-RIXS spectral intensity strictly follows the selection rules for the electronic transitions (cf.Fig. 2b).
The effect of the ligand and the oxidation state on the U bonding properties
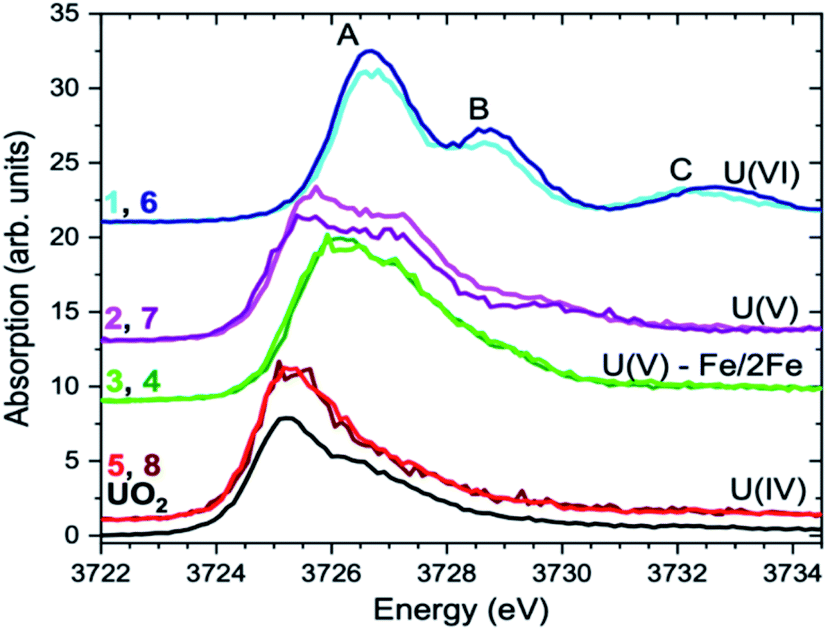 | ||
| Fig. 6 The U M4 edge HR-XANES spectra of 1–8 compounds and the UO2 reference. | ||
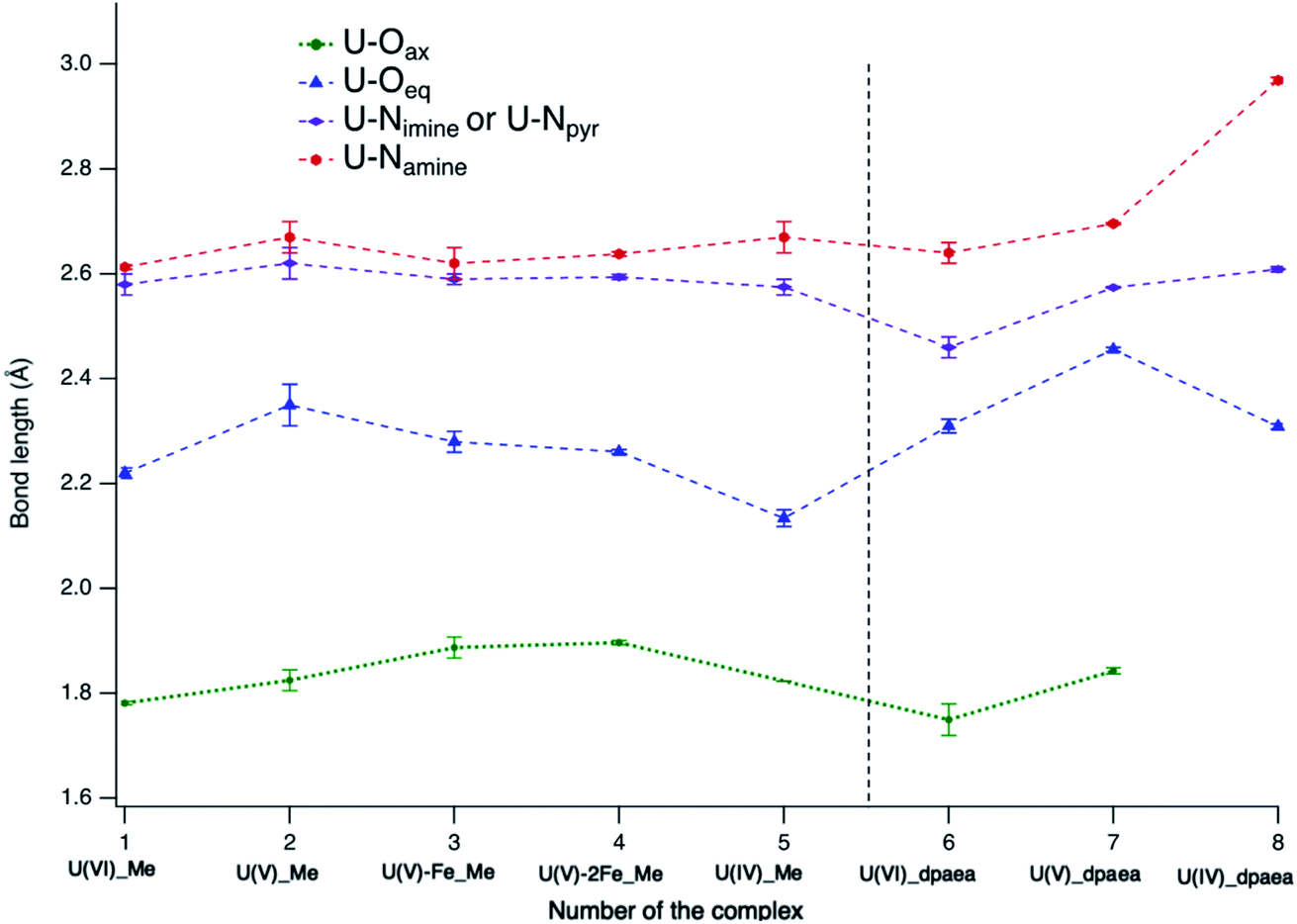 | ||
| Fig. 7 Comparison of the bond lengths in complexes 1–8 obtained from single crystal X-ray diffraction including errors bars. The dotted lines connecting the data points are a useful guide for the eyes. | ||
The effect of Fe on the U bonding properties
The U(V)-yl complexes {[UVO2(Mesaldien)]K}n (2) and [Fe(TPA)(Py)UVO2(Mesaldien)]I (3) differ only in the nature of the bound counter-cation, being mono-cationic K+ for complex 2 and di-cationic Fe2+ for complex 3. The higher ability of the di-cation to reduce the U(V)-yl oxo electron density results in the elongation of both U–O(yl) bonds (1.84(2) Å to 1.935(19) Å and 1.787(1) Å to 1.84(2) Å) and in the consequent shortening of all bonds between U and the equatorial donor atoms from the Mesaldien ligand (cf.Fig. 7).The reduction of electron density on U, caused by the bonding of Fe to the axial O atom, is also manifested in the U M4 and M5 (only for 3) edge HR-XANES spectra of 3 and 4 by an energy shift of the absorption edge by 0.4 ± 0.05 eV to higher energies relative to the spectrum of 2 (cf.Fig. 4 and 6). The same trend is observed for the U L3 edge XANES spectra (cf. Fig. S26†). These findings confirm that part of the electron density is removed from U(V). Furthermore, the energy shifts between A–B and A–C peaks decreases so much that peaks B and C are barely visible (cf.Fig. 4 and 6). This result gives clear experimental evidence that the average covalency of the U–O(yl) bond decreases (cf.Fig. 7, Table S4, EXAFS for 1, 2 and 3 in Fig. S27, Table S7†).The larger integral intensity