
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA theoretical roadmap for the best oxygen reduction activity in two-dimensional transition metal tellurides†
Xin
Yang
 ab,
Hanyu
Liu
abcd,
Zexing
Qu
e,
Yu
Xie
*abc and
Yanming
Ma
abd
ab,
Hanyu
Liu
abcd,
Zexing
Qu
e,
Yu
Xie
*abc and
Yanming
Ma
abd
aState Key Laboratory of Superhard Materials, College of Physics, Jilin University, Changchun 130012, China. E-mail: xieyu@jlu.edu.cn
bInternational Center of Computational Method and Software, College of Physics, Jilin University, Changchun 130012, China
cKey Laboratory of Physics and Technology for Advanced Batteries (Ministry of Education), College of Physics, Jilin University, Changchun 130012, China
dInternational Center of Future Science, Jilin University, Changchun 130012, China
eInstitute of Theoretical Chemistry, College of Chemistry, Jilin University, Changchun, 130023, China
First published on 25th August 2022
Abstract
Developing highly active and cost-effective electrocatalysts to replace Pt-based catalysts for the sluggish oxygen reduction reaction (ORR) is a major challenge in the commercialization of fuel cells. Although two-dimensional (2D) transition-metal tellurides have recently been proposed as alternative low-cost ORR catalysts, a fundamental study on the origin of the activity is required to further optimize their composition and performance. Herein, we investigated the electronic properties and ORR catalytic performances of a series of exfoliable 2D transition-metal tellurides to uncover the underlying mechanisms by means of density functional theory simulations. Our in-depth analysis shows that the activation of the ORR mainly depends on the partially filled pz state of active Te atoms, which can simultaneously accept and donate electrons behaving similarly to both the occupied and unoccupied d orbitals of Pt atoms. This results in a linear relationship between the pz-band center and the adsorption free energies of O2 and intermediates, indicating that the pz-band center might be used as an effective descriptor to probe the performance of telluride catalysts. On this basis, we predicted several 2D transition-metal tellurides with promising catalytic performance and reduced precious-metal contents, where NbRhTe4 reaches the top of the activity volcano with a limiting potential of 0.96 V. This study provides theoretical guidance to design high-performing 2D telluride ORR catalysts, and its principle might be applicable to other electrochemical reactions in 2D chalcogenides.
Introduction
Proton exchange membrane fuel cells (PEMFCs) are recognized as one of the most effective technologies for growing clean and sustainable energy demands, as they can directly convert chemical energy into electricity.1–3 Generally, the overall performance of PEMFCs depends largely on the kinetics of the cathodic oxygen reduction reaction (ORR), as it is much slower than the anodic hydrogen oxidation reaction (HOR), and other reactions including the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER).4 The key to overcoming this bottleneck is finding highly efficient catalysts to boost the sluggish ORR kinetics, which are currently dominated by Pt-based catalysts (e.g., Pt nano-frameworks and Pt alloys) owing to their high activity and good stability.5–7 However, the high cost of Pt-based catalysts significantly hampers large-scale applications of PEMFCs.8,9 Thus, there is an urgent need to design highly efficient ORR catalysts with a reduced Pt (precious metal) content for the sustainable development of fuel cells.Emerging two-dimensional (2D) materials have shown great potential as efficient catalysts owing to their high specific surface area and minimum migration distance for carriers.10–14 Among them, doped transition metal dichalcogenides (TMDs) and metal(M)–N–C single-atom catalysts (SACs), produced by embedding atomically dispersed metal species into graphene, were found to exhibit good ORR catalytic activity.15–19 Unfortunately, the advantages of 2D morphology for most of these materials are not well utilized, because their pristine basal planes are catalytically inactive. To fully leverage the potential of 2D materials for the catalytic ORR, it is essential to seek 2D catalysts with high basal plane activity, where there has recently been increasing attention focused on metallic transition metal telluride monolayers.20–28 For instance, layered PtTe2 was demonstrated to show ORR activity on par with the commercial Pt/C catalyst.21 Monolayer PtTe was predicted to have a limiting potential of 0.86 V,22 which outperforms Pt (111) electrodes (0.78 V). The precious metal free NiTe and TaTe2 monolayers were soon after demonstrated with similar catalytic performance compared to Pt electrodes.23 The activity of 2D Ni2SbTe2 was also recently examined,27 where the endothermic O2 adsorption might prohibit the ORR process on the basal plane. Although 2D transition metal tellurides indeed exhibit excellent catalytic activity towards the ORR, PtTe, with currently the best overall performance, still contains a large amount of the precious metal (50% formula weight) hindering its practical applications. Rational design of transition metal tellurides is desired to achieve better performance in both cost and catalytic activity. This remains untouched, primarily due to the lack of knowledge of the ORR activity origin of tellurides. In particular, unlike Pt-based catalysts, the electronic characteristics of Te atoms vary in different tellurides, as the electronegativity difference between Te and the transition metal is different. Thus, elucidating the catalytic activity origin of the Te atom is fundamental in the design of high-performing 2D telluride ORR catalysts that may extend to other chalcogenides.
In this study, we theoretically explore the ORR catalytic performance of a series of 2D stable nanosheets, NbTe2, WTe2, MoTe2, and MxXyTez nanosheets (M = Nb and Ta and X = transition metals), which can readily be exfoliated from their bulk phases.29–37 By combining the electronic density of states and molecular orbital analysis, we found that the partially filled pz state of Te atoms can simultaneously accept electrons from the orbital and donate electrons to the π* orbital of the O2 molecule, endowing telluride nanosheets with activities for O2 fixation and protonation. The adsorption energies of O2, O, and OH are thus linearly related to the pz-band center of Te atoms, where the closer the band center to the Fermi level, the stronger the binding of the adsorbates. The constructed ORR activity volcano plot allows the pz-band center derivation between −2.5 and −1.8 eV for telluride monolayers with potentially high activity. Following this guidance, several telluride monolayers, such as MXTe4 and Ta4Pd3Te16, were identified with limiting potentials of 0.73–0.96 V in the four-electron (4e−) pathway and precious-metal contents of less than 24%, implying that they are potentially highly performing ORR catalysts.
Results and discussion
Geometric and electronic properties
We first selected nine telluride monolayers with various stoichiometries and compositions in conjunction with previously studied 2D telluride catalysts to probe the nature and origin of the electrocatalytic ORR activity of telluride nanosheets. The selected nine tellurides are NbTe2, WTe2, NbNiTe2, TaCo2Te2, TaNi2Te3, NbIrTe4, NbPdTe5, Ta2Pd3Te5, and Ta3Pd3Te14 (Fig. 1). Because all of these 2D tellurides have experimentally synthesized bulk counterparts,29–37 the cleavage energies (Ecl) were evaluated to assess the experimental exfoliation feasibility by using a five-layer slab. As presented in Fig. S1,† the Ecl of NbTe2, WTe2, TaNi2Te3, NbIrTe4, and NbPdTe5 are comparable to that of exfoliated 2D MoS2 (0.42 J m−2),38 which can be classified as easily exfoliated according to the report of Mounet and coworkers.39 Other telluride nanosheets have slightly higher Ecl and can be regarded as potentially exfoliated. Therefore, isolation of these 2D tellurides from their parent phases via exfoliation is experimentally feasible. Structurally, each nanosheet comprises transition metal atomic layers sandwiched between two Te atomic layers, where NbTe2, WTe2, NbIrTe4, NbPdTe5, and Ta3Pd3Te14 comprise one metal layer, while others comprise two or three metal layers. Specifically, NbTe2, WTe2, and NbIrTe4 are composed of MTe6 (XTe6) octahedra. Similarly, TaNi2Te3 consists of XTe4 tetrahedra and MTe6 octahedra, which are also contained in Ta2Pd3Te5, where one of the octahedral apexes is X (MTe5X octahedra). NbPdTe5 and Ta3Pd3Te14 are formed of MTe8 decahedra and MTe6 (XTe6) octahedra. The rest of the two tellurides, NbNiTe2 and TaCo2Te2, do not possess the above polyhedra. The dynamic stability of the monolayers was confirmed from their computed phonon frequencies, as reported in Materials Cloud two-dimensional crystals databases39 and database C2DB40 of 2D materials. The calculated band structures and density of states (DOS) of the above nanosheets reveal that NbIrTe4 is semi-metallic with a zero bandgap that exhibits metallic behaviors,41 and the other nanosheets are well-defined metals (Fig. S2†). Moreover, we also examined the charge distribution of these tellurides through the Bader charge analysis. As shown in Table S1,† most Te atoms are negatively charged due to the charge transfer from inner transition metal atoms to outer Te atoms. Only Te3 atoms in NbIrTe4 are positively charged with around 0.25 e transferred from Te to Ir. | ||
| Fig. 1 (a) Side views of optimized structures of monolayer NbTe2, WTe2, NbNiTe2, TaCo2Te2, TaNi2Te3, NbIrTe4, NbPdTe5, Ta2Pd3Te5, and Ta3Pd3Te14. The blue, grey and yellow balls represent M, X, and Te atoms, respectively. (b) The O2 adsorption energy of monolayer tellurides. | ||
Origin of O2 activation
After determining the stability and electronic properties of these nanosheets, we proceeded to explore the adsorption of the O2 molecule, which is the prerequisite for an efficient ORR process. The most favorable O2 adsorption configurations and calculated adsorption energies (ΔGO2) are shown and summarized in Fig. 1b and S3, and Table S2–3.† The ΔGO2 of NbTe2 and NbIrTe4 monolayers is −0.13 and −0.09 eV, respectively, suggesting that O2 can be chemisorbed. The moderate ΔGO2 of 0.05 eV indicates that O2 might be physisorbed on the Ta3Pd3Te14 monolayer that is similar to the PtTe monolayer. Other telluride monolayers possess ΔGO2 larger than 0.20 eV, where the endothermic nature suggests that O2 molecules are hardly adsorbed on these telluride monolayers to trigger the ORR process. Moreover, the O–O bond length is significantly stretched from 1.23 Å in the gas phase to 1.38 and 1.39 Å when O2 is adsorbed on NbTe2 and NbIrTe4, respectively. The weakening of O–O bonds is expected to facilitate the subsequent ORR steps. From the nudged-elastic-band (NEB) calculations, the energy barriers of O2 dissociation into two adsorbed O* are around 0.42 and 0.54 eV for NbTe2 and NbIrTe4, respectively (Fig. S4†), which are comparable to the O2 dissociation barrier on Pt (111) (0.38–0.50 eV),42,43 indicating that O2 has been effectively activated.Since O2 activation plays a crucial role in the ORR process, uncovering the origin of O2 adsorption is beneficial for designing efficient ORR catalysts. Previously, an electron activation mechanism was proposed for the positively charged Te in the PtTe monolayer.22 According to this mechanism, the Te–O bond strength depends on the number of electrons the Te atom gains/loses (Ne), where the smaller Ne leads to stronger binding of O2. Thus, we examined the correlation between Ne and ΔGO2 of the studied telluride monolayers (Fig. S5†). Unlike the observed linear relationship between Ne and the binding energy of adsorbates in other systems, we found that ΔGO2 does not depend on Ne. Either the positively or negatively charged Te atom can activate the O2 molecule. Moreover, for most telluride monolayers, a larger Ne causes a stronger adsorption of O2 that is opposite to the proposed mechanism. Therefore, O2 adsorption cannot be simply described by the electron activation mechanism.
To gain a better understanding of the underlying O2 adsorption mechanism, we further analyzed the orbital configuration of Te atoms in comparison with Pt-based catalysts. For the Pt atom (Fig. 2a), the coexistence of unoccupied and occupied d orbitals can facilitate O2 adsorption and activation through a two-way charge transfer, where the unoccupied eg orbital accepts lone-pair electrons from O2, and the occupied t2g orbital donates electrons back to O2 antibonding orbitals.44,45 This donation and back-donation concept is standard in molecular chemistry (Blyholder model) and has been successfully employed to describe the interaction between small molecules (e.g., N2, C2H4, and CO) and transition metal and boron-based catalysts.46–53 Interestingly, the Te atom possesses an orbital configuration analogous to that of Pt and B atoms in transition metal stuffed boron nitride nanotubes. As illustrated in Fig. 2b, the valence electronic configuration of the Te atom is 5s25p4 regardless of the charge transfer between Te and transition metal atoms, resulting in one fully occupied p orbital and two partially filled p orbitals. Therefore, the Te atom is eligible to drive a similar “donation and back-donation” process to adsorb the O2 molecule, as shown in Fig. 2b.
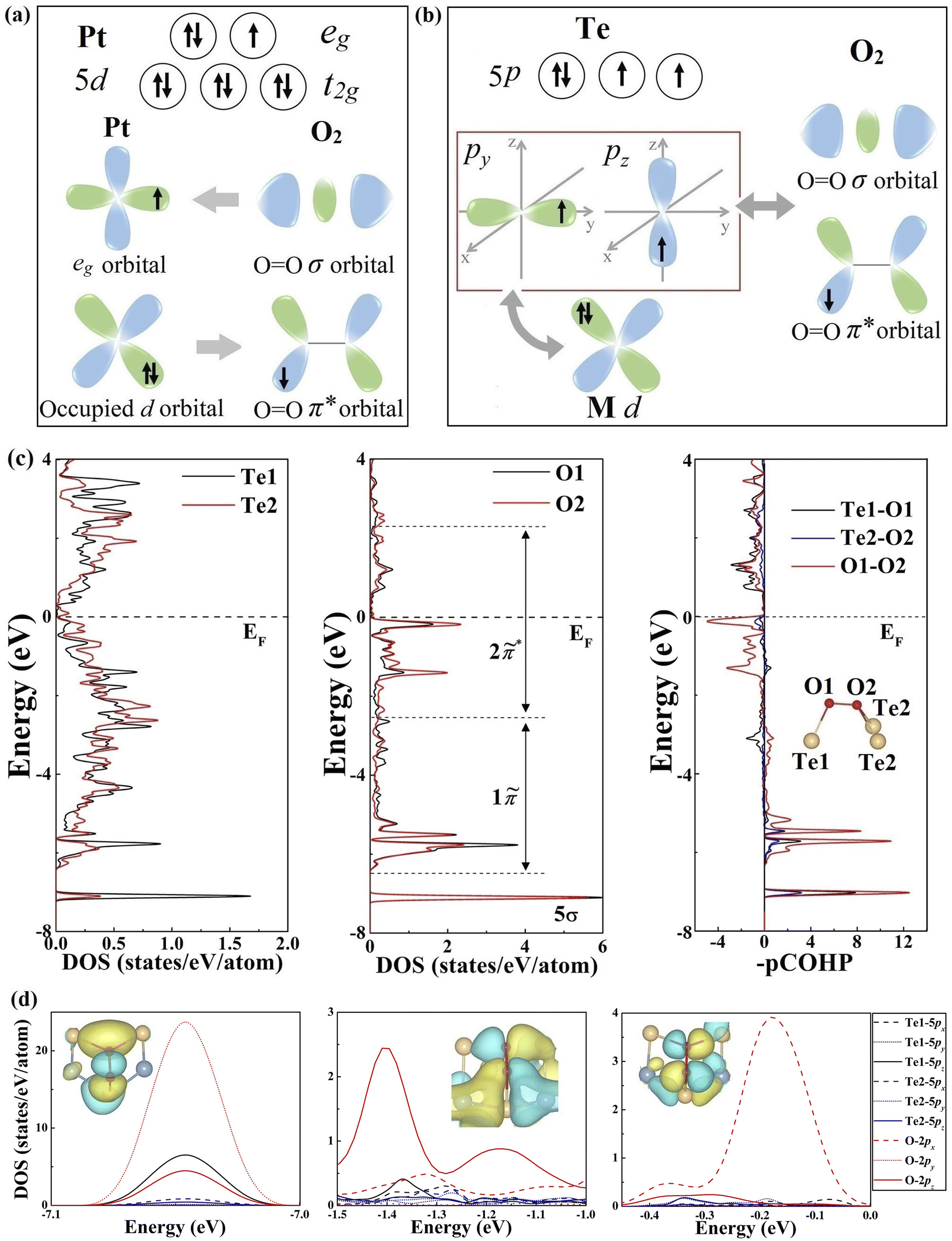 | ||
| Fig. 2 (a) Simplified schematic of O2 bonding to Pt. (b) Mechanism of activation of monolayer NbIrTe4 for O2 adsorption (c) DOS and pCOHP curves of O2-adsorbed tellurides. The orbitals of O2 in O2-adsorbed tellurides are marked here. (d) pDOS and the corresponding molecular orbital of the O2-adsorbed NbIrTe4 monolayer. | ||
To verify our inference, the charge density difference between O2* and telluride monolayers was calculated to reveal the underlying charge transfer. As shown in Fig. S6–9,† O2* exhibits a clear picture of the two-way charge transfer exhibiting charge accumulation (yellow) and depletion (blue). Interestingly, Te atoms seem to deviate from this mechanism as electrons barely accumulate around them.