of the U M4 VB-RIXS for 3 compared to 2 (cf.Fig. 5) illustrates higher total U 5f and 6p electron density in the occupied valence band for 3. These spectra probe both axial and equatorial U(V) bonds. The U M4 edge VB-RIXS and HR-XANES combined results reveal that upon binding of Fe(II) to U(V)-yl, part of the electron density is redrawn from the axial bond and is redistributed to the equatorial U(V) – ligand bond leading to increase of U(V) – equatorial ligand bond covalency.
In addition, the computations reveal (cf. above) that there is a stronger covalent interaction of U(V) with the equatorial ligands for 3 compared to 2. The presence of an additional Fe bound to the second axial O in 4 does not lead to substantial changes of the HR-XANES spectrum signifying that the U electronic structure, similar to the geometric structure (cf.Fig. 6, Table S4†), is not further notably influenced. This result is in accordance with our previous results since also the U(V) stability against reduction was not increased by binding Fe to the second axial O bound to U(V) as demonstrated by the cyclic voltammetry studies.36
Overall the calculations and spectroscopic results (HR-XANES and VB-RIXS) show that binding one Fe(II) atom to U(V)-yl in 3 (U(V)-yl-Fe) leads to reduction of the covalency of the U–O(yl) bond coordinated to Fe but stabilizes and makes the second U–O(yl) bond, the Fe–O and the equatorial bonds more covalent. This significant changes of the electronic structure yield an effective stabilization of the +V oxidation state of U (1) against reduction that we observed by cyclic voltammetry studies and (2) and against proton induced disproportionation.36 The U(V) U M4 edge HR-XANES spectrum of 3 closely resembles the U M4 edge HR-XANES reported in ref. 10 of U(V) incorporated in the structure of magnetite and coordinated by Fe suggesting alike U(V) stabilisation mechanism in natural system.