To decipher this phenomenon, we then evaluated the O–O and O2–Te interactions by a combined electronic structure and projected crystal orbital Hamilton populations (pCOHP) analysis, taking the NbIrTe4 monolayer as an example. The quantity of −(pCOHP) was used to directly correspond the positive and negative values with bonding and antibonding states, respectively. The molecular orbital (MO) diagram, projected density of states (pDOS), and pCOHP of the free O2 molecule were examined first, as presented in Fig. S10.† By analyzing the pDOS and pCOHP, it is clear that the fully occupied 5σ and 1π orbitals contribute to the bonding of the O2 molecule, while the partially filled 2π* orbital is antibonding.
After O2 adsorption on the NbIrTe4 monolayer, the sharp localized MOs of O2 in a vacuum hybridize with surface Te p states that broaden into rather delocalized states and shift down to lower energy (Fig. 2c). The new hybrid states are marked as “tildes” of the original orbitals to indicate the difference (Fig. S11†). The 5![[small sigma, Greek, tilde]](https://www.rsc.org/images/entities/char_e10d.gif) band showed up in a narrow energy range at around −7.05 eV relative to the Fermi level, and the electron density of the 5 band is reduced compared to the 5σ orbital of free O2. This agrees with the Blyholder picture of σ forward donation, as reflected by the flow of 0.23 electrons from O2 to Te atoms, resulting in the formation of a novel Te DOS peak, mainly composed of the pz state of the Te1 atom in the same energy range and σ-type Te–O bonds (Fig. S6–9).† The degenerated 1π MO split into two narrow 1π bands at around −6.4 and −5.5 eV, respectively, and broad bands
band showed up in a narrow energy range at around −7.05 eV relative to the Fermi level, and the electron density of the 5 band is reduced compared to the 5σ orbital of free O2. This agrees with the Blyholder picture of σ forward donation, as reflected by the flow of 0.23 electrons from O2 to Te atoms, resulting in the formation of a novel Te DOS peak, mainly composed of the pz state of the Te1 atom in the same energy range and σ-type Te–O bonds (Fig. S6–9).† The degenerated 1π MO split into two narrow 1π bands at around −6.4 and −5.5 eV, respectively, and broad bands ![[P with combining tilde]](https://www.rsc.org/images/entities/char_0050_0303.gif) 1π and 2π* between −5.3 and −1.1 eV upon hybridization with Te states and 2π* MO. Importantly, the unoccupied 2π* MO of free O2 became partially populated and transformed into 2
1π and 2π* between −5.3 and −1.1 eV upon hybridization with Te states and 2π* MO. Importantly, the unoccupied 2π* MO of free O2 became partially populated and transformed into 2![[small pi, Greek, tilde]](https://www.rsc.org/images/entities/char_e1e9.gif) * bands in the energy range between −1.0 and 2.3 eV, where 0.5 electrons were back-donated from the Te atom to the 2π* orbital of O2 (Fig. S6†). The net charge transfer from Te to O2 agrees with the Bader charge analysis, as O has a higher electronegativity, implying that the back-donation is a more dominant Te–O interaction. Interestingly, pz of the Te1 atom was also the main contribution for the back-donation. Therefore, Te atoms could accept and donate electrons from and to O2 molecules simultaneously, while the pz orbital of the Te1 atom contributes the most. The electron depletion of Te can thus be understood, as the two-way charge transfer mainly takes place in the same orbital, and the net charge transfer between Te and O2 is negative. Other studied telluride monolayers, such as PtTe, NbTe2, and Ta3Pd3Te14, also present the same phenomenon (Fig. S7–9).† Therefore, the origin of O2 activation on 2D tellurides is a unique “donation and back-donation” mechanism that stretches and weakens the O–O bond.
* bands in the energy range between −1.0 and 2.3 eV, where 0.5 electrons were back-donated from the Te atom to the 2π* orbital of O2 (Fig. S6†). The net charge transfer from Te to O2 agrees with the Bader charge analysis, as O has a higher electronegativity, implying that the back-donation is a more dominant Te–O interaction. Interestingly, pz of the Te1 atom was also the main contribution for the back-donation. Therefore, Te atoms could accept and donate electrons from and to O2 molecules simultaneously, while the pz orbital of the Te1 atom contributes the most. The electron depletion of Te can thus be understood, as the two-way charge transfer mainly takes place in the same orbital, and the net charge transfer between Te and O2 is negative. Other studied telluride monolayers, such as PtTe, NbTe2, and Ta3Pd3Te14, also present the same phenomenon (Fig. S7–9).† Therefore, the origin of O2 activation on 2D tellurides is a unique “donation and back-donation” mechanism that stretches and weakens the O–O bond.
ORR catalytic performance
We assessed the catalytic performance of telluride monolayers for the reduction of O2 to H2O. The typical association and dissociation mechanisms (Fig. 3), including all possible intermediate reactants (Fig. S12 and 13)† were widely investigated for NbTe2, NbIrTe4, and Ta3Pd3Te14 monolayers. For other telluride monolayers, we only calculated the adsorption energies of O and OH for comparison. As illustrated in Fig. 3, the NbTe2 monolayer shows similar catalytic behaviors to the TaTe2 monolayer,23 where the association pathway has a higher limiting potential (UL) of 0.80 V (OH* reduction to H2O) than the dissociation pathway of 0.63 V (Fig. S12†), suggesting that the association mechanism exhibits higher efficiency. Alternatively, the dissociation mechanism is more efficient in NbIrTe4 and Ta3Pd3Te14 monolayers, as their UL of 0.88 and 0.80 V (O* reduction to OH*) are higher than the association ones of 0.65 and 0.45 V (O2 to OOH*), respectively (Fig. 3 and S13†). In addition to the 4e electrochemical processes, we also examined the 2e reduction pathways for the dominant reduction mechanism. The free energy diagrams show that the UL of the 2e association pathway is 0.6 V for the NbTe2 monolayer, which is lower than that of the 4e association pathway. The H2O2 formation is endothermic for the dissociation pathway of NbIrTe4 and Ta3Pd3Te14 monolayers, indicating that it is difficult to occur during the ORR process. These results suggest that NbTe2, NbIrTe4, and Ta3Pd3Te14 monolayers are rather good ORR catalysts with high 4e pathway selectivity. Considering that monolayers are generally more difficult to obtain than multilayers, we also explored the electrocatalytic performance of bilayer and trilayer NbIrTe4 (Fig. S14–16†). The electrocatalytic performance of bilayer and trilayer NbIrTe4 is similar to that of the monolayer with limiting potentials of 0.89 and 0.81 V.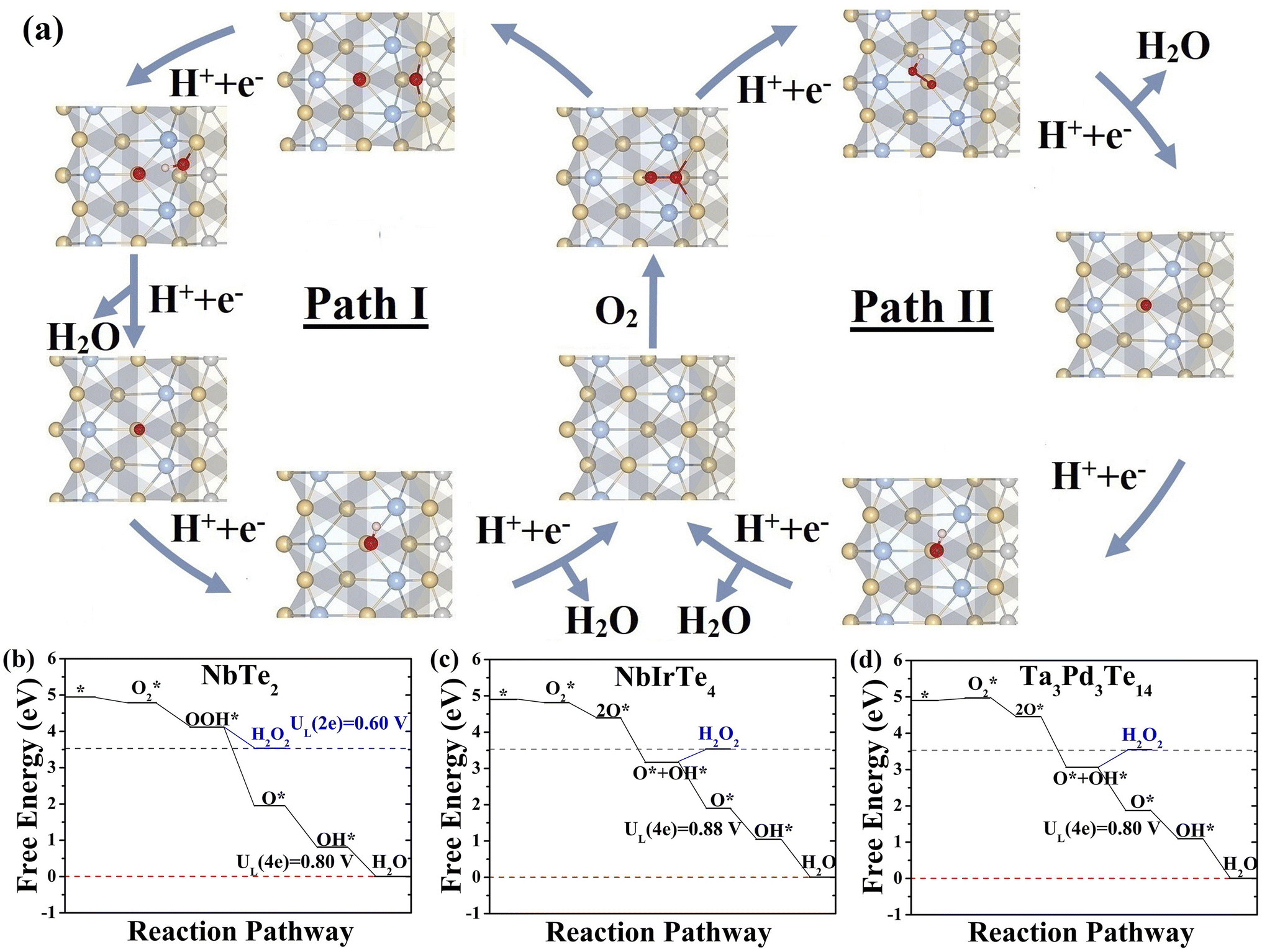 | ||
| Fig. 3 (a) Scheme of the possible two ORR reaction pathways of monolayer NbIrTe4. Calculated free energy diagrams for the ORR pathway on monolayer (b) NbTe2 in an association pathway and (c) NbIrTe4 and (d)Ta3Pd3Te14 in a dissociation pathway. | ||
A previously established theory for transition metal-based catalysts suggested that the adsorption energies of O-containing intermediates, governing the catalytic performance, are linearly correlated.54–60 Thus, the catalytic activity can be described by the adsorption energies of a single intermediate reactant. Similar to transition metal-based catalysts, linear scaling relationships between ΔGOH* and other reactants were also identified in telluride monolayers (Fig. 4a and S17†). For example, ΔGOH* and ΔGOOH* are related to each other by a constant of approximately 3.3 eV that is close to 3.2 eV, observed in transition metal-based catalysts (Fig. S17b†). Given the excellent linear relationship, we plotted UL as a function of ΔGOH* for telluride monolayers, where the transition metal-based catalysts were also included for comparison (Fig. 4b).22,23,27,61,62 This leads to the observation of a universal volcano relationship that follows the century-old Sabatier principle, where an ideal catalyst must bind the intermediate reactants with an intermediate strength: not too weak to activate the reactants, and not too strong to desorb the products. The estimated highest limiting potential is approximately 0.94 V, which demonstrates that the dissociation pathway can elevate the limiting potential close to 0.1 V compared with the previously suggested UL of 0.86 V for an ideal ORR catalyst. For an efficient catalyst with a limiting potential higher than that of Pt (111), one should search for tellurides with a ΔGOH* between 0.8 and 1.2 eV.