Conclusions
We presented the first U M4 and M5 edge HR-XANES and VB-RIXS study combined with DFT and CASSCF calculations of U(VI), U(V) and U(IV) complexes with two different O2N3 ligands. We showed that the stability of 7 (U(V)-dpaea) can be explained with strong interaction of U(V) with the equatorial ligand with ionic character. Our results showed that stabilization of U(V) requires strong bonding interactions of U(V) with the equatorial ligands, which can be of covalent or ionic nature. Here we also investigated how changes in the electronic structure upon Fe(II) coordination to the uranyl(V) oxo group result in a different chemical behaviour. The stabilisation effect of Fe(II) on U(V)-yl was previously demonstrated by (1) environmental studies, (2) by protonation reaction stabilising U(V)-yl in presence of Fe2+ against disproportionation, (3) by cyclic voltammetry showing higher redox stability of U(V)-yl in presence of Fe2+.Here, we provided evidence that the binding of Fe(II) to the uranyl oxygen results in the weakening the uranyl bond and in the redistribution of electron density leading to an increase of the bond covalency in the interaction of U(V) with the equatorial ligands. In short, the Fe(II) weakening of the uranyl bond and the redistribution of electron density, is the driving mechanism behind the stabilisation of U(V)-yl upon coordination with Fe(II). The U M4,5 edge HR-XANES technique provides a decoupled measure of two different electronic effects in the same spectrum: reduction of electron density on U (the energy shift of the main edge) and reduction of U–Oax bond covalency (loss of crystal field splitting measured by change of A–C shift). We can exactly see that, not only the charge is removed from U(V) by Fe(II) but also that the U(V)–Oax is less covalent.
We introduced a new spectroscopic instrument, notably the U M4,5 VB-RIXS, for detailed characterisation of the uranium complexes electronic structure and bond covalency which could easily be extended to other actinide elements (cf.Fig. 3a and 5). The U M4,5 VB-RIXS spectroscopic technique probes the U–Oax bond covalency (energy shift between the two peaks). But it also measures the total U 5f + 6p electron density in the occupied valence band and thereby it is sensitive to variations of bond covalency in both equatorial and axial U – ligand bonds. Such a complete picture of the changes of the chemical bonding, electronic and geometric structure upon systematic change of U oxidation state and bonding environment (ligand or effect of Fe2+) demonstrates the high potential of the approach and paves the way for its future applications in actinide science. Specifically, it can be pivotal in understanding the role of the 5f electrons in the covalency of actinide bonding and the interconnection between actinide bond covalency, reactivity and complex stability.
Experimental
The experimental details are provided in the ESI.†Data availability
We will provide the computational and experimental data on a reasonable request.Author contributions
T. V. designed the study, analysed the data, prepared some of the plots and wrote the manuscript; R. F. prepared the compounds, prepared some of the plots and wrote the manuscript; J. I. A. E. performed the DFT and CASSCF calculations ; L. M. designed the study, analyzed the DFT and CASSCF computations, wrote the manuscript; T. P. performed the FDMNES computations and performed some of the experiments; T. N. performed some of the experiments and the analyses of the EXAFS data; A. B. performed some of the experiments and some of the data analyses; B. S. performed some of the experiments and some of the data analyses; F.-T. F. carried out the X-ray single crystal structure analyses; M. M. designed the study, analysed the experimental data and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge funding from the European Research Council (ERC) Consolidator Grant 2020 under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 101003292). We acknowledge the synchrotron light source KARA for provision of instrumentation and beamtime. We acknowledge support by the Ecole Polytechnique Fédérale de Lausanne (EPFL) and by the Swiss National Science Foundation grant (number CR23I2_16645). L. M. is a senior member of the Institut Universitaire de France. CalMip is gratefully acknowledged for a generous grant of computing time.References
- H. Geckeis, J. Lutzenkirchen, R. Polly, T. Rabung and M. Schmidt, Chem. Rev., 2013, 113, 1016–1062 CrossRef CAS PubMed.
- M. Altmaier, X. Gaona and T. Fanghanel, Chem. Rev., 2013, 113, 901–943 CrossRef CAS PubMed.
- R. C. Ewing, Nat. Mater., 2015, 14, 252–257 CrossRef CAS PubMed.
- A. Morgenstern, C. Apostolidis, F. Bruchertseifer, R. Capote, T. Gouder, F. Simonelli, M. Sin and K. Abbas, Appl. Radiat. Isot., 2008, 66, 1275–1280 CrossRef CAS PubMed.
- M. Peplow, ACS Cent. Sci., 2017, 3, 510–511 CrossRef CAS PubMed.
- M. Pepper and B. E. Bursten, Chem. Rev., 1991, 91, 719–741 CrossRef CAS.
- M. Mazzanti, R. Wietzke, J. Pecaut, J. M. Latour, P. Maldivi and M. Remy, Inorg. Chem., 2002, 41, 2389–2399 CrossRef CAS PubMed.
- N. Kaltsoyannis, J. P. Hay, J. Li, J. P. Blaudeau and B. E. Bursten, Theoretical Studies of the Electronic Structure of Compounds of Actinide Elements, Springer, Dordrecht, 2006 Search PubMed.
- M. B. Jones, A. J. Gaunt, J. C. Gordon, N. Kaltsoyannis, M. P. Neu and B. L. Scott, Chem. Sci., 2013, 4, 1189–1203 RSC.
- N. Kaltsoyannis, Inorg. Chem., 2013, 52, 3407–3413 CrossRef CAS PubMed.
- M. L. Neidig, D. L. Clark and R. L. Martin, Coord. Chem. Rev., 2013, 257, 394–406 CrossRef CAS.
- A. Kovacs, R. J. Konings, J. K. Gibson, I. Infante and L. Gagliardi, Chem. Rev., 2015, 115, 1725–1759 CrossRef CAS PubMed.
- J. Su, E. R. Batista, K. S. Boland, S. E. Bone, J. A. Bradley, S. K. Cary, D. L. Clark, S. D. Conradson, A. S. Ditter, N. Kaltsoyannis, J. M. Keith, A. Kerridge, S. A. Kozimor, M. W. Loble, R. L. Martin, S. G. Minasian, V. Mocko, H. S. La Pierre, G. T. Seidler, D. K. Shuh, M. P. Wilkerson, L. E. Wolfsberg and P. Yang, J. Am. Chem. Soc., 2018, 140, 17977–17984 CrossRef CAS PubMed.
- T. Cheisson, K. D. Kersey, N. Mahieu, A. McSkimming, M. R. Gau, P. J. Carroll and E. J. Schelter, J. Am. Chem. Soc., 2019, 141, 9185–9190 CrossRef PubMed.
- R. G. Denning, Struct. Bonding, 1992, 79, 215–276 CrossRef CAS.
- N. Kaltsoyannis, Chem. Soc. Rev., 2003, 32, 9–16 RSC.
- R. G. Denning, J. Phys. Chem. A, 2007, 111, 4125–4143 CrossRef CAS PubMed.
- P. L. Arnold, M. S. Dutkiewicz, M. Zegke, O. Walter, C. Apostolidis, E. Hollis, A. F. Pecharman, N. Magnani, J. C. Griveau, E. Colineau, R. Caciuffo, X. Zhang, G. Schreckenbach and J. B. Love, Angew. Chem., Int. Ed., 2016, 55, 12797–12801 CrossRef CAS PubMed.
- M. Zegke, X. Zhang, I. Pidchenko, J. A. Hlina, R. M. Lord, J. Purkis, G. S. Nichol, N. Magnani, G. Schreckenbach, T. Vitova, J. B. Love and P. L. Arnold, Chem. Sci., 2019, 10, 9740–9751 RSC.
- B. Teyar, S. Boucenina, L. Belkhiri, B. Le Guennic, A. Boucekkine and M. Mazzanti, Inorg. Chem., 2019, 58, 10097–10110 CrossRef CAS PubMed.
- L. Kohler, M. Patzschke, S. Bauters, T. Vitova, S. M. Butorin, K. O. Kvashnina, M. Schmidt, T. Stumpf and J. Marz, Chem.–Eur. J., 2022, 28, e202200119 CrossRef CAS PubMed.
- P. L. Arnold, J. B. Love and D. Patel, Coord. Chem. Rev., 2009, 253, 1973–1978 CrossRef CAS.