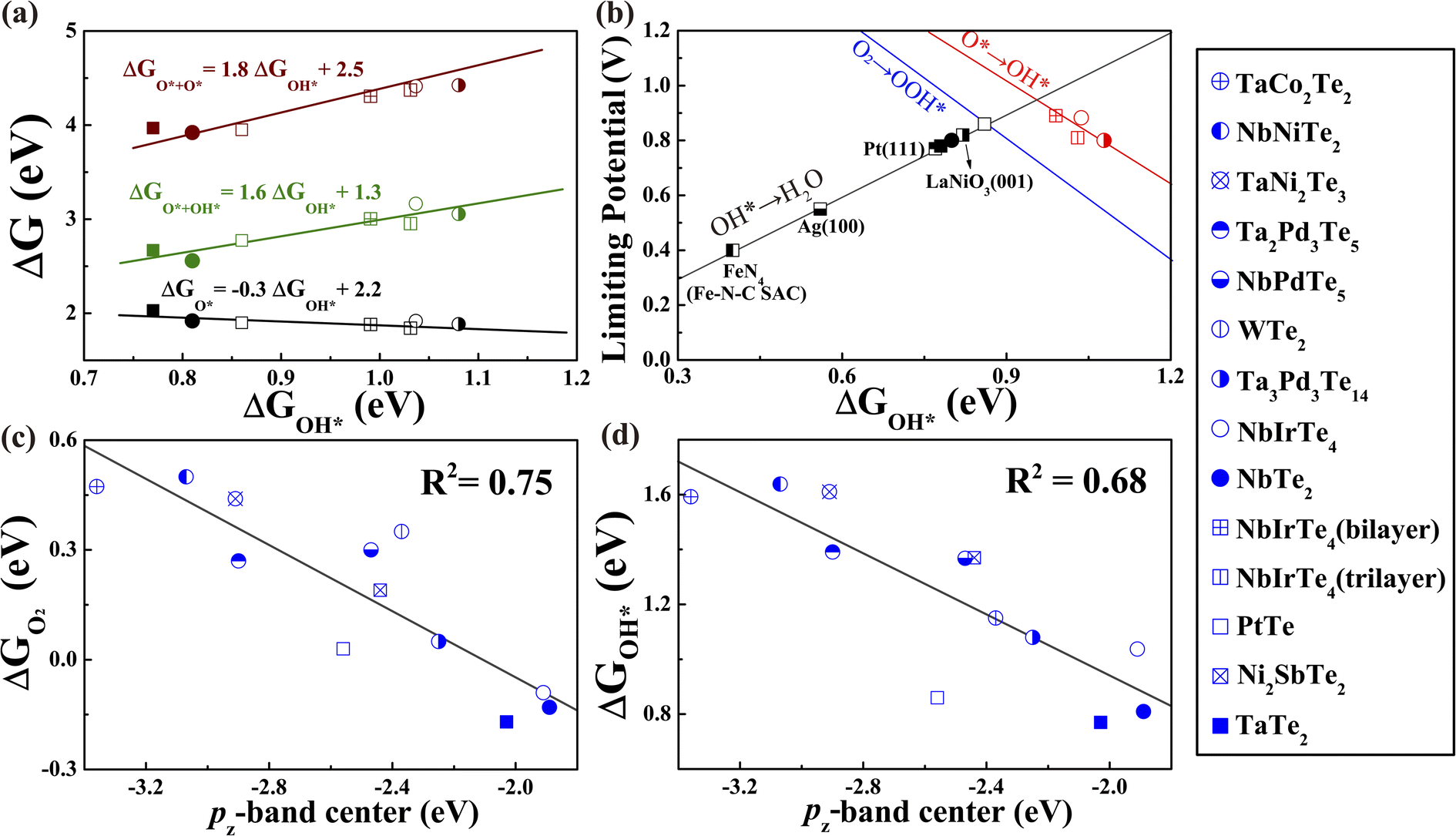 | ||
| Fig. 4 (a) The variation of adsorption energy of intermediate adsorbates in the ORR dissociation pathway as a function of ΔGOH* (b) Volcano plot of the ORR limiting potentials for studied tellurides and other known catalysts.15,22,23,63,64 The blue line (O2 to OOH*) was taken from previous work.64 Other details are shown in Fig. S17.† (c) The O2 adsorption energy and (d) OH adsorption energy versus the pz-band center of active Te sites for initially studied monolayer tellurides. | ||
Activity descriptor
Since the simulated limiting potential of 2D tellurides relates well to the binding energy of intermediate adsorbates, similarly to that of transition metal-based catalysts, we proceed to seek a simple descriptor that can correlate the adsorption energy with the intrinsic properties of tellurides like the d-band center model for transition metals.50,65–70 As we already know that the partially filled pz orbital is responsible for the Te–O interactions upon O2 adsorption, we introduced a descriptor εpz defined as the centroid of the projected density of states of the pz orbital relative to the Fermi level analogous to the d-band center.71–75 As presented in Fig. 4c and d, εpz of the active Te atoms shows a reasonable linear relationship to the adsorption energies of O2 and intermediate oxygen-containing adsorbates. The linear relationships between ΔGO2 and other possible descriptors, such as the p-band center εp, are also calculated as shown in Fig. S18,† all of which are inferior to that of εpz. Similar to the d-band center model, the closer the εpz to the Fermi level, the stronger the binding strength of the adsorbates on telluride monolayers.63 This can be understood through the bond order, which is half of the difference between the electron number of the bonding orbital and the antibonding orbital according to molecular orbital theory. A high bond order corresponds to strong binding and vice versa. When O-containing groups are adsorbed on telluride monolayers, the O 2p orbitals hybridize with the Te 5pz orbital forming a fully filled bonding state and partially filled antibonding state. A high εpz results in a low filling of the antibonding state and hence a large bond order that could lead to enhanced Te–O binding strength. On the contrary, a low bond order corresponding to low εpz and high filling of the antibonding state weakens the telluride-adsorbate interaction, destabilizing the Te–O bond. In this sense, the pz-band center, εpz, can serve as a simple catalytic activity descriptor for telluride monolayers. An efficient 2D telluride catalyst with a ΔGOH* between 0.8 and 1.2 eV and the ability for O2 chemisorption are predicted to possess a εpz between −2.5 and −1.8 eV. Thus, we obtained the screening criterion.Rational screening of efficient ORR catalysts
With the established activity descriptor, we moved on to search for potential 2D telluride catalysts that may possess a better catalytic performance. We further evaluated the electronic properties of Td-MoTe2, TaNiTe2, NbRhTe4, TaRhTe4, TaIrTe4, Ta2Ni3Te5, and Ta4Pd3Te16 monolayers. Except for NbRhTe4, the bulk phases of these monolayers have also been experimentally synthesized.29,76–78 The thermodynamic stability of bulk NbRhTe4 was confirmed by constructing the Nb–Rh–Te ternary phase diagram as shown in Fig. S19(a),† which is in line with previous theoretical studies.79,80 As shown in Fig. S19(b),† the isolation of these 2D tellurides from their bulk phases via exfoliation is experimentally feasible. Among them, NbRhTe4, TaRhTe4, TaIrTe4, and Ta4Pd3Te16 have εpz values between −2.5 and −1.8 eV that meet the criteria for efficient ORR catalysts. Thus, as previously, we focused on the catalytic activity of these four tellurides for both dissociation and association pathways and calculated the adsorption free energies of the rest of the tellurides for comparison and construction of the correlation. The calculated ΔGO2 shows that O2 molecules can be chemisorbed on NbRhTe4, TaRhTe4, TaIrTe4, and Ta4Pd3Te16 to trigger the ORR process as expected. The free energy diagrams (Fig. S20–22)† show that the dissociative pathway generally exhibits higher limiting potential than the associative pathway. The rate limiting step is OH* to H2O, O* to OH*, O* to OH*, and O + OH* to O* for NbRhTe4, TaRhTe4, TaIrTe4, and Ta4Pd3Te16, respectively. The corresponding limiting potentials are all above 0.78 eV, which are comparable to that of Pt (111) and the PtTe monolayer. Encouragingly, these MXTe4 and Ta4Pd3Te16 tellurides contain small amounts of precious metals of 17% and 13% formula weight. In particular, the NbRhTe4 monolayer has a limiting potential as high as 0.96 V, that is close to the predicted UL of 0.94 V for an ideal catalyst, indicating that the NbRhTe4 monolayer could reach the top of the 4e ORR activity volcano (Fig. 5b). Herein, we also explored the electrocatalytic performance of bilayer and trilayer NbRhTe4 (Fig. S23†). The limiting potential of bilayer and trilayer NbRhTe4 is similar to that of the monolayer with 0.95 and 0.85 V.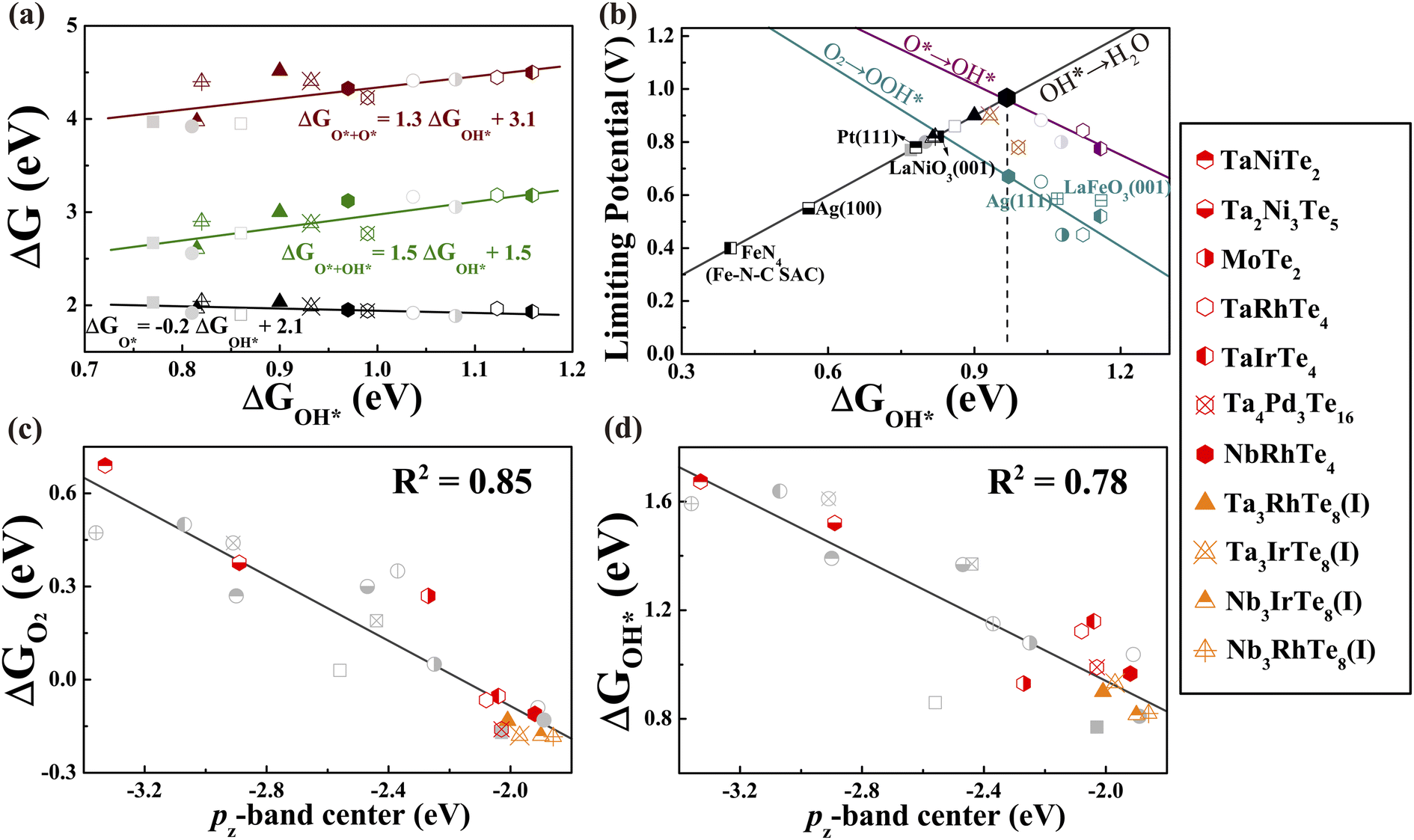 | ||
| Fig. 5 (a) The variation of adsorption energy of intermediate adsorbates in the ORR dissociation pathway as a function of ΔGOH*. (b) Volcano plot of the ORR limiting potentials for all studied monolayer tellurides and other known catalysts. (c) The O2 adsorption energy and (d) OH adsorption energy versus the pz-band center of active Te sites for all studied monolayer tellurides. The grey circle and square points represent initially studied tellurides and known catalysts in other studies15,22,23,63,64 shown in Fig. 4. The star and triangle points represent newly studied tellurides and M-doped variants, respectively. | ||
Notably, the newly identified promising MXTe4 type telluride catalysts can be viewed as X substituted MTe2 (50% substitution). Thus, by varying the substitution ratio, we may obtain potential telluride catalysts with an even lower precious-metal content, while maintaining high activity, as they should possess εpz close to that of MXTe4 or MTe2 monolayers. Hence, we designed two hypothetical M3XTe8 (25% substitution) structures, phase I and II that possess similar energies, as a tentative attempt (Fig. S24a and b†). The calculated formation energy of the bulk phases of these hypothetical monolayers was between 20 and 25 meV/atom, suggesting that the hypothetical M3XTe8 phases are metastable and might be realized in the experiments (Fig. S24†). Similarly, we considered both dissociation and association pathways. Fig. S25 and 26† presented the detailed ORR process and the calculated free energy diagrams of phase I Nb3RhTe8, Ta3RhTe8, Ta3IrTe8, and phase II Nb3IrTe8, which showed better ORR performance in these two phases (the details of another phase are summarized in Table S4 and S5†). As shown in the free energy diagrams, the limiting-potential of phase I of Nb3RhTe8 and Ta3RhTe8, and phase II of Nb3IrTe8 is predicted to be 0.82, 0.90, and 0.81 V, respectively, with the limiting step of OH* → H2O. Differently, the limiting step of phase I of Ta3IrTe8 is predicted to be O + OH* → O*, with a ΔG of −0.90 eV. These hypothetical substituted monolayers show superior ORR activity with a decreased precious-metal content (8%).
Finally, we re-examined the correlation between the adsorption energies of oxygen-containing groups and εpz by including all studied telluride monolayers. A better linear relationship between ΔGadsorbates and εpz was also observed (Fig. 5c, d and S27†), indicating that all these 2D telluride catalysts should have a similar underlying mechanism, and the pz orbital is primarily responsible, so that εpz could be used to describe the interaction between reactants and substrates. Therefore, once the optimal εpz is established, one can rationally design highly active 2D telluride-based ORR catalysts through this simple activity descriptor. Notably, as we only considered transition metal tellurides in the current study, a large discrepancy may occur for the catalytic activity prediction of tellurides containing main-group elements or with different activation mechanisms, where the descriptor should be corrected accordingly.
Conclusions
In summary, systematical density functional theory calculations were employed to investigate the nature and origin of ORR activities of a series of experimentally synthesizable 2D transition-metal telluride catalysts. Our results suggested that the partially occupied pz state of Te atoms can effectively drive the “donation-back-donation” process through a two-way charge transfer, playing an important role in the activation of O2. The subsequent O2 reduction processes proceed through the four-electron pathway, where most of the tellurides take the dissociative pathway. The binding strength of O2 and intermediates is linearly dependent on the pz-band center, implying that the latter can serve as an indicator to probe the catalytic activity of tellurides. By adopting this concept to justify the volcano plot between the limiting potential and OH adsorption free energy, we predicted that MXTe4, Ta3Pd3Te14, and Ta4Pd3Te16 are potential ORR catalysts, where NbRhTe4 shows the highest limiting potential of 0.96 V among studied transition metal telluride catalysts, reaching the top of the activity volcano with a precious-metal content of only 17%. The hypothetical substitution of MXTe4 leads to Ta3IrTe8 with a limiting potential of 0.90 V and a precious-metal content of 8%. Our study demonstrates that transition metal telluride nanosheets are highly compelling ORR electrocatalysts and proves the generality of the physically derived descriptor for screening active tellurides. We further expect the presented strategy to be extended to other electrocatalysis processes to identify descriptors to rationally and efficiently design highly active catalysts among 2D chalcogenides.Methods
Density functional theory computations were performed by using the Vienna Ab initio simulation package (VASP) within the projected augmented wave method.81,82 The Perdew−Burke−Ernzerhof (PBE) functional in the generalized gradient approximation (GGA) was used to describe the exchange-correlation potential.83 A plane-wave energy cut-off of 500 eV was used, and all atoms were fully relaxed using the conjugate gradient algorithm with a criterion of convergence of 10−5 eV and 0.03 eV per atom.84 For multilayer systems, the DFT-D3 empirical correction was used to describe van der Waals (vdW) interactions, which has been proven reliable for describing long-range vdW interactions.85 The phonon spectra of 2D tellurides were computed using the finite displacement method, as implemented in the Phonopy code.86 Bader's quantum theory of atoms in molecules (QTAIM) analysis was employed for the charge transfer calculations.87 The bonding was further analyzed by calculating the projected crystal orbital Hamilton populations (pCOHPs) using the LOBSTER package.88–90 The energy barriers were calculated using the climbing image nudged-elastic-band (CI-NEB) methods implemented in the VASP.91 The ORR processes were calculated on the basis of the computational hydrogen electrode (CHE) model.92Data availability
The data supporting the findings of this study are available in the manuscript and ESI† and from the corresponding author upon reasonable request.Author contributions
Yu Xie conceived the presented idea. Xin Yang, Zexing Qu, and Yu Xie performed the first-principles calculations. Xin Yang, Hanyu Liu, Yu Xie and Yanming Ma wrote the manuscript. All authors discussed the results and assisted during manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant no. 12022408), the Interdisciplinary Integration and Innovation Project of JLU, Fundamental Research Funds for the Central Universities and the Program for JLU Science and Technology Innovative Research Team (JLUSTIRT). This paper is dedicated to the 70th anniversary of College of Physics of Jilin University. Part of the calculations were performed at the High Performance Computing Center of Jilin University.Notes and references
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414(6861), 345–352 CrossRef CAS PubMed.