- T. W. Hayton, Chem. Commun., 2013, 49, 2956–2973 RSC.
- R. Faizova, R. Scopelliti, A. S. Chauvin and M. Mazzanti, J. Am. Chem. Soc., 2018, 140, 13554–13557 CrossRef CAS PubMed.
- P. C. Burns and R. J. Finch, Am. Mineral., 1999, 84, 1456–1460 CrossRef CAS.
- F. C. Hawthorne, R. J. Finch and R. C. Ewing, Can. Mineral., 2006, 44, 1379–1385 CrossRef CAS.
- E. S. Ilton, A. Haiduc, C. L. Cahill and A. R. Felmy, Inorg. Chem., 2005, 44, 2986–2988 CrossRef CAS PubMed.
- E. S. Ilton, J. F. Boily, E. C. Buck, F. N. Skomurski, K. M. Rosso, C. L. Cahill, J. R. Bargar and A. R. Felmy, Environ. Sci. Technol., 2010, 44, 170–176 CrossRef CAS PubMed.
- F. N. Skomurski, E. S. Ilton, M. H. Engelhard, B. W. Arey and K. M. Rosso, Geochim. Cosmochim. Acta, 2011, 75, 7277–7290 CrossRef CAS.
- K. Yuan, E. S. Ilton, M. R. Antonio, Z. Li, P. J. Cook and U. Becker, Environ. Sci. Technol., 2015, 49, 6206–6213 CrossRef CAS PubMed.
- I. Pidchenko, K. O. Kvashnina, T. Yokosawa, N. Finck, S. Bahl, D. Schild, R. Polly, E. Bohnert, A. Rossberg, J. Gottlicher, K. Dardenne, J. Rothe, T. Schafer, H. Geckeis and T. Vitova, Environ. Sci. Technol., 2017, 51, 2217–2225 CrossRef CAS PubMed.
- E. S. Ilton, J. S. Pacheco, J. R. Bargar, Z. Shi, J. Liu, L. Kovarik, M. H. Engelhard and A. R. Felmy, Environ. Sci. Technol., 2012, 46, 9428–9436 CrossRef CAS PubMed.
- G. F. Vettese, K. Morris, L. S. Natrajan, S. Shaw, T. Vitova, J. Galanzew, D. L. Jones and J. R. Lloyd, Environ. Sci. Technol., 2020, 54, 2268–2276 CrossRef CAS PubMed.
- P. L. Arnold, D. Patel, C. Wilson and J. B. Love, Nature, 2008, 451, 315–317 CrossRef CAS PubMed.
- L. Chatelain, J. Pecaut, F. Tuna and M. Mazzanti, Chem.–Eur. J., 2015, 21, 18038–18042 CrossRef CAS PubMed.
- R. Faizova, S. White, R. Scopelliti and M. Mazzanti, Chem. Sci., 2018, 9, 7520–7527 RSC.
- V. Mougel, P. Horeglad, G. Nocton, J. Pecaut and M. Mazzanti, Chem.–Eur. J., 2010, 16, 14365–14377 CrossRef CAS PubMed.
- K. Takao, S. Tsushima, S. Takao, A. C. Scheinost, G. Bernhard, Y. Ikeda and C. Hennig, Inorg. Chem., 2009, 48, 9602–9604 CrossRef CAS PubMed.
- P. Horeglad, G. Nocton, Y. Filinchuk, J. Pecaut and M. Mazzanti, Chem. Commun., 2009, 1843–1845, 10.1039/b821398d.
- V. Mougel, P. Horeglad, G. Nocton, J. Pecaut and M. Mazzanti, Angew. Chem., Int. Ed., 2009, 48, 8477–8480 CrossRef CAS PubMed.
- G. Nocton, P. Horeglad, V. Vetere, J. Pecaut, L. Dubois, P. Maldivi, N. M. Edelstein and M. Mazzanti, J. Am. Chem. Soc., 2010, 132, 495–508 CrossRef CAS PubMed.
- P. L. Arnold, A. F. Pecharman, E. Hollis, A. Yahia, L. Maron, S. Parsons and J. B. Love, Nat. Chem., 2010, 2, 1056–1061 CrossRef CAS PubMed.
- V. Mougel, J. Pecaut and M. Mazzanti, Chem. Commun., 2012, 48, 868–870 RSC.
- L. Chatelain, V. Mougel, J. Pecaut and M. Mazzanti, Chem. Sci., 2012, 3, 1075–1079 RSC.