- M. K. Debe, Nature, 2012, 486(7401), 43–51 CrossRef CAS PubMed.
- A. Rabis, P. Rodriguez and T. J. Schmidt, ACS Catal., 2012, 2(5), 864–890 CrossRef CAS.
- M. Shao, Q. Chang, J. P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116(6), 3594–3657 CrossRef CAS PubMed.
- L. Huang, S. Zaman, X. Tian, Z. Wang, W. Fang and B. Y. Xia, Acc. Chem. Res., 2021, 54(2), 311–322 CrossRef CAS PubMed.
- C. Cui, X. Hu and L. Wen, J. Semicond., 2020, 41(9), 091705 CrossRef CAS.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56(1–2), 9–35 CrossRef CAS.
- X. Huang, Z. Zhao, L. Cao, Y. Chen, E. Zhu, Z. Lin, M. Li, A. Yan, A. Zettl and Y. M. Wang, Science, 2015, 348(6240), 1230–1234 CrossRef CAS PubMed.
- Y. Kang, X. Ye, J. Chen, Y. Cai, R. E. Diaz, R. R. Adzic, E. A. Stach and C. B. Murray, J. Am. Chem. Soc., 2013, 135(1), 42–45 CrossRef CAS PubMed.
- D. Deng, K. S. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11(3), 218–230 CrossRef CAS PubMed.
- X. Chia and M. Pumera, Nat. Catal., 2018, 1(12), 909–921 CrossRef CAS.
- K. Lasek, J. Li, S. Kolekar, P. M. Coelho, L. Guo, M. Zhang, Z. Wang and M. Batzill, Surf. Sci. Rep., 2021, 76(2), 100523 CrossRef CAS.
- Z. Pei, J. Gu, Y. Wang, Z. Tang, Z. Liu, Y. Huang, Y. Huang, J. Zhao, Z. Chen and C. Zhi, ACS Nano, 2017, 11(6), 6004–6014 CrossRef CAS PubMed.
- H. Zhang, Y. Tian, J. Zhao, Q. Cai and Z. Chen, Electrochim. Acta, 2017, 225, 543–550 CrossRef CAS.
- Y. Wang, Y. J. Tang and K. Zhou, J. Am. Chem. Soc., 2019, 141(36), 14115–14119 CrossRef CAS PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332(6028), 443–447 CrossRef CAS PubMed.
- S. Lin, H. Xu, Y. Wang, X. C. Zeng and Z. Chen, J. Mater. Chem. A, 2020, 8(11), 5663–5670 RSC.
- C. Deng, Y. Su, F. Li, W. Shen, Z. Chen and Q. Tang, J. Mater. Chem. A, 2020, 8(46), 24563–24571 RSC.
- H. Jiang, J. Gu, X. Zheng, M. Liu, X. Qiu, L. Wang, W. Li, Z. Chen, X. Ji and J. Li, Energy Environ. Sci., 2019, 12(1), 322–333 RSC.
- N. Karmodak, L. Bursi and O. Andreussi, J. Phys. Chem. Lett., 2022, 13(1), 58–65 CrossRef CAS PubMed.
- N. F. Rosli, C. C. Mayorga-Martinez, N. M. Latiff, N. Rohaizad, Z. Sofer, A. C. Fisher and M. Pumera, ACS Sustainable Chem. Eng., 2018, 6(6), 7432–7441 CrossRef CAS.
- Y. Wang, Y. Li and T. Heine, J. Am. Chem. Soc., 2018, 140(40), 12732–12735 CrossRef CAS PubMed.
- Y. Wang and K. Zhou, Green Energy Environ., 2022, 7(3), 525–532 CrossRef.
- S. Zhao, K. Wang, X. Zou, L. Gan, H. Du, C. Xu, F. Kang, W. Duan and J. Li, Nano Res., 2019, 12(4), 925–930 CrossRef CAS.
- N. Zhang, F. Zheng, B. Huang, Y. Ji, Q. Shao, Y. Li, X. Xiao and X. Huang, Adv. Mater., 2020, 32(22), e1906477 CrossRef PubMed.
- L. H. Li, J. H. Yuan, K. H. Xue, M. Xu, M. Xu, J. F. Wang and X. S. Miao, J. Phys. Chem. C, 2020, 124(6), 3671–3680 CrossRef CAS.
- L. Zhao, G. Yu, X. Huang and W. Chen, Inorg. Chem., 2022, 61(4), 2284–2291 CrossRef CAS PubMed.
- T. Liu, Y. Wang and Y. Li, J. Phys. Chem. C, 2021, 125(35), 19164–19170 CrossRef.
- A. Mar, S. Jobic and J. A. Ibers, J. Am. Chem. Soc., 1992, 114(23), 8963–8971 CrossRef CAS.
- A. Mar and J. A. Ibers, J. Solid State Chem., 1992, 97(2), 366–376 CrossRef CAS.
- B. E. Brown, Acta Crystallogr., 1996, 20(2), 264–267 Search PubMed.
- E. W. Liimatta and J. A. Ibers, J. Solid State Chem., 1988, 77(1), 141–147 CrossRef CAS.
- E. W. Liimatta and J. A. Ibers, J. Solid State Chem., 1989, 78(1), 7–16 CrossRef CAS.
- W. Tremel, Angew. Chem., Int. Ed., 1992, 31(2), 217–220 CrossRef.
- J. Neuhausen, V. K. Evstaf'iev, T. Block, E. W. Finckh, W. Tremel, L. Augustin, H. Fuchs, D. Voss and P. Krüger, Chem. Mater., 1998, 10(12), 3870–3878 CrossRef CAS.
- J. Neu, K. Wei, X. He, O. Delaire, R. Baumbach, Z. Feng, Y. Fu, Y. Zhang, D. J. Singh and T. Siegrist, Phys. Rev. B, 2019, 100(14), 144102 CrossRef CAS.
- Y. Liu, C. Q. Xu, W. H. Jiao, P. G. Cai, B. Li, W. Zhou, B. Qian, X. F. Jiang, R. Sankar and X. L. Ke, Tungsten, 2021, 1–7 CrossRef.
- T. Bjorkman, A. Gulans, A. V. Krasheninnikov and R. M. Nieminen, Phys. Rev. Lett., 2012, 108(23), 235502 CrossRef CAS PubMed.
- N. Mounet, M. Gibertini, P. Schwaller, D. Campi, A. Merkys, A. Marrazzo, T. Sohier, I. E. Castelli, A. Cepellotti, G. Pizzi and N. Marzari, Nat. Nanotechnol., 2018, 13(3), 246–252 CrossRef CAS PubMed.
- S. Haastrup, M. Strange, M. Pandey, T. Deilmann, P. S. Schmidt, N. F. Hinsche, M. N. Gjerding, D. Torelli, P. M. Larsen and A. C. Riis-Jensen, 2D Materials, 2018, 5(4), 042002 CrossRef CAS.
- M. Jin, P. Yu, C. Fan, Q. Li, P. Kong, Z. Shen, X. Qin, Z. Chi, C. Jin, G. Liu, G. Zhong, G. Xu, Z. Liu and J. Zhu, Adv. Sci., 2021, 8(24), e2103250 CrossRef PubMed.
- V. Tripković, E. Skúlason, S. Siahrostami, J. K. Nørskov and J. Rossmeisl, Electrochim. Acta, 2010, 55(27), 7975–7981 CrossRef.
- H. A. Hansen, V. Viswanathan and J. K. Nørskov, J. Phys. Chem. C, 2014, 118(13), 6706–6718 CrossRef CAS.
- F. Lu, W. Xie, D. Yi, Y. Wang, F. Zhang, Y. Xu, B. Zhou, S. Liu, X. Wang and J. Yao, CCS Chem., 2021, 3(11), 180–188 CrossRef CAS.
- Y. Zhou, Q. Gu, K. Yin, Y. Li, L. Tao, H. Tan, Y. Yang and S. Guo, Angew. Chem., Int. Ed., 2022, e202201416 CAS.
- A. Föhlisch, M. Nyberg, P. Bennich, L. Triguero, J. Hasselström, O. Karis, L. G. M. Pettersson and A. Nilsson, J. Chem. Phys., 2000, 112(4), 1946–1958 CrossRef.
- A. Föhlisch, M. Nyberg, J. Hasselström, O. Karis, L. G. M. Pettersson and A. Nilsson, Phys. Rev. Lett., 2000, 85(15), 3309 CrossRef PubMed.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3(7), 546–550 CrossRef CAS PubMed.
- J. K. Norskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1(1), 37–46 CrossRef CAS PubMed.
- C. Ling, X. Niu, Q. Li, A. Du and J. Wang, J. Am. Chem. Soc., 2018, 140(43), 14161–14168 CrossRef CAS PubMed.
- H. B. Tao, L. Fang, J. Chen, H. B. Yang, J. Gao, J. Miao, S. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138(31), 9978–9985 CrossRef CAS PubMed.
- S. Zhou, X. Yang, X. Xu, S. X. Dou, Y. Du and J. Zhao, J. Am. Chem. Soc., 2020, 142(1), 308–317 CrossRef CAS PubMed.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136(11), 4394–4403 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1(1), 37–46 CrossRef PubMed.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3(7), 1159–1165 CrossRef CAS.
- M. M. Montemore and J. W. Medlin, Catal. Sci. Technol., 2014, 4(11), 3748–3761 RSC.
- M. Andersen, S. V. Levchenko, M. Scheffler and K. Reuter, ACS Catal., 2019, 9(4), 2752–2759 CrossRef CAS.
- E. M. Fernandez, P. G. Moses, A. Toftelund, H. A. Hansen, J. I. Martinez, F. Abild-Pedersen, J. Kleis, B. Hinnemann, J. Rossmeisl, T. Bligaard and J. K. Norskov, Angew. Chem., Int. Ed., 2008, 47(25), 4683–4686 CrossRef CAS PubMed.
- A. Vojvodic, A. Hellman, C. Ruberto and B. I. Lundqvist, Phys. Rev. Lett., 2009, 103(14), 146103 CrossRef CAS PubMed.
- F. Abild-Pedersen, J. Greeley, F. Studt, J. Rossmeisl, T. R. Munter, P. G. Moses, E. Skulason, T. Bligaard and J. K. Norskov, Phys. Rev. Lett., 2007, 99(1), 016105 CrossRef CAS PubMed.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Norskov, Chem. Rev., 2018, 118(5), 2302–2312 CrossRef CAS PubMed.
- A. Mahata, A. S. Nair and B. Pathak, Cataly. Sci. Technol., 2019, 9(18), 4835–4863 RSC.
- Y. L. Lee, M. J. Gadre, Y. Shao-Horn and D. Morgan, Phys. Chem. Chem. Phys., 2015, 17(33), 21643–21663 RSC.
- V. Viswanathan, H. A. Hansen, J. Rossmeisl and J. K. Norskov, J. Phys. Chem. Lett., 2012, 3(20), 2948–2951 CrossRef CAS PubMed.
- J. R. Kitchin, J. K. Norskov, M. A. Barteau and J. G. Chen, J. Chem. Phys., 2004, 120(21), 10240–10246 CrossRef CAS PubMed.
- V. Pallassana, M. Neurock, L. B. Hansen, B. Hammer and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60(8), 6146 CrossRef CAS.
- F. H. B. Lima, J. Zhang, M. H. Shao, K. Sasaki, M. B. Vukmirovic, a. E. A. Ticianelli and R. R. Adzic, J. Phys. Chem. C, 2007, 111(1), 404–410 CrossRef CAS.
- N. Liu, S. Zhou and J. Zhao, Mater. Today Phys., 2021, 16, 100312 CrossRef CAS.
- Y.-L. Lee, J. Kleis, J. Rossmeisl, Y. Shao-Horn and D. Morgan, Energy Environ. Sci., 2011, 4(10), 3966–3970 RSC.
- B. Wang and F. Zhang, Angew. Chem., Int. Ed., 2022, 61(4), e202111026 CAS.
- X. Zhu, X. Tan, K. H. Wu, S. C. Haw, C. W. Pao, B. J. Su, J. Jiang, S. C. Smith, J. M. Chen, R. Amal and X. Lu, Angew. Chem., Int. Ed., 2021, 60(40), 21911–21917 CrossRef CAS PubMed.
- Y. Chang, Y. Cheng, Y. Feng, K. Li, H. Jian and H. Zhang, ACS Appl. Mater. Interfaces, 2019, 11(13), 12224–12231 CrossRef CAS PubMed.
- S. Zhou, W. Pei, Y. Zhao, X. Yang, N. Liu and J. Zhao, npj Comput. Mater., 2021, 7(1), 1–34 CrossRef.
- C. F. Dickens, J. H. Montoya, A. R. Kulkarni, M. Bajdich and J. K. Nørskov, Surf. Sci., 2019, 681, 122–129 CrossRef CAS.
- S. Zhou, X. Yang, W. Pei, N. Liu and J. Zhao, Nanoscale, 2018, 10(23), 10876–10883 RSC.
- A. Tamai, Q. S. Wu, I. Cucchi, F. Y. Bruno, S. Riccò, T. K. Kim, M. Hoesch, C. Barreteau, E. Giannini, C. Besnard, A. A. Soluyanov and F. Baumberger, Phys. Rev. X, 2016, 6(3), 031021 Search PubMed.
- W. Tremel, Angew. Chem., 1991, 103(7), 900–903 CrossRef CAS.
- A. Mar and J. A. Ibers, J. Chem. Soc. Dalton Trans., 1991, 4, 639 RSC.
- J. Liu, H. Wang, C. Fang, L. Fu and X. Qian, Nano Lett., 2017, 17(1), 467–475 CrossRef CAS PubMed.
- A. Sufyan, G. Macam, C. Hsu, Z. Huang, S. Huang, H. Lin and F. Chuang, Chinese J. Phys., 2021, 73, 95 CrossRef CAS.
- Y. Wang and J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43(11), 8911–8916 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169 CrossRef CAS PubMed.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45(23), 13244–13249 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13(12), 5188–5192 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32(7), 1456–1465 CrossRef CAS PubMed.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78(13), 134106 CrossRef.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21(8), 084204 CrossRef CAS PubMed.
- R. Dronskowski and P. E. Bloechl, J. Phys. Chem., 1993, 97(33), 8617–8624 CrossRef CAS.
- V. L. Deringer, A. L. Tchougreeff and R. Dronskowski, J. Phys. Chem. A, 2011, 115(21), 5461–5466 CrossRef CAS PubMed.
- S. Maintz, V. L. Deringer, A. L. Tchougreeff and R. Dronskowski, J. Comput. Chem., 2016, 37(11), 1030–1035 CrossRef CAS PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113(22), 9901–9904 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108(46), 17886–17892 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Details of computational methods, structures, electronic properties, adsorption energies, energy barriers of O2 dissociation, free energy diagrams and linear relationships. See https://doi.org/10.1039/d2sc03686j |
| This journal is © The Royal Society of Chemistry 2022 |