- M. B. Jones and A. J. Gaunt, Chem. Rev., 2013, 113, 1137–1198 CrossRef CAS PubMed.
- P. L. Arnold, E. Hollis, G. S. Nichol, J. B. Love, J. C. Griveau, R. Caciuffo, N. Magnani, L. Maron, L. Castro, A. Yahia, S. O. Odoh and G. Schreckenbach, J. Am. Chem. Soc., 2013, 135, 3841–3854 CrossRef CAS PubMed.
- P. L. Arnold, B. E. Cowie, M. Suvova, M. Zegke, N. Magnani, E. Colineau, J. C. Griveau, R. Caciuffo and J. B. Love, Angew. Chem., Int. Ed., 2017, 56, 10775–10779 CrossRef CAS PubMed.
- B. E. Cowie, J. M. Purkis, J. Austin, J. B. Love and P. L. Arnold, Chem. Rev., 2019, 119, 10595–10637 CrossRef CAS PubMed.
- T. Vitova, M. A. Denecke, J. Göttlicher, K. Jorissen, J. J. Kas, K. Kvashnina, T. Prüßmann, J. J. Rehr and J. Rothe, Int. J. Pharmacol. Clin. Sci., 2013, 430, 012117 CAS.
- T. Vitova, J. C. Green, R. G. Denning, M. Loble, K. Kvashnina, J. J. Kas, K. Jorissen, J. J. Rehr, T. Malcherek and M. A. Denecke, Inorg. Chem., 2015, 54, 174–182 CrossRef CAS PubMed.
- S. M. Butorin, K. O. Kvashnina, A. L. Smith, K. Popa and P. M. Martin, Chem.–Eur. J., 2016, 22, 9693–9698 CrossRef CAS PubMed.
- S. Bahl, S. Peuget, I. Pidchenko, T. Pruessmann, J. Rothe, K. Dardenne, J. Delrieu, D. Fellhauer, C. Jégou, H. Geckeis and T. Vitova, Inorg. Chem., 2017, 56, 13982–13990 CrossRef CAS PubMed.
- T. Vitova, I. Pidchenko, D. Schild, T. Prussmann, V. Montoya, D. Fellhauer, X. Gaona, E. Bohnert, J. Rothe, R. J. Baker and H. Geckeis, Inorg. Chem., 2020, 59, 8–22 CrossRef CAS PubMed.
- A. Zimina, K. Dardenne, M. A. Denecke, D. E. Doronkin, E. Huttel, H. Lichtenberg, S. Mangold, T. Pruessmann, J. Rothe, T. Spangenberg, R. Steininger, T. Vitova, H. Geckeis and J. D. Grunwaldt, Rev. Sci. Instrum., 2017, 88, 113113 CrossRef CAS PubMed.
- Y. Podkovyrina, I. Pidchenko, T. Prüßmann, S. Bahl, J. Göttlicher, A. Soldatov and T. Vitova, Int. J. Pharmacol. Clin. Sci., 2016, 712, 012092 Search PubMed.
- T. Vitova, I. Pidchenko, D. Fellhauer, P. S. Bagus, Y. Joly, T. Pruessmann, S. Bahl, E. Gonzalez-Robles, J. Rothe, M. Altmaier, M. A. Denecke and H. Geckeis, Nat. Commun., 2017, 8, 16053 CrossRef CAS PubMed.
- T. Vitova, I. Pidchenko, S. Biswas, G. Beridze, P. W. Dunne, D. Schild, Z. Wang, P. M. Kowalski and R. J. Baker, Inorg. Chem., 2018, 57, 1735–1743 CrossRef CAS PubMed.
- S. Matsika, Z. Zhang, S. R. Brozell, J. P. Baudeau, Q. Wang and R. M. Pitzer, J. Phys. Chem. A, 2001, 105, 3825–3828 CrossRef CAS.
- R. Polly, B. Schacherl, J. Rothe and T. Vitova, Inorg. Chem., 2021, 60, 18764–18776 CrossRef CAS PubMed.
- D. C. Sergentu, T. J. Duignan and J. Autschbach, J. Phys. Chem. Lett., 2018, 9, 5583–5591 CrossRef CAS PubMed.
- K. O. Kvashnina, S. M. Butorin, P. Martin and P. Glatzel, Phys. Rev. Lett., 2013, 111, 253002 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc03416f |
| This journal is © The Royal Society of Chemistry 2022 